
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fluorous biphase drug delivery system triggered by low frequency ultrasound: controlled release from perfluorous discoidal porous silicon particles†
Jing
Liu
 ,
Shuo
Li
,
Lina
Liu
and
Zhiling
Zhu
*
,
Shuo
Li
,
Lina
Liu
and
Zhiling
Zhu
*
College of Materials Science and Engineering, Qingdao University of Science and Technology, Qingdao, Shandong, China 266042. E-mail: zlzhu@qust.edu.cn
First published on 30th June 2020
Abstract
Conventional drug delivery systems face unsatisfactory loading efficiency, poor biological bypass, and uncontrollable release, which are great barriers for improving the treatment of many diseases. Herein, a proof-of-concept of a fluorous biphase drug delivery system (FB-DDS) trigged by low frequency ultrasound (LFUS) is proposed for the first time, where promoted incorporation and stabilization of therapeutic agents in nanocarriers was achieved through fluorine–fluorine interactions, and the encapsulated drugs were controllably released by external sources, resulting in minimized nonspecific toxicity and enhanced therapeutic efficacy. The FB-DDS was constructed from monodisperse, discoidal porous silicon particles (PSP) and was functionalized with 1H,1H,2H,2H-perfluorodecyltrimethoxysilane (FAS17) for loading perfluoropentane (PFP) and fluorinated drugs through fluorine–fluorine interactions. This delivery system was demonstrated by utilizing model compounds including a fluorous-tagged fluorescein and a fluorine containing antibiotic ciprofloxacin. Loading of the model molecules into fluorocarbon-coated carriers was facilitated by fluorous interactions, whereas ejection of the model molecules was promoted by applying LFUS to rapidly evaporate PFP. In the in vitro test, these carriers loaded with fluorine containing ciprofloxacin exhibited excellent antimicrobial activity against Pseudomonas aeruginosa biofilm formation. Overall, this innovative stimulus-responsive fluorous biphase drug delivery system will be a promising candidate for practical applications as well as encouraging further investigation of drug delivery and controlled release strategies.
Introduction
Clinical use of many drugs suffers from several important drawbacks, such as poor solubility, nonspecific distribution, rapid metabolism, low bioavailability, and toxic side effects during delivery.1,2 Owing to the impressive progress in materials science and pharmaceutics, nanoparticle-based drug delivery platforms have emerged as intriguing candidates for overcoming these limitations associated with conventional drug formulations, thus increasing drug efficacy and intracellular penetration.3–8 Unfortunately, great efforts and vast investments over the past thirty years have yielded only a few nanoformulations that were approved by the US Food and Drug Administration (FDA) for clinical applications, e.g., Doxil®9 and Abraxane®.10 Moreover, most of the conventional drug delivery systems (DDS) depend on either the conjugation and functionalization methodologies for incorporation of therapeutic drugs or the unique porous architectures of nanocarriers for drug encapsulation, which are not yet satisfactory in clinical usage and call for the development of potential alternative strategies.“Fluorous biphasic system (FBS)” was a concept initially proposed by Horváth et al. in the 1990s for the selective separation or delivery of materials based on their difference in solubility.11–13 A FBS consists of a fluorous phase that carries dissolved reagents, and a second phase that could be any solvent with limited or no solubility in the fluorous phase. The fluorous phase is mostly perfluorocarbons (PFC). The FBS compatible reagents comprise fluorous moieties and will be soluble only or preferentially in the fluorous phase. By incorporation of fluorous tags, therapeutic agents can be stabilized in fluorous carriers filled with PFC through noncovalent fluorine–fluorine interactions during transportation and storage.14–17 Inspired by the FBS, we hypothesize that a fluorous biphase drug delivery system (FB-DDS) can dramatically enhance the drug loading efficiency and biological stability, particularly for the vast majority of fluorine containing pharmaceuticals (Table S1, ESI†),18,19 which have ever been reported.
Delivery of drugs from the carrier to the target is mainly dependent on diffusion that can be blocked by physical barriers in the body system. To achieve a rapid and controlled release in targeted tissues or cells where the particles accumulate, as well as to reduce systemic toxicity, stimulus-responsive nanoparticles are often employed.20–22 These nanoparticles present an intriguing combination of properties that release drugs on demand through external energy sources or triggers at a desired time, location and/or dose, i.e., temperature,23,24 pH,25 magnetic field,26,27 illumination,28 or ultrasound.29,30 Low frequency ultrasound (LFUS) triggered release has features such as being non-invasive, permeable, precisely controllable using frequency, power density, duty cycles, and time, and can be focused on the targeted regions with a resolution of 1–2 mm for release.31 A variety of LFUS responsive carriers such as liposomes,32,33 micelles,34 hydrogels,35,36 and emulsion droplets37–39 have been used to encapsulate poorly soluble drugs, protect them from premature degradation in the body, and decrease their interaction with healthy tissues to reduce side effects.40–44 Among them, biocompatible porous silicon particles (PSP) have drawn particular attention,45,46 as an excellent candidate47–50 for controlled drug delivery.51,52
In the present work, a proof-of-concept of a FB-DDS trigged by LFUS is demonstrated. We filled fluorous PSP with volatile PFC, i.e., perfluoropentane (PFP),53–55 an FDA approved biomedical agent.56–58 Although PFP has a low boiling point at 28 °C, its micro and nanodroplet emulsions remain in a metastable state even above the boiling point, attributed to a higher internal pressure in these droplets caused by surface tension, or namely Laplace pressure.59,60 Thus, fluorine containing drugs can be stabilized in fluorous nanocarriers, which can form a fluorous biphase system during delivery. Furthermore, upon interaction with LFUS, spontaneous vaporization of these droplets can occur, producing gas bubbles of equilibrium volume hundred times larger than that of the original droplets.61 The acoustic cavitation produces local extremes of temperature and pressure of these droplets confined in the nanochannels, which facilitates drug delivery across the endothelial barrier. To highlight the promising practical applications of the FB-DDS, we examined two model compounds including a short fluorous-tagged fluorescein and a fluorine containing antibiotic ciprofloxacin. These model compounds can be easily loaded into the fluorous discoidal carriers and the LFUS delivery system exhibits a desired controlled release profile.
Experimental
Materials
1H,1H,2H,2H-Perfluorodecyltrimethoxysilane (FAS17, 97%) was purchased from Apollo Scientific. Decafluorobutane (98%), dodecafluoro-n-pentane (99%), and perfluoro-n-hexane (99%) were purchased from SynQuest Labs. 1H,1H-Perfluorooctyl-amine (97%) was purchased from Alfa Aesar. Fluorescein isothiocyanate (FITC, 97%) and Dulbecco's Modified Eagle Medium (DMEM) were purchased from Thermo Scientific. Ciprofloxacin (98%), perfluoropentanoyl chloride (98%), triethylamine (99%), and phosphate buffered saline (10× PBS) were purchased from Sigma-Aldrich. LB broth base (powder) and LB agar (powder) were purchased from Becton-Dickinson. Cell counting kit-8 (CCK-8) was purchased from Beyotime Biotechnology. All reactants were used as received without further purification.Preparation of discoidal porous silicon particles
The fabrication of discoidal porous silicon particles (PSP) was modified from a reported procedure.50 Briefly, a heavily doped P-type (100) silicon wafer with a resistivity of 0.005 ohm × cm (Silicon Quest, Inc.) was employed as a substrate. The substrate was exposed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 HF/ethanol solution and electrochemically etched with an applied current density of 10 mA cm−2 to form a porous silicon film with a mean pore size of 40 nm. Afterwards, a high electrical current (76 mA cm−2) was applied to form a high porosity release layer. An 80 nm low temperature oxide (LTO) layer was deposited on the porous silicon film in a low pressure chemical vapor deposition (LPCVD) furnace. A standard photolithography process was used to pattern the arrays of 600 nm and 1000 nm circles with a contact aligner (K. Suss MA6 mask aligner) and NR9-500P photoresist (Futurrex Franklin). The patterns were transferred to LTO and porous silicon films by reactive ion etching in tetrafluoromethane plasma (Plasmatherm Batchtop VII). After stripping the photoresist and the LTO, the as-prepared particles were retained on the substrate as an array allowing for further surface functionalization. Noticeably, this fabrication strategy provides sufficient flexibility to fabricate multistage vectors62 that can be directly translated to large scale production within the framework of established good manufacturing practice semiconductor processes.
:3 HF/ethanol solution and electrochemically etched with an applied current density of 10 mA cm−2 to form a porous silicon film with a mean pore size of 40 nm. Afterwards, a high electrical current (76 mA cm−2) was applied to form a high porosity release layer. An 80 nm low temperature oxide (LTO) layer was deposited on the porous silicon film in a low pressure chemical vapor deposition (LPCVD) furnace. A standard photolithography process was used to pattern the arrays of 600 nm and 1000 nm circles with a contact aligner (K. Suss MA6 mask aligner) and NR9-500P photoresist (Futurrex Franklin). The patterns were transferred to LTO and porous silicon films by reactive ion etching in tetrafluoromethane plasma (Plasmatherm Batchtop VII). After stripping the photoresist and the LTO, the as-prepared particles were retained on the substrate as an array allowing for further surface functionalization. Noticeably, this fabrication strategy provides sufficient flexibility to fabricate multistage vectors62 that can be directly translated to large scale production within the framework of established good manufacturing practice semiconductor processes.
Functionalization of porous silicon particles with perfluorosilane
The as-made porous silicon particles on the wafer were rinsed with ethanol, dried with N2, and subsequently immersed in a piranha solution (H2SO4/H2O2, v/v = 2:1, caution: hot and corrosive) for 10 min to oxidize the silicon surface to silanol. After cooling down to room temperature, the array was rinsed with copious Milli-Q water and dried under reduced pressure. The oxidized array was then immersed in 1 mL of 1 mM 1H,1H,2H,2H-perfluorododecyltrimethoxysilane (FAS17) in dichloromethane and was incubated statically for 2 hours at room temperature. Afterwards, the array was rinsed with ethanol and Milli-Q water successively, and dried with N2. Finally, the array was placed in a 25 mL Schlenk flask and annealed in a N2 atmosphere at 100 °C for 1 hour. The resultant fluorous particles were denoted as F-PSP.
Loading of perfluorocarbon (PFC) and fluoro-compounds
Controlled release of FITC-C8F15
The FITC-C8F15 loaded fluorous porous silicon particles on the 5 × 5 mm2 array were immersed in a 5 mL vial with 0.01 M PBS solution (1 mL). Low frequency ultrasound (LFUS) was applied to the sample for 5 seconds by using an ultrasound bath (Branson model 1510) operating at a frequency of 40 kHz. After that, the array was withdrawn from the solution and dried with N2. The concentration of the released FITC-C8F15 in the solution was quantitated against a calibration curve of FITC-C8F15 in the range of 0–50 μM (Fig. S1, ESI†), through the measurement of FITC fluorescence intensity at λex/λem 495/535 nm using a microplate reader (Tecan Spark®). The wafer was also examined by reflected bright field and fluorescence microscopy imaging with a Nikon eclipse 80i microscope at the excitation and emission wavelengths of 494/517 nm for FITC-C8F15. All images were processed using NIS-Elements AR3.0 software (Nikon).Biofilm inhibition assay
Evaluation of cytotoxicity
The in vitro cytotoxicity was evaluated using a cell counting kit-8 (CCK-8). Briefly, HeLa cells were seeded at a concentration of 1 × 104 cells per well in a 96-well plate containing DMEM (100 μL) that was supplied with 10% FBS, streptomycin (100 μg mL−1), and penicillin (100 μg mL−1). The culture medium was then replaced with fresh medium supplemented with C5F12 loaded fluorous particles (C5F12-F-PSP, 1 mg mL−1), fluorous particles (F-PSP, 1 mg mL−1), and unmodified particles (1 mg mL−1) after culturing for 24 hours (37 °C, 5% CO2) and was continued to incubate for additional 24–48 h. The untreated cells served as the negative control. At the selected time point, the medium was replaced by CCK-8 solution and statically incubated at 37 °C for 1 hour. The absorbance at 450 nm was read using a microplate reader. The percentage of cell viability was calculated by normalizing the absorbance of the treated samples with that of the untreated sample.Results and discussion
In this study, we investigated ultrasound triggered release by utilizing discoidal porous silicon particles (PSP) as the biocompatible carriers. The particle surface was modified with perfluorosilane molecules to allow the incorporation of perfluorocarbon (PFC) and fluorous-tagged compounds into the pores through fluorine–fluorine interactions.63,64 As illustrated in Fig. 1A, the discoidal particles deposited on a silicon wafer were functionalized with 1H,1H,2H,2H-perfluorodecyltri-methoxysilane (FAS17) to generate a fluorinated surface.65 The amplified area represents a pore containing PFC and fluorous-tagged compounds. Remarkably, loading of PFC and fluorous-tagged compounds is facilitated by fluorine–fluorine interactions, and after evaporation of PFC under LFUS, the fluorous-tagged compounds released rapidly from the pores.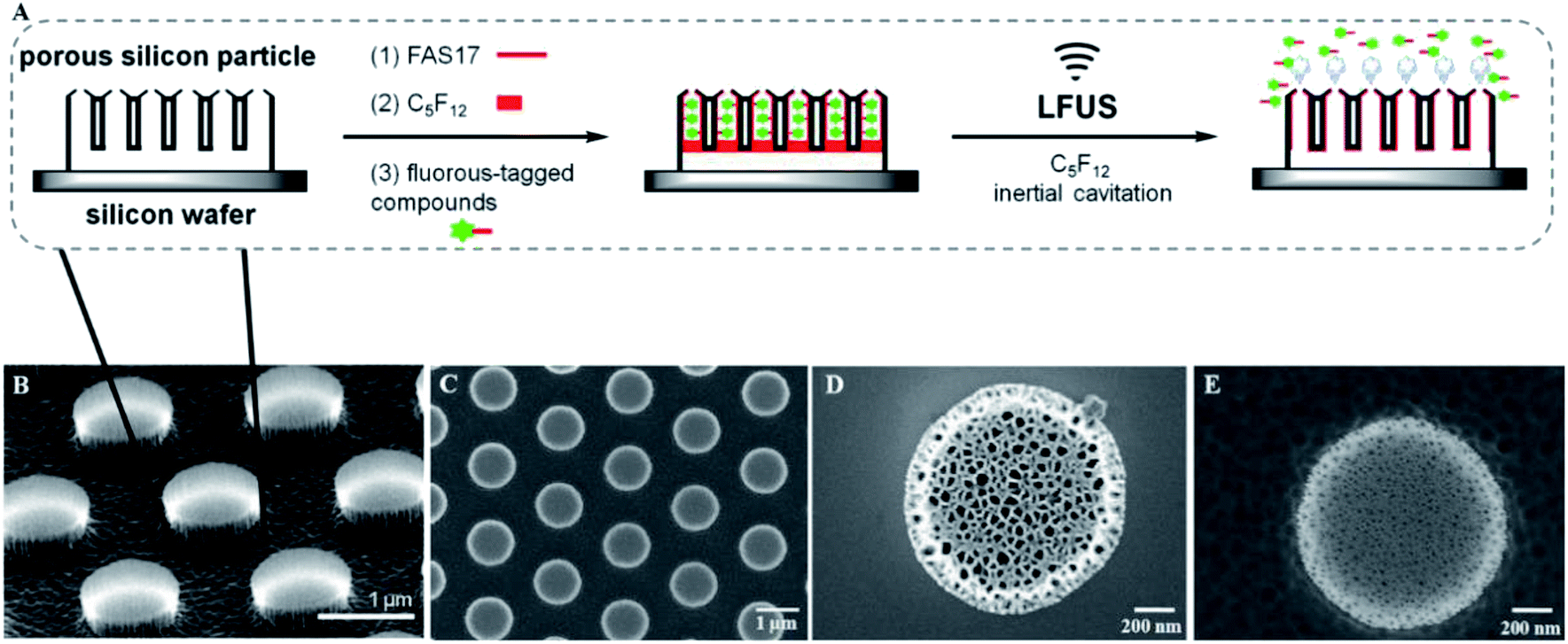 | ||
| Fig. 1 (A) Schematic illustration of the general procedure for the low frequency ultrasound triggered release of fluorous-tagged compounds. The tilted view (B) and top view (C) of the SEM images of the discoidal porous silicon particles deposited on a silicon wafer. SEM images of the porous morphology of the release layer (D) and the device layer (E). | ||
Characterization of discoidal porous silicon particles
The discoidal particles were fabricated by the combination of photolithography and controlled electrochemical etching.50Fig. 1B shows the formation of the ordered discoidal particle array on a silicon wafer. The average particle size was determined as 1.11 ± 0.01 μm with a thickness of 320 ± 17 nm and a constant distance between particles of 0.75 ± 0.02 μm (Fig. 1C). The particles consisted of two layers of different pore sizes. The pore sizes of the top layer – release layer (Fig. 1D) and the bottom layer – device layer (Fig. 1E) were determined as 75.4 ± 9.3 nm and 13.5 ± 3.5 nm, respectively. Noticeably, this bilayer structure is anticipated to form internal cavities capable of efficient encapsulation and reduce the diffusion of the payload.Prior to the fluorination, a systematic optimization of perfluorosilane concentration in the range of 0.1 mM to 10 mM indicated that 1 mM was optimal to generate a high density fluorine film on the particles (Fig. S3, ESI†). Successively, the fluorous particles were characterized by SEM, TEM, EDS, FTIR, and XPS. Both TEM (Fig. 2A–C) and SEM (Fig. 2D) analyses confirmed the preservation of the bilayer porous morphology in the particles after fluorination, with the pore sizes of the release layer (Fig. 2A and S4A, ESI†) and the device layer (Fig. 2B and S4B, ESI†) being 57.5 ± 15.2 nm and 14.4 ± 5.6 nm. The tilt view (Fig. 2C) of the fluorous particles shows dense cavities inside the particles. The EDS analysis revealed the presence of fluorine groups within the porous structure, together with silicon, oxygen, and carbon as the particle compositional elements (Fig. 2E). The detected copper was assumed to be from the copper grid.
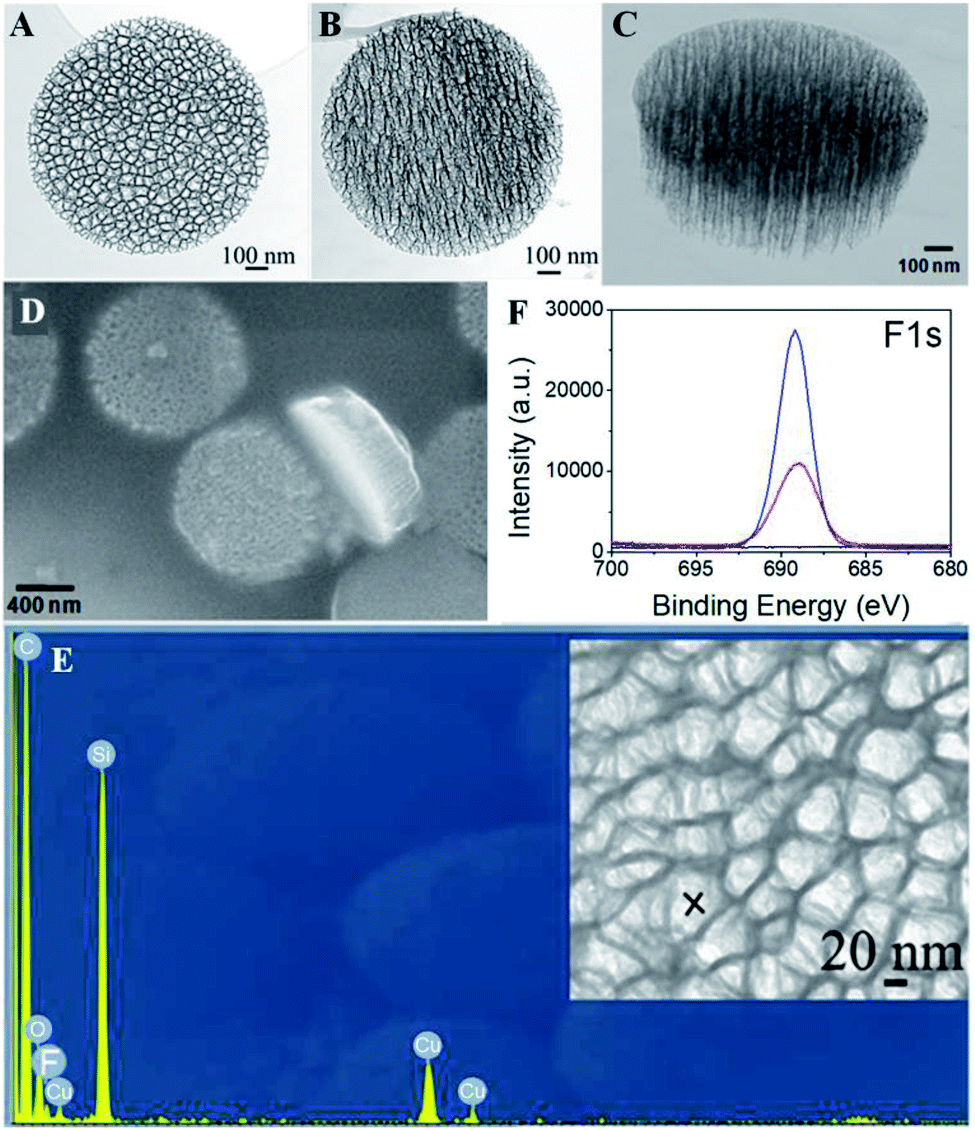 | ||
| Fig. 2 TEM images of the release layer (A) and device layer (B), and tilt view (C) and SEM image (D) of the fluorous discoidal porous silicon particles. (E) EDS spectrum of the area marked with an “X” on the fluorous porous silicon particles. (F) High-resolution F1s scans of the unmodified porous silicon particles (black line), fluorous porous silicon particles (red line), and FITC-C8F15 incorporated fluorous porous silicon particles (blue line). | ||
Further, the fluorine film density on the particles was determined by XPS analysis by using a self-assembled monolayer (SAM) of CF3(CH2)13SH on Au(111) as a reference.66 The F1s peak at 688 eV in the high resolution XPS indicated the presence of fluorine groups in the fluorous particles (Fig. 2F, red line) and the absence of the F1s signal in the unmodified particles (Fig. 2F, black line). The F1s peak areas of the fluorous particles and the CF3-terminated SAM surface were AF-PSP = 16717 ± 2459 and AF-SAM = 7577 ± 286. The numbers of fluorine atoms on the FAS17 and the CF3(CH2)13SH are NF-PSP = 17 and NF-SAM = 3, respectively. Considering the known density of the SAM on Au(111) as a standard (DF-SAM = 4.6 × 1014 molecules per cm2),66 the fluorine density on the particles can be calculated as (1.8 ± 0.1) × 1013 molecules per cm2, and the perfluorosilane coverage is approximately 39%.
The FTIR spectra (Fig. 3A) of the pure FAS17 molecules (red line) and the fluorous particles (black line) were collected, and the FTIR spectra (Fig. 3B) of the fluorous particles (red line) and the unmodified particles (black line) were also recorded. In the fluorous particle spectrum (Fig. 3A), the peak at ∼1154 cm−1 is assigned to the asymmetric stretching of –CF2–, and the peaks at 1248 and 1231 cm−1 are attributed to the symmetric stretching of –CF2– and –CF3. The peaks at ∼2884 and 2927 cm−1 are attributed to the symmetric and asymmetric stretching of –CH2–, respectively. The peak at ∼1350 cm−1 is attributed to immobilized C–F groups.48,67 The peaks at ∼1360 cm−1 and ∼1079 cm−1 are the stretching of Si–O–Si and Si–O groups.68
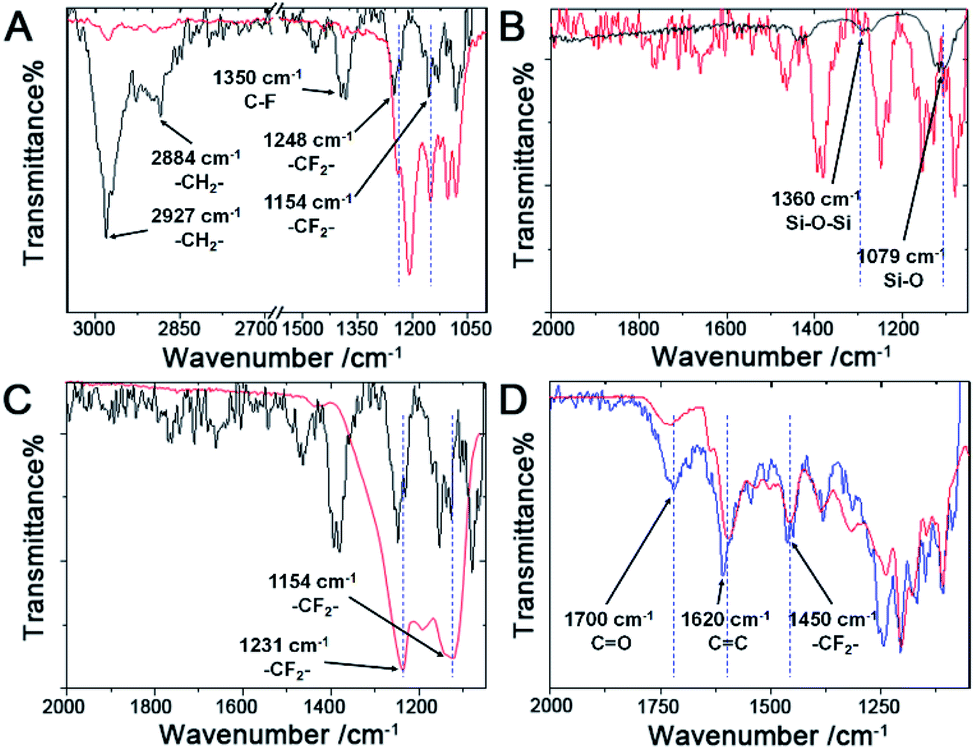 | ||
| Fig. 3 (A) FTIR spectra of the fluorous porous silicon particles (black line) and pure FAS17 molecules (red line). (B) FTIR spectra of the unmodified porous silicon particles (black line) and the fluorous porous silicon particles (red line). (C) FTIR spectra of the fluorous porous silicon particles (black line) and the C5F12 incorporated fluorous porous silicon particles (red line). (D) FTIR spectra of the fluorous porous silicon particles after incorporation of FITC-C8F15 (blue line) and the pure FITC-C8F15 molecules (red line). | ||
Loading of perfluorocarbon (PFC)
Fluorous chemistry is dependent on unique fluorine–fluorine interactions with which fluorocarbons are attracted to each other, much more than to other media.15 On this basis, fluorous interactions can be applied for loading of PFC and cargoes with a fluorous tag.The boiling points of C4F10 (PFB), C5F12 (PFP), and C6F14 (PFH) are −1.7 °C, 28 °C, and 59 °C, but they remain as superheated liquids in the porous structure due to the Laplace pressure. To investigate the loading efficiency of PFC in the fluorous particles, FTIR was employed. In Fig. 3C, two signature peaks at ∼1154 and 1231 cm−1, attributed to the asymmetric and symmetric stretching of –CF2–, appeared after the loading of PFC. The higher intensity peak at 1231 cm−1 was utilized to monitor the PFC loading in the fluorous particles. The release profiles at room temperature (Fig. 4) were plotted using absorbance at 1231 cm−1vs. time and were fitted using the first-order release equation 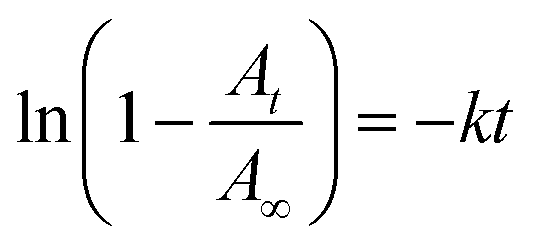 . As a result, the release rate constant (k) followed the order: k (C4F10) ≫ k (C5F12) ≈ k (C6F14), which was consistent with the corresponding boiling point of each PFC. In order to achieve both a steady release profile and a rapid explosion effect, C5F12 was therefore chosen as a model PFC in the following experiments.
. As a result, the release rate constant (k) followed the order: k (C4F10) ≫ k (C5F12) ≈ k (C6F14), which was consistent with the corresponding boiling point of each PFC. In order to achieve both a steady release profile and a rapid explosion effect, C5F12 was therefore chosen as a model PFC in the following experiments.
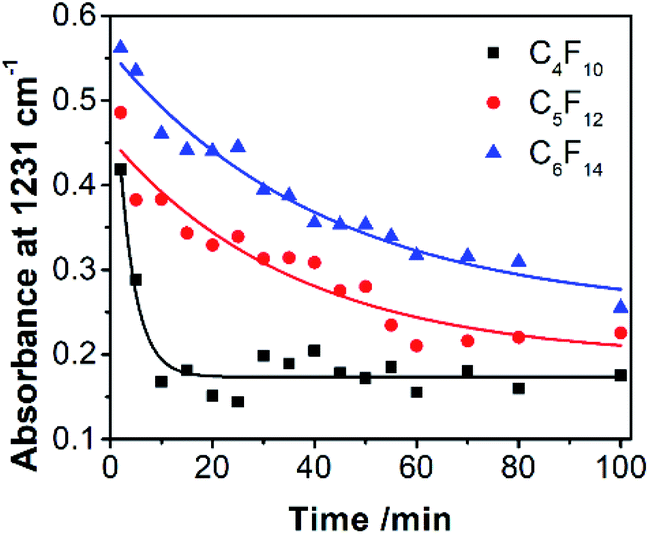 | ||
Fig. 4 The release profiles of PFC at room temperature were plotted using absorbance at 1231 cm−1vs. time and were fitted using the first-order release equation 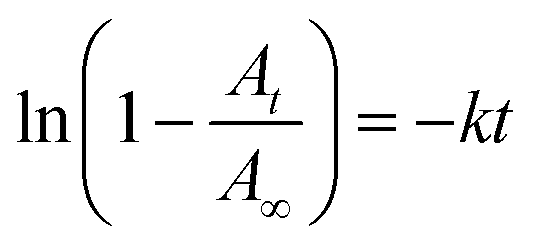 , where A is the absorbance of PFC at 1231 cm−1, and t is the release time. The release rate constants (k) are 0.3092 (C4F10), 0.02753 (C5F12), and 0.02372 (C6F14). The correlation coefficient (r2) is 0.9575 (C4F10), 0.8897 (C5F12), and 0.9148 (C6F14), respectively. , where A is the absorbance of PFC at 1231 cm−1, and t is the release time. The release rate constants (k) are 0.3092 (C4F10), 0.02753 (C5F12), and 0.02372 (C6F14). The correlation coefficient (r2) is 0.9575 (C4F10), 0.8897 (C5F12), and 0.9148 (C6F14), respectively. | ||
Loading of fluorous-tagged compounds
A fluorinated fluorescein isothiocyanate (FITC) derivative was exploited as a model compound in this study. Fluorinated FITC-C8F15 molecules were incorporated into the fluorous particles as indicated in the Experimental section. The successful incorporation of fluorinated FITC molecules in the fluorous particles was demonstrated by XPS and FTIR. In the XPS spectra (Fig. 2F), an increase in the F1s peak intensity of the FITC-C8F15 incorporated fluorous particles (blue line) in comparison with the fluorous particles (red line) and unmodified particles (black line) indicated the presence of FITC-C8F15 in the fluorous particles. In the FTIR spectra (Fig. 3D), the feature peaks of the fluorinated FITC molecules (red line) at 1450 cm−1, 1620 cm−1, and 1700 cm−1 appeared for the FITC-C8F15 incorporated fluorous particles (blue line).The incorporation of cargoes was also visualized by fluorescence microscopy. A monodisperse array of particles on the silicon wafer was observed in the reflected bright-field image (Fig. 5A). After treatment with FITC-C8F15, each particle exhibited green fluorescence as shown in the fluorescence microscopy image (Fig. 5B), indicating the presence of FITC-C8F15 in the fluorous particles. Moreover, the surface plot image at three different angles shown in Fig. S5 (ESI†) confirmed the presence of FITC-C8F15 in each fluorous particle. A control experiment was carried out by using a monodisperse array of unmodified particles after the same treatment with FITC-C8F15 (Fig. 5C). In contrast, an overwhelming absence of fluorescence (Fig. 5D) implied the failure of loading of fluorinated molecules into the particle pores. This result validated the relevance of perfluorosilane functionalization for a successful incorporation of fluorinated molecules into the fluorous particles.
 | ||
| Fig. 5 Reflected bright-field (A) and fluorescence microscopy images (B) for the fluorous porous silicon particles deposited on a silicon wafer after treatment with FITC-C8F15. Reflected bright-field (C) and fluorescence microscopy images (D) for the unmodified porous silicon particles deposited on a silicon wafer after treatment with FITC-C8F15. | ||
Low frequency ultrasound triggered controlled release of fluorous-tagged compounds
The primary goal of the fabrication of this ultrasound triggered structure is to apply it as a delivery system for the controlled release of therapeutic molecules in biomedical applications. The low frequency ultrasound (10–60 kHz) is known to enhance the permeability of biological membranes and improve the release efficacy of therapeutic agents.44 The ultrasonic power density was determined calorimetrically as described by Hill et al.69 In this study, the application of fabricated fluorous particles with LFUS triggered controlled release was explored. The release experiments were performed with the fluorous particles on a silicon wafer aiming to establish a convenient observation by optical microscopy. In order to simulate physiological conditions, the array was immersed in a vial containing 0.01 M phosphate buffered saline (PBS), and sonicated for 5 seconds in an ultrasonic bath, operating at 40 kHz at room temperature.Before applying LFUS, each particle incorporated with fluorescent molecules displayed a green dot on the surface (Fig. 6A). After applying LFUS, the fluorescent molecules were ejected out of the particles and two different regions were observed in the microscopy images. In region (I), the FITC molecules were released simultaneously from the porous particles and the silicon wafer surface (Fig. 6B), and transferred to the PBS solution. In region (II), the FITC molecules were released selectively from the porous particles but remained on the silicon wafer surface (Fig. 6C). Fig. 6D presents a mixed region of (I) and (II). The presence of FITC molecules on the silicon wafer accounted for the multilayer structure of the pores. The mean fluorescence intensity (MFI) was measured as 11106 before sonication and was decreased to 1476 in region (I) and 5666 in region (II) after sonication. The MFI of the mixed regions of (I) and (II) was 2411. The inertial cavitation during ultrasound produced the release of FITC-C8F15 molecules through the release layers. The result suggests that a multi-level controlled release of cargoes can be tuned by adjusting the sonication time.
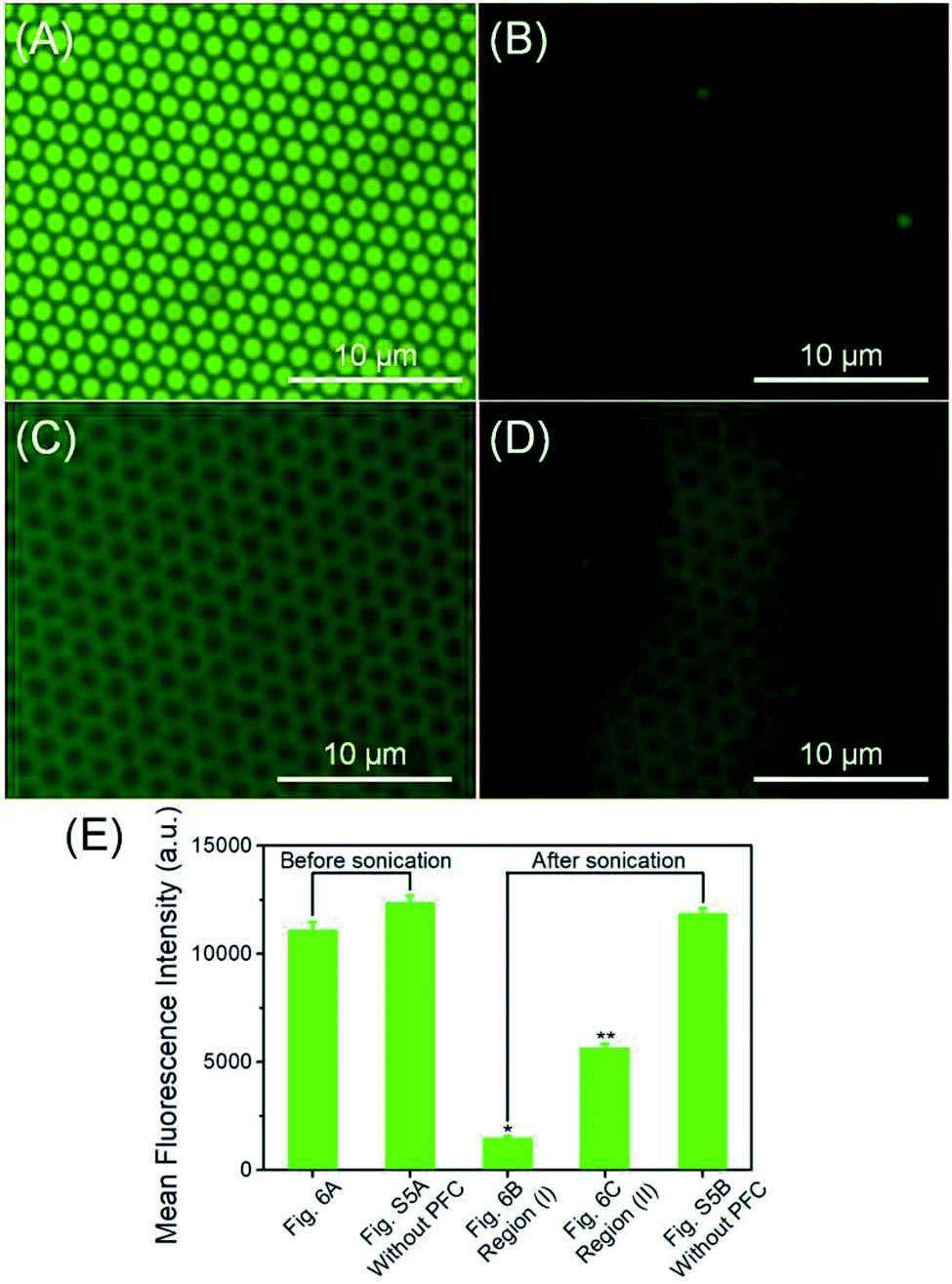 | ||
| Fig. 6 Fluorescence microscopy images of the fluorinated FITC loaded fluorous particles deposited on a silicon wafer before (A) and after (B), (C), and (D) after 5 seconds of 40 kHz ultrasound application (note: the acquisition time was 200 ms in all the images). The mean fluorescence intensity (MFI) was extracted from these images (A = 642 μm2). (E) Comparison of the MFI before and after applying LFUS. The control groups are the fluorinated FITC loaded fluorous particles deposited on a silicon wafer without C5F12 incorporation. *P < 0.001 and **P < 0.001 from the MFI extracted from (A). | ||
In addition, the fluorinated FITC released into the PBS solution was analyzed using a fluorescence spectrometer to estimate the fluorinated FITC concentration. As a precaution to avoid artifacts from the detached particles, the solution was centrifuged at 12000 rpm for 40 min prior to the measurement. The fluorescence intensity of the supernatant was measured as 42546, corresponding to 48 μM fluorinated FITC in 400 μL, indicating that all the FITC molecules originated from the sonication application.
Finally, the relevance of the treatment with PFC was confirmed by a control experiment developed by sonication of the fluorous particles incorporated only with fluorinated FITC molecules. The fluorescence microscopy images of the fluorous particles on a silicon wafer before (Fig. S6A, ESI†) and after (Fig. S6B, ESI†) applying LFUS without C5F12 incorporation are presented. As expected, the MFI was 12353 and 11878, demonstrating that the controlled release of the fluorinated FITC molecules was produced as a consequence of rapid PFC evaporation during ultrasound application (Fig. 6E).
Application for biofilm inhibition
The same methodology was used to design an antibiotic controlled release system on the fluorous particles, aiming to prevent pathogenic biofilm formation. Ciprofloxacin (Cip) is a well-known fluorine containing antibiotic and was recently reported as an active antibiotic to modify contact lenses against the notorious “superbug” pathogenic Pseudomonas aeruginosa strain (PAO1) via fluorine chemistry.17 A genetically modified PAO1-GFP strain to express the green fluorescent protein was used in this study. The PAO1-GFP suspension was inoculated into a 48-well plate containing Cip and C5F12 incorporated fluorous particles. The load rate of Cip on the fluorous particles was determined to be 58%. The control groups contained the fluorous particles without Cip or LFUS application, or bare LFUS application. The bacterial culture without any treatment was the negative control. As shown in Fig. 7A, no biofilm was formed on the Cip loaded fluorous particles subjected to LFUS. In contrast, densely packed biofilms were observed on the control groups, including the bare LFUS application (Fig. 7B), the fluorous particles without Cip (Fig. 7C), the Cip loaded fluorous particles without LFUS application (Fig. 7D), and the negative control groups (Fig. 7E). Further, the viable bacterial cells in these biofilms were counted (Fig. 7F). The sharp reduction by >5 orders of magnitude in the bacterial cell number in the experiment group compared to that of control groups demonstrated the effectiveness of this delivery system.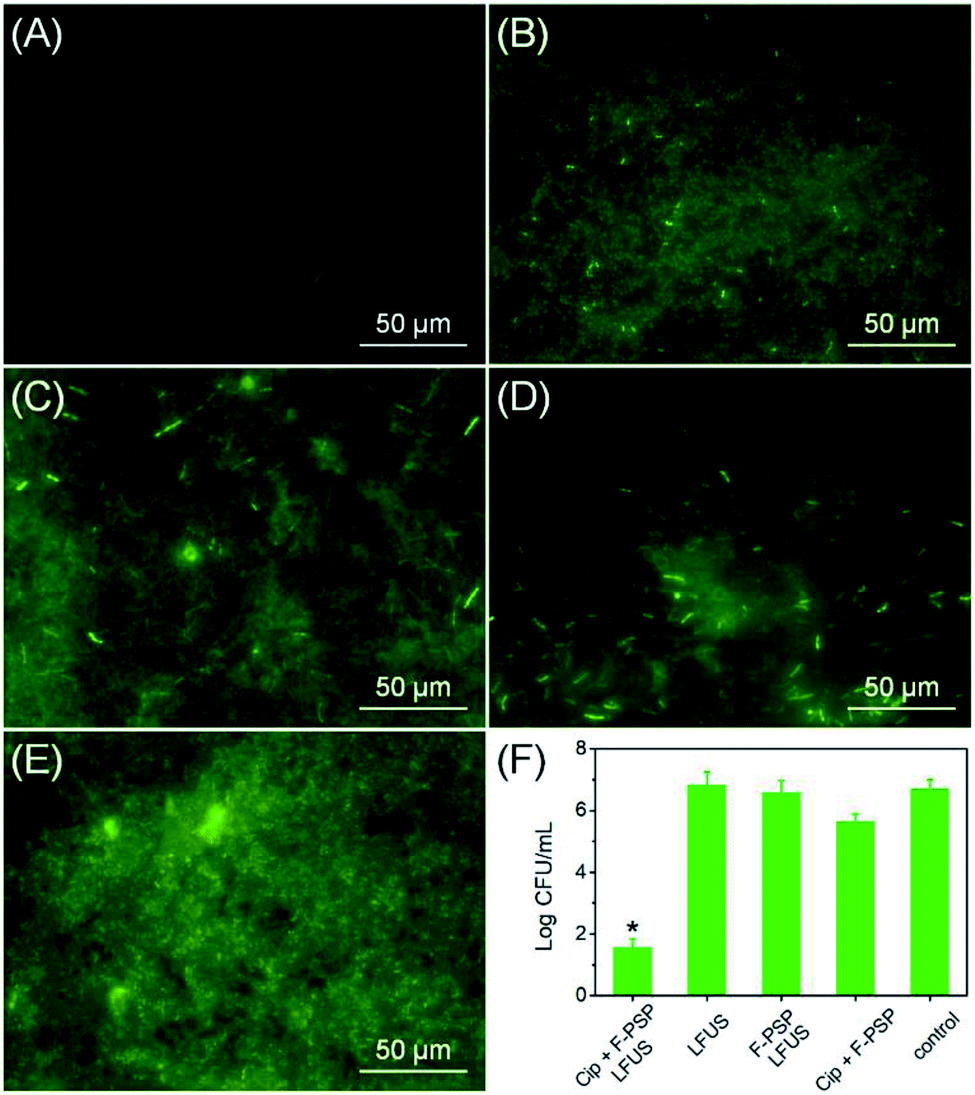 | ||
| Fig. 7 (A) Representative fluorescence image of GFP-transformed Pseudomonas aeruginosa (PAO1-GFP) cultured in a 48-well plate with Cip and C5F12 incorporated fluorous particles after LFUS application for 24 h at 37 °C. The representative fluorescence images of the biofilms of PAO1-GFP cultured in a 48-well plate without the Cip loaded fluorous particles (B), Cip (C) and LFUS (D) treatment, and the negative control (E) after 24 h incubation at 37 °C. (F) Viable bacterial CFU counts of PAO1-GFP remaining on the bottom of the 48-well plate. *P < 0.001 from data obtained for the control. | ||
Cytotoxicity
The cytotoxicity of the C5F12 loaded fluorous particles (C5F12-F-PSP, 1 mg mL−1), fluorous particles (F-PSP, 1 mg mL−1) and unmodified particles (1 mg mL−1) was evaluated using Hela cells as a model. The in vitro cell viability was determined using a commercial cell counting kit-8 (CCK-8). No apparent impact on cell viability was observed up to 1 mg mL−1 concentration of all these particles within 48 h of incubation, which is similar to that of the control group (Fig. 8). The results suggested that the C5F12 loaded fluorous particles were biosafe to mammalian cells.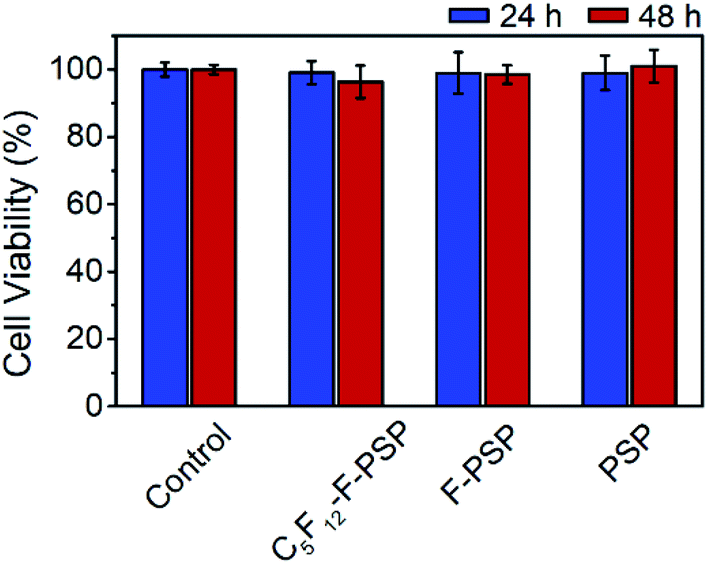 | ||
| Fig. 8 Hela cell viability evaluated by CCK-8 assay after treatment with the indicated conditions for 24 to 48 h. Each value represents the relative viability compared to the control group. Each data point represents an average of five replicates and the error bars represent one standard deviation from the mean. | ||
Conclusions
In conclusion, we have constructed a biphase fluorous drug delivery system (FB-DDS) triggered by low frequency ultrasound (LFUS). The discoidal porous silicon particles were synthesized by a combined procedure of photolithography and controlled electrochemical HF etching, and then were functionalized with perfluorosilane (FAS17). This delivery system was demonstrated by utilizing model compounds including a fluorous-tagged fluorescein and a fluorine containing antibiotic ciprofloxacin. Loading of the model molecules into fluorocarbon-coated carriers was facilitated by fluorous interactions, whereas ejection of the model molecules was promoted by applying LFUS to rapidly evaporate perfluoropentane. In the in vitro test, these carriers loaded with fluorine containing ciprofloxacin exhibited excellent antimicrobial activity against Pseudomonas aeruginosa biofilm formation. Overall, this innovative stimulus-responsive fluorous biphase drug delivery system will be a promising candidate for practical applications as well as encouraging further investigation of drug delivery and controlled release strategies.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 31800800) and Natural Science Foundation of Shandong Province, China (Grant No. ZR2019BC101).Notes and references
- M. M. Rubiana and P. S. Luciano, Curr. Drug Targets, 2004, 5, 449–455 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef.
- S. T. Gao, G. S. Tang, D. W. Hua, R. H. Xiong, J. Q. Han, S. H. Jiang, Q. L. Zhang and C. B. Huang, J. Mater. Chem. B, 2019, 7, 709–729 RSC.
- L. Wang, M. Zheng and Z. G. Xie, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17 Search PubMed.
- H. Wang, Q. W. Chen and S. Q. Zhou, Chem. Soc. Rev., 2018, 47, 4198–4232 RSC.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Y. Barenholz, J. Control. Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Deliv. Rev., 2008, 60, 876–885 CrossRef CAS PubMed.
- I. T. Horvath and J. Rabai, Science, 1994, 266, 72–75 CrossRef CAS.
- I. T. Horváth, Acc. Chem. Res., 1998, 31, 641–650 CrossRef.
- B. Barabás, O. Fülöp, R. Molontay and G. Pályi, ACS Sustain. Chem. Eng., 2017, 5, 8108–8118 CrossRef.
- F. A. Jaipuri, B. Y. M. Collet and N. L. Pohl, Angew. Chem., Int. Ed., 2008, 47, 1707–1710 CrossRef CAS PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- C. M. Santos, A. Kumar, S. S. Kolar, R. Contreras-Caceres, A. McDermott and C. Cai, ACS Appl. Mater. Interfaces, 2013, 5, 12789–12793 CrossRef CAS.
- G. T. Qin, Z. L. Zhu, S. H. Li, A. M. McDermott and C. Z. Cai, Biomaterials, 2017, 124, 55–64 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- O. C. Farokhzad and L. Robert, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- R. R. Costa, C. A. Custódio, F. J. Arias, J. C. Rodríguez-Cabello and J. F. Mano, Nanomedicine, 2013, 9, 895–902 CrossRef CAS.
- X. Wu, Z. Wang, D. Zhu, S. Zong, L. Yang, Y. Zhong and Y. Cui, ACS Appl. Mater. Interfaces, 2013, 5, 10895–10903 CrossRef CAS PubMed.
- J. Cui, Y. Yan, Y. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- A. A. McBride, D. N. Price, L. R. Lamoureux, A. A. Elmaoued, J. M. Vargas, N. L. Adolphi and P. Muttil, Mol. Pharm., 2013, 10, 3574–3581 CrossRef CAS PubMed.
- S.-H. Hu, T.-Y. Liu, H.-Y. Huang, D.-M. Liu and S.-Y. Chen, Langmuir, 2008, 24, 239–244 CrossRef CAS PubMed.
- A. Schroeder, M. S. Goldberg, C. Kastrup, Y. Wang, S. Jiang, B. J. Joseph, C. G. Levins, S. T. Kannan, R. Langer and D. G. Anderson, Nano Lett., 2012, 12, 2685–2689 CrossRef CAS PubMed.
- V. Frenkel, Adv. Drug Deliv. Rev., 2008, 60, 1193–1208 CrossRef CAS PubMed.
- R. Deckers and C. T. W. Moonen, J. Contr. Release, 2010, 148, 25–33 CrossRef CAS PubMed.
- T. G. Leighton, Prog. Biophys. Mol. Biol., 2007, 93, 3–83 CrossRef PubMed.
- S. L. Huang, Adv. Drug Deliv. Rev., 2008, 60, 1167–1176 CrossRef CAS PubMed.
- J. Tang, R. F. Liu and Z. F. Dai, Prog. Chem., 2018, 30, 1669–1680 Search PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. H. Zhao, J. Kim, C. A. Cezar, Z. G. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS.
- M. L. Fabiilli, C. G. Wilson, F. Padilla, F. M. Martin-Saavedra, J. B. Fowlkes and R. T. Franceschi, Acta Biomater., 2013, 9, 7399–7409 CrossRef CAS PubMed.
- M. L. Fabiilli, J. A. Lee, O. D. Kripfgans, P. L. Carson and J. B. Fowlkes, Pharm. Res., 2010, 27, 2753–2765 CrossRef CAS PubMed.
- W. J. Duncanson, L. R. Arriaga, W. L. Ung, J. A. Kopechek, T. M. Porter and D. A. Weitz, Langmuir, 2014, 30, 13765–13770 CrossRef CAS PubMed.
- K. Stark, J. P. Hitchcock, A. Fiaz, A. L. White, E. A. Baxter, S. Biggs, J. R. McLaughlan, S. Freear and O. J. Cayre, ACS Appl. Mater. Interfaces, 2019, 11, 12272–12282 CrossRef CAS PubMed.
- D. Kagan, M. J. Benchimol, J. C. Claussen, E. Chuluun-Erdene, S. Esener and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 7519–7522 CrossRef CAS PubMed.
- S.-T. Kang and C.-K. Yeh, Langmuir, 2011, 27, 13183–13188 CrossRef CAS PubMed.
- N. Y. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K.-H. Nam, J. Control. Release, 2009, 138, 268–276 CrossRef CAS PubMed.
- M. Javadi, W. G. Pitt, C. M. Tracy, J. R. Barrow, B. M. Willardson, J. M. Hartley and N. H. Tsosie, J. Control. Release, 2013, 167, 92–100 CrossRef CAS PubMed.
- A. Schroeder, R. Honen, K. Turjeman, A. Gabizon, J. Kost and Y. Barenholz, J. Control. Release, 2009, 137, 63–68 CrossRef CAS PubMed.
- J. L. Coffer, M. A. Whitehead, D. K. Nagesha, P. Mukherjee, G. Akkaraju, M. Totolici, R. S. Saffie and L. T. Canham, Phys. Status Solidi A, 2005, 202, 1451–1455 CrossRef CAS.
- S. H. C. Anderson, H. Elliott, D. J. Wallis, L. T. Canham and J. J. Powell, Phys. Status Solidi A, 2003, 197, 331–335 CrossRef CAS.
- A. P. Mann, T. Tanaka, A. Somasunderam, X. Liu, D. G. Gorenstein and M. Ferrari, Adv. Mater., 2011, 23, H278–H282 CrossRef CAS PubMed.
- G. Osei-Prempeh, H.-J. Lehmler, S. E. Rankin and B. L. Knutson, Ind. Eng. Chem. Res., 2011, 50, 5510–5522 CrossRef CAS.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V.-P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS PubMed.
- B. Godin, C. Chiappini, S. Srinivasan, J. F. Alexander, K. Yokoi, M. Ferrari, P. Decuzzi and X. Liu, Adv. Funct. Mater., 2012, 22, 4225–4235 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- J. Zhu, Y. Niu, Y. Li, Y. Gong, H. Shi, Q. Huo, Y. Liu and Q. Xu, J. Mater. Chem. B, 2017, 5, 1339–1352 RSC.
- N. Reznik, O. Shpak, E. C. Gelderblom, R. Williams, N. de Jong, M. Versluis and P. N. Burns, Ultrasonics, 2013, 53, 1368–1376 CrossRef CAS PubMed.
- R. Singh, G. A. Husseini and W. G. Pitt, Ultrason. Sonochem., 2012, 19, 1120–1125 CrossRef CAS PubMed.
- O. D. Kripfgans, J. B. Fowlkes, D. L. Miller, O. P. Eldevik and P. L. Carson, Ultrasound Med. Biol., 2000, 26, 1177–1189 CrossRef CAS PubMed.
- I. Gorelikov, A. L. Martin, M. Seo and N. Matsuura, Langmuir, 2011, 27, 15024–15033 CrossRef CAS PubMed.
- O. Couture, P. D. Bevan, E. Cherin, K. Cheung, P. N. Burns and F. S. Foster, Ultrasound Med. Biol., 2006, 32, 73–82 CrossRef PubMed.
- M. M. Kaneda, S. Caruthers, G. M. Lanza and S. A. Wickline, Ann. Biomed. Eng., 2009, 37, 1922–1933 CrossRef PubMed.
- J. W. Mo, J. J. Sha, D. K. Li, Z. G. Li and Y. F. Chen, Nanoscale, 2019, 11, 8408–8415 RSC.
- L. Wu, Z. C. Dong, F. Y. Li and Y. L. Song, Langmuir, 2018, 34, 639–645 CrossRef CAS PubMed.
- N. Y. Rapoport, A. L. Efros, D. A. Christensen, A. M. Kennedy and K. H. Nam, Bubble Sci. Eng. Technol., 2009, 1, 31–39 CrossRef CAS PubMed.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M.-C. Cheng, P. Decuzzi, J. M. Tour, F. Robertson and M. Ferrari, Nat. Nanotechnol., 2008, 3, 151–157 CrossRef CAS PubMed.
- C. Hirayama, H. Ihara, S. Nagaoka and T. Wada, Polym. J., 1994, 26, 499–503 CrossRef CAS.
- R. J. Baker, P. E. Colavita, D. M. Murphy, J. A. Platts and J. D. Wallis, J. Phys. Chem. A, 2012, 116, 1435–1444 CrossRef CAS PubMed.
- S. Suzuki, A. Nakajima, N. Yoshida, M. Sakai, A. Hashimoto, Y. Kameshima and K. Okada, Langmuir, 2007, 23, 8674–8677 CrossRef CAS PubMed.
- S. Li, P. Cao, R. Colorado, X. Yan, I. Wenzl, O. E. Shmakova, M. Graupe, T. R. Lee and S. S. Perry, Langmuir, 2005, 21, 933–936 CrossRef CAS PubMed.
- J.-D. Brassard, D. K. Sarkar and J. Perron, ACS Appl. Mater. Interfaces, 2011, 3, 3583–3588 CrossRef CAS PubMed.
- D. B. Mawhinney, J. A. Glass and J. T. Yates, J. Phys. Chem. B, 1997, 101, 1202–1206 CrossRef CAS.
- C. R. Hill, J. C. Bamber and G. R. ter Haar, Detection and Measurement of Acoustic Fields, Physical Principles of Medical Ultrasonics, John Wiley & Sons, 2004, pp. 69–91 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of fluorous-tagged compounds, bacterial cell culture, plate counting assay, particle characterization, 1H, 13C, and 19F NMR spectra of perfluorooctanyl fluorescein isothiocyanate (FITC-C8F15), the plot of fluorescence intensity vs. concentration of FITC-C8F15, the surface plot images at three different angles for the fluorous particles, fluorescence microscopy images of the fluorinated FITC molecules incorporated in fluorous particles. See DOI: 10.1039/d0na00324g |
| This journal is © The Royal Society of Chemistry 2020 |