
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly-D-lysine coated nanoparticles to identify pro-inflammatory macrophages†
Derek S.
Hernandez
a,
Hattie C.
Schunk
 ab,
Karan M.
Shankar
a,
Adrianne M.
Rosales
b and
Laura J.
Suggs‡
*a
ab,
Karan M.
Shankar
a,
Adrianne M.
Rosales
b and
Laura J.
Suggs‡
*a
aDepartment of Biomedical Engineering, University of Texas at Austin, Austin, TX 78712, USA. E-mail: suggs@utexas.edu
bMcKetta Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA
First published on 13th July 2020
Abstract
Identifying pro-inflammatory macrophages (M1) is of immense importance to diagnose, monitor, and treat various pathologies. In addition, adoptive cell therapies, where harvested cells are isolated, modified to express an M1-like phenotype, then re-implanted to the patient, are also becoming more prevalent to treat diseases such as cancer. In a step toward identifying, labeling, and monitoring macrophage phenotype for adoptive cell therapies, we developed a reactive oxygen species (ROS)-sensitive, gold nanoparticle (AuNP) that fluorescently labels M1 macrophages. AuNPs are electrostatically coated with a proteolysis resistant, fluorescein isothiocyanate-conjugated, poly-D-lysine (PDL-FITC) that is susceptible to backbone cleavage by ROS. When PDL-FITC is bound to AuNPs, fluorescence is quenched via a combination of nanoparticle surface (NSET) and Forster resonance (FRET) energy transfer mechanisms. Upon ROS-induced cleavage of PDL-FITC, up to a 7-fold change in fluorescence is demonstrated. PDL-FITC AuNPs were loaded into RAW 264.7 macrophages (RAWs) and primary bone marrow- derived macrophages (BMDMs) prior to in vitro polarization. For both cell types, detectable differences in intracellular fluorescence were observed between M1 polarized and non-stimulated (M0) control groups after 24 h using both confocal imaging and flow cytometry. PDL-FITC AuNPs can potentially be useful in identifying M1 macrophages within diverse cell populations and provide longitudinal macrophage response data to external cues.
Introduction
Macrophages are critically important in managing injury, disease, and regeneration.1–4 Natively, macrophages can exist along a spectrum of phenotypes, from pro-inflammatory M1 cells to regenerative M2 phenotypes.5–7 Unlike many other cell types, they exhibit tremendous plasticity to reprogram, or switch between phenotypes, under certain conditions.8–10 Recently, macrophages and their monocyte precursors are being used in adoptive cell therapies (ACTs) to target and treat cancer, inflammation, and other diseases.11–15 ACTs typically use isolated cells from blood or bone marrow, modify them ex vivo to promote recognition and/or phenotypic differentiation, then reintroduce them to patients as a treatment. Given their established plasticity, using monocytes or macrophages as cell therapies necessitates the ability to track and visualize implanted cells and their phenotype in vivo for proper evaluation of treatment efficacy.The most distinguishable functional feature of M1 macrophages is their production of intra- and extra-cellular reactive oxygen species (ROS) at elevated concentrations. Extracellularly, ROS act to kill pathogens and break down cellular debris.16–18 Intracellularly, ROS play an important role in regulating a variety of biological processes including cell metabolism, proliferation, apoptosis and immune function.19 Targeting these particular intracellular characteristics, numerous methods have been developed to detect ROS in vitro and in vivo.20,21 ROS-sensitive bioluminescent and fluorescent dyes have been used to label pro-inflammatory M1 macrophages.22,23 However, these dyes are often limited in their specificity towards any one oxidant, and stability against numerous competing chemical interactions in biological environments.24 HyPer probes have also shown promise for detecting intracellular ROS production in M1 macrophages, but these probes require genetic modifications that can be expensive and time-consuming.25,26 Some of the more promising approaches for translational application use gold nanoparticles (AuNPs) modified with ROS-responsive functionalities.27,28 For example, Lee and colleagues used fluorescein-conjugated, hyaluronic acid (HA) coated AuNPs to fluorescently label M1 macrophages in vitro caused by oxidative degradation of HA.29 However, HA is susceptible to both enzymatic (hyaluronidase) and oxidative degradation. In fact, many reaction-based probes suffer from similar cross-reactivity issues,27 making a material with higher oxidative selectivity necessary for in vivo translation.30
In this study, we used a fluorophore-conjugated, ROS-selective polypeptide, poly-D-lysine (PDL) to create a AuNP-based M1 macrophage sensor. PDL was fluorescently tagged using fluorescein isothiocyanate (FITC) prior to electrostatic conjugation to AuNPs, creating PDL-FITC AuNPs, which present significant fluorescence quenching. When PDL-FITC is degraded by ROS and liberated from the AuNP core, fluorescence increases. PDL-FITC AuNPs were demonstrated to be responsive to hydrogen peroxide (H2O2) and applied to identify M1 macrophages in vitro using fluorescence via confocal microscopy and flow cytometry.
Results and discussion
PDL degradation
The resistance of PDL to enzymatic degradation has been demonstrated in literature, and its susceptibility to ROS degradation has been suggested.31,32 To confirm selectivity of PDL to oxidative degradation, we conducted degradation studies in solution using either trypsin, an enzyme known to degrade proteins at lysine residues, or a metal catalyzed oxidation (MCO) reaction, commonly used to mimic production of superoxide and hydroxyl radicals by macrophages.33 For this controlled study, 18-mer oligomers of PDL were synthesized (MW = 2322.74) and exposed for 24 h at 37 °C to 50 μM trypsin or 1 M H2O2 combined with 50 μM CuSO4 for the MCO. PBS buffer was used as a negative control, and oligomer degradation was assessed using a combination of high-pressure liquid chromatography (HPLC) and mass spectrometry (MS). Results displayed in Fig. 1 reveal that PDL is degraded in the presence of ROS and is resistant to enzymatic degradation. In Fig. 1A, primary peak retention times are unchanged between trypsin and a PBS control group. However, PDL exposed to MCO leads to a significantly shorter retention time and complete disappearance of the control peak. When these same formulations were run on MS (Fig. 1B), the MCO exposed PDL was the only group lacking an intact PDL oligomer mass peak (m/z = 2322.74), suggesting the polymer was degraded. These degradative properties of PDL were deemed appropriate for M1 macrophage sensing because M1 macrophages produce significantly higher levels of ROS than non-stimulated macrophages.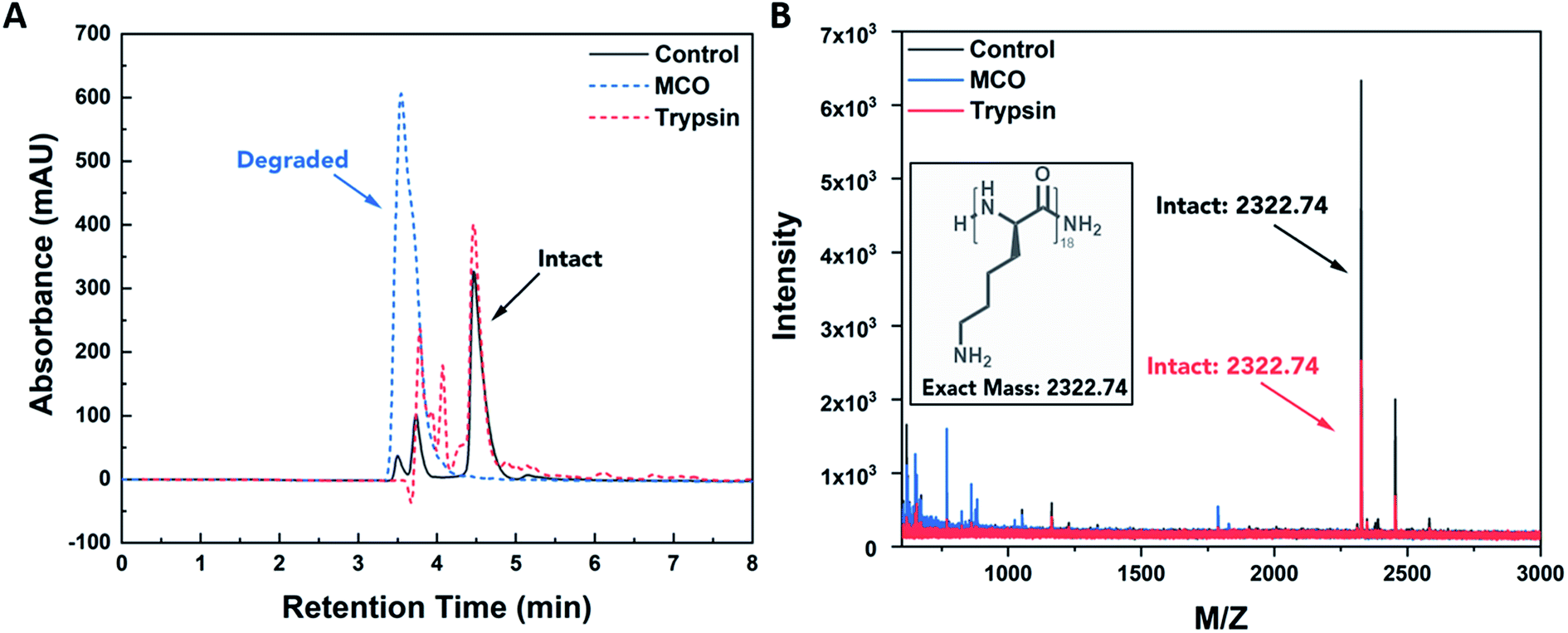 | ||
| Fig. 1 PDL oligomer degradation. (A) HPLC UV trace (214 nm) of D-lysine oligomer upon exposure to MCO (1 M H2O2 + 50 μM CuSO4) and enzymatic (50 μM trypsin) stimuli. Oxidative degradation is observed by shifting retention times for MCO chromatogram peaks in comparison to the control and enzymatic treatment. (B) MALDI spectra confirming degradation in the case of MCO treatment, as observed by disappearance of mass peak characteristic of the intact peptide. Notably, the PDL oligomer maintains resistance to proteolysis by trypsin, as confirmed by remaining intact substrate. | ||
PDL-FITC AuNP synthesis and characterization
After confirming ROS selectively degrade PDL, we synthesized a nanoparticle-based sensor to fluorescently label M1 macrophages. Relying on a combination of FRET and NSET mechanisms (Fig. S1†), we developed a polymer conjugated gold nanoparticle (AuNP) where fluorescence would ‘turn on’ when the polymer is degraded by ROS (Fig. 2A). PDL was first conjugated to amine-reactive FITC, forming isothiourea linkages, at a molar ratio of approximately 10–13 FITC molecules per PDL polymer. UV-Vis absorbance was used to calculate the FITC/PDL ratio. The blue-shifted, double-peak of FITC absorbance shown in Fig. 2B is characteristic of different protonation states of fluorescein caused by molecular crowding.34,35 After PDL-FITC was synthesized and purified using a centrifugal filter, it was electrostatically bound to citrate-capped AuNPs. Specifically, the positively charged primary amines on PDL side chains and negatively charged AuNP surface bind to create a PDL-FITC AuNP conjugate.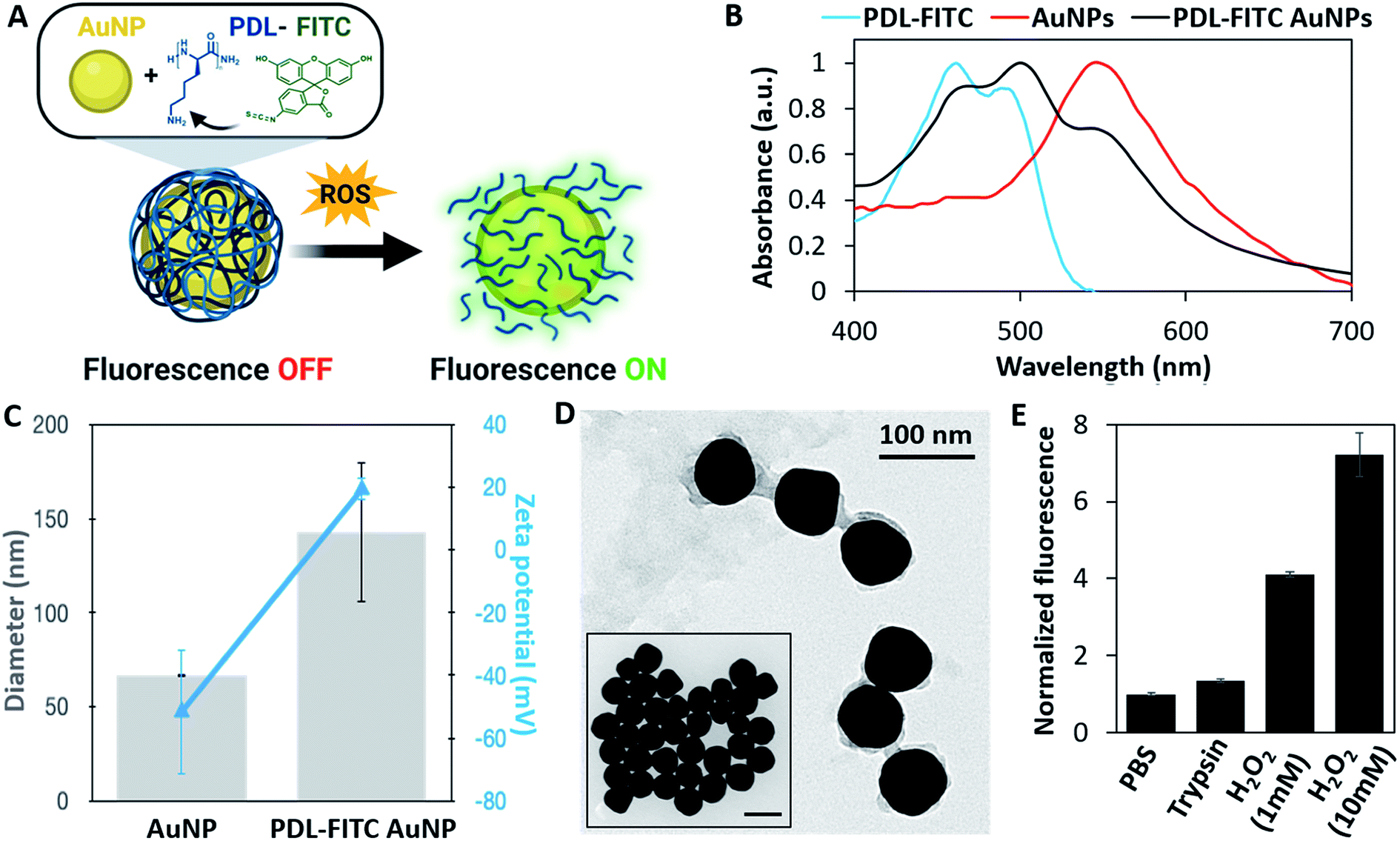 | ||
| Fig. 2 PDL-FITC AuNP characterization. (A) Schematic of PDL-FITC AuNP synthesis and ROS-induced degradation. (B) Normalized absorbance spectra of PDL-FITC, AuNPs, and PDL-FITC AuNPs confirming conjugation. (C) Diameter (gray bars) and zeta potential (blue triangles) for AuNPs and PDL-FITC AuNPs. Diameter and zeta potential were determined by DLS for PDL-FITC AuNPs and provided by the supplier for AuNPs. (D) TEM of AuNPs (inset) and PDL-FITC coated AuNPs. Scale bar = 100 nm for both images. (E) PDL-FITC AuNP degradation-induced fluorescence after exposure to PBS, trypsin, and H2O2 for 24 h. Error bars represent ± standard deviation, p < 0.05, two-tailed, paired t-test. | ||
PDL-FITC AuNP conjugation was confirmed using a variety of characterization methods, including UV-VIS spectroscopy, dynamic light scattering (DLS), zeta potential, and transmission electron microscopy (TEM). An absorbance spectrum of PDL-FITC AuNPs (Fig. 2B) show both AuNP and PDL-FITC peaks are present. Furthermore, DLS (Fig. 2C) reveals an increase in diameter of PDL-FITC AuNPs that is combined with a charge flip in zeta potential from (−) to (+), indicating PDL coats the AuNP surface. An image captured using TEM (Fig. 2D) confirms a PDL-FITC coating surrounding each AuNP, as compared to the TEM of AuNPs prior to conjugation (Fig. 2D, inset).
PDL-FITC AuNP degradation via ROS
Solution-based degradation studies were conducted with PDL-FITC AuNPs using similar methods as above. PDL-FITC AuNPs were mixed with 1× PBS, 0.05% trypsin/EDTA, or H2O2 at different concentrations (1 mM or 10 mM) in microcentrifuge tubes and stored at 37 °C for 24 h. All groups were centrifuged, and supernatant was collected then dispensed in a 96 well plate to measure fluorescence. Fluorescence plotted relative to the PBS control group is shown in Fig. 2E, where a 4-fold and 7-fold change in fluorescence was achieved in the presence of 1 mM and 10 mM H2O2, respectively. A lower 1.35-fold increase was observed between trypsin and PBS groups, aligning with our expectations that PDL is insensitive to enzymatic degradation. These results demonstrate PDL-FITC AuNPs are more selective for ROS than trypsin, making them useful sensors to label ROS-producing, M1 macrophages.In vitro studies with ROS-sensitive AuNPs
In vitro studies were first conducted in RAW 264.7 macrophages (RAWs), a murine cell line, to verify PDL-FITC AuNPs react with endogenously produced ROS resulting in a detectable increase in fluorescence in M1 macrophages. As would be the procedure for use in ACTs, PDL-FITC AuNPs were pre-loaded into non-stimulated cells for 4 h. During this time, the cells were observed to spontaneously take up the particles. The media was then replaced with growth media (M0 control) or growth media + 1000 ng mL−1 LPS + 100 ng mL−1 IFN-γ (M1 polarization). Z-Stacks of cells were acquired using a confocal microscope 24 h after stimulation. Summed Z-projections of representative fluorescent and bright-field images are displayed in Fig. 3A. The bright-field images show LPS and IFN-γ stimulated macrophages exhibit a large, rounded and flat shape characteristic of the M1 phenotype.36 The M1 macrophages also show an observably higher fluorescence intensity in M1 macrophages over M0 macrophages. To further confirm M1 polarization, a Griess assay was performed, and shows that M1 macrophages produced significantly more NO when compared to M0 macrophages (Fig. 3B). The M0 macrophage signal for this assay was comparable to a well without cells, indicating negligible NO was produced. Cells were also collected (via scraping) and analyzed using flow cytometry to quantify the differences in fluorescence between groups. Notably, M1 macrophages exhibited a 71% increase in median fluorescence intensity (MFI) over M0 macrophages (Fig. 3C).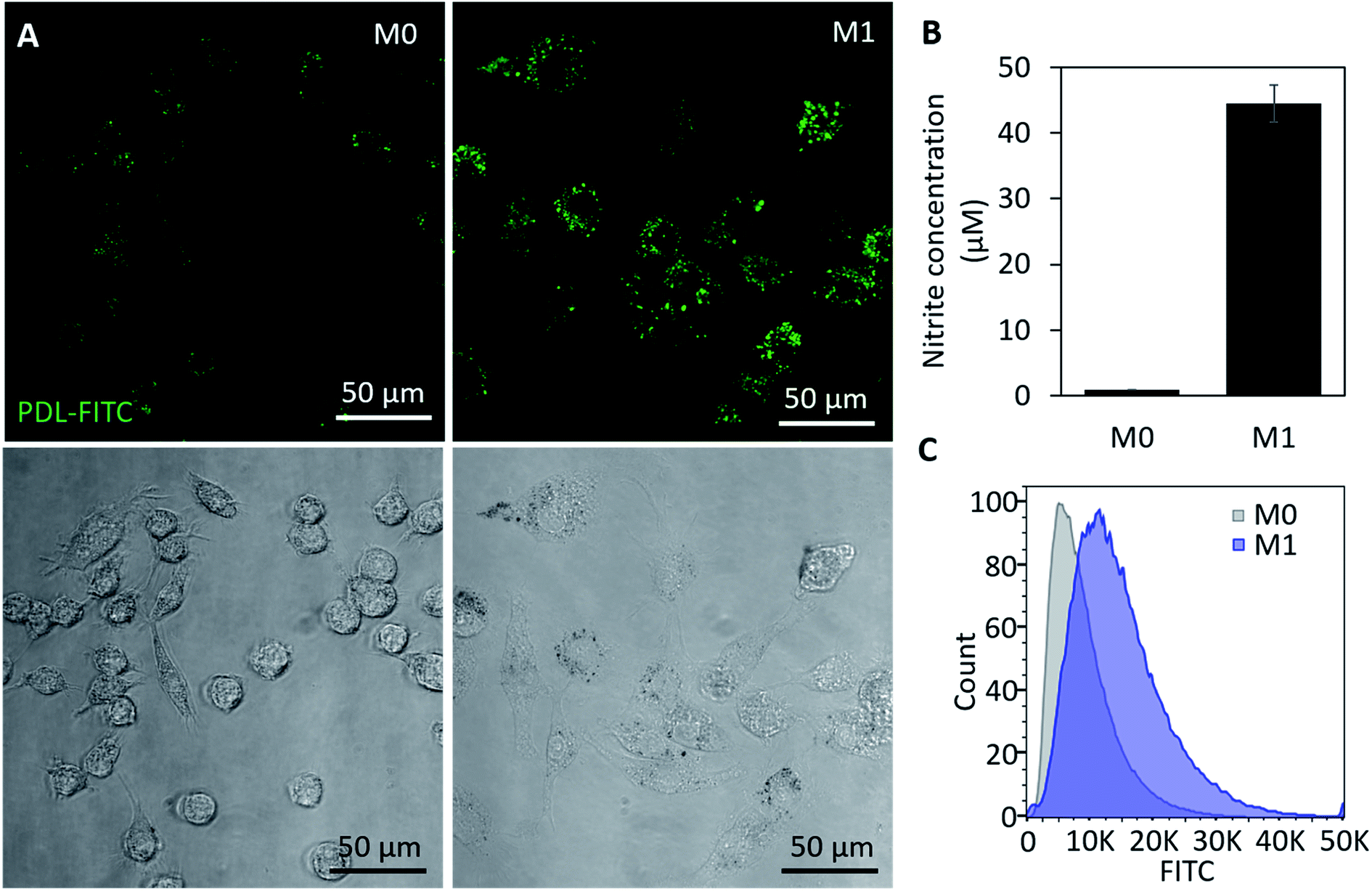 | ||
| Fig. 3 PDL-FITC AuNPs in RAWs. (A) Confocal Z-projections (sum) of representative fluorescent and bright-field images of PDL-FITC AuNP loaded RAWs acquired 24 h after loading. M1 macrophages were polarized using 1 μg mL−1 LPS and 100 ng mL−1 IFN-γ, and non-stimulated M0 macrophages were used as a control. M1 macrophages displayed higher levels of fluorescence in images. (B) Griess assay confirms M1 cell polarization. Error bars represent ± standard deviation, p < 0.05, two-tailed, paired t-test. (C) Flow cytometry of M0 and M1 cells acquired 24 h post-stimulation also shows an increase in FITC fluorescence in M1 macrophages. | ||
As a more clinically relevant model of macrophage behavior, additional in vitro studies were conducted using bone marrow derived macrophages (BMDMs). Cells were isolated from a mouse then differentiated to BMDMs over a 7 day period prior to beginning each experiment. Experimental conditions were similar to our study using RAWs, except cells were polarized using 100 ng mL−1 of IFN-γ and LPS. To track with the timeline of cell signaling and morphology changes, images were acquired 3 h (Fig. S2†) and 6 h post-stimulation. This also corresponds to the earliest time point in which NO becomes detectable using a Griess assay. Cells were then fixed 24 h post-stimulation and stained using DAPI (nuclear stain) and MHCII (M1 macrophage marker) before acquiring additional confocal images. At both time points, significantly higher fluorescence is visible in M1 macrophages when compared to M0 groups (Fig. 4A). MHCII+ staining also confirms that cells were polarizing toward an M1 phenotype in stimulated groups. Griess (Fig. 4B) and flow cytometry data (Fig. 4C) show comparable increases in NO production (5×) and FITC fluorescence (43% increase in MFI), respectively, to what was observed in our previous studies using RAW macrophages. Together, these results support use of this particle as an intracellular probe that could be pre-loaded into macrophages for use as a real-time, phenotypic indicator in ACTs.
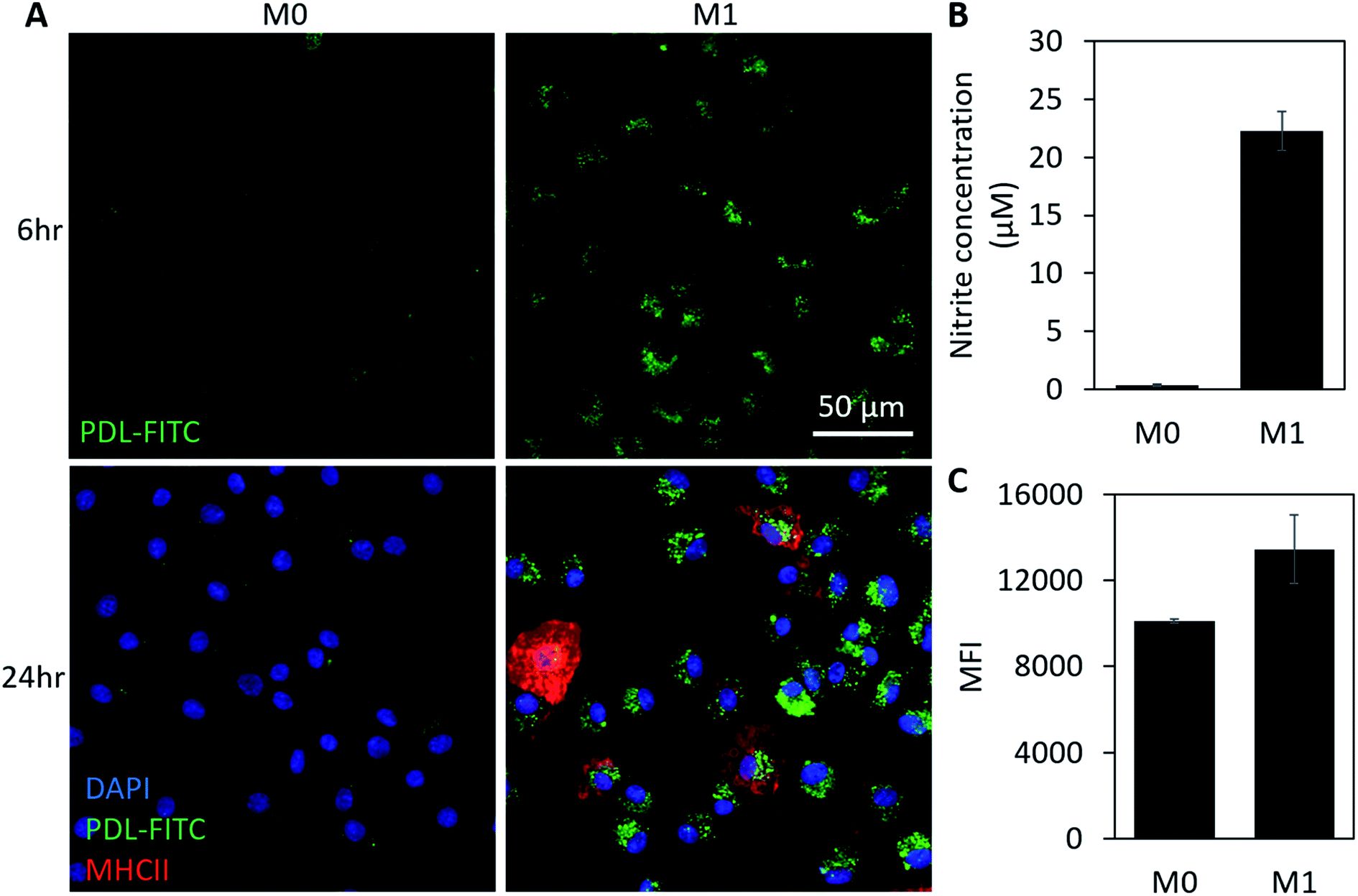 | ||
| Fig. 4 PDL-FITC AuNPs in BMDMs. (A) Confocal Z-projections (sum) of PDL-FITC AuNP loaded BMDMs acquired 6 and 24 h after loading. M1 macrophages were polarized using 100 μg mL−1 LPS and 100 ng mL−1 IFN-γ, and non-stimulated M0 macrophages were used as a control. M1 macrophages display higher levels of fluorescence at both time points. For images acquired at 24 h, cells were fixed and stained with DAPI (nuclear stain) and MHCII to confirm M1 polarization. (B) Griess assay confirms M1 cell polarization. Error bars represent ± standard deviation, p < 0.05, two-tailed, paired t-test. (C) Median fluorescence intensities acquired using flow cytometry of M0 and M1 cells 24 h post-stimulation show a higher FITC fluorescence in M1 macrophages. Error bars represent standard deviation of n = 2 independent runs, p < 0.1, one-way ANOVA. | ||
Conclusion
Current work detecting macrophage phenotypes has primarily focused on the use of gene expression profiling for determining cell status. The limitation of this approach is that it misses contributions of downstream species, such as ROS, which are critical to disease pathology. Here, we have demonstrated synthesis and application of a fluorescence-based ROS sensor that can differentiate between M0 and M1 macrophages on the basis of intracellular ROS-production. Not only is the PDL-FITC AuNP synthesis rapid and simple, but also analyses can be performed quickly using common analytical modalities (fluorescence microscopy, flow cytometry). Our ROS-sensitive probe is both cell-permeable, and remains in the cell after its reaction, enabling researchers to detect phenotypical changes in macrophages attributed to local ROS production. Detecting macrophage phenotypes could lead to a better understanding of how these cells target and respond to local environments when used in adoptive therapies. While the stability of this electrostatically constructed particle may be a concern for ultimate application in vivo, our particle provides an attractive nanoparticle-based platform for ultimate use as a real-time, phenotypic indicator of macrophages for use in ACTs, and we are working on a next-generation thiolated version to improve stability. Future studies will aim to incorporate NIR dyes for in vivo imaging,37 or translate this system to non-invasive trackable platforms, such as photoacoustic imaging,38 for a more clinic-ready approach.Experimental
PDL degradation
To investigate degradation of D-lysine in a controlled setting, PDL oligomers were synthesized and purified using standard techniques. Specifically, a D-lysine oligomer peptide (18 residues) was synthesized using a Rink Amide polystyrene resin (0.72 mmol g−1 from Gyros Protein Technologies) on a Prelude X automated peptide synthesizer (Gyros Protein Technologies). Traditional Fmoc-mediated coupling methods were used. Once complete, the synthesis was cleaved from resin with 10 mL of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 trifluoroacetic acid:water:triisopropylsilane for 4 h. Prior to purification, the peptides were dissolved at 10 mg mL−1 in 10:90 acetonitrile:water (ACN:H2O) with 0.1% trifluoroacetic acid (TFA). The crude peptide was then purified using high-performance liquid chromatography (HPLC) with a C18 column on a Dionex UltiMate 3000 UHPLC using a 25 min gradient of ACN:H2O (10–100%, 10 mL min−1). After lyophilization, masses were verified via Matrix Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF) MS.
:2.5:2.5 trifluoroacetic acid:water:triisopropylsilane for 4 h. Prior to purification, the peptides were dissolved at 10 mg mL−1 in 10:90 acetonitrile:water (ACN:H2O) with 0.1% trifluoroacetic acid (TFA). The crude peptide was then purified using high-performance liquid chromatography (HPLC) with a C18 column on a Dionex UltiMate 3000 UHPLC using a 25 min gradient of ACN:H2O (10–100%, 10 mL min−1). After lyophilization, masses were verified via Matrix Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF) MS.
Following synthesis and purification, the substrate was dissolved in PBS buffer at a concentration of 1 mg mL−1 and exposed to either 1 M H2O2 + 50 μM CuSO4 or 50 μM trypsin. A control containing 1 mg mL−1 substrate in 1× PBS buffer alone was also performed. Following an incubation period of 24 h, samples were flash frozen in liquid nitrogen to quench the reaction and lyophilized for further analysis using HPLC and MALDI-TOF MS.
Degradation was monitored by shifting retention time of analytical HPLC chromatogram peaks in comparison to controls. Lyophilized samples were dissolved at 0.1 mg mL−1 in 10:90 ACN:H2O with 0.1% (v/v) TFA and analyzed on HPLC using an analytical Thermo BioBasic™ C18 column (250 mm × 4.6 mm, i.d. 5 μm). HPLC analysis conditions included a mobile phase containing ACN and H2O with 0.1% (v/v) TFA in a 10:90 ratio at 1.0 mL min−1. A 15 minute gradient was employed (5 minute equilibration period followed by increasing ACN ratio from 10–50% over 15 min) with the eluents measured at 214 nm. MS was used to confirm degradation. Specifically, samples associated with each eluted HPLC peak were prepped in α-cyano-4-hydroxycinnamic acid (CHCA) matrix and analyzed using MALDI-TOF MS (Applied Biosystems-Voyager-DE™ PRO) to confirm if any intact substrate remained.
PDL-FITC AuNP synthesis
PDL-FITC was prepared by dissolving 0.4 mg mL−1 PDL (30–70 kDa, Sigma Aldrich) in sodium bicarbonate buffer (4.2 mg/50 mL DI H2O, pH = 8.5–9, Sigma Aldrich), then combined with an equal volume of 0.8 mg mL−1 FITC (Sigma Aldrich) in dimethylformamide. The reaction vial was wrapped in foil and proceeded for 2 h at room temperature (RT) on a vortex shaker. Conjugated PDL-FITC was purified using centrifugal filtration (10 kDa molecular weight cutoff) at 3000g for 20 min then diluted in DI H2O. The filtration process was repeated a minimum of four times. PDL-FITC solution was collected after the final centrifugation and transferred to a microcentrifuge tube for long-term storage at 4 °C. PDL-FITC concentration was estimated using an A205 method. A FITC/PDL ratio was calculated using the formula: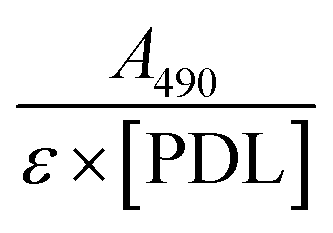 , where ε = 70000 cm−1 M−1 for fluorescein, and all PDL-FITC batches used ranged from 10–13 moles FITC per mole PDL. Absorbance spectra were acquired using a Nanodrop (ThermoFisher Scientific) spectrophotometer.
, where ε = 70000 cm−1 M−1 for fluorescein, and all PDL-FITC batches used ranged from 10–13 moles FITC per mole PDL. Absorbance spectra were acquired using a Nanodrop (ThermoFisher Scientific) spectrophotometer.
For PDL-FITC AuNP synthesis, PDL-FITC solution was diluted in DI H2O to a concentration of ∼0.1 mg mL−1. Then, citrate-capped AuNPs (63 nm, Nanohybrids) were added dropwise to the solution until a final optical density (OD) of 2.5 was reached. The solution was wrapped in foil and mixed on a vortex shaker for 30 min at RT. PDL-FITC AuNPs were purified using centrifugation (6000g, 10 min) and re-suspended in DI H2O a total of three times.
PDL-FITC AuNP characterization
PDL-FITC AuNPs were characterized using a combination of spectrophotometry (Cytation 3, Biotek), dynamic light scattering (DLS) and zeta potential (Malvern Zetasizer Nano-ZS), and transmission electron microscopy (TEM; FEI Tecnai). All PDL-FITC AuNP suspensions were sonicated for 20–30 seconds prior to analysis. Absorbance spectra were acquired to confirm PDL-FITC conjugation to AuNPs. DLS and zeta potential measurements, performed in DI water, were used to obtain size and charge information using standard methods. TEM samples were prepared by drop-casting PDL-FITC AuNPs on a plasma irradiated, carbon-coated copper grid (Electron Microscopy Sciences). The suspension was blotted with filter paper and the grid was allowed to fully air dry prior to imaging.PDL-FITC AuNP in situ degradation
PDL-FITC AuNPs (25 μL) were added to microcentrifuge tubes with 1× PBS (negative control), 1× PBS + H2O2 (1 or 10 mM), or 1× PBS + 0.05% trypsin–EDTA (ThermoFisher Scientific) and stored in the dark at 37 °C for 24 h. Afterwards, tubes were centrifuged (6000g for 10 min) and 100 μL of supernatant was collected and dispensed into a 96-well plate (n = 3 wells per group). Fluorescence intensity (Ex/Em: 485/532 nm) was acquired using a plate reader (Infinite® 200 Pro, Tecan), and all groups were normalized to control fluorescence.NSET contribution of PDL-FITC AuNP
To determine the relative contribution of NSET, a protocol adapted from Decher et al.39 was performed. Briefly, AuNPs were added to tubes containing 1× PBS or 32 mM potassium cyanide (KCN) for 30 min at RT. AuNP dissolution was verified in the KCN group using absorbance spectroscopy. Tubes were centrifuged at 6000g for 10 min to pellet any remaining AuNPs. Equal volumes of supernatant were diluted in 1× PBS and relative fluorescence intensities were obtained using a plate reader.RAW cell culture, loading, and polarization
RAW 264.7 murine macrophages (Lonza) were cultured using standard protocols. Briefly, cells were seeded in T75 flasks using high glucose DMEM (Caisson Labs) supplemented with 10% FBS, 1% penicillin–streptomycin (PS), and 1% sodium pyruvate. Flasks were incubated at 37 °C with 5% CO2, and media was replenished every 2–3 days. When RAWs reached 80% confluence, cells were passaged using a cell scraper and reseeded into a fresh flask for culture propagation. For use in experiments, cells were counted using a hemocytometer and trypan blue stain. All RAWs used for experiments were under passage #12.RAWs were seeded at 50k cells per well or 500k cells per well in 8 chambered coverglass and 6 well plates, respectively, and allowed to adhere overnight. In 8-chambered coverglass, 20 μL of PDL-FITC AuNPs were added to each well (PDL-FITC AuNP OD = 0.1). For flow cytometry studies, 100 μL of PDL-FITC AuNPs were added to each well of a 6-well plate (PDL-FITC AuNP OD = 0.01). Cells were loaded for 4 h, then media was replaced with fresh growth media for M0 maintenance or growth media with 1000 ng mL−1 of LPS and 100 ng mL−1 IFN-γ for M1 polarization for the entire duration of the experiment (24 h).
BMDM cell isolation, culture, loading, and polarization
Primary monocytes were harvested from femurs and fibula of FVB mice using a previously published method.40,41 Briefly, a mouse was sacrificed and its hind limb bones were harvested, cleaned of tissue, and placed into cold 1× PBS. Each bone was sterilized in a 70% ethanol (EtOH) bath before proceeding. The ends of each bone were cut using sterilized surgical tools to expose bone marrow. A pre-filled syringe with 5 mL of 1× PBS was used to flush bone marrow from each bone. Marrow was collected in a 50 mL centrifugal filter, then centrifuged at 500g for 5 min to pellet cells. Cells were re-suspended in 47 mL of culture media (RPMI + 10% heat inactivated FBS + 1% PS + 10 ng mL−1 macrophage colony stimulating factor (MCSF)) and divided evenly among 3–6 well plates. Cells were stored in an incubator (37 °C, 5% CO2) and media was replaced on days 3 and 5. BMDMs were passaged on day 7 using 1× trypsin (5–7 min incubation), then re-suspended in media containing a lower concentration of MCSF (1 ng mL−1) before seeding into experimental wells. For use in experiments, cells were counted using a hemocytometer and trypan blue stain. BMDMs were cultured overnight in 8-chambered coverglass or 6-well plates after seeding at 50k or 500k cells per well, respectively, before loading with PDL-FITC AuNPs.In 8-chambered coverglass, 30 μL of PDL-FITC AuNPs were added to each well (PDL-FITC AuNP OD = 0.07). For flow cytometry studies, 80 μL of PDL-FITC AuNPs were added to each well of a 6-well plate (PDL-FITC AuNP OD = 0.04). BMDMs were loaded for 4 h, then media was replaced with MCSF-free growth media for M0 maintenance or MCSF-free growth media with 100 ng mL−1 of LPS and IFN-γ for M1 polarization for the entire duration of the experiment (24 h).
Griess assay
To confirm M1 polarization, 50 μL of culture media was collected 24 h after cytokine addition and transferred to a 48-well plate. Each sample was combined with 25 μL of sulfanilamide then 25 μL of (N-1-napthylethylenediamine dihydrochloride) NED. After a 10 min incubation in the dark at RT, absorbance at 540 nm was measured. Nitrite standards were used to create a calibration curve and calculate the nitrite produced per group.Confocal imaging
For imaging experiments, 50k cells were plated in each well of an 8-well chambered coverglass. Cells were treated as previously described, and imaged (without exchanging media) at 6 or 24 h after stimulation using a confocal microscope (Olympus FluoView FV10i) and 60×, oil objective. Z-Stacks (1 μm steps, 12 total steps) were acquired at 3–4 independent locations in each well. For stained cells, cells were fixed using paraformaldehyde (4% for 10 min) and immunostained prior to image acquisition. Staining was performed using the following protocol: 30 min blocking solution at RT using 1% BSA in PBS, 2 h incubation with MHC class II-AlexaFluor® 647 antibody (1:200 dilution; BioLegend) at RT, 5 min incubation with DAPI (1:1000 dilution). Three 1× PBS rinses were performed after each dye addition.
Flow cytometry
Cells were cultured in 6-well plates and seeded at 500k cells per well. After 24 h of cytokine stimulation, cells were collected via scraping for RAWs and trypsinization (1× trypsin, 5–10 min at 37 °C) for BMDMs, stained with ethidium homodimer III (1:1000 dilution in PBS, 10 min incubation on ice) to identify dead cells, then centrifuged and re-suspended in 1× PBS for analysis. Flow data was acquired using an Accuri C6 (BD). A minimum of 1400 events was collected for each sample.
Statistical analysis
Numeric data is reported as the mean ± standard deviation, with at least 3 replicates per group unless otherwise indicated. PDL-FITC AuNP characterization, degradation-induced fluorescence quantification, MFI quantification in RAWs and NO production were statistically assessed using a two-tailed, paired t-test. MFI quantification in BMDMs was statistically assessed with a one-way ANOVA.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PDL-FITC AuNP schematic and Table of Contents figure were created using Biorender.com. This work was supported by the National Institute of Health (NIH EB015007). Hattie Schunk acknowledges the support of a National Science Foundation Graduate Research Fellowship as well as a fellowship from the Graduate School at UT Austin.References
- A. Shapouri-Moghaddam, S. Mohammadian, H. Vazini, M. Taghadosi, S.-A. Esmaeili, F. Mardani, B. Seifi, A. Mohammadi, J. T. Afshari and A. Sahebkar, Macrophage Plasticity, Polarization, and Function in Health and Disease, J. Cell. Physiol., 2018, 233(9), 6425–6440, DOI:10.1002/jcp.26429.
- C. E. Lewis and J. W. Pollard, Distinct Role of Macrophages in Different Tumor Microenvironments, Cancer Res., 2006, 66(2), 605–612, DOI:10.1158/0008-5472.CAN-05-4005.
- T. J. Koh and L. A. DiPietro, Inflammation and Wound Healing: The Role of the Macrophage, Expert Rev. Mol. Med., 2011, 13, e23, DOI:10.1017/S1462399411001943.
- S. Y. Kim and M. G. Nair, Macrophages in Wound Healing: Activation and Plasticity, Immunol. Cell Biol., 2019, 97(3), 258–267, DOI:10.1111/imcb.12236.
- D. M. Mosser and J. P. Edwards, Exploring the Full Spectrum of Macrophage Activation, Nat. Rev. Immunol., 2008, 958–969, DOI:10.1038/nri2448.
- L. C. Davies, S. J. Jenkins, J. E. Allen and P. R. Taylor, Tissue-Resident Macrophages, Nat. Immunol., 2013, 14(10), 986–995, DOI:10.1038/ni.2705.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J.-L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines, Immunity, 2014, 41(1), 14–20, DOI:10.1016/j.immuni.2014.06.008.
- K. Liddiard and P. R. Taylor, Understanding Local Macrophage Phenotypes in Disease: Shape-Shifting Macrophages, Nat. Med., 2015, 21(2), 119–120, DOI:10.1038/nm.3798.
- I. Malyshev and Y. Malyshev, Current Concept and Update of the Macrophage Plasticity Concept: Intracellular Mechanisms of Reprogramming and M3 Macrophage “Switch” Phenotype, BioMed Res. Int., 2015, 2015, 341308, DOI:10.1155/2015/341308.
- S. K. Biswas, A. Sica and C. E. Lewis, Plasticity of Macrophage Function during Tumor Progression: Regulation by Distinct Molecular Mechanisms, J. Immunol., 2008, 180(4), 2011–2017, DOI:10.4049/jimmunol.180.4.2011.
- R. Andreesen, B. Hennemann and S. W. Krause, Adoptive Immunotherapy of Cancer Using Monocyte-Derived Macrophages: Rationale, Current Status, and Perspectives, J. Leukocyte Biol., 1998, 64(4), 419–426, DOI:10.1002/jlb.64.4.419.
- A. Q. Ford, P. Dasgupta, I. Mikhailenko, E. M. P. Smith, N. Noben-Trauth and A. D. Keegan, Adoptive Transfer of IL-4Rα+ Macrophages Is Sufficient to Enhance Eosinophilic Inflammation in a Mouse Model of Allergic Lung Inflammation, BMC Immunol., 2012, 13(1), 6, DOI:10.1186/1471-2172-13-6.
- G. Escobar, D. Moi, A. Ranghetti, P. Ozkal-Baydin, M. L. Squadrito, A. Kajaste-Rudnitski, A. Bondanza, B. Gentner, M. De Palma, R. Mazzieri and L. Naldini, Genetic Engineering of Hematopoiesis for Targeted IFN-Alpha Delivery Inhibits Breast Cancer Progression, Sci. Transl. Med., 2014, 6(217), 217ra3, DOI:10.1126/scitranslmed.3006353.
- A. C. Anselmo, J. B. Gilbert, S. Kumar, V. Gupta, R. E. Cohen, M. F. Rubner and S. Mitragotri, Monocyte-Mediated Delivery of Polymeric Backpacks to Inflamed Tissues: A Generalized Strategy to Deliver Drugs to Treat Inflammation, J. Controlled Release, 2015, 199, 29–36, DOI:10.1016/j.jconrel.2014.11.027.
- V. Rybalko, P.-L. Hsieh, M. Merscham-Banda, L. J. Suggs and R. P. Farrar, The Development of Macrophage-Mediated Cell Therapy to Improve Skeletal Muscle Function after Injury, PLoS One, 2016, 10(12), e0145550 CrossRef PubMed.
- H. J. Forman and M. Torres, Signaling by the Respiratory Burst in Macrophages, IUBMB Life, 2001, 51(6), 365–371, DOI:10.1080/152165401753366122.
- H. Y. Tan, N. Wang, S. Li, M. Hong, X. Wang and Y. Feng, The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases, Oxid. Med. Cell. Longevity, 2016, 1–16, DOI:10.1155/2016/2795090.
- J. Canton, R. Khezri, M. Glogauer and S. Grinstein, Contrasting Phagosome pH Regulation and Maturation in Human M1 and M2 Macrophages, Mol. Biol. Cell, 2014, 25(21), 3330–3341, DOI:10.1091/mbc.E14-05-0967.
- M. B. Grisham, Methods to Detect Hydrogen Peroxide in Living Cells: Possibilities and Pitfalls, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2013, 429–438, DOI:10.1016/j.cbpa.2013.02.003.
- S. G. Rhee, T. S. Chang, W. Jeong and D. Kang, Methods for Detection and Measurement of Hydrogen Peroxide inside and Outside of Cells, Mol. Cells, 2010, 29(6), 539–549, DOI:10.1007/s10059-010-0082-3.
- K.-C. Yan, A. C. Sedgwick, Y. Zang, G.-R. Chen, X.-P. He, J. Li, J. Yoon and T. D. James, Sensors, Imaging Agents, and Theranostics to Help Understand and Treat Reactive Oxygen Species Related Diseases, Small Methods, 2019, 1900013, DOI:10.1002/smtd.201900013.
- X. Bai, K. K.-H. Ng, J. J. Hu, S. Ye and D. Yang, Small-Molecule-Based Fluorescent Sensors for Selective Detection of Reactive Oxygen Species in Biological Systems, Annu. Rev. Biochem., 2019, 88(1), 605–633, DOI:10.1146/annurev-biochem-013118-111754.
- P. R. Escamilla, Y. Shen, Q. Zhang, D. S. Hernandez, C. J. Howard, X. Qian, D. Y. Filonov, A. V. Kinev, J. B. Shear, E. V. Anslyn and Y. Yang, 2-Amino-3′-Dialkylaminobiphenyl-Based Fluorescent Intracellular Probes for Nitric Oxide Surrogate N2O3, Chem. Sci., 2020, 11(5), 1394–1403, 10.1039/c9sc04304g.
- C. C. Winterbourn, The Challenges of Using Fluorescent Probes to Detect and Quantify Specific Reactive Oxygen Species in Living Cells, Biochim. Biophys. Acta, Gen. Subj., 2014, 730–738, DOI:10.1016/j.bbagen.2013.05.004.
- E. C. Greenwald, S. Mehta and J. Zhang, Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks, Chem. Rev., 2018, 118(24), 11707–11794, DOI:10.1021/acs.chemrev.8b00333.
- D. S. Bilan and V. V. Belousov, In Vivo Imaging of Hydrogen Peroxide with HyPer Probes, Antioxid. Redox Signaling, 2018, 29(6), 569–584, DOI:10.1089/ars.2018.7540.
- H. C. Schunk, D. S. Hernandez, M. J. Austin, A. M. Rosales, L. J. Suggs and K. S. Dhada, Assessing the Range of Enzymatic and Oxidative Tunability for Biosensor Design, J. Mater. Chem. B, 2020, 8(16), 3460–3487, 10.1039/c9tb02666e.
- L. M. Uusitalo and N. Hempel, Recent Advances in Intracellular and in Vivo ROS Sensing: Focus on Nanoparticle and Nanotube Applications, Int. J. Mol. Sci., 2012, 13(9), 10660–10679, DOI:10.3390/ijms130910660.
- H. Lee, K. Lee, I. K. Kim and T. G. Park, Fluorescent Gold Nanoprobe Sensitive to Intracellular Reactive Oxygen Species, Adv. Funct. Mater., 2009, 19(12), 1884–1890, DOI:10.1002/adfm.200801838.
- H. Lee, K. Lee, I. K. Kim and T. G. Park, Synthesis, Characterization, and in Vivo Diagnostic Applications of Hyaluronic Acid Immobilized Gold Nanoprobes, Biomaterials, 2008, 29(35), 4709–4718, DOI:10.1016/j.biomaterials.2008.08.038.
- S. K. Lee, L. J. Mortensen, C. P. Lin and C. H. Tung, An Authentic Imaging Probe to Track Cell Fate from Beginning to End, Nat. Commun., 2014, 5, 1–8, DOI:10.1038/ncomms6216.
- F. Jia, J. Wang, J. Peng, P. Zhao, Z. Kong, K. Wang, W. Yan and R. Wang, D-Amino Acid Substitution Enhances the Stability of Antimicrobial Peptide Polybia-CP, Acta Biochim. Biophys. Sin., 2017, 49(10), 916–925, DOI:10.1093/abbs/gmx091.
- J. Ulbricht, R. Jordan and R. Luxenhofer, On the Biodegradability of Polyethylene Glycol, Polypeptoids and Poly(2-Oxazoline)S, Biomaterials, 2014, 35(17), 4848–4861, DOI:10.1016/j.biomaterials.2014.02.029.
- D. Margulies, G. Melman and A. Shanzer, Fluorescein as a Model Molecular Calculator with Reset Capability, Nat. Mater., 2005, 4(10), 768–771, DOI:10.1038/nmat1469.
- J. Zhou, C. Wei, G. Jia, X. Wang, Z. Feng and C. Li, Formation and Stabilization of G-Quadruplex in Nanosized Water Pools, Chem. Commun., 2010, 46(10), 1700–1702, 10.1039/B925000J.
- H. M. Rostam, P. M. Reynolds, M. R. Alexander, N. Gadegaard and A. M. Ghaemmaghami, Image Based Machine Learning for Identification of Macrophage Subsets, Sci. Rep., 2017, 7(1), 1–11, DOI:10.1038/s41598-017-03780-z.
- T. Yildiz, R. Gu, S. Zauscher and T. Betancourt, Doxorubicin-Loaded Protease-Activated Near-Infrared Fluorescent Polymeric Nanoparticles for Imaging and Therapy of Cancer, Int. J. Nanomed., 2018, 13, 6961–6986, DOI:10.2147/IJN.S174068.
- K. S. Dhada, D. S. Hernandez, W. Huang and L. J. Suggs, Gold Nanorods as Photoacoustic Nanoprobes to Detect Pro-Inflammatory Macrophages and Inflammation, ACS Appl. Nano Mater., 2020 DOI:10.1021/acsanm.0c01324.
- G. Schneider, G. Decher, N. Nerambourg, R. Praho, M. H. V. Werts and M. Blanchard-Desce, Distance-Dependent Fluorescence Quenching on Gold Nanoparticles Ensheathed with Layer-by-Layer Assembled Polyelectrolytes, Nano Lett., 2006, 6(3), 530–536, DOI:10.1021/nl052441s.
- V. Rybalko, P.-L. Hsieh, L. M. Ricles, E. Chung, R. P. Farrar and L. J. Suggs, Therapeutic Potential of Adipose-Derived Stem Cells and Macrophages for Ischemic Skeletal Muscle Repair, Regener. Med., 2017, 12(2), 153–167, DOI:10.2217/rme-2016-0094.
- C. A. Kraynak, D. J. Yan and L. J. Suggs, Modulating Inflammatory Macrophages with an Apoptotic Body-Inspired Nanoparticle, Acta Biomater., 2020, 1–11, DOI:10.1016/j.actbio.2020.03.041.
Footnotes |
| † Electronic supplementary information (ESI) available: Analysis of NSET vs. FRET contributions and images of BMDMs captured at early time points (3 h post stimulation). See DOI: 10.1039/d0na00373e |
| ‡ Present address: Department of Biomedical Engineering, The University of Texas at Austin, 1 University Station, C0800 Austin, TX 78712-023, USA. |
| This journal is © The Royal Society of Chemistry 2020 |