
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into N, P, S multi-doped Mo2C/C composites as highly efficient hydrogen evolution reaction catalysts†
Jieyu
Xu
a,
Lin
Ge
b,
Yajun
Zhou
*a,
Guangyu
Jiang
a,
Liang
Li
 a,
Yunheng
Li
a and
Yongsheng
Li
*a
a,
Yunheng
Li
a and
Yongsheng
Li
*a
aLab of Low-Dimensional Materials Chemistry, Key Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: zhouyj@ecust.edu.cn; ysli@ecust.edu.cn
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
First published on 3rd June 2020
Abstract
Heteroatom doping has been proved to be an effective strategy to optimize the activity of hydrogen evolution reaction (HER) catalysts. Herein, we report N, P, S multi-doped Mo2C/C composites exhibiting highly efficient HER performance in acidic solution, which are facilely fabricated via annealing of N, P, S-containing MoOx–polyaniline (MoOx–PANI) hybrid precursors. The optimized N, P, S multi-doped Mo2C/C catalyst with a moderate P dopant level (NPS-Mo2C/C-0.5) exhibits excellent performance with an overpotential of 53 mV to achieve a current density of 20 mA cm−2, a Tafel slope of 72 mV dec−1 and good stability in acidic electrolytes. Based on the study of XPS, EPR and 31P MAS NMR, the excellent electrocatalytic performance could be attributed to the effective electronic configuration modulation of both Mo2C nanorods and the carbon matrix, derived from stronger synergistic N, P, S multi-doping coupling effects. This work provides a promising methodology for the design and fabrication of multi-doped transition metal based electrocatalysts via electronic structure engineering.
1. Introduction
Electro-driven water splitting represents an appealing strategy to produce clean and renewable hydrogen for energy conversion and storage.1,2 It can also mitigate intermittent energy issues associated with wind and sunlight.3,4 In fact, sustainable hydrogen fuel production requires active catalysts to facilitate HER kinetics and make the electrolyzer practically feasible.5 In line with the Sabatier principle, Pt-based compounds with ideal hydrogen adsorption–desorption energies have been demonstrated as the state-of-the-art HER catalysts.6 However, the high cost and scarcity of noble Pt metals have limited their large-scale commercial applications. Thus, for the sake of practical water splitting applications, ideal catalysts featuring excellent catalytic activities, good stability and low-cost are highly desired.7,8 Over the past few years, transition metal catalysts, such as alloys (Ni–Mo9 and Ni–Fe10), phosphides (Ni2P,11 FeP,12 and CoP13), sulfides (MoS2 (ref. 14) and WS2 (ref. 15)) and carbides (Mo2C16 and WC17), have attracted worldwide attention because of their potential applications as water-splitting catalysts. Among them, molybdenum carbide (Mo2C) is highlighted as one of the most promising candidates with the merits of Pt-like electronic configuration, high chemical stability and tunable surface/bulk properties.18 Nevertheless, both the excessive aggregation and the unoccupied d-orbitals with a large density of Mo2C have greatly caused intrinsic limitations on the HER kinetics.19,20Recently, as demonstrated by a series of theoretical and experimental investigations, heteroatomic dopants, including transition-metal atoms (e.g., Fe,21 Co,22 Ni,23 Mn,24 and W25) and nonmetal atoms (e.g., N,26,27 P,28 and S29), have been verified to be feasible to modulate the electronic properties and lower the density of d-orbital vacancies of Mo2C, owing to the enriched electrons or relatively lower electronegativity. For example, Leonard and co-workers first introduced controllable Fe dopants into a β-Mo2C lattice, which led to a broader valence band for Mo2C and favored HER activity.21 Gao et al. adopted Co-doping to effectively optimize nanostructures and electronic features.22 It has also been reported that the introduced metals could form a new phase providing other effects beyond doping.30 However, metal dopants may suffer from mass loss during electrolysis in acidic media and excessive metals (>25 wt%) would obstruct the active sites.4 From this point of view, nonmetallic dopants with relatively low electronegativity seem to be a better choice. Generally, N, P, and S dopants have been widely employed to modulate the electronic state of transition metal carbides (TMCs). The promotion by N-doping on Mo2C has been proved by producing more active sites and activating the neighbouring C atoms.31 Compared to N atoms, S dopants with a similar electronegativity to C atoms are more positively charged and tend to modify the spin density rather than charge density, which can enrich the electron population on the Mo centers.32 Moreover, considering the lower electronegativity of P (2.19) than that of C (2.55), N (3.04), and S (2.58), P-doping is believed to be a more attractive way to optimize the electronic configuration of Mo2C for accelerating HER kinetics.33 Previous P-doping methods of adopting a P-containing salt (e.g. sodium hypophosphite34) or organic compounds (e.g. phosphomolybdic acid27,35,36) were limited by the uncontrolled dopant concentration. Fortunately, Shi et al. designed a new strategy to synthesize P–Mo2C with controlled P contents by adopting phytic acid (PA) as the P dopant, which showed excellent HER performance at a moderate P-doping level.37 Furthermore, it is expected that dual- or multi-heteroatom-doping is more active than its singly doped counterparts.38 Cai et al. have demonstrated that the N–P chemical interaction after N incorporation could increase the atomic wave function overlap and decrease the hydrogen adsorption energy.39 Based on this, it is anticipated that multi-doping may co-activate the adjacent atoms with much stronger synergistic effects than single and dual-doping. However, as far as we know, multiple-heteroatom-doping of Mo2C for the hydrogen evolution reaction is rarely reported and the underlying synergistic effect is also ambiguous.
Herein, we report novel N, P, S multi-doped Mo2C/C composites as highly effective and stable catalysts for the HER, which were fabricated via annealing of MoOx–polyaniline (MoOx–PANI) precursors. The N, P, S multi-doped Mo2C nanorods tightly anchored on the carbon matrix can expose abundant active sites. Moreover, the N, P, S multi-doping, especially the strong N–P interaction, can modulate the electronic properties of both Mo2C and the carbon matrix and contribute to stronger synergistic coupling effects, leading to improved HER activity. As a result, the N, P, S multi-doped Mo2C/C composites with a moderate P dopant level show outstanding electrocatalytic performance in acidic media with an ultra-small overpotential of 53 mV at 20 mA cm−2, a low Tafel slope of 72 mV dec−1 and high stability, which is remarkably comparable to commercial Pt/C catalysts.
2. Experimental
Chemicals
Ammonium molybdate tetrahydrate (AMT, (NH4)6Mo7O24·4H2O, 99%) was obtained from Alfa Aesar. Aniline (C6H7N, 99.5%), phytic acid (PA, C6H18O24P6, 90%), ammonium persulfate (APS, (NH4)2S2O8, 98%), and hydrochloric acid (HCl, 36–38%) were purchased from Shanghai Lingfeng Chemical Reagent, Shanghai Macklin Biochemical, Aladdin Industrial Corporation and Shanghai Titan Scientific Co., LTD, respectively. The commercial Pt/C (20 wt%) catalyst was obtained from Johnson Matthey. All the reagents were used as received without further treatment.Synthesis of N, P, S multi-doped Mo2C/C composites
N, P, S multi-doped Mo2C/C composites were synthesized via annealing of MoOx–polyaniline (MoOx–PANI) precursors. In a typical synthesis, 1.25 mL aniline and a calculated amount of phytic acid (PA) were dissolved in 20 mL distilled water under constant stirring followed by 2.5 g (NH4)2S2O8 to initiate the polymerization of aniline, and the reaction lasted for 12 h. Then, the obtained polyaniline (PANI) was washed with distilled water several times to remove excessive (NH4)2S2O8. After that, the wet PANI was mixed with 1 g (NH4)6Mo7O24·4H2O in 20 mL distilled water. The mixture was subsequently dried at 100 °C in an oil bath under magnetic stirring to remove water. Then, the dried MoOx–PANI powder was ground and transferred to a tube furnace. The calcination was performed at 700 °C for 3 h at a heating rate of 3 °C min−1 under a 10% H2/Ar atmosphere. The obtained product was marked as NPS-Mo2C/C-x (x = 0.2, 0.5, 0.8), and x referred to the molar ratio of PA and aniline, which was varied to control the P content in the final products. In addition, the powder with a feeding ratio of 0.5 of PA and aniline was calcined at 800 °C and donated as NPS-Mo2C/C-0.5-800.Synthesis of NS-Mo2C/C-0.5 and NPS-C-0.5 composites
The fabrication process was similar to that of N, P, S multi-doped Mo2C/C composites (NPS-Mo2C/C-0.5). The NS-Mo2C/C-0.5 catalyst was synthesized by replacing PA with HCl solution during the polymerization process of aniline, which possessed the same H+ concentration as PA. The NPS-C-0.5 catalyst was synthesized without adding (NH4)6Mo7O24·4H2O.Physical measurements
X-ray diffraction (XRD) characterization was conducted on a Bruker D8 Focus diffractometer with Cu Kα radiation. Raman spectroscopy was performed on a laser micro-Raman spectrometer (inVia Reflex, 532 nm). The morphologies, crystalline structures and elemental information of the catalysts were detected comprehensively by adopting field emission scanning electron microscopy (FE-SEM, Gemini SEM 500) and transmission electron microscopy (TEM, FEI Tecnai G2 F20). Selected area electron diffraction (SAED) was performed on a FEI Tecnai G2 F20 electron microscope. The HAADF and corresponding elemental mapping images were recorded on a scanning electron microscopy instrument (SEM, FEI Magellan 400) using the STEM model. The chemical states and bonding energies of surface elements were investigated by X-ray photoelectron spectroscopy (XPS, Escalab 250Xi, British Thermo Fisher Corporation). The 31P solid-state nuclear magnetic resonance under magic angle spinning conditions (MAS-NMR) was obtained using a Bruker super conducting Fourier NMR spectrometer (500 MHz/Avance III) to detect the chemical environment of P element in the prepared samples. The P contents of the catalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 725) and the C, N, and S contents were measured on an element analyzer (Vario EL III, Elementar). The electronic properties of the samples were analyzed by electron paramagnetic resonance (EPR) techniques on a Bruker EMX-8/2.7 spectrometer at room temperature. The g value was calculated according to Bohr-frequency conditions: ΔE = hν = gβHr, where h is Planck's constant, ν is the microwave frequency, β is the Bohr magnetron, and Hr is the applied magnetic field strength.2.5 Electrochemical measurements
All the HER measurements were carried out on an electrochemical workstation (CHI 760E) using a conventional three-electrode setup in 0.5 M H2SO4 and all the potentials were converted to the reversible hydrogen electrode (RHE) scale (ERHE = EAg/AgCl + 0.059 × pH + 0.197). The three-electrode setup involves a saturated KCl Ag/AgCl electrode as the reference electrode, and a carbon rod as the counter electrode and the working electrode. In a typical test, to prepare a working electrode, 4 mg catalyst, 0.5 mg carbon black (Vulcan XC-72) and 60 μL Nafion were added into 2 mL isopropanol and the mixture was treated with sonication for over 30 minutes to obtain evenly dispersed catalyst ink. Then, 80 μL ink was dropped on a glassy carbon electrode (GCE, 6 mm diameter) and the loading mass was ca. 0.55 mg cm−2. Linear sweep voltammetry (LSV) was conducted at a scan rate of 5 mV s−1 to obtain polarization curves of the catalysts. Based on LSV curves, overpotential (η) in contrast to log current density (log|j|) gives the Tafel plots and the Tafel slope (b) represents the slope values of tangent lines to Tafel plots according to the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log|j| + a). Electrochemical impedance spectroscopy (EIS) was performed at an overpotential of 53 mV with a frequency range between 100 kHz and 0.01 Hz and an amplitude of 5 mV. The electrochemically active surface area (ECSA) of the catalysts can be qualitatively analyzed from electrochemical double layer capacitances (EDLC, Cdl), which were calculated according to cyclic voltammogram tests at a potential of 0–0.3 V with scan rates ranging from 20 to 200 mV s−1 without a faradaic process. In addition, polarization curves before and after 10000 cycles at a scan rate of 100 mV s−1 were recorded to investigate the stability of the catalysts. All the electrochemical data were used without IR correction.
log|j| + a). Electrochemical impedance spectroscopy (EIS) was performed at an overpotential of 53 mV with a frequency range between 100 kHz and 0.01 Hz and an amplitude of 5 mV. The electrochemically active surface area (ECSA) of the catalysts can be qualitatively analyzed from electrochemical double layer capacitances (EDLC, Cdl), which were calculated according to cyclic voltammogram tests at a potential of 0–0.3 V with scan rates ranging from 20 to 200 mV s−1 without a faradaic process. In addition, polarization curves before and after 10000 cycles at a scan rate of 100 mV s−1 were recorded to investigate the stability of the catalysts. All the electrochemical data were used without IR correction.
3. Results and discussion
As shown in Scheme 1, the N, P, S multi-doped Mo2C/C composites were facilely synthesized via annealing of N, P, S-containing MoOx–polyaniline (MoOx–PANI) hybrid precursors. Firstly, PANI was synthesized by a traditional chemical oxidative polymerization method,40 using phytic acid (PA) as a dopant and ammonium persulfate (APS) as an oxidant. Here APS was also the S source, and PA provided an acidic environment as well as P source. Then, the PANI and ammonium molybdate tetrahydrate (AMT) were mixed in aqueous solution at 100 °C to yield MoOx–PANI hybrid precursors through electrostatic adsorption. The NPS-Mo2C/C composites were finally obtained after calcination in a 10% H2/Ar flow. The P contents in the NPS-Mo2C/C composites were controllable by varying the feeding molar ratio of PA and aniline from 0.2 to 0.8 (Table S1†). In order to better understand the roles of PA in the synthetic process, counterpart samples were fabricated by replacing PA with HCl solution (denoted as NS-Mo2C/C-0.5) or without adding ammonium molybdate tetrahydrate (denoted as NPS-C-0.5), respectively. The X-ray diffraction (XRD) technique was applied to characterize the phase and composition of the as-synthesized catalysts. In Fig. 1a, the N, P, S-doped C sample exhibits two diffraction peaks at 25° and 44° in the XRD pattern, which belong to the (002) and (100) diffractions of graphitic carbon. As shown in Fig. 1b, both of the N, P, S multi-doped and N, S dual-doped Mo2C/C composites exhibit similar XRD patterns with the diffraction peaks located at 37.7°, 39.5°, 61.8° and 74.7°, corresponding to the (002), (101), (110) and (112) planes of hexagonal β-Mo2C (PDF#35–0787). The wider shape of the diffraction peaks indicates an ultra-small size of β-Mo2C, and the peaks become obvious when the calcination temperature increases to 800 °C (Fig. S1†). These XRD results confirm that both of the N, S dual-doped and N, P, S multi-doped composites can well retain the typical crystal structure of Mo2C.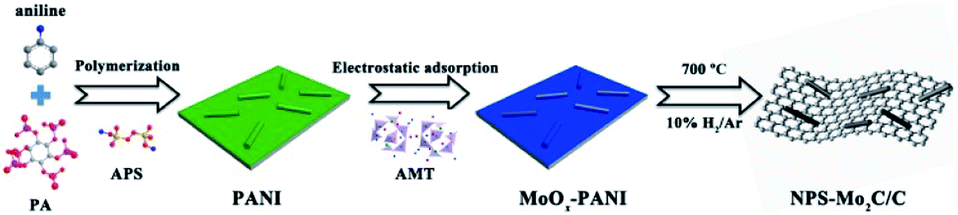 | ||
| Scheme 1 Schematic illustration for the synthesis of the N, P, S multi-doped Mo2C/C catalysts. | ||
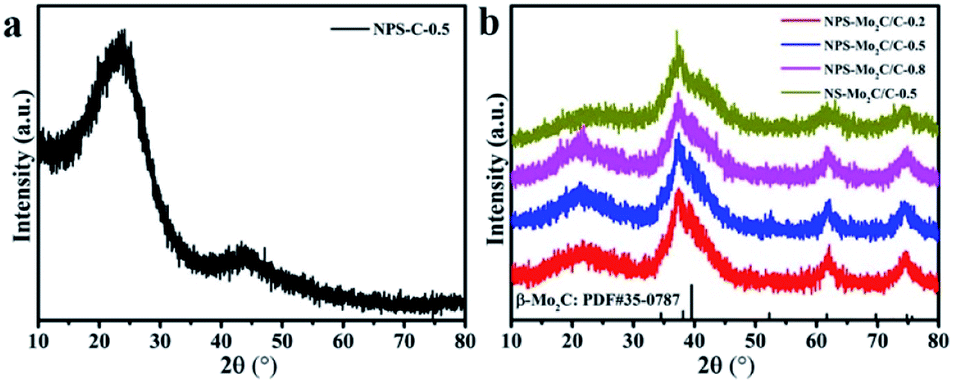 | ||
| Fig. 1 XRD patterns of (a) NPS-C-0.5, (b) NPS-Mo2C/C-0.2, NPS-Mo2C/C-0.5, NPS-Mo2C/C-0.8 and NS-Mo2C/C-0.5. | ||
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were adopted to study the morphological properties of the N, P, S multi-doped Mo2C/C composites. For NPS-Mo2C/C-0.5, both of the SEM (Fig. 2a) and TEM (Fig. 2b) images show that the nanorods with a length of ca. 200–300 nm are tightly anchored on carbon sheets. The HAADF-STEM and corresponding elemental mapping images in Fig. 2f show that Mo, P, and S elements distributed on the nanorods are brighter than other areas, indicating that the nanorods are mainly Mo2C, and N, P, S heteroatoms have been successfully incorporated into Mo2C and the carbon matrix simultaneously. A close observation in Fig. 2c further reveals that the Mo2C nanorods are composed of ultrafine nanoparticles of 5–10 nm, which can expose more active sites for catalysis. The apparent lattice fringes with a distance of 0.23 nm and 0.24 nm of NPS-Mo2C/C-0.5 are attributed to the (011) and (002) planes of hexagonal Mo2C, respectively in the HRTEM image (Fig. 2d). The selected area electron diffraction (SAED) patterns in Fig. 2e further confirm the polycrystalline nature of Mo2C. The N, S dual-doped Mo2C/C composites also show a similar morphology to NPS-Mo2C/C-0.5 (Fig. S3†), suggesting that the dopants have less impact on the morphological properties of heteroatom-doped Mo2C/C catalysts. Such a structure is beneficial to exposing more active sites, leading to enhanced electron transportation and catalytic activities.
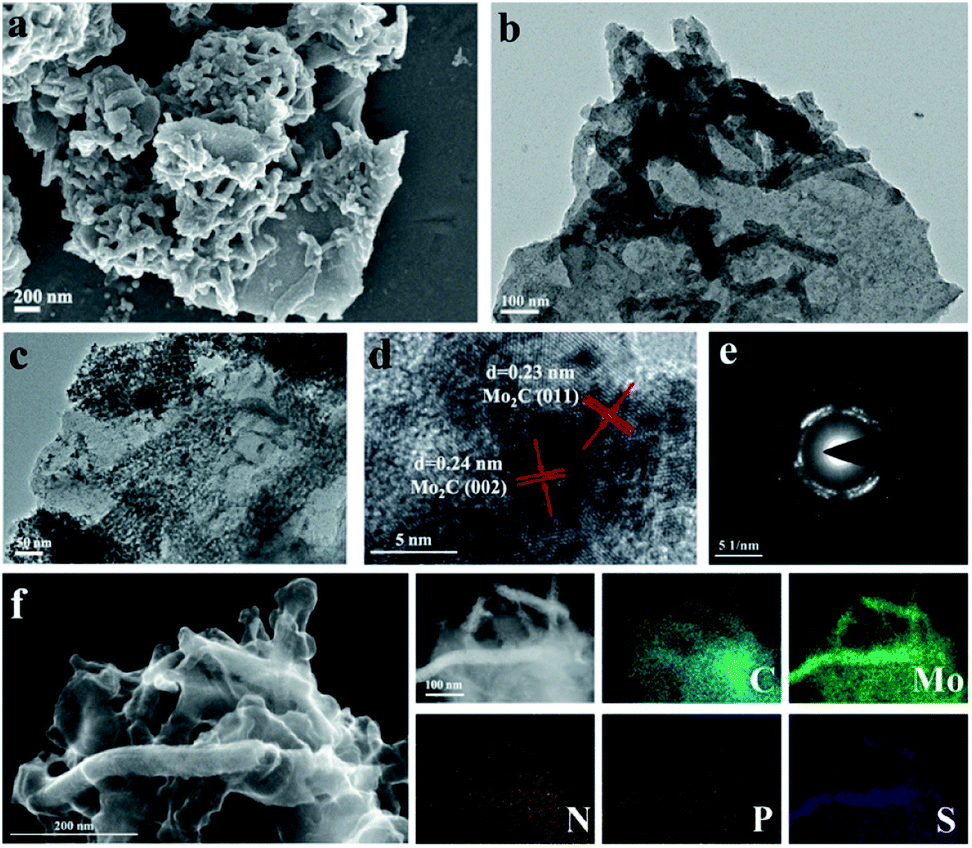 | ||
| Fig. 2 (a) SEM, (b and c) TEM, (d) HRTEM, (e) selected-area electron diffraction (SAED) images, (f) HAADF-STEM and the corresponding elemental mapping images of C, Mo, N, P, and S elements for NPS-Mo2C/C-0.5. | ||
The surface chemical states and electronic features of the as-synthesized catalysts were detected by the X-ray photoelectron spectroscopy (XPS) technique. The survey spectrum of NPS-Mo2C/C-0.5 in Fig. 3a demonstrates the presence of C, N, Mo and O elements. However, the signals of P and S elements are not obvious, due to their low contents in NPS-Mo2C/C-0.5 (Table S1†). As displayed in Fig. 3b, the high-resolution C 1s spectrum of NPS-Mo2C/C-0.5 can be resolved and attributed to C–Mo (283.9 eV), C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.6 eV), C–N (285.4 eV), and CO (286.8 eV).41 The broad peak of C–N demonstrates the successful incorporation of N into the carbon matrix. Besides, the high-resolution Mo 3d spectrum (Fig. 3c) confirms four states of Mo on the surface: Mo2+ (228.9, 232.1 eV), Mo3+ (229.6, 232.7 eV), Mo4+ (230.6, 233.7 eV) and Mo6+ (233.0, 236.1 eV).42 The Mo2+ is ascribed to the Mo2C species. The presence of Mo3+ should be related to heteroatom-Mo bonding (P–Mo and N–Mo), which indicates the successful incorporation of N and P dopants into Mo2C.38 Besides, the Mo4+ and Mo6+ signals are attributed to molybdenum oxides due to unavoidable surface oxidation of Mo2C.42 In Fig. 3d, the N 1s profile contains five main peaks at 395.2, 397.1, 398.5, 399.8 and 401.1 eV, corresponding to Mo 3p, Mo–N, pyridinic N, pyrrolic N and graphitic N, respectively.43 The C 1s (C–N, 285.4 eV), Mo 3d (N–Mo, 229.6 and 232.7 eV) and N 1s (Mo–N, 397.1 eV) confirm the N incorporation in both Mo2C and the carbon matrix. The S 2p spectrum in Fig. 3e can be deconvoluted and assigned to S–Mo (162.2 and 163.3 eV) and C–S–C (164.3 eV),29,38 verifying the achievement of S dopants in the catalyst. With respect to the P 2p spectrum in Fig. 3f, the doublet peaks with spin–orbit at 129.2 eV and 130.2 eV are attributed to the P 2p3/2 and P 2p1/2 states of P–Mo.44 The peak at 134.3 eV corresponds to P–O bonding.28 The other two peaks located at 133.6 and 132.8 eV are typical for P–N and P–C bonding, respectively, since the binding energy of P–C is ∼1–2 eV lower than that of P–N bonding.28 Notably, unlike the dominant P–C species in NPS-C-0.5, the high intensity of P–N bonding implies a strong interaction between N and P dopants, which is expected to regulate the electronic structure and enhance the performance of NPS-Mo2C/C-0.5.
C (284.6 eV), C–N (285.4 eV), and CO (286.8 eV).41 The broad peak of C–N demonstrates the successful incorporation of N into the carbon matrix. Besides, the high-resolution Mo 3d spectrum (Fig. 3c) confirms four states of Mo on the surface: Mo2+ (228.9, 232.1 eV), Mo3+ (229.6, 232.7 eV), Mo4+ (230.6, 233.7 eV) and Mo6+ (233.0, 236.1 eV).42 The Mo2+ is ascribed to the Mo2C species. The presence of Mo3+ should be related to heteroatom-Mo bonding (P–Mo and N–Mo), which indicates the successful incorporation of N and P dopants into Mo2C.38 Besides, the Mo4+ and Mo6+ signals are attributed to molybdenum oxides due to unavoidable surface oxidation of Mo2C.42 In Fig. 3d, the N 1s profile contains five main peaks at 395.2, 397.1, 398.5, 399.8 and 401.1 eV, corresponding to Mo 3p, Mo–N, pyridinic N, pyrrolic N and graphitic N, respectively.43 The C 1s (C–N, 285.4 eV), Mo 3d (N–Mo, 229.6 and 232.7 eV) and N 1s (Mo–N, 397.1 eV) confirm the N incorporation in both Mo2C and the carbon matrix. The S 2p spectrum in Fig. 3e can be deconvoluted and assigned to S–Mo (162.2 and 163.3 eV) and C–S–C (164.3 eV),29,38 verifying the achievement of S dopants in the catalyst. With respect to the P 2p spectrum in Fig. 3f, the doublet peaks with spin–orbit at 129.2 eV and 130.2 eV are attributed to the P 2p3/2 and P 2p1/2 states of P–Mo.44 The peak at 134.3 eV corresponds to P–O bonding.28 The other two peaks located at 133.6 and 132.8 eV are typical for P–N and P–C bonding, respectively, since the binding energy of P–C is ∼1–2 eV lower than that of P–N bonding.28 Notably, unlike the dominant P–C species in NPS-C-0.5, the high intensity of P–N bonding implies a strong interaction between N and P dopants, which is expected to regulate the electronic structure and enhance the performance of NPS-Mo2C/C-0.5.
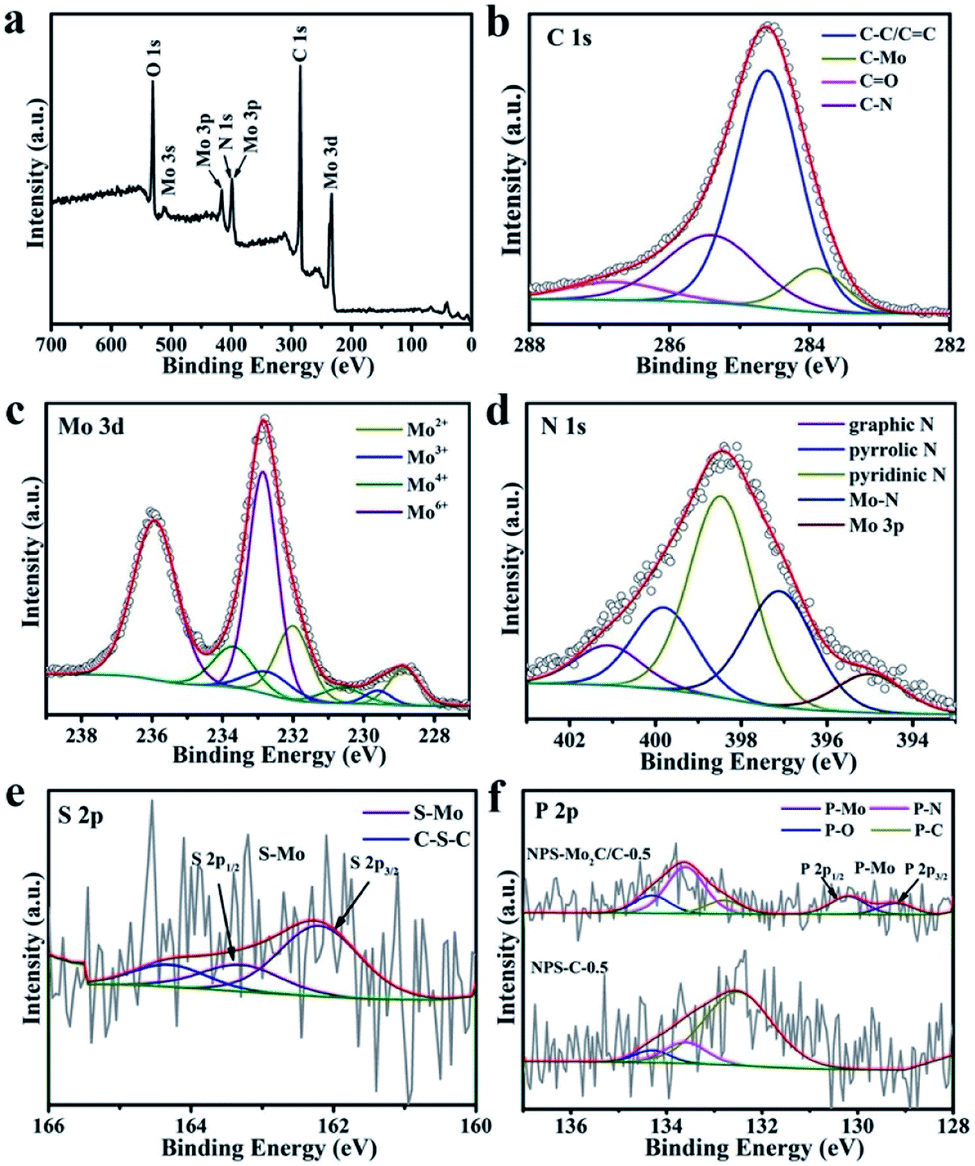 | ||
| Fig. 3 (a) XPS survey spectra of NPS-Mo2C/C-0.5 and the corresponding high-resolution spectra of (b) C 1s, (c) Mo 3d, (d) N 1s, and (e) S 2p. (f) High-resolution spectra of NPS-Mo2C/C-0.5 and NPS-C-0.5. | ||
Electron paramagnetic resonance (EPR) spectroscopy was used to study the electronic properties of the multi-doped catalysts.45 As shown in Fig. 4a, the prominent signals of NPS-Mo2C/C-0.5 (g1 = 1.9223) and NS-Mo2C/C-0.5 (g2 = 1.9262) are assigned to the Mo–N coordination, since pure Mo2C (4d4, Mo2+) has no unpaired electrons and EPR signals.24,46 The EPR spectrum of NPS-C-0.5 has a strong signal of the g value at 1.9978, which is probably due to the unpaired electron spins introduced by heteroatom dopants.47 According to the fingerprint-like feature of EPR signals, the weak signals of NPS-Mo2C/C-0.5 (g4 = 1.9989) and NS-Mo2C/C-0.5 (g5 = 2.0020) are attributed to the heteroatom-doped carbon matrix. The Mo–S signals can also be observed for NPS-Mo2C/C-0.5 (g6 = 1.9824) and NS-Mo2C/C-0.5 (g7 = 1.9855).48 Notably, compared to N, S dual-doped Mo2C/C, the presence of an extra relatively weak signal in NPS-Mo2C/C-0.5 (g8 = 1.9598) implies more unpaired electrons and active sites after the incorporation of P dopants. The paramagnetic signals demonstrate that N, P, and S dopants can optimize the electronic configuration of Mo2C synergistically, which is believed to reduce the hydrogen adsorption energy and improve the intrinsic activities. 31P solid-state MAS NMR spectroscopy was employed to further explore the chemical environment of P elements in the catalysts. As shown in Fig. 4b, the NPS-C-0.5 catalyst exhibits three peaks at −10.4, −16.8 and 7.6 ppm, which correspond to three different positions in the carbon structure. The 7.6 ppm signal is the resonance for P–O.49,50 Based on the XPS result of NPS-C-0.5, the −10.4 and −16.8 ppm signals are ascribed to P–C and P–N, respectively. So the broad dominant peak at −16.8 ppm of NPS-Mo2C/C is attributed to the P–N signal, demonstrating the strong interaction of P–N in NPS-Mo2C/C. From the study of EPR and 31P MAS NMR, the N, P, and S multiple dopants, especially the strong N–P interaction, contribute to much stronger synergistic effects to modulate the electronic properties of Mo2C and the carbon matrix.
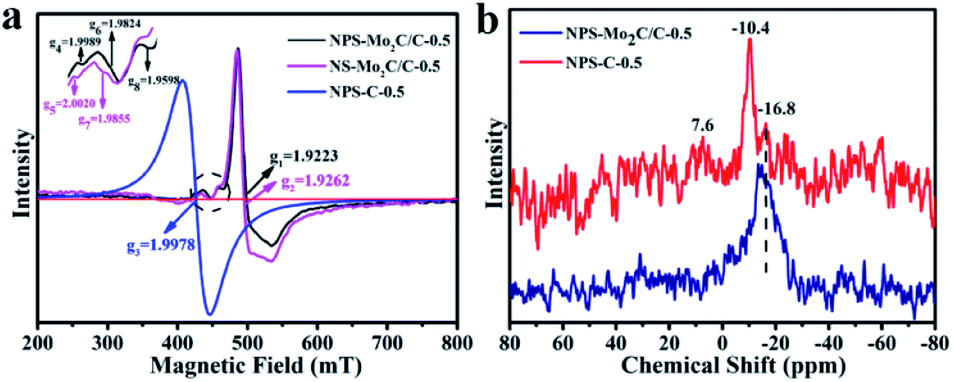 | ||
| Fig. 4 (a) Room temperature EPR spectra of NPS-Mo2C/C-0.5, NS-Mo2C/C-0.5 and NPS-C-0.5. (b) 31P solid-state MAS NMR spectra of NPS-Mo2C/C-0.5 and NS-Mo2C/C-0.5. | ||
Electrochemical measurements of the N, P, S multi-doped Mo2C/C composites were performed in N2-purged 0.5 M H2SO4 solution using a standard three-electrode configuration. The HER activities were first evaluated by linear scanning voltammetry (LSV) at a scan rate of 5 mV s−1. The corresponding polarization curves without IR compensation are shown in Fig. 5a. As displayed, compared with the N, S dual-doped Mo2C/C catalyst (NS–Mo2C/C-0.5), the HER activities of N, P, S multi-doped catalysts have been largely enhanced, which indicates that the N, P, S multi-doping is more active than the N, S dual-doping. Especially, the NPS-Mo2C/C-0.5 catalyst shows the lowest overpotential of only 53 mV to deliver a current density of 20 mA cm−2, which is much lower than that of NPS-Mo2C/C-0.2 (75 mV) and NPS-Mo2C/C-0.8 (114 mV), and even outperforms that of the commercial Pt/C catalyst (62 mV). Notably, the NPS-C-0.5 catalyst is almost inactive for the HER, implying that the N, P, S multi-doped Mo2C should be the dominant active constituent. Moreover, the Tafel slope is calculated to explore the HER mechanism. According to the HER kinetic model, hydrogen evolution in acidic solution consists of two steps. The first one is the Volmer process (H+ + e− → Hads, Hads represents H atoms adsorbed on the active sites of the catalyst), and the second one is related to the Heyrovsky reaction (H+ + e− + Hads → H2) or the Tafel reaction (Hads + Hads → H2), which deliver Tafel slopes of 118 mV dec−1, 39 mV dec−1 and 29 mV dec−1 at room temperature respectively.2 As shown in Fig. 5b, all of the heteroatom-doped Mo2C/C catalysts involve a Volmer–Heyrovsky mechanism. As summarized in Table 1, the NPS-Mo2C/C-0.5 catalyst delivers the smallest Tafel slope (72 mV dec−1) among the Mo2C/C catalysts, suggesting faster HER kinetics. However, this value is still much larger than that of the commercial Pt/C catalyst (33 mV dec−1), which may be the reason for the inferior performance of NPS-Mo2C/C-0.5 to Pt/C at a larger current density. Remarkably, the excellent HER performance of NPS-Mo2C/C-0.5, with a low overpotential (22 mV) at a current density of 10 mA cm−2 and a small Tafel slope (72 mV dec−1), is still among the best performances of Mo-based electrocatalysts (Fig. 5e).
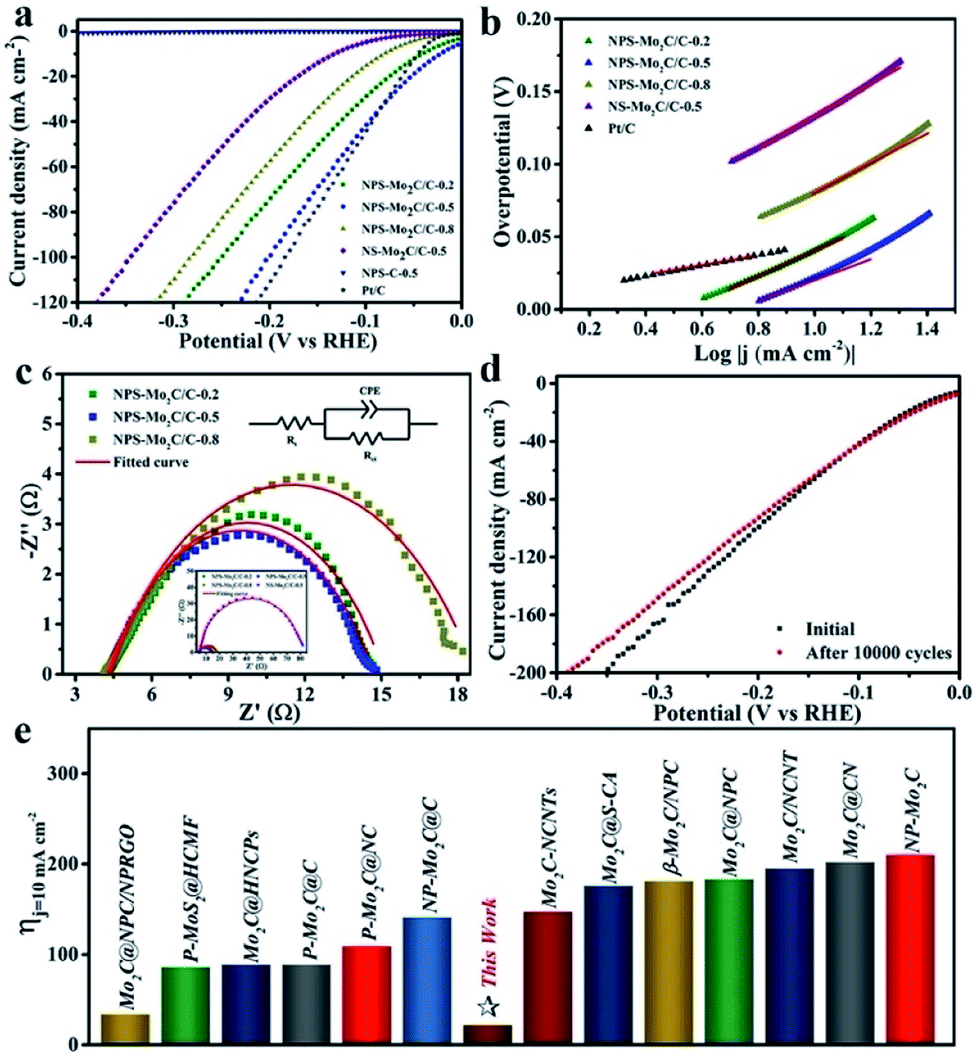 | ||
| Fig. 5 (a) Polarization curves and (b) the corresponding Tafel plots of the N, P, S multi-doped electrocatalysts and Pt/C in 0.5 M H2SO4. (c) Nyquist plots (η = 53 mV) of the N, P, S multi-doped catalysts. Inset: Nyquist plots of NS-Mo2C/C-0.5 and the N, P, S multi-doped catalysts. (d) Polarization curves before and after 10000 cycles of NPS-Mo2C/C-0.5. (e) Comparison of η10 between NPS-Mo2C/C-0.5 and the reported heteroatom-doped Mo-based electrocatalysts in acidic media: Mo2C@NPC/NPRGO,36 P–MoS2@HCMF,51 Mo2C@HNCPs,43 P–Mo2C@C,37 P–Mo2C@NC,52 NP-Mo2C@C,28 Mo2C-NCNTs,31 Mo2C@S-CA,29 β-Mo2C/NPC,53 Mo2C@NPC,44 Mo2C/NCNT,54 Mo2C@CN,27 and NP-Mo2C.38 | ||
| Sample | η 20 [mV] | Tafel slope [mV dec−1] | R ct [Ω] | C dl [mF cm−2] |
|---|---|---|---|---|
| NPS-Mo2C/C-0.2 | 75 | 87 | 11 | 41.4 |
| NPS-Mo2C/C-0.5 | 53 | 72 | 10 | 67.0 |
| NPS-Mo2C/C-0.8 | 114 | 102 | 14 | 27.8 |
| NS-Mo2C/C-0.5 | 182 | 111 | 76 | 14.0 |
Electrochemical impedance spectroscopy (EIS) was also performed to analyse the charge transfer behavior of the catalysts. The diameter of the pseudo-semicircle in the low frequency region of the Nyquist plot is associated with the charge transfer resistance (Rct), which can be used to evaluate charge transfer efficiency during the HER process. According to the equivalent circuit in Fig. 5c, the NPS-Mo2C/C-0.5 catalyst exhibits a relatively small Rct value of 10 Ω, in comparison to that of NPS-Mo2C/C-0.2 (11 Ω) and NPS-Mo2C/C-0.5 (14 Ω), corresponding to a higher electron transportation rate. Moreover, the Rct values of N, P, S multi-doped Mo2C/C catalysts are much smaller than that of the N, S dual-doped Mo2C/C catalyst. The enhanced conductivity may result from the N, P, S multi-doping into the carbon matrix, which can further optimize the intrinsic electronic structure of the carbon matrix when compared to N, S dual-doping. Similarly, as shown in Fig. S7† and Table 1, a much larger double layer capacitance (Cdl) of NPS-Mo2C/C than that of NS-Mo2C/C-0.5 (14.0 mF cm−2) can be detected, indicating more active sites derived from N, P, S multi-doping. In addition, the Cdl value has a maximum at a moderate P dopant level, suggesting that too many dopants may result in structural disorder and reduce the electrochemically active sites. The stability of a catalyst in acidic solution is another important index. The stability curve of NPS-Mo2C/C-0.5 is shown in Fig. 5d. As observed, the almost overlapped polarization curves before and after 10000 cycles, except a slight decay by ca. 15 mV at a current density of 120 mA cm−2, demonstrate the excellent stability of NPS-Mo2C/C-0.5.
The excellent HER performance of NPS-Mo2C/C-0.5 should be due to the following merits. (1) The Mo2C nanorods tightly anchored on carbon sheets can provide abundant active sites and improve the conductivity; (2) Due to the nitrophilicity of P properties, there's a strong N–P interaction in NPS-Mo2C/C-0.5, which can effectively regulate the electronic structure of Mo2C. Furthermore, the S dopants together with the strong N–P interaction contribute to much stronger synergistic coupling effects and facilitate HER kinetics, thus leading to enhanced HER activity.
4. Conclusions
In summary, N, S, P multi-doped Mo2C/C composites as effective and stable HER catalysts have been successfully fabricated via annealing of MoOx–polyaniline (MoOx–PANI) precursors. Compared with N, S dual-doped samples, the N, P, S multi-doping in Mo2C/C composites, especially the formation of P–N bonding, could modulate the electronic properties with stronger synergistic coupling effects. As a result, the optimal NPS-Mo2C/C-0.5 catalyst displays highly efficient HER performance with a small overpotential (53 mV) at a current density of 20 mA cm−2, a low Tafel slope (72 mV dec−1) and good stability in acidic solution. This work provides a novel pathway for the design and construction of multi-doped transition metal based electrocatalysts via electronic structure engineering.Conflicts of interest
There are no conflicts for this work to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51672083), program of Shanghai Academic/Technology Research Leader (18XD1401400), Basic Research Program of Shanghai (17JC1404702), Leading talents in Shanghai in 2018, 111 project (B14018), Research Foundation of China Postdoctoral Science (2017M621384) and Fundamental Research Funds for the Central Universities (222201718002).Notes and references
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem.–Eur. J., 2017, 23, 10947–10961 CrossRef CAS PubMed.
- Z. Zhuang, J. Huang, Y. Li, L. Zhou and L. Mai, ChemElectroChem, 2019, 6, 3570–3589 CrossRef CAS.
- S. T. Hunt, M. Milina, Z. Wang and Y. Román-Leshkov, Energy Environ. Sci., 2016, 9, 3290–3301 RSC.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- C. Zhang, Y. Shi, Y. Yu, Y. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- R. Wu, J. Zhang, Y. Shi, D. Liu and B. Zhang, J. Am. Chem. Soc., 2015, 137, 6983–6986 CrossRef CAS PubMed.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- X. Jin, J. Li, Y. Cui, X. Liu, K. Wang, Y. Zhou, W. Yang, X. Zhang, C. Zhang, X. Jiang and B. Liu, Int. J. Hydrogen Energy, 2019, 44, 5739–5747 CrossRef CAS.
- G. Cho, Y. Park, H. Kang, Y.-k. Hong, T. Lee and D.-H. Ha, Appl. Surf. Sci., 2020, 510, 145427 CrossRef CAS.
- S. Liu, A. Jiang, Z. Wang, M. Lin, D. Xia, Q. Li and M. Dong, Int. J. Hydrogen Energy, 2020, 45, 1729–1737 CrossRef CAS.
- X. Meng, L. Yu, C. Ma, B. Nan, R. Si, Y. Tu, J. Deng, D. Deng and X. Bao, Nano Energy, 2019, 61, 611–616 CrossRef CAS.
- T. P. Nguyen, D. L. T. Nguyen, V.-H. Nguyen, T.-H. Le, Q. V. Ly, D.-V. N. Vo, Q. V. Nguyen, H. S. Le, H. W. Jang, S. Y. Kim and Q. V. Le, Appl. Surf. Sci., 2020, 505, 144574 CrossRef.
- Q. Yan, X. Yang, T. Wei, C. Zhou, W. Wu, L. Zeng, R. Zhu, K. Cheng, K. Ye, K. Zhu, J. Yan, D. Cao and G. Wang, J. Colloid Interface Sci., 2020, 563, 104–111 CrossRef CAS PubMed.
- J. Huang, W. Hong, J. Li, B. Wang and W. Liu, Sustainable Energy Fuels, 2020, 4, 1078–1083 RSC.
- T. Meng and M. Cao, Chem.–Eur. J., 2018, 24, 16716–16736 CrossRef CAS PubMed.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- W. Han, L. Chen, B. Ma, J. Wang, W. Song, X. Fan, Y. Li, F. Zhang and W. Peng, J. Mater. Chem. A, 2019, 7, 4734–4743 RSC.
- C. Wan and B. M. Leonard, Chem. Mater., 2015, 27, 4281–4288 CrossRef CAS.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590–5598 CrossRef CAS.
- D. Wang, X. Zhang, Y. Shen and Z. Wu, RSC Adv., 2016, 6, 16656–16661 RSC.
- Y. Zhou, J. Xu, C. Lian, L. Ge, L. Zhang, L. Li, Y. Li, M. Wang, H. Liu and Y. Li, Inorg. Chem. Front., 2019, 6, 2464–2471 RSC.
- X. Wang, Y. Chen, B. Yu, Z. Wang, H. Wang, B. Sun, W. Li, D. Yang and W. Zhang, Small, 2019, 15, 1902613 CrossRef PubMed.
- Y. Wang, R. A. Senthil, J. Pan, Y. Sun, S. Osman, A. Khan and X. Liu, Ionics, 2019, 25, 4273–4283 CrossRef CAS.
- Y. Li, Q. Huang, H. Wu, L. Cai, Y. Du, S. Liu, Z. Sheng and M. Wu, J. Solid State Electrochem., 2019, 23, 2043–2050 CrossRef CAS.
- Y.-Y. Chen, Y. Zhang, W.-J. Jiang, X. Zhang, Z. Dai, L.-J. Wan and J.-S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef CAS PubMed.
- X. Kong, S. Chen, Y. Zou, S. Lyu, X. She, Y. Lu, J. Sun, H. Zhang and D. Yang, Int. J. Hydrogen Energy, 2018, 43, 13720–13726 CrossRef CAS.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- K. Zhang, Y. Zhao, D. Fu and Y. Chen, J. Mater. Chem. A, 2015, 3, 5783–5788 RSC.
- M. A. R. Anjum, M. D. Bhatt, M. H. Lee and J. S. Lee, Chem. Mater., 2018, 30, 8861–8870 CrossRef CAS.
- J. Li, M. Zhang, H. Zang, B. Yu, Y. Ma and Y. Qu, ChemCatChem, 2019, 11, 4998–5012 CrossRef CAS.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. I. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS PubMed.
- L. Ji, J. Wang, X. Teng, H. Dong, X. He and Z. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 14632–14640 CrossRef CAS PubMed.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- Z. Shi, K. Nie, Z.-J. Shao, B. Gao, H. Lin, H. Zhang, B. Liu, Y. Wang, Y. Zhang, X. Sun, X.-M. Cao, P. Hu, Q. Gao and Y. Tang, Energy Environ. Sci., 2017, 10, 1262–1271 RSC.
- D. Wang, T. Liu, J. Wang and Z. Wu, Carbon, 2018, 139, 845–852 CrossRef CAS.
- J. Cai, Y. Song, Y. Zang, S. Niu, Y. Wu, Y. Xie, X. Zheng, Y. Liu, Y. Lin, X. Liu, G. Wang and Y. Qian, Sci. Adv., 2020, 6, eaaw8113 CrossRef PubMed.
- S. Cho, S. J. Lee and H. Joo, Polymers, 2019, 11 CAS.
- Z. Wei, X. Hu, S. Ning, X. Kang and S. Chen, ACS Sustainable Chem. Eng., 2019, 7, 8458–8465 CrossRef CAS.
- R. Jiang, L. Pi, B. Deng, L. Hu, X. Liu, J. Cui, X. Mao and D. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 38606–38615 CrossRef CAS PubMed.
- L. Chai, L. Zhang, X. Wang, Z. Ma, T.-T. Li, H. Li, Y. Hu, J. Qian and S. Huang, Electrochim. Acta, 2019, 321, 134680 CrossRef CAS.
- K. An, X. Cui, X. Xu and Y. Wang, J. Porous Mater., 2019, 26, 1035–1042 CrossRef CAS.
- J. R. González, R. Alcántara, J. L. Tirado, A. J. Fielding and R. A. W. Dryfe, Chem. Mater., 2017, 29, 5886–5895 CrossRef.
- A. R. Yacob, M. K. A. Amat Mustajab and N. H. Suhaimi, Adv. Mater. Res., 2015, 1107, 91–95 Search PubMed.
- W. P. Wright, V. D. Marsicano, J. M. Keartland, R. M. Erasmus, S. M. A. Dube and N. J. Coville, Mater. Chem. Phys., 2014, 147, 908–914 CrossRef CAS.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, J. Mater. Chem. A, 2015, 3, 3862–3867 RSC.
- J. Xiong, J. Li, J. Shi, X. Zhang, W. Cai, Z. Yang and H. Cheng, Appl. Catal., B, 2019, 243, 614–620 CrossRef CAS.
- G. Yan, X. Feng, S. U. Khan, L. Xiao, W. Xi, H. Tan, Y. Ma, L. Zhang and Y. Li, Chem.–Asian J., 2018, 13, 158–163 CrossRef CAS PubMed.
- J. Tan, X. He, F. Yin, X. Liang, B. Chen, G. Li and H. Yin, J. Mater. Sci., 2019, 54, 4589–4600 CrossRef CAS.
- Y.-J. Song, J.-T. Ren, G. Yuan, Y. Yao, X. Liu and Z.-Y. Yuan, J. Energy Chem., 2019, 38, 68–77 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00335b |
| This journal is © The Royal Society of Chemistry 2020 |