
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-based atomic/molecular layer deposition of lithium ethylene carbonate thin films†
Juho
Heiska
 ,
Milad
Madadi
and
Maarit
Karppinen
*
,
Milad
Madadi
and
Maarit
Karppinen
*
Department of Chemistry and Materials Science, Aalto University, FI-00076 Espoo, Finland. E-mail: maarit.karppinen@aalto.fi
First published on 7th May 2020
Abstract
When a conventional lithium-ion battery (LIB) is cycled, a solid electrolyte interphase (SEI) forms on the surface of a negative electrode, passivating it but also depleting the capacity of the battery. Most commercial LIBs utilize a carbonate-based electrolyte, which at least temporarily leads to the formation of lithium alkyl carbonates (ROCO2Li) as the main organic SEI component. Here, we pioneer the use of atomic/molecular layer deposition (ALD/MLD) for the fabrication of lithium ethyl glycoxide (LiEG) and lithium ethylene carbonate (LiEGCO) thin films, to mimic the lithium alkyl carbonate component of the SEI. For the in situ growth of LiEGCO, we employ for the first time CO2 as an ALD/MLD precursor. The films are characterized using XRR, GIXRD, FTIR, AFM and SEM.
Introduction
The lithium-ion battery (LIB) is one of the most influential technologies of the 21st century. In most LIBs, a solid electrolyte interphase (SEI) spontaneously forms on the negative electrode during the initial charge/discharge cycles. The SEI is a result of interfacial reactions between a solid electrode and a liquid electrolyte; it consists of inorganic and organic parts and is often regarded as both the most important and the least-understood component of a LIB.1,2 Recently, it was shown that the organic part of the SEI on graphite electrodes with ethyl carbonate electrolyte initially consists mainly of lithium ethylene mono-carbonate (LEMC; see Chart 1), while the inorganic part consists of Li2O, Li2CO3 and LiF.3 The composition of SEI is constantly evolving, though, such that upon aging the LEMC may decompose into more stable inorganic compounds, and more alkyl carbonates may form.2,4 It is clear from numerous studies that an artificially pre-fabricated SEI could improve battery performance by preventing cracks and stopping the consumption of the electrolyte and Li-ions.5–16 An artificial SEI could also help prevent damage caused by mechanical stress and by volume expansion during (de)insertion of Li-ions, but this feature is dependent on the cell chemistry.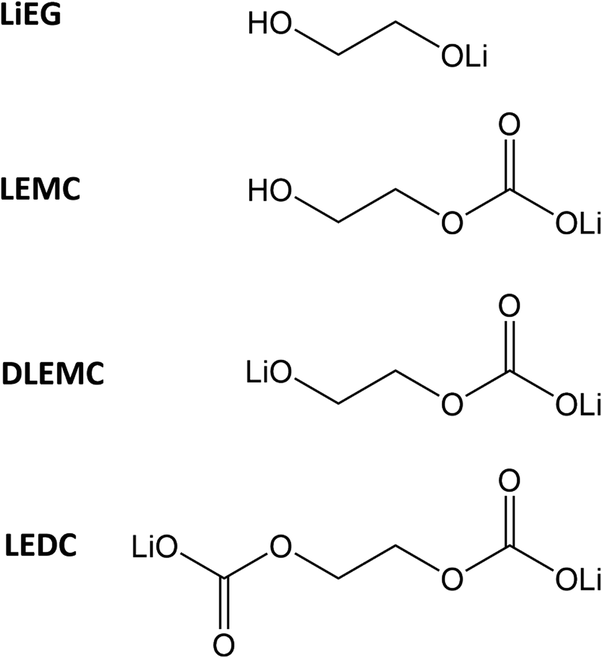 | ||
| Chart 1 Molecular structures and abbreviations of the Li–organic compounds referenced in this work: LiEG (mono-lithium ethylene glycoxide), LEMC (lithium ethylene mono-carbonate), DLEMC (di-lithium ethylene mono-carbonate) and LEDC (lithium ethylene di-carbonate). | ||
Atomic/molecular layer deposition (ALD/MLD) is an emerging thin-film deposition technique for producing hybrid inorganic–organic materials with a high degree of controllability,17–19 and could thus provide us with an intriguing means of manufacturing artificial SEIs with atomic/molecular level accuracy. It is a branch of the well-established ALD technology, originally invented and now extensively employed for high-quality ultrathin films of simple inorganic materials of commercial importance.18,20,21 Recent advances in ALD/MLD have already demonstrated its potential in a number of front-line energy-related applications, e.g. thermoelectrics,22,23 Li–organic battery,24–26 and electrocatalysis/hydrogen evolution.27,28
In ALD/MLD – mimicking the parent ALD – the substrate is sequentially exposed to gaseous metal and organic precursors that undergo self-limiting reactions on the surface. As the precursors cannot react with themselves and only react with the surface, this results in a growth of (at most) one monolayer per precursor pulsing cycle. Thus, the produced films are highly conformal and pinhole-free, and their thickness can be controlled precisely.18,19,29 Depositing the hybrid material directly from the gas phase, rather than utilizing traditional solution-based synthesis techniques, would give unmatched control over the deposition process and enable the coating of high-aspect-ratio structures. An interesting group of these hybrid materials are the simple aliphatic metal alkoxides, commonly referred to in literature as metalcones, e.g. alucone.6,10 The alkoxide itself is a very reactive species, especially so towards ambient moisture. Alkoxides have been used as precursors in organic synthesis for centuries, and also as scaffolds for making porous inorganic oxides in ALD/MLD literature.30 In addition, even a lithium alkoxide coating was recently reported to greatly improve the cyclability of a lithium anode by preventing dendrite formation.31 But most importantly, metal alkyl carbonates are known to readily form in the reaction of metal alkoxide with CO2.32–34
Here we demonstrate for the first time the ALD/MLD process of lithium ethylene glycoxide (LiEG), and incorporation of CO2 into this process to form metal alkyl carbonate species of LiEG. CO2 has previously been used in ALD processes – but not in ALD/MLD – with lithium precursors to deposit films of Li2CO3,35 this usually leading to much smoother and more stable films.36 Also, very few ALD/MLD processes have so far been developed in which lithium is directly incorporated as the inorganic constituent.24,25,37,38 In this work, we systematically optimize the ALD/MLD growth of our LiEG and LiEGCO thin films and investigate their morphology, chemical structure and stability, as well as propose a possible reaction mechanism. We foresee these coatings presenting an elegant and convenient way of studying the chemical and mechanical properties of the organic portion of the SEI, and possibly functioning as an artificial one.
Materials and methods
The vast majority of LiEG and LIEGCO thin films were deposited on test silicon wafers (Okmetic Oy) and carbon nanotubes (Thin MWCNT 95+% C purity Nanocyl Belgium) in a commercial flow-type hot-wall F-120 ALD reactor (ASM Microchemistry Ltd.) using lithium bis(trimethylsilyl)amide (LiHMDS, Sigma-Aldrich, 97%) and ethylene glycol (EG, TCI, 99.5%) as precursors. Carbon dioxide (99.99%) was pulsed into the reactor through a silica-gel-filled tube (∼600 cm3) to remove residual moisture, and the flow speed was controlled quantitatively with a needle valve. The precursors, LiHMDS and EG, were heated to 55 and 30 °C, respectively, and held in open glass boats inside the reactor to ensure efficient transport of the precursor. Nitrogen (99.999%), generated from air with Parker HPN 5000, was used as both carrier and purging gas. The pressure inside the reactor was below 5 mbar. The purging times were kept mostly constant, while the pulsing time was varied to study the growth saturation behavior. As the materials in question were highly sensitive to humidity, extra care was taken in the sample handling. The air exposure was always minimized, although it could not be completely avoided, e.g. when the samples were placed in various apparatuses and during the measurements. The samples were always stored in a desiccator unless otherwise stated.All X-ray patterns were collected with a PANanalytical X'Pert Pro diffractometer (Cu Kα1; λ = 1.540598 Å). The thickness and the crystallinity of the films were determined from X-ray reflection (XRR) patterns and grazing incidence X-ray diffraction (GIXRD) diffractograms, respectively. The density of the films was calculated from the dependency of the critical angle (θc) on the mean electron density (ρe) and the mass density (ρm) of the material. Here we must assume the elemental composition, which is just a rough approximation, as it was not exactly determined. The formulae are: ρe = (θc2π)/(λ2re), and ρm = (ρeA)/(NAZ), where λ is the X-ray wavelength, re is the classical electron radius, A is the average molar mass, NA is the Avogadro constant, and Z is the average atomic number. This calculation will not result in precise density values, but it can be used to detect density changes in the film within a series.
The chemical composition/state of the films was studied with Fourier-transform infrared spectroscopy (FTIR, Bruker ALPHA II Transmittance Spectrometer) in a range of 400 to 4000 cm−1 with a resolution of 4 cm−1. All films were measured in transmission mode (32 scans), and the background absorbance of silicon wafer was subtracted from the spectrum. The bulk samples were measured (64 scans) using the same apparatus with a Platinum-ATR-sampling module, with a diamond crystal. Atomic force microscopy images were taken in tapping mode with a Veeco Dimension 5000. Tips used were Mikromasch HQ: NSC14/AlBS with a typical radius of 8 nm and a 5 N m−1 force constant. Scanning electron microscopy (SEM) images were recorded with a Hitachi S-4700 FE-SEM with a 5.0 kV acceleration voltage using secondary electrons.
Results and discussion
We first investigated the surface-reaction behavior of our LiHMDS and EG precursors by varying the pulse lengths of the precursors and monitoring the resultant film growth-per-cycle (GPC) value based on the film thickness value from XRR (Fig. 1a), while the deposition temperature was set to 80 °C. The pulse saturation of EG was faster (3 s) than that of LiHMDS (5 s). Unlike EG, LiHMDS is a solid precursor, which may explain the longer pulsing time needed to achieve saturation. The resultant saturated growth rate of ∼2.8 Å C−1 is similar to that of other metal-EG processes.10,39 Many ALD/MLD processes do not exhibit the so-called “ALD window” where the process would have an essentially constant GPC in a certain temperature range. Indeed, this process is one of them, as the GPC continuously decreases from 2.8 to 0.7 Å C−1 when ascending through the investigated temperature range of 80 to 140 °C (Fig. 1b).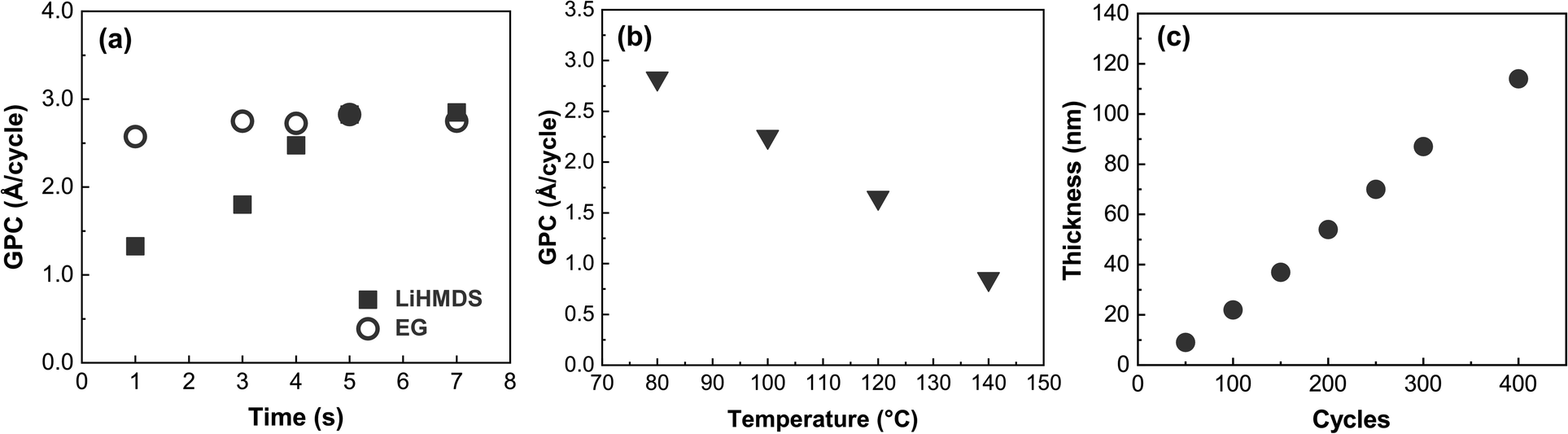 | ||
| Fig. 1 Growth per cycle as a function of (a) precursor pulse lengths at 80 °C and 200 cycles, and (b) deposition temperature; (c) film thickness against the number of ALD/MLD cycles for the LiHMDS + EG process deposited on Si. | ||
Using the optimized precursor pulse lengths, we examined the thickness of the films relative to the number of ALD/MLD cycles (Fig. 1c). By definition, an ALD/MLD process should have a linear relationship between the film thickness and the number of ALD/MLD cycles. With LiEG, the growth rate is initially slower, but it stabilizes for thicker films. The slower growth rate in the beginning is often observed with ALD/MLD processes, probably due to nucleation issues.24,26 In addition, very similar behavior was observed with a Li-propanediol ALD/MLD process.38 It is also worth noting here that the LiEG films were quite rough, which made thickness determination from XRR data challenging – and impossible for films thicker than appr. 140 nm, for which no fringes were visible.
Assuming that our LiEG film is chemically similar to the lithium salt of ethylene glycoxide, it should be possible to have it reacted with CO2 to form lithium ethylene mono-carbonate (LEMC) or, in harsher synthesis conditions, even lithium ethylene di-carbonate (LEDC).3 We carried out preliminary tests with long pulses of CO2 at the end of the deposition, but – as later discussed in the FTIR section – we believe that the transformation was not complete, at least not within reasonable pulsing times. Next, we introduced shorter CO2 pulses regularly during the film growth, testing different frequencies. Tentatively, the results indicated that the CO2 pulse may not be needed during every ALD/MLD cycle to make the desired reaction happen, e.g. pulsing CO2 every 10th cycle seemed to be enough to convert the entire film to the carbonate phase (ESI Fig. 1†). In the rest of the experiments, we included the CO2 pulse in every cycle to guarantee complete conversion; the product of this ternary process was named LiEGCO.
Next, we optimized the pulse length of CO2 while keeping the already optimized parameters for LiHMDS and EG. The saturation of the CO2 pulse, linearity of growth and temperature behavior are shown in Fig. 2. The addition of CO2 to the process had little effect on the growth rate. This can be explained by a notable change in the density of the films, as revealed from XRR data (Fig. 2). In addition, in the XRR patterns of thicker LiEG films, Kiessig fringes were barely visible and often decayed quickly, indicating a high surface roughness of the film. On the other hand, the LiEGCO films almost always displayed clear fringes within the entire measurement range. From the fitted XRR data, the differences can easily be seen. The fitting was straightforward for both LiEGCO and LiEG, aside from the uncertainty about the exact molecular formulae of the materials. Taking all these factors into account, the simulated density and roughness values for LiEG were 1.28 g cm−3 (1.3 g cm−3 when calculated from the critical angle) and 5 nm, and for LiEGCO 1.58 g cm−3 (1.5 g cm−3 from the critical angle) and 0.54 nm, respectively. The roughness was remarkably lower for LiEGCO than for LiEG.
 | ||
| Fig. 2 In (a) growth per cycle relative to deposition temperature (300 cycles) and in (b) film thickness relative to ALD/MLD cycles and inset with CO2 pulse saturation in LiHMDS + EG + CO2 process; in (c) fitted XRR patterns for LiEGCO and LiEG films grown on Si with 300 and 400 ALD/MLD cycles, respectively. | ||
The AFM images confirmed the XRR results, as the LiEG film surfaces were found to be rough and wavy, while the LiEGCO films were much smoother (Fig. 3a). The calculated roughness values for the LiEG and LiEGCO films were 18 and 7 nm, respectively. Based on Fig. 3, the LiEG surface consists of 1D rods, as often seen for metal ethylene glycoxide complexes, which have been used as templates for creating nanomaterials with specific form factors.40,41 The LiEGCO surface also consists of small particles, but overall it is much smoother.
 | ||
| Fig. 3 (a) AFM images (∼100 nm on Si), and (b) SEM images (∼50 nm on CNT) of LiEG and LiEGCO films. | ||
In the growth-rate versus deposition-temperature plot for the ternary LiHMDS + EG + CO2 process, a clear turning point is seen at 140 °C. The same is seen in FTIR spectra, where the characteristic peaks of alkyl carbonate complexes (1654 and 825 cm−1) disappear and new vibrations arise at 1450 and 2850 cm−1 (ESI Fig. 2†). We believe that the reasons behind these observations are the formation of Li2CO3, as well as the decomposition of the alkyl carbonate complex and regeneration of some forms of aliphatic alkoxides. This implies that the LiEGCO process is quite well controlled up to 120 °C.
To investigate the conformality of our processes, we deposited LiEG and LiEGCO on top of cleaned multi-walled carbon nanotubes (CNT) prepared by spray painting on top of Cu foil (Fig. 3b). Carbon nanotubes were chosen as a model substrate because of their decently high aspect ratio and because they represent an actual electrode surface. The CNTs were coated by both LiEG and LiEGCO very evenly and continuously, as expected in the case of an ideal ALD/MLD process. The coatings do not necessary penetrate very deep into the CNTs – but this was expected since, during the deposition, the precursor flow does not travel through the CNT sample. However, it is very clear that the films effortlessly cover the whole easily accessible surface area. Moreover, no visual differences between the two samples were observed.
So, what is the structure of our LiEG and LiEGCO films? We performed GIXRD measurements for both films, but no peaks were seen when the measurement was done directly after deposition. Luckily, FTIR turned out to be a valuable tool for gaining understanding about the chemical state of the films, as there exists plenty of information about the vibrations of lithium alkyl carbonates and lithium alkoxides to compare the data to.3,42 In Fig. 4, we have annotated the FTIR spectra of our LiEG and LiEGCO films to our best ability, according to previous results and literature.3,42–46
 | ||
| Fig. 4 FTIR spectra of LiEG with 400 ALD/MLD cycles and LIEGCO with 300 ALD/MLD cycles. The vibration is color-coded (blue for LiEGCO and red for LiEG) if it is only visible in either of the materials, and in black if it is a shared vibrational mode. The sharp peak at 667 cm−1 is caused by the silicon substrate. | ||
The spectrum of LiEG shares a lot of characteristics with those of other metal ethylene glycoxides,10,40–42,44,46,47 and in particular with the FTIR spectra reported by Aurbach et al.,48 where the most intense peaks are caused by C–H stretches below 3000 cm−1 and a C–stretch vibration around 1080 cm−1. In early studies reporting the synthesis of LiEG, its structure was assumed to be dilithium ethylene glycoxide.48 Recently, it was shown that monolithium ethylene glycoxide is virtually insoluble in ethereal solvents and thus always precipitates before it has time to deprotonate for a second time.3 Also importantly, a similar trend was seen with other alkali metal alkoxides, where only mono-deprotonated EG formed, even if a highly activated form of metal was used.49 In addition, the crystal structure of monolithium ethylene glycoxide is reported and known from single crystal data.50 In this structure, EG forms a bidentate complex with lithium and strong hydrogen bonds to an adjacent complex. Therefore, it is a reasonable assumption that dilithium ethylene glycoxide does not actually form very easily, if at all in normal synthesis conditions. Normally, one would expect to see strong absorption peaks for O–H at high frequencies, but they are absent. However, with strong and coordinated hydrogen bonds present, we believe that the O–H peaks are superimposed with C–H peaks below 3000 cm−1.49,51–54 This would explain the abnormally strong absorption intensity in this range, and why these peaks are dampened when the material is reacted with CO2. The same conclusion was made in the case of other alkali metal glycoxides.49 To test this, we synthesized a conventional bulk LiEG sample by immersing Li-foil in ethyl glycol and, after evaporating the solvent, measured the resulting white solid using ATR-IR. The IR spectrum matched well with the one measured for our thin film and, most importantly, lacked any O–H peaks at frequencies above 3000 cm−1. This supports our initial hypothesis of the O–H and C–H peaks being superimposed (ESI Fig. 3†). Comparing the spectrum of our LiEG film to that of liquid EG, one can clearly see the difference in the C–H vibration range, where EG only has a doublet due to C–H, while LiEG has at least four distinguishable peaks (ESI Fig. 4†).
The LiEG films were found to be very sensitive towards moisture: they rapidly began to deteriorate in a humid atmosphere, as seen from a clear peak caused by LiOH (a characteristic O–H vibration at ∼3700 cm−1), in line with observations by Aurbach et al.46 In addition, the C–H peaks dramatically changed in intensity and position, further supporting their proposed superimposition with O–H vibrations (ESI Fig. 5†).
When the additional CO2 pulse was introduced in the LiHMDS + EG process, the changes in the FTIR spectrum were quite drastic. Intense peaks appeared at 1650 and 1305 cm−1. Such peaks are characteristic for various types of carbonyl vibrations and match well with those previously reported for lithium alkyl carbonates,3,42,46,55 and they are clearly not caused by Li2CO3 (ESI Fig. 4†). Another clear medium-intensity peak appeared at 825 cm−1; it could readily be assigned to a CO3²⁻ bending mode.3,42,55 It should also be mentioned that the pulsing time of CO2 – 10 or 30 s – had no effect on the spectrum, which we took as a sign that the carbonation reaction saturates at 10 s (ESI Fig. 6†). When comparing the spectrum to one previously obtained for bulk LEMC,3 it appears that the two spectra share many features but do not exactly match (ESI Fig. 7†). LEMC has a characteristic strong O–H peak at ∼3400 cm−1, but we do not observe anything in that region for our LiEGCO film, which is a strong indication of our LiEGCO material being different from LEMC. We also compared our LiEGCO spectrum to one reported for bulk LEDC (though with additional DMSO solvent molecules); again, there are some common features but no perfect match. We also find that it is improbable that LEDC would actually be stable during the ALD/MLD cycles, as Wang et al.3 reported that there exists a delicate balance in solution, where monolithiated ethylene glycoxide will react with LEDC, generating LEMC and dilithium ethylene mono-carbonate (DLEMC). This leads us to believe that there are two plausible scenarios. Either the conversion reaction with CO2 is never complete and there always exists some LiEG in the film, making the comparison of IR spectra unreliable; or the structure is in fact DLEMC, for which no IR spectrum has previously been reported. In conclusion, due to the amorphous state of the films, it is nearly impossible to accurately determine the structure of LiEGCO; however, it most probably contains alkyl carbonate moieties without –OH groups.
Tentatively, we propose that the growth of our LiEGCO films could, within one ALD/MLD cycle, proceed as follows (Fig. 5): (i) LiHMDS reacts with the –OH-covered surface to form Li–O; (ii) a following EG pulse reacts with the Li–O to create a bidentate LiEG complex with coordinated hydrogen bonds; (iii) during the subsequent CO2 pulse, the alkoxide bond breaks and the alkyl carbonate LEMC forms, possibly also disturbing the tight hydrogen bond, such that (iv) the remaining –OH group can then be lithiated with another pulse of lithium, generating one layer of DLEMC. Naturally, this is a simplified scheme (with e.g. the effects of ligands being ignored), and the actual surface chemistry of LiHMDS can be significantly more complicated.56 It should also be noted that the scheme does not explain the full growth behavior of LiEG.
 | ||
| Fig. 5 Schematic of a possible growth behaviour during the cycling of LIEGCO. | ||
From previous literature,46,55,57 it could be anticipated that both LiEG (through hydrolysis to LiOH) and LiEGCO (directly) would decompose into Li2CO3 (possibly also generating ethene) upon storage in ambient conditions.2,57 This was indeed seen – both as emerging strong peaks at ∼1450 cm−1 in IR spectra, and as small peaks at 30.5° and 32° in GIXRD data, matching with Li2CO3 (ESI Fig. 8 and 9†). We also heat-treated our as-deposited films within the ALD reactor at 200 °C for 2 hours immediately after deposition (carried out at 80 °C). As expected, the LiEG films remained unchanged, while the LiEGCO films clearly started to decompose into Li2CO3 (ESI Fig. 10†).
Finally, it should be mentioned that other groups have reported ALD/MLD metal-alkoxide thin films for which FTIR peaks were seen to arise in the 1700–1400 cm−1 region after extended storage in ambient atmosphere; this was attributed to dehydration of the film and generation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vinyl and CO carbonyl vibrations, as the CO vibrations were also seen with XPS.10,28,30 However, one should also consider the possibility of formation of alkyl carbonates, especially with alkaline and earth-alkaline metal alkoxides. This was nicely shown to be the case for calcium alkoxide, for which two different pathways to CaCO3 were found – one through hydrolysis of calcium alkoxide and the second through calcium carbonate species.58 This is also the case for Li-propanediol thin films, where the carbonate species were initially formed upon ambient exposure.38 However, with our LiEG films, we only observed the hydrolysis route, but this might be highly sensitive to ambient atmospheric conditions.
C vinyl and CO carbonyl vibrations, as the CO vibrations were also seen with XPS.10,28,30 However, one should also consider the possibility of formation of alkyl carbonates, especially with alkaline and earth-alkaline metal alkoxides. This was nicely shown to be the case for calcium alkoxide, for which two different pathways to CaCO3 were found – one through hydrolysis of calcium alkoxide and the second through calcium carbonate species.58 This is also the case for Li-propanediol thin films, where the carbonate species were initially formed upon ambient exposure.38 However, with our LiEG films, we only observed the hydrolysis route, but this might be highly sensitive to ambient atmospheric conditions.
Conclusions
We have developed two new ALD/MLD processes, based on precursors LiHMDS and EG, for depositing thin films of two intriguing aliphatic lithium–organic materials. The key advancement was the reaction of our LiEG lithium alkoxide films with CO2 as the third precursor to the more stable and application-wise more attractive alkyl carbonate (LiEGCO) films. Carbon dioxide has been previously used in ALD for the deposition of metal carbonates, but – to our best knowledge – never before in ALD/MLD for the production of alkyl carbonates.Both processes exhibited typical ALD/MLD behavior, with growth rates in the range of 2.5 to 3 Å per cycle. They yielded uniform coatings on CNTs, with the only notable difference being film roughness upon exposure to ambient atmosphere. The films were amorphous, so only the chemical bonding structure could be investigated through FTIR analysis; this revealed features related to monolithium ethylene glycoxide (LiEG) and dilithium ethylene mono-carbonate (DLEMC).
Lithium alkyl carbonates have been shown to constitute the major organic component in the important SEI layers in Li-ion batteries. Crucially, we have now developed reaction schemes for the ALD/MLD fabrication of artificial SEI coatings that closely mimic the naturally forming SEI layers. We strongly believe that depositing these coatings on top of e.g. carbon electrodes would allow us to better understand the behavior of SEIs in LIBs. Moreover, since the reaction scheme with CO2 should proceed similarly for all group 1 and 2 alkoxides, our approach could open up completely new avenues for depositing other organic alkyl carbonates as well.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from Academy of Finland (296299), and the use of the RawMatTERS Finland Infrastructure (RAMI) and the OtaNano – Nanomicroscopy Center (Aalto-NMC) at Aalto University.Notes and references
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- L. Wang, A. Menakath, F. Han, Y. Wang, P. Y. Zavalij, K. J. Gaskell, O. Borodin, D. Iuga, S. P. Brown, C. Wang, K. Xu and B. W. Eichhorn, Nat. Chem., 2019, 11, 789–796 CrossRef CAS PubMed.
- K. Leung, F. Soto, K. Hankins, P. B. Balbuena and K. L. Harrison, J. Phys. Chem. C, 2016, 120, 6302–6313 CrossRef CAS.
- Y. Zhao, K. Zheng and X. Sun, Joule, 2018, 2, 2583–2604 CrossRef CAS.
- B. H. Lee, B. Yoon, A. I. Abdulagatov, R. A. Hall and S. M. George, Adv. Funct. Mater., 2013, 23, 532–546 CrossRef CAS.
- K. R. Adair, C. Zhao, M. N. Banis, Y. Zhao, R. Li, M. Cai and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 1–7 CrossRef PubMed.
- J. M. Wallas, B. C. Welch, Y. Wang, J. Liu, S. E. Hafner, R. Qiao, T. Yoon, Y. T. Cheng, S. M. George and C. Ban, ACS Appl. Energy Mater., 2019, 2, 4135–4143 CrossRef CAS.
- X. Li, M. Banis, A. Lushington, X. Yang, Q. Sun, Y. Zhao, C. Liu, Q. Li, B. Wang, W. Xiao, C. Wang, M. Li, J. Liang, R. Li, Y. Hu, L. Goncharova, H. Zhang, T. K. Sham and X. Sun, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- A. A. Dameron, D. Seghete, B. B. Burton, S. D. Davidson, A. S. Cavanagh, J. A. Bertrand and S. M. George, Chem. Mater., 2008, 20, 3315–3326 CrossRef CAS.
- Y. Zhao and X. Sun, ACS Energy Lett., 2018, 3, 899–914 CrossRef CAS.
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S.-H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S.-H. Lee, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS PubMed.
- Y. Zhao, M. Amirmaleki, Q. Sun, C. Zhao, A. Codirenzi, L. V. Goncharova, C. Wang, K. Adair, X. Li, X. Yang, F. Zhao, R. Li, T. Filleter, M. Cai and X. Sun, Matter, 2019, 1, 1215–1231 CrossRef.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735–18758 RSC.
- D. Molina Piper, S.-B. Son, J. J. Travis, Y. Lee, S. S. Han, S. C. Kim, K. H. Oh, S. M. George, S.-H. Lee and C. Ban, J. Power Sources, 2015, 275, 605–611 CrossRef CAS.
- Y. Sun, Y. Zhao, J. Wang, J. Liang, C. Wang, Q. Sun, X. Lin, K. R. Adair, J. Luo, D. Wang, R. Li, M. Cai, T.-K. K. Sham and X. Sun, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- O. Nilsen, K. Klepper, H. Nielsen and H. Fjellvåg, ECS Trans., 2008, 16, 3–14 CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- T. Suntola, Mater. Sci. Rep., 1989, 4, 261–312 CrossRef CAS.
- T. Suntola and J. Antson, US Pat. 4058430, 1977.
- T. Tynell, A. Giri, J. Gaskins, P. E. Hopkins, P. Mele, K. Miyazaki and M. Karppinen, J. Mater. Chem. A, 2014, 2, 12150–12152 RSC.
- T. Tynell, I. Terasaki, H. Yamauchi and M. Karppinen, J. Mater. Chem. A, 2013, 1, 13619 RSC.
- M. Nisula and M. Karppinen, Nano Lett., 2016, 16, 1276–1281 CrossRef CAS PubMed.
- M. Nisula and M. Karppinen, J. Mater. Chem. A, 2018, 6, 7027–7033 RSC.
- J. Heiska, M. Nisula, E.-L. Rautama, A. J. Karttunen and M. Karppinen, Dalton Trans., 2020, 49, 1591–1599 RSC.
- C. MacIsaac, J. R. Schneider, R. G. Closser, T. R. Hellstern, D. S. Bergsman, J. Park, Y. Liu, R. Sinclair and S. F. Bent, Adv. Funct. Mater., 2018, 28, 1800852 CrossRef.
- D. S. Bergsman, J. G. Baker, R. G. Closser, C. MacIsaac, M. Lillethorup, A. L. Strickler, L. Azarnouche, L. Godet and S. F. Bent, Adv. Funct. Mater., 2019, 29, 1904129 CrossRef CAS.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- K. Van de Kerckhove, M. K. S. Barr, L. Santinacci, P. M. Vereecken, J. Dendooven and C. Detavernier, Dalton Trans., 2018, 47, 5860–5870 RSC.
- D. Kang, N. Hart, J. Koh, L. Ma, W. Liang, J. Xu, S. Sardar and J. P. Lemmon, Energy Storage Materials, 2020, 24, 618–625 CrossRef.
- M. Kunert, P. Wiegeleben, H. Görls and E. Dinjus, Inorg. Chem. Commun., 1998, 1, 131–133 CrossRef CAS.
- T. P. Whaley, The Chemistry of Lithium, Sodium, Potassium, Rubidium, Cesium and Francium, Elsevier, 1973 Search PubMed.
- J. K. Varghese, T. P. Gonçalves, K. W. Huang, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2017, 50, 6752–6761 CrossRef CAS.
- E. Østreng, P. Vajeeston, O. Nilsen and H. Fjellvåg, RSC Adv., 2012, 2, 6315 RSC.
- O. Nilsen, V. Miikkulainen, K. B. Gandrud, E. Østreng, A. Ruud and H. Fjellvåg, Phys. Status Solidi, 2014, 211, 357–367 CrossRef CAS.
- M. Nisula, J. Linnera, A. J. Karttunen and M. Karppinen, Chem.–Eur. J., 2017, 23, 2988–2992 CrossRef CAS PubMed.
- H. Wang, K. E. Gregorczyk, S. B. Lee, G. W. Rubloff and C. Lin, J. Phys. Chem. C, 2020, 124, 6830–6837 CrossRef CAS.
- P. C. Lemaire, C. J. Oldham and G. N. Parsons, J. Vac. Sci. Technol., A, 2016, 34, 01A134 CrossRef.
- X. Wang, X. Bi, S. Zheng, S. Wang, Y. Zhang, H. Du and J. Lu, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed.
- D. Wang, R. Yu, N. Kumada and N. Kinomura, Chem. Mater., 1999, 11, 2008–2012 CrossRef CAS.
- S. Matsuta, T. Asada and K. Kitaura, J. Electrochem. Soc., 2000, 147, 1695–1702 CrossRef CAS.
- P. Larkin, Infrared and Raman Spectroscopy, Elsevier, 2011, vol. 9 Search PubMed.
- A. P. Simonov, D. N. Shigorin, G. V. Tsareva, T. V. Talalaeva and K. A. Kocheshkov, J. Appl. Spectrosc., 1965, 3, 398–403 CrossRef.
- K. Xu, G. V. Zhuang, J. L. Allen, U. Lee, S. S. Zhang, P. N. Ross and T. R. Jow, J. Phys. Chem. B, 2006, 110, 7708–7719 CrossRef CAS PubMed.
- D. Aurbach, J. Electrochem. Soc., 1989, 136, 3198 CrossRef CAS.
- B. Ksapabutr, E. Gulari and S. Wongkasemjit, Mater. Chem. Phys., 2004, 83, 34–42 CrossRef CAS.
- D. Aurbach, J. Electrochem. Soc., 1989, 136, 1606 CrossRef CAS.
- L. M. C. Silva, P. G. Rosado, J. B. Branco, M. A. Antunes and J. P. Leal, J. Chem. Thermodyn., 2017, 115, 332–341 CrossRef CAS.
- M. Mamak, P. Y. Zavalij and M. S. Whittingham, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 937–939 CrossRef.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Springer Netherlands, Dordrecht, 1980, vol. 3 Search PubMed.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, in The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Elsevier, 1991, vol. 42, pp. 117–154 Search PubMed.
- Z. Dega-Szafran, M. Petryna, E. Tykarska and M. Szafran, J. Mol. Struct., 2002, 643, 69–75 CrossRef CAS.
- E. Selli, A. Isernia and L. Forni, Phys. Chem. Chem. Phys., 2000, 2, 3301–3305 RSC.
- D. Aurbach, J. Electrochem. Soc., 1989, 136, 1611–1614 CrossRef CAS.
- Y. Tomczak, K. Knapas, M. Sundberg, M. Leskelä and M. Ritala, J. Phys. Chem. C, 2013, 117, 14241–14246 CrossRef CAS.
- W. Behrendt, G. Gattow and M. Dräger, Z. Anorg. Allg. Chem., 1973, 397, 237–246 CrossRef CAS.
- M. Favaro, P. Tomasin, F. Ossola and P. A. Vigato, Appl. Organomet. Chem., 2008, 22, 698–704 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00254b |
| This journal is © The Royal Society of Chemistry 2020 |