
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile green synthesis of silicon nanoparticles from Equisetum arvense for fluorescence based detection of Fe(III) ions†
T. V. S.
Adinarayana
 a,
Ayushi
Mishra
b,
Ishu
Singhal
*b and
D. V. Rama
Koti Reddy
a
a,
Ayushi
Mishra
b,
Ishu
Singhal
*b and
D. V. Rama
Koti Reddy
a
aDepartment of Instrument Technology, Andhra University College of Engineering, Andhra University, Andhra Pradesh, India
bSchool of Biotechnology, Jawaharlal Nehru University, New Delhi, India. E-mail: ishu_singhal@yahoo.com
First published on 4th August 2020
Abstract
Fluorescent silicon nanoparticles (SiNPs) might be one of the excellent candidates for use as optical markers in biological profiling and diagnostic applications. To exploit this perspective, they ought to be essentially synthesized from any green precursor rich in silicon. Stable dispersibility in water along with prolonged luminescence under different conditions is also desired. Moreover, one of the main challenges is to produce such optically (photoluminescence) stable and water-dispersible SiNPs. In our present work, we have reported the synthesis of a highly stable silicon nanoparticle aqueous suspension via a single-step microwave-assisted facile green route. Our as-prepared SiNPs exhibit inherent stable dispersibility, strong fluorescence, and photo-stable behavior. The experimental results demonstrate that the synthesized SiNPs are highly suitable for the detection of Fe(III) ions. This optical sensing study opens a new avenue for use of SiNPs as a valuable optical probe in chemosensory applications. Our results provide a single-step methodology for the synthesis of highly stable SiNPs from a biological precursor, which can be used as a promising tool for various chemical and biological applications.
1. Introduction
Light sensitive nanoparticles have garnered great attention owing to their unique optical properties.1 When compared to organic fluorophores, which are currently being used in extensive applications, they are robust, have broader absorption cross-sections and their emission wavelength can be tuned with their size, and they also offer optical and chemical advantages.2 They have been mostly studied for biomedical applications. The limiting factor that may hinder their use in biology and medicine is the toxicity associated with heavy metal nanoparticles such as cadmium, selenium, etc.3 Silicon (Si), being non-toxic and abundant, eradicates most of these shortcomings found in direct bandgap materials and provides a far cheaper approach.4 Majorly the most important drawback with indirect bandgap semiconductors is the low probability of radiative rate, however, this shortcoming is partially overcome in SiNPs as they have spatial confinement of charge carriers.5 Even though silicon nanoparticles are expected to be far less toxic than the existing semiconductor NPs, they have not been used broadly in practice yet because of the inherent challenges encountered in making them water-soluble and biologically compatible.6 Thus, synthesis of optically stable and water-soluble SiNPs is still an important challenge for their utilization in various research studies and applications.Considerable advancement has been made in the area of chemical synthesis of SiNPs including functionalization, particle size control, surface passivation, etc. Other synthesis techniques being followed are laser ablation,7 mechanical ball-milling and ultrasonication,8 and etching of Si powders.9 Among bottom-up techniques, frequently used syntheses are gas phase laser pyrolysis,10 plasma synthesis,11 and chemical synthesis.12 Of these, chemical synthesis offers better prospects of control over the particle size and shape,13 macroscopic yields,14 and adaptable surface termination.15 Furthermore, these techniques provide silicon nanoparticles capable of bright emission in the UV/blue/green electromagnetic spectrum. For example, a rapid ‘one-pot’ chemical synthesis using trimethoxysilane derivatives has been successfully demonstrated for synthesizing bright blue-emitting photo-stable SiNPs.13 In the known literature available, the report on the first luminescent nano-form of silicon synthesized by the electrochemical etching approach has gained widespread attention among researchers.16 SiNPs synthesized by various approaches show particle size dependent and tunable luminescence17 and efficient electroluminescence.18
However, almost all of these techniques are based on chemical precursors, which are generally costly and toxic reducing agents. Furthermore, multi-step purification procedures are another drawback which are time-consuming and costly and cannot be adapted for scale-up synthesis procedures. For deposition and assembly applications, such as doctor blade, drop-casting, and spin coating methods, the most desirable candidate is water-soluble silicon NPs.19 Water-soluble SiNPs can also be used for up-scale industrial applications and are suitable for applications such as ink-jet printing, stamping, spraying, etc.
Dong et al. prepared two highly luminescent materials by covalent co-binding using multiple materials such as CdTe quantum dots (QDs) and europium ions in silica nanoparticles. They have successfully demonstrated the application of these composite nanoparticles as ratiometric luminescent probes for the detection of Cu2+ and Fe2+ ions.20 Recently, selective sensing of copper ions has also been done using molybdenum disulfide quantum dots.21 Similarly, other researchers have also provided successful selective methods for the detection of other metal ions using different fluorescent probes such as graphene oxide QDs, core–shell QDs and other carbon-based nanomaterials.21–25 Chen and co-workers successfully synthesized and utilized water-soluble SiNPs as a nanosensor for the determination of potassium ferrocyanide and it can also be used to monitor the change in the pH of the solution.26
SiNPs have also been employed as optical sensing probes in the recent past.12,27,28 Regardless of these advances, additional research work is still required to devise new environmentally friendly and low-cost procedures for the synthesis of SiNPs with desired properties for broad applications. Herein, we report the preparation of SiNPs via a single-step microwave-assisted facile green synthesis route. The as-prepared SiNPs are water-soluble with high dispersibility and exhibit strong photoluminescence. Furthermore, the synthesized SiNPs were found to be highly suitable for the sensitive and selective detection Fe(III) ions.
2. Experimental
2.1. Materials/chemicals
Potassium chloride (KCl) and different standard stock solutions containing 1000 ppm of Ni(II), Mg(II), Co(II), Pb(II), Cu(II), Fe(III), F−, Br−, PO42− and NO3−, were purchased from Merck India Ltd. Potassium ferrocyanide and potassium ferricyanide were purchased from Loba Chemie Private Limited. For the synthesis of SiNPs, a commonly available green precursor, i.e. horsetail “Equisetum arvense” plant was used. Preparation of the reagents for all experimental procedures was done using Millipore deionized water. All the experiments were carried out at room temperature in an inert nitrogen environment.2.2. Synthesis of SiNPs
Silicon nanoparticles were synthesized by using a silicon-rich green precursor Equisetum arvense, commonly known as horsetail plant.29 The extract of the plant was obtained in water and it was filtered using a syringe filter of pore size 0.45 μm. The filtered extract was then pyrolyzed at a high temperature (473 K) in a typical household microwave oven. After 30 minutes of microwave irradiation, the sample was collected and a pale-yellow solution was obtained. The synthesized NPs were stored at 4 °C and were stable for more than 6 months. The obtained SiNPs showed bright blue luminescence in a neutral pH environment in an ultraviolet illuminator.2.3. Instrumentation
A house-hold microwave oven was used for the synthesis of SiNPs. All the fluorescence measurements were taken using a photoluminescence (PL, Shimadzu, RF-5301PC) spectrophotometer. For UV-visible and Fourier transform infrared spectroscopy Hitachi Ltd, Japan, and 670 IR, Varian, USA were used respectively. Raman spectra were collected at λex = 514 nm with an inVia instrument (Renishaw). XRD was recorded using a PANalytical X'pert PRO. A PANalytical Epsilon 5 Energy Dispersive X-ray Fluorescence (EDXRF) spectrometer was used for the determination of elemental composition. Lifetime measurements were made using an Edinburgh FL920 fluorescence life time spectrometer. All the electrochemical measurements were taken using an AUTPGSTAT128N Autolab potentiostat (Metrohm) with the three-electrode arrangement. In our three-electrode potentiostat system, we used a glassy carbon electrode as the working electrode, platinum wire as an auxiliary electrode and Ag/AgCl saturated with 3 M KCl was used as the reference electrode. For cyclic voltammetry analysis, we have used a 5 mM ferri–ferro solution along with 0.25 M KCl solution due to its low internal resistance, as required for reversible pseudocapacitance. All the electrochemical measurement results are provided in the ESI.†2.4. Time resolved fluorescence spectroscopy and quantum yield calculations
Time resolved fluorescence spectroscopy of the purified proteins was carried out using an FLS1000 fluorescence spectrometer by Edinburgh Instruments. Time correlated single photon counting (TCSPC) using a pulsed laser diode was used for studying the fluorescence decay as a function of time. A 467 nm laser with 50 ps pulse width and repetition frequency 20 MHz was used for illuminating the samples. Fluorescence emission was recorded at 495 nm and graphs were plotted along with Instrument Response Function (IRF). Analysis and fitting was done using iterative re-convolution of IRF with decay curve f(t) using eqn (1). FAST program by Edinburgh Instruments was used for the all analysis. The decay curves were fitted with eqn (2):| I(t) = IRF(t) × f(t) | (1) |
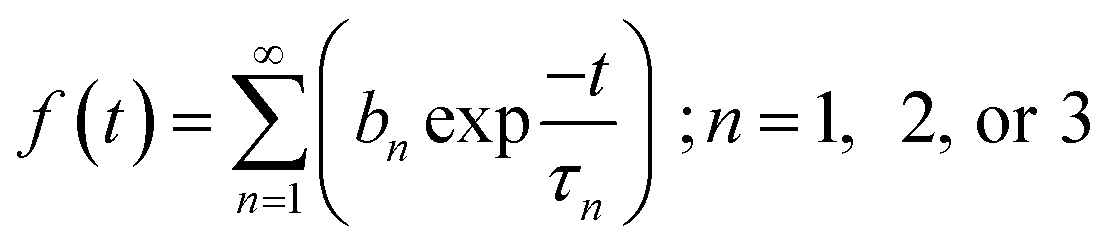 | (2) |
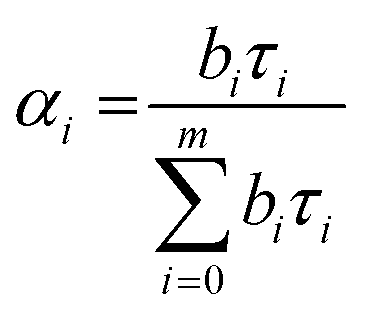 | (3) |
The average lifetime value was calculated using eqn (3):
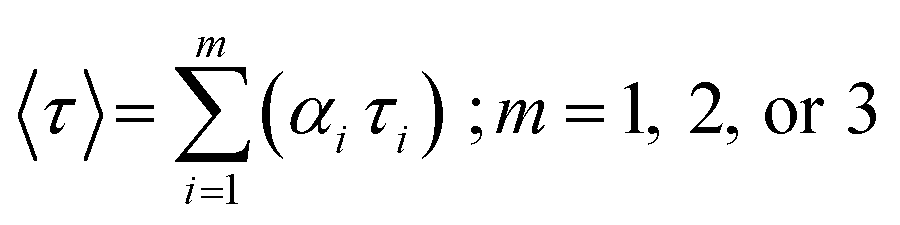 | (4) |
The quantum yield (QY) was investigated by comparing quinine sulfate reference dye in 0.01 M sulphuric acid (QY = 54%) with SiNPs at 350 nm absorbance value. The quantum yield is given by
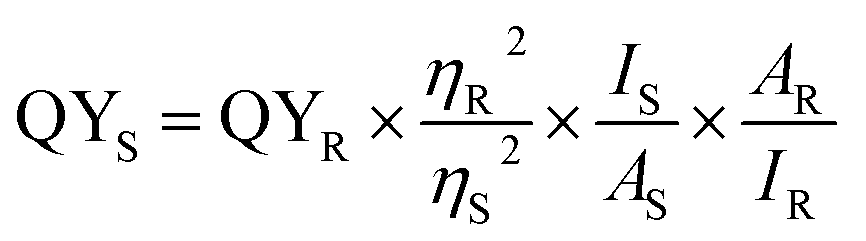 | (5) |
2.5. Optical determination of Fe(III) with SiNP probes
For Fe(III) sensing, SiNPs were further purified by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and investigated against different concentrations of Fe(III). The detection efficiency of SiNPs was also studied and analyzed by adjusting the pH values. A fixed volume of SiNPs was taken in the reaction cell and Fe(III) ions were added consecutively in order to determine the detection range and limit. On increase of Fe(III) ion concentration, a uniform decrease in emission intensity was reported due to quenching without any shift in the peak. To study the selective detection of Fe(III), standard stock solutions of different ions such as Ni(II), Mg(II), Co(II), Pb(II), Cu(II), F−, Br−, PO42− and NO3− were added separately and documented.
000 rpm and investigated against different concentrations of Fe(III). The detection efficiency of SiNPs was also studied and analyzed by adjusting the pH values. A fixed volume of SiNPs was taken in the reaction cell and Fe(III) ions were added consecutively in order to determine the detection range and limit. On increase of Fe(III) ion concentration, a uniform decrease in emission intensity was reported due to quenching without any shift in the peak. To study the selective detection of Fe(III), standard stock solutions of different ions such as Ni(II), Mg(II), Co(II), Pb(II), Cu(II), F−, Br−, PO42− and NO3− were added separately and documented.
3. Results and discussion
3.1. Structural & morphological characterization
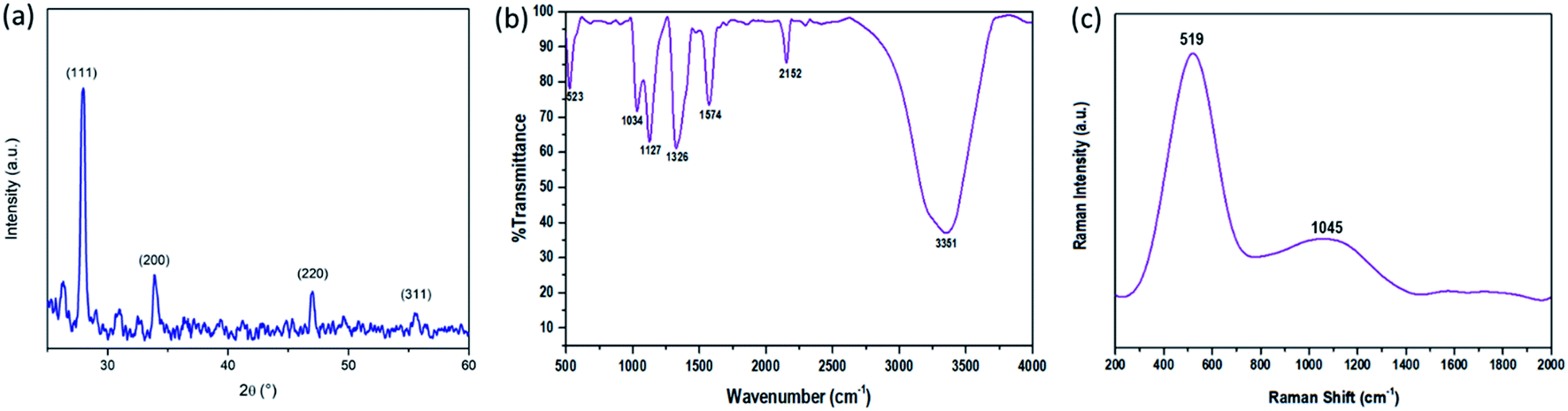
Structural characterization of the synthesized SiNPs was performed using a PANalytical X'pert PRO X-ray diffraction (XRD) instrument and is shown in Fig. 1(a). The graph shows the peaks corresponding to the (111), (200), (220), and (311) planes of the silicon nanoparticles. Crystallization of SiNPs is evidenced by the presence of sharp peaks in the XRD graph.30,31 Surface characteristics of SiNPs were investigated using FTIR spectra. The FTIR spectra were recorded by drop-casting the SiNP sample onto KBr pellets and are shown in Fig. 1(b). It reveals the major Si–Si characteristics at approximately 523 cm−1. The peaks at 1034 cm−1 and 1127 cm−1 are due to the presence of vibrational stretching because of oxygen moieties, i.e. Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/Si–OR, also, the other two observed peaks at 1326 cm−1 and 1576 cm−1 are due to the vibrational scissoring and symmetric bending of Si–CH2. Si–H stretching, with the symmetric Si–Hx and antisymmetric Si–Hx stretching vibrations, is confirmed by the peak at 2152 cm−1 and the peak at 3351 cm−1 is due to the stretching vibration and deformation of hydroxy (–OH) groups.
O/Si–OR, also, the other two observed peaks at 1326 cm−1 and 1576 cm−1 are due to the vibrational scissoring and symmetric bending of Si–CH2. Si–H stretching, with the symmetric Si–Hx and antisymmetric Si–Hx stretching vibrations, is confirmed by the peak at 2152 cm−1 and the peak at 3351 cm−1 is due to the stretching vibration and deformation of hydroxy (–OH) groups.
 | ||
| Fig. 1 (a) X-ray diffraction (XRD) graph, (a and b) FT-IR spectra, and (c) Raman spectra of the synthesized SiNPs. | ||
The Raman spectra of SiNPs were taken with a non-resonant light source at a longer wavelength range of ∼514.5 nm and are shown in Fig. 1(c). According to literature studies, the peak of bulk silicon is observed at 521 cm−1.32 Here a peak at 519 cm−1 was observed which signifies the reduced particle size of SiNPs and the redshift towards the lower energy can be explained by the phonon confinement effect. The strong resonance at 519 cm−1 characterized by the shift in resonance and broadening of the FWHM is observed in silicon nanoparticles of small size.33–36 One more peak at 1045 cm−1 was observed in the Raman spectra as can be seen in Fig. 1(c). This peak signifies the Si–O–Si modes and hence can be attributed to the presence of oxygen moieties at the surface of silicon nanoparticles.31,37,38
3.2. Transmission electron microscopy (TEM), dynamic light scattering (DLS), and energy-dispersive X-ray fluorescence (EDXRF)
Surface morphology and particle size distribution of the synthesized SiNPs were investigated using TEM. Transmission electron micrographs were taken using a Hitachi H-7500 and are shown in Fig. 2(a). The TEM images clearly show the pseudo-spherical surface morphology with a smooth surface and uniformly distributed nanoparticles with an average diameter of approximately 2–3 nm. High resolution TEM (HRTEM) was also performed and the d-spacing was found to be 0.3 nm as can be seen in Fig. 2(b). The HRTEM image clearly shows the high crystallinity of the synthesized silicon nanoparticles.39 The average particle size distribution curve was calculated from more than 300 nanoparticles and is shown in Fig. 2(c). The size distribution histogram was fitted with a Gaussian curve and the average size from the curve was calculated to be 2.5 nm. The average particle size of the SiNPs was also measured using dynamic light scattering (DLS) as shown in Fig. 2(d). The hydrodynamic diameter of the particles as calculated from the DLS histogram is found to be ∼6 nm. Two different diameters are calculated using TEM and DLS because the sample and measurement conditions are different for both the techniques. The DLS measurement gives the hydrodynamic radius while the effects of the solvent are ignored in the case of TEM analysis. Therefore, the diameter revealed from TEM particle size distribution is smaller than the one calculated from DLS.40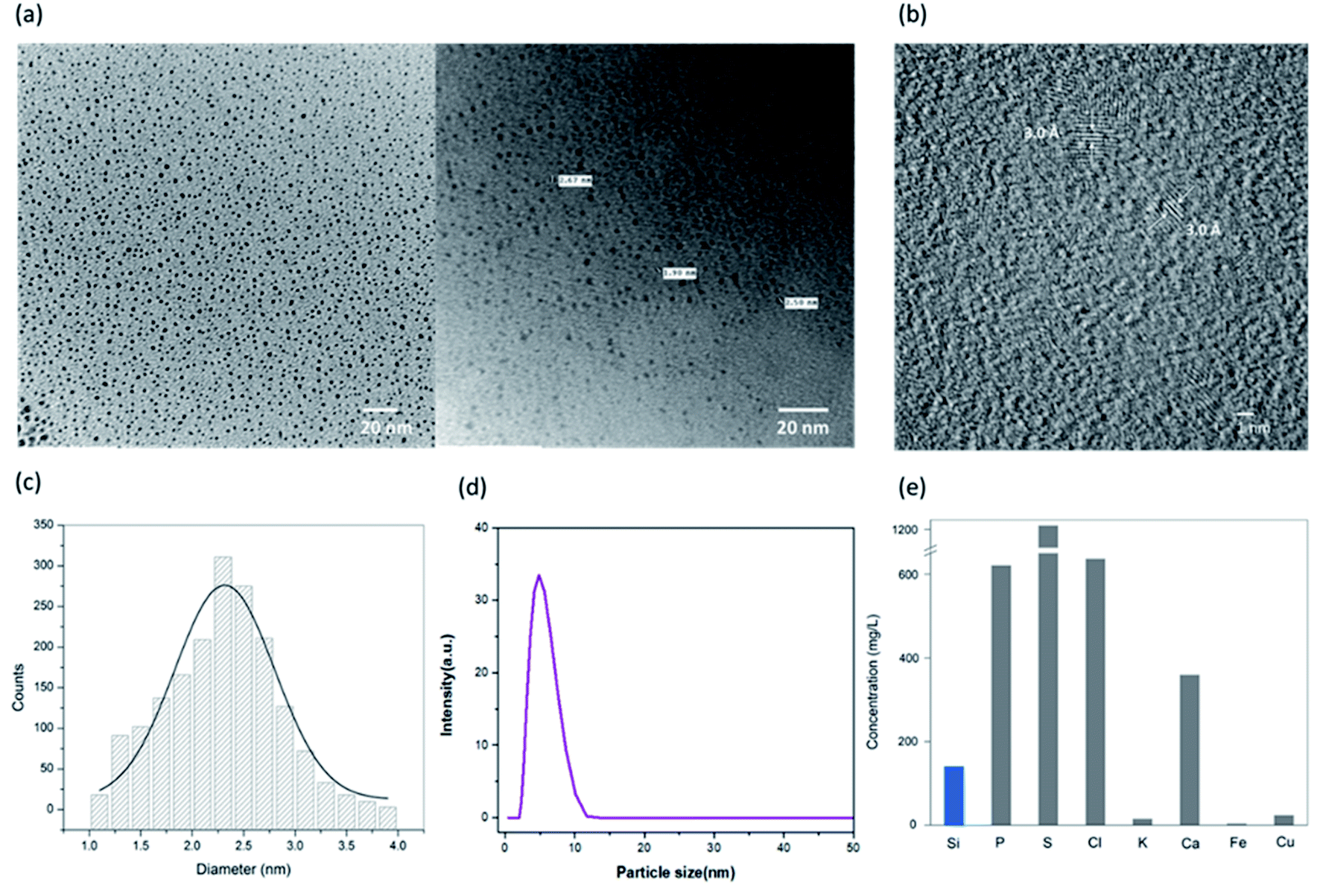 | ||
| Fig. 2 (a) TEM images of the synthesized SiNPs, (b) HRTEM of the synthesized SiNPs, (c) TEM particle size distribution, (d) DLS graph for the hydrodynamic size, and (e) EDXRF of the SiNPs for elemental composition. | ||
The as-synthesized SiNPs were studied for elemental composition to quantify the presence of silicon in comparison with the other elements. The elemental composition was studied using a PANalytical Epsilon 5 Energy Dispersive X-ray Fluorescence (EDXRF) spectrometer and is shown in Fig. 2(e). The EDXRF graph plot shows high concentrations of sulphur, chlorine, calcium and phosphorus as expected, which might be attributed to the plant cell content as per the previous studies.41 The elemental quantification depends on the season and geographical region from which the plant is harvested. The current data confirm the presence of silicon with a concentration of 140 mg L−1 approximately.
3.3. Optical characterization
The optical properties of SiNPs were studied from their emission and absorbance spectra. UV-visible absorption and fluorescence emission spectra of the SiNPs are shown in Fig. 3(a). The absorption spectra show a peak between 325 and 400 nm with a maximum around 355 nm. This absorption peak at 355 nm is attributed to a direct band gap transition at the Γ point.14,42 The presence of this distinct peak indicates the uniform shape and size of the nanoparticles. Fig. 3(a) also shows the emission spectra of SiNPs upon excitation with 340 nm wavelength (shown in blue). The emission spectrum is in the UV/blue region of the electromagnetic spectrum with a maximum at 420 nm with full width half maximum (FWHM) of approximately 60 nm.43,44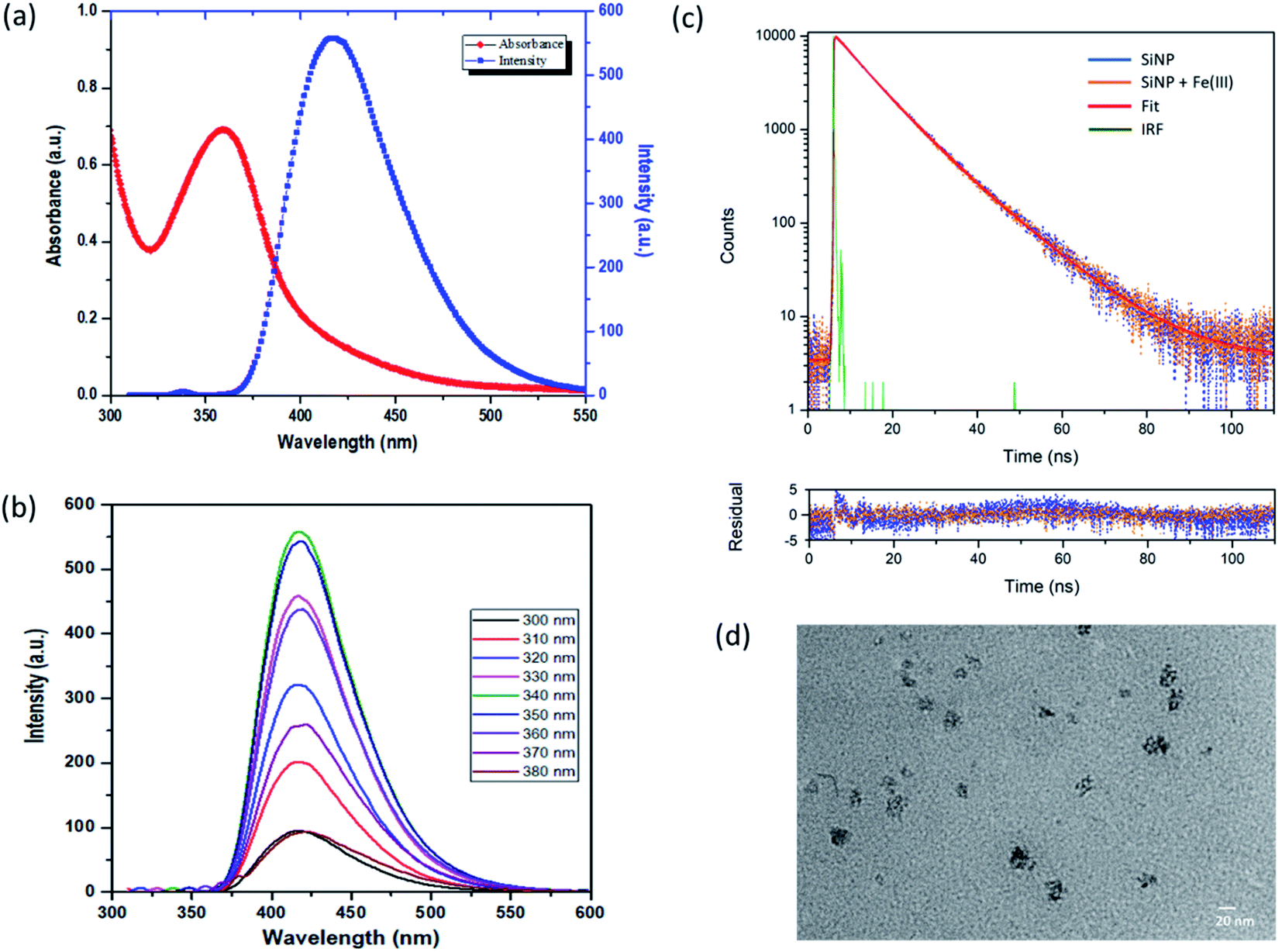 | ||
| Fig. 3 Optical properties of the synthesized SiNPs; (a) absorption spectra (red curve) and emission spectra (blue curve), and (b) excitation wavelength-independent photoluminescence behavior along with excitation wavelength ranging from 300 nm to 380 nm. (c) PL lifetime measurement of the synthesized SiNPs using time resolved spectroscopy, and (d) TEM image of SiNPs with Fe(III) ions. | ||
Furthermore, the emission spectra of the synthesized SiNPs were recorded for excitation wavelength ranging from 300 nm to 380 nm as shown in Fig. 3(b). The photoluminescence spectra shown here are independent of the excitation wavelength in this range, although it was observed that the intensity of photoluminescence changes with different excitation wavelengths. The maximum emission intensity was found when the nanoparticles were excited with 340 nm wavelength. This type of excitation wavelength-independent behaviour has been reported previously.42,45–47 The photoluminescence in silicon nanoparticles is usually a combination of direct as well as indirect bandgap transitions. The PL behaviour of SiNPs is generally explained by the quantum confinement effect or surface states with reports suggesting that the presence of surface passivation groups gives rise to photoluminescence in the UV/blue region. Here we propose that the excitation wavelength-independent PL behaviour is due to oxide related surface states, rather than quantum confinement effects.42 The quantum yield (QY) of SiNPs was determined and was found to be 15% with respect to quinine sulfate as a reference dye.
3.4. Lifetime measurement and morphology of SiNPs
Furthermore, to study the nature of photoluminescence of the synthesized SiNPs, time dependent photoluminescence decay was carried out using time correlated single photon counting (TCSPC) using a pulsed laser diode. Fig. 3(c) shows the photoluminescence decay profile of the silicon nanoparticles after excitation with 460 nm.Decay profiles for both bare SiNPs and SiNPs with Fe(III) ions were taken. Both lifetimes were found to be in the order of nanoseconds and exhibited good biexponential fitting. The overall decay time for bare silicon nanoparticles was found to be approximately 9.37 ns. Lifetimes in the case of indirect band gap materials like silicon are usually in the order of micro or milliseconds exhibiting slow electron hole recombination. However, in the case of direct band gap materials like CdSe and GaAS, this electron–hole recombination is faster resulting in lifetimes usually in the 1 to 10 ns range. Here as we can see the e–h recombination rate is fast and it can be attributed to the direct band gap transition resulting from oxide related surface effects.14,39,42,47,48
The photoluminescence lifetime after addition of Fe(III) ions was also measured and the average lifetime was calculated to be 9.02 ns. The photoluminescence lifetime was found to decrease with the introduction of Fe(III) ions. However, the change in lifetime was not in proportion to the change in intensity as observed from steady state fluorescence spectroscopy and hence it can be due to the combined effect of dynamic and static quenching.37,38,49 All the fitting parameters are given in Table 1. The change in morphology after addition of Fe(III) ions was also studied using transmission electron microscopy (TEM) and the image is shown in Fig. 3(d). Some cluster formation was observed in the nanoparticles after addition of iron(III) ions. The size of the cluster was found to be around 20 nm. This cluster formation may be attributed to the aggregation caused by adsorption of charged ions on the surface of the silicon nanoparticles.48
| Sample | χ 2 | α 1 (%) | τ 1 (ns) | α 2 (%) | τ 2 (ns) | 〈τ〉 (ns) |
|---|---|---|---|---|---|---|
| SiNPs | 1.33 | 56.65 | 7.3 | 43.34 | 12.1 | 9.37 |
| SiNPs + Fe(III) | 1.31 | 57.89 | 6.93 | 42.21 | 11.89 | 9.02 |
3.5. Fluorescence based sensing studies
Silicon nanoparticles exhibit fluorescence quenching in the presence of analytes. Due to this property they have been utilized as fluorescence sensors, where the fluorescence of SiNPs is quenched upon surface adsorption of analytes.50,51 Here in our work, we have used the synthesized SiNPs as an Fe(III) sensor by measuring the quenching of fluorescence. A schematic representation of this is shown in Fig. 4(a). Electrochemical response, cyclic voltammetry, capacitance, and frequency response analysis (FRA) of SiNPs were also studied and are shown in the ESI.† Initially, the fluorescence response of SiNPs at pH 7 was studied for three concentration variations of Fe(III), i.e. 50 ppb, 100 ppb, and 1000 ppb. The graph is represented in Fig. 4(b) and shows a uniform decrease in the fluorescence intensity of SiNPs on consecutive increase in the concentration of Fe(III) due to the quenching.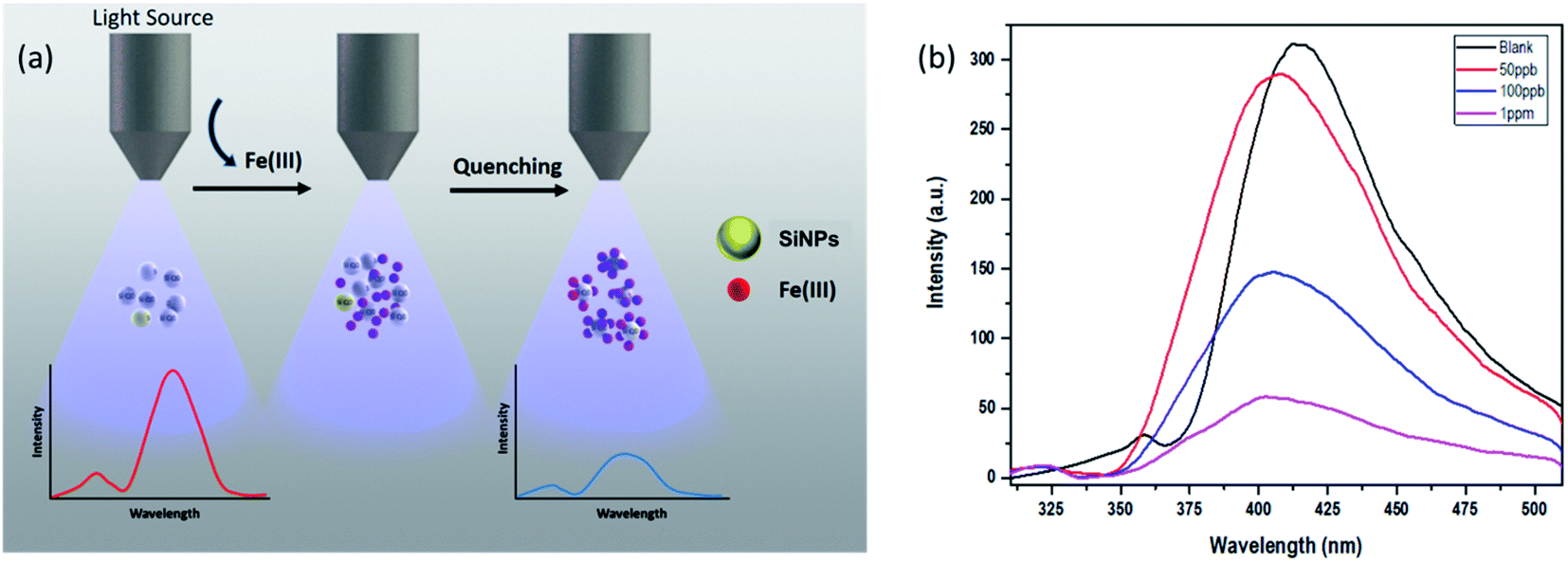 | ||
| Fig. 4 (a) Schematic illustration representing fluorescence quenching of SiNPs upon addition of Fe(III) ions, (b) sensing behaviour of SiNPs towards Fe(III) ions. | ||
The sensing response of the synthesized SiNPs towards Fe(III) ions for concentrations ranging from 50 ppb to 1000 ppb is shown in Fig. 5(a). The steady decrease in luminescence intensity can be attributed to the strong bonding between oxygen groups and Fe(III) ions. The corresponding regression curve for the linear range 50 to 1000 ppb is shown in Fig. 5(b). Under well-optimized experimental conditions, the quenching of luminescence is caused by the Fe(III) analyte and is well described by the Stern–Volmer equation:
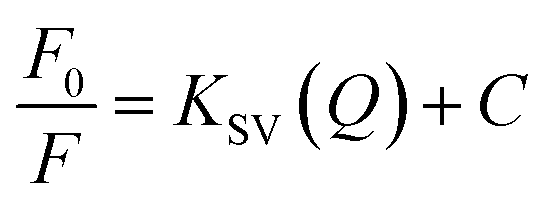 | (6) |
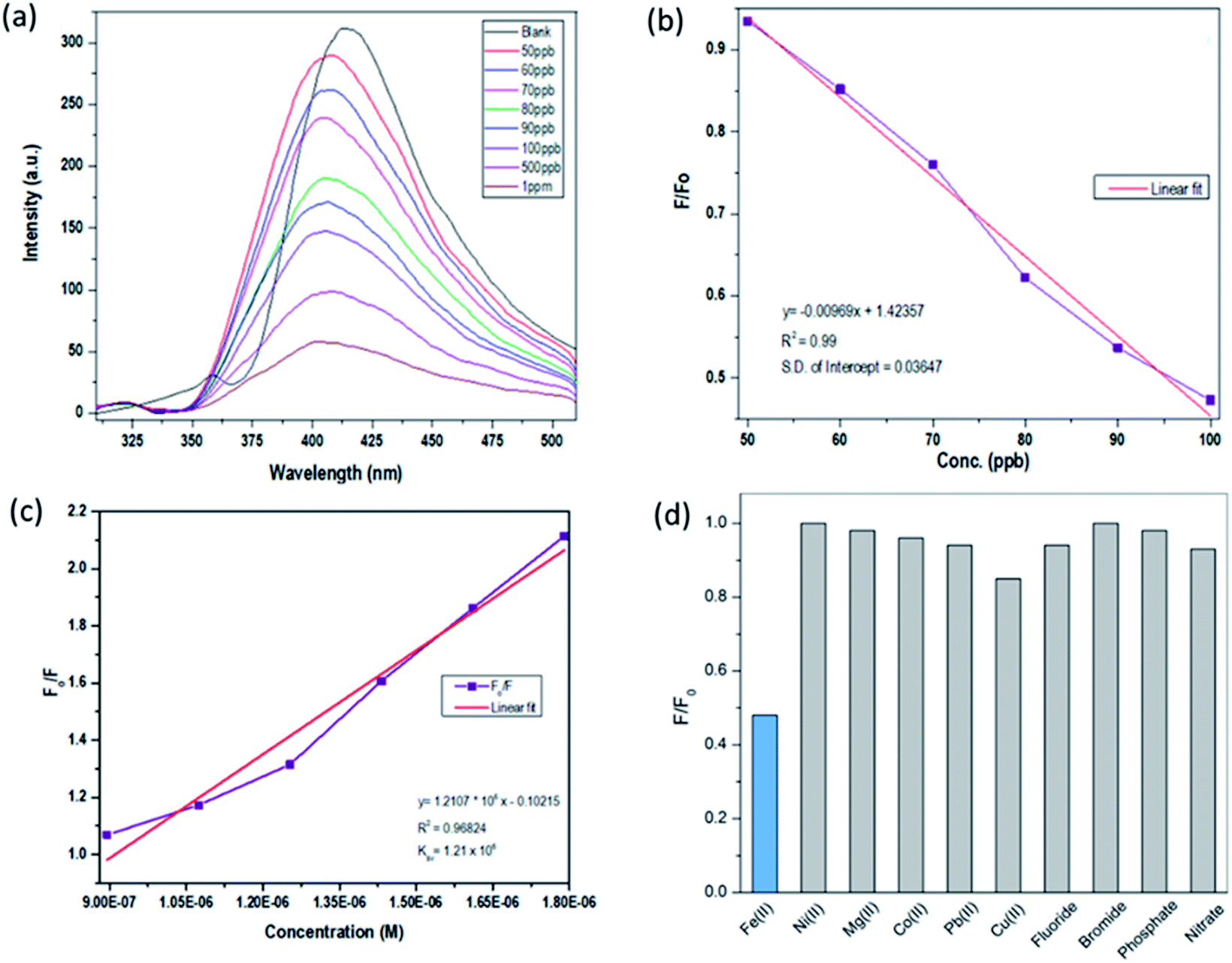 | ||
| Fig. 5 (a) Sensing study of SiNPs towards Fe(III), (b) linear regression curve, (c) Stern–Volmer plot, and (d) interference studies with respect to a library of anions and cations. | ||
| Electrode | Linear range (ppm) | LOD (ppb) | Ref. |
|---|---|---|---|
| Graphene quantum dots (GQD) | 1.11–20 | 403.2 | 52 |
| Organic–inorganic hybrid (LQS) | 0–1.11 | 234.5 | 53 |
| Adenosine monophosphate-gold nanoclusters (AMP-Au NCs) | 0.5–5.5 | 111.6 | 54 |
| 2,5-Bis[2,3,3-trimethyl-3H-indole-5-sulfonic acid]-croconaine (TISC) | 0–3.8 | 21.38 | 55 |
| Carbon dots | 1.11–27.9 | 22.89 | 56 |
| Calix[4]resorcinarene polyhydrazide-silver nanopartilces (CPH-AgNPs) | 0.005–0.5 | 5.58 | 57 |
| N-Acetyl-L-cysteine (NALC)-stabilized Ag nanoparticles (NALC-Ag NPs) | 4.4–4467 | 4.46 | 58 |
| Metal–organic framework (MOF) | 0.16–11.1 μM | 502.6 | 59 |
| Silicon nanoparticles (SiNPs) | 0.05–1 | 12.4 | This work |
3.6. Interference studies
The selectivity performance of SiNP was investigated with 100 ppb Fe(III) ions, and standard stock solutions of common interfering ions such as Ni(II), Mg(II), Co(II), Pb(II), Cu(II), Fe(III), F−, Br−, PO42− and NO3− were used. The interference study was carried out with 1 ppm concentration of interfering ions and the results are shown in Fig. 5(d) in the form of a bar graph. The results clearly show that Fe(III) induces maximum quenching of the synthesized luminophore, but not other interfering ionic species. The selectivity is ensured at even ten-fold concentrations, as the Fe(III) induced maximum quenching, which is not the case for other interfering ions at higher concentrations.4. Conclusions
Here, we have reported the successful synthesis of highly stable aqueous soluble SiNPs via a simple and effective single-step microwave-assisted facile green synthesis route. We have demonstrated the use of a silicon-rich extract of “Equisetum arvense” as a biological precursor for the synthesis procedure. The as-prepared SiNPs feature excellent water solubility, strong fluorescence along with photo-stable behavior. Under well-optimized experimental parameters, blue luminescence is shown by SiNPs along with a comparable quantum yield of 15%. The experiments demonstrate that the SiNPs are suitable for the detection of Fe3+ ions and monitoring in water up to the WHO recommended level of 50 ppb. These optical sensing results provide a new insight for SiNPs as an excellent optical probe in chemosensory applications. The results presented and discussed here provide an easy and environmentally friendly approach for the green synthesis of SiNPs, which can be utilized for other scientific applications.Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
The authors would like to acknowledge the Department of Instrument Technology, Andhra University, Vishakhapatnam for their financial support. The authors would also like to acknowledge the financial assistance provided by the Council of Scientific and Industrial Research.References
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- R. E. Bailey, A. M. Smith and S. Nie, Phys. E, 2004, 25, 1–12 CrossRef CAS.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef PubMed.
- J. H. Warner, A. Hoshino, K. Yamamoto and R. D. Tilley, Angew. Chem., 2005, 44, 4550–4554 CrossRef CAS PubMed.
- M. V. Wolkin, J. Jorne, P. M. Fauchet, G. Allan and C. Delerue, Phys. Rev. Lett., 1999, 82, 197–200 CrossRef CAS.
- F. Erogbogbo, K. T. Yong, I. Roy, G. Xu, P. N. Prasad and M. T. Swihart, ACS Nano, 2008, 2, 873–878 CrossRef CAS PubMed.
- D. Tan, B. Xu, P. Chen, Y. Dai, S. Zhou, G. Ma and J. Qiu, RSC Adv., 2012, 2, 8254 RSC.
- K. Dohnalová, L. Ondič, K. Kůsová, I. Pelant, J. L. Rehspringer and R. R. Mafouana, J. Appl. Phys., 2010, 107, 053102 CrossRef.
- S. Sato and M. T. Swihart, Chem. Mater., 2006, 18, 4083–4088 CrossRef CAS.
- R. K. Baldwin, K. A. Pettigrew, J. C. Garno, P. P. Power, G.-y. Liu and S. M. Kauzlarich, J. Am. Chem. Soc., 2002, 124, 1150–1151 CrossRef CAS PubMed.
- B. N. Jariwala, N. J. Kramer, M. C. Petcu, D. C. Bobela, M. C. M. v. d. Sanden, P. Stradins, C. V. Ciobanu and S. Agarwal, J. Phys. Chem. C, 2011, 115, 20375–20379 CrossRef CAS.
- B. B. Campos, M. Algarra, B. Alonso, C. M. Casado, J. Jimenez-Jimenez, E. Rodriguez-Castellon and J. C. Esteves da Silva, Talanta, 2015, 144, 862–867 CrossRef CAS PubMed.
- M. Rosso-Vasic, E. Spruijt, B. van Lagen, L. De Cola and H. Zuilhof, Small, 2008, 4, 1835–1841 CrossRef CAS PubMed.
- J. P. Wilcoxon, G. A. Samara and P. N. Provencio, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 2704–2714 CrossRef CAS.
- N. S. Bhairamadgi, S. Gangarapu, M. A. Caipa Campos, J. M. Paulusse, C. J. van Rijn and H. Zuilhof, Langmuir, 2013, 29, 4535–4542 CrossRef CAS PubMed.
- L. T. Canham, Appl. Phys. Lett., 1990, 57, 1046–1048 CrossRef CAS.
- D. Kovalev, H. Heckler, G. Polisski, J. Diener and F. Koch, Opt. Mater., 2001, 17, 35–40 CrossRef CAS.
- B. Gelloz and N. Koshida, J. Appl. Phys., 2000, 88, 4319 CrossRef CAS.
- R. D. Tilley, J. H. Warner, K. Yamamoto, I. Matsui and H. Fujimori, Chem. Commun., 2005, 1833–1835, 10.1039/b416069j.
- H. Dong, Y. Liu, D. Wang, W. Zhang, Z. Ye, G. Wang and J. Yuan, Nanotechnology, 2010, 21, 395504 CrossRef PubMed.
- E. Soheyli, R. Sahraei, G. Nabiyouni, F. Nazari, R. Tabaraki and B. Ghaemi, Nanotechnology, 2018, 29, 445602 CrossRef PubMed.
- H. Zhu, W. Zhang, K. Zhang and S. Wang, Nanotechnology, 2012, 23, 315502 CrossRef PubMed.
- A. P. Demchenko and M. O. Dekaliuk, Methods Appl. Fluoresc., 2013, 1, 042001 CrossRef PubMed.
- W. Xuan, L. Ruiyi, F. Saiying, L. Zaijun, W. Guangli, G. Zhiguo and L. Junkang, Sens. Actuators, B, 2017, 243, 211–220 CrossRef CAS.
- A. Dutta Chowdhury and R. A. Doong, ACS Appl. Mater. Interfaces, 2016, 8, 21002–21010 CrossRef CAS PubMed.
- M. Na, Y. Chen, Y. Han, S. Ma, J. Liu and X. Chen, Food Chem., 2019, 288, 248–255 CrossRef CAS PubMed.
- Y. Yi, G. Zhu, C. Liu, Y. Huang, Y. Zhang, H. Li, J. Zhao and S. Yao, Anal. Chem., 2013, 85, 11464–11470 CrossRef CAS PubMed.
- N. Dhenadhayalan, H. L. Lee, K. Yadav, K. C. Lin, Y. T. Lin and A. H. Chang, ACS Appl. Mater. Interfaces, 2016, 8, 23953–23962 CrossRef CAS PubMed.
- P. Labun, D. Grulova, I. Salamon and F. Šeršeň, Food Nutr. Sci., 2013, 04, 510–514 CAS.
- P. Zaumseil, J. Appl. Crystallogr., 2015, 48, 528–532 CrossRef CAS PubMed.
- A. R. Jose, U. Sivasankaran, S. Menon and K. G. Kumar, Anal. Methods, 2016, 8, 5701–5706 RSC.
- H. Xia, Y. L. He, L. C. Wang, W. Zhang, X. N. Liu, X. K. Zhang, D. Feng and H. E. Jackson, J. Appl. Phys., 1995, 78, 6705–6708 CrossRef CAS.
- S. K. Gupta and P. K. Jha, Solid State Commun., 2009, 149, 1989–1992 CrossRef CAS.
- G. Faraci, S. Gibilisco, A. R. Pennisi and C. Faraci, J. Appl. Phys., 2011, 109, 074311 CrossRef.
- V. Paillard, P. Puech, M. A. Laguna, R. Carles, B. Kohn and F. Huisken, J. Appl. Phys., 1999, 86, 1921–1924 CrossRef CAS.
- M. D. Efremov, V. A. Volodin, D. V. Marin, S. A. Arzhannikova, S. V. Goryainov, A. I. Korchagin, V. V. Cherepkov, A. V. Lavrukhin, S. N. Fadeev, R. A. Salimov and S. P. Bardakhanov, Journal of Experimental and Theoretical Physics Letters, 2004, 80, 544–547 CrossRef CAS.
- L. Liu, J. Fan, L. Ding, B. Zhu, X. Huang, W. Gong, Y. Xin and Y. Fang, J. Photochem. Photobiol., A, 2016, 328, 1–9 CrossRef CAS.
- H. Chen, L. Wu, Y. Wan, L. Huang, N. Li, J. Chen and G. Lai, Analyst, 2019, 144, 4006–4012 RSC.
- J. Wu, J. Dai, Y. Shao and Y. Sun, RSC Adv., 2015, 5, 83581–83587 RSC.
- Y. Zhong, F. Peng, F. Bao, S. Wang, X. Ji, L. Yang, Y. Su, S. T. Lee and Y. He, J. Am. Chem. Soc., 2013, 135, 8350–8356 CrossRef CAS PubMed.
- A. Sola-Rabada, J. Rinck, D. J. Belton, A. K. Powell and C. C. Perry, JBIC, J. Biol. Inorg. Chem., 2016, 21, 101–112 CrossRef CAS PubMed.
- D. Tan, Z. Ma, B. Xu, Y. Dai, G. Ma, M. He, Z. Jin and J. Qiu, Phys. Chem. Chem. Phys., 2011, 13, 20255–20261 RSC.
- J. W. Fan, R. Vankayala, C. L. Chang, C. H. Chang, C. S. Chiang and K. C. Hwang, Nanotechnology, 2015, 26, 215703 CrossRef PubMed.
- J. Zou, R. K. Baldwin, K. A. Pettigrew and S. M. Kauzlarich, Nano Lett., 2004, 4, 1181–1186 CrossRef CAS.
- D. Dobrovolskas, J. Mickevičius, G. Tamulaitis and V. Reipa, J. Phys. Chem. Solids, 2009, 70, 439–443 CrossRef CAS.
- J. D. Holmes, K. J. Ziegler, R. C. Doty, L. E. Pell, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2001, 123, 3743–3748 CrossRef CAS PubMed.
- M. J. Portoles, F. R. Nieto, D. B. Soria, J. I. Amalvy, P. J. Peruzzo, D. O. Martire, M. Kotler, O. Holub and M. C. Gonzalez, J. Phys. Chem. C, 2009, 113, 13694–13702 CrossRef PubMed.
- R. Borgohain, P. Kumar Boruah and S. Baruah, Sens. Actuators, B, 2016, 226, 534–539 CrossRef CAS.
- U. Alexiev and D. L. Farrens, Biochim. Biophys. Acta, 2014, 1837, 694–709 CrossRef CAS PubMed.
- L. Mu, W. Shi, J. C. Chang and S. T. Lee, Nano Lett., 2008, 8, 104–109 CrossRef CAS PubMed.
- R. Miao, L. Mu, H. Zhang, G. She, B. Zhou, H. Xu, P. Wang and W. Shi, Nano Lett., 2014, 14, 3124–3129 CrossRef CAS PubMed.
- A. Ananthanarayanan, X. Wang, P. Routh, B. Sana, S. Lim, D.-H. Kim, K.-H. Lim, J. Li and P. Chen, Adv. Funct. Mater., 2014, 24, 3021–3026 CrossRef CAS.
- M. Karimi, A. Badiei, N. Lashgari, J. Afshani and G. Mohammadi Ziarani, J. Lumin., 2015, 168, 1–6 CrossRef CAS.
- D. Ungor, E. Csapo, B. Kismarton, A. Juhasz and I. Dekany, Colloids Surf., B, 2017, 155, 135–141 CrossRef CAS PubMed.
- S. Ye, C. Zhang, J. Mei, Z. Li, S. Xu, X. Li and C. Yao, J. Photochem. Photobiol., A, 2017, 347, 130–137 CrossRef CAS.
- G. Venkatesan, V. Rajagopalan and S. N. Chakravarthula, J. Environ. Chem. Eng., 2019, 7, 103013 CrossRef CAS.
- B. A. Makwana, D. J. Vyas, K. D. Bhatt, V. K. Jain and Y. K. Agrawal, Spectrochim. Acta, Part A, 2015, 134, 73–80 CrossRef CAS PubMed.
- X. Gao, Y. Lu, S. He, X. Li and W. Chen, Anal. Chim. Acta, 2015, 879, 118–125 CrossRef CAS PubMed.
- C. X. Yang, H. B. Ren and X. P. Yan, Anal. Chem., 2013, 85, 7441–7446 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00307g |
| This journal is © The Royal Society of Chemistry 2020 |