
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConnectability of protein cages
Karolina
Majsterkiewicz
 ab,
Yusuke
Azuma
a and
Jonathan G.
Heddle
*a
ab,
Yusuke
Azuma
a and
Jonathan G.
Heddle
*a
aMałopolska Centre of Biotechnology, Jagiellonian University, Gronostajowa 7A, 30-387 Krakow, Poland. E-mail: jonathan.heddle@uj.edu.pl
bPostgraduate School of Molecular Medicine, Trojdena 2a, 02-091 Warsaw, Poland
First published on 18th May 2020
Abstract
Regular, hollow proteinaceous nanoparticles are widespread in nature. The well-defined structures as well as diverse functions of naturally existing protein cages have inspired the development of new nanoarchitectures with desired capabilities. In such approaches, a key functionality is “connectability”. Engineering of interfaces between cage building blocks to modulate intra-cage connectability leads to protein cages with new morphologies and assembly–disassembly properties. Modification of protein cage surfaces to control inter-cage connectability enables their arrangement into lattice-like nanomaterials. Here, we review the current progress in control of intra- and inter-cage connectability for protein cage-based nanotechnology development.
1. Introduction
Protein cages are hollow, well-defined structures formed by self-assembly of repetitive protein building blocks. In nature, such proteinaceous compartments are widely spread across many different species, and have broad functionalities and morphologies (Fig. 1).1–5 The most well-known example are viral capsids that package their own genomic information and deliver it to host cells. Virus-like particles, capsids lacking genetic material, are typically composed of one to a few kinds of protein that spontaneously assemble into icosahedral symmetrical cages of a variety of morphologies, where the lumenal volume depends on their genome type and size (Fig. 1I and II).6,7 Another abundant protein cage ferritin, which acts as an iron storage compartment, typically forms 24-meric cages with an octahedral symmetry (Fig. 1III).8 Protein cages also serve as reaction chambers for metabolic enzymes such as encapsulins9 and lumazine synthases.10 Both form dodecahedral assemblies consisting of 12 pentameric subunits (Fig. 1IV). These molecular containers can transport and protect specific cargoes and/or provide isolated spaces for biological processes. | ||
| Fig. 1 Protein cage comparison. (I and II) Cryo-EM structures of the prolate head of bacteriophage T4 (EMD: 6323) (I)1 and the procapsid shell of bacteriophage P22 (EMD: 5149) (II).2 (III and IV) Crystal structures of human heavy chain ferritin (PDB: 2FHA) (III)3 and Aquifex aeolicus lumazine synthase (PDB: 1HQK) (IV).4 Note the difference in the scale bars, 100 Å for (I), and (II) and 10 Å for (III) and (IV). | ||
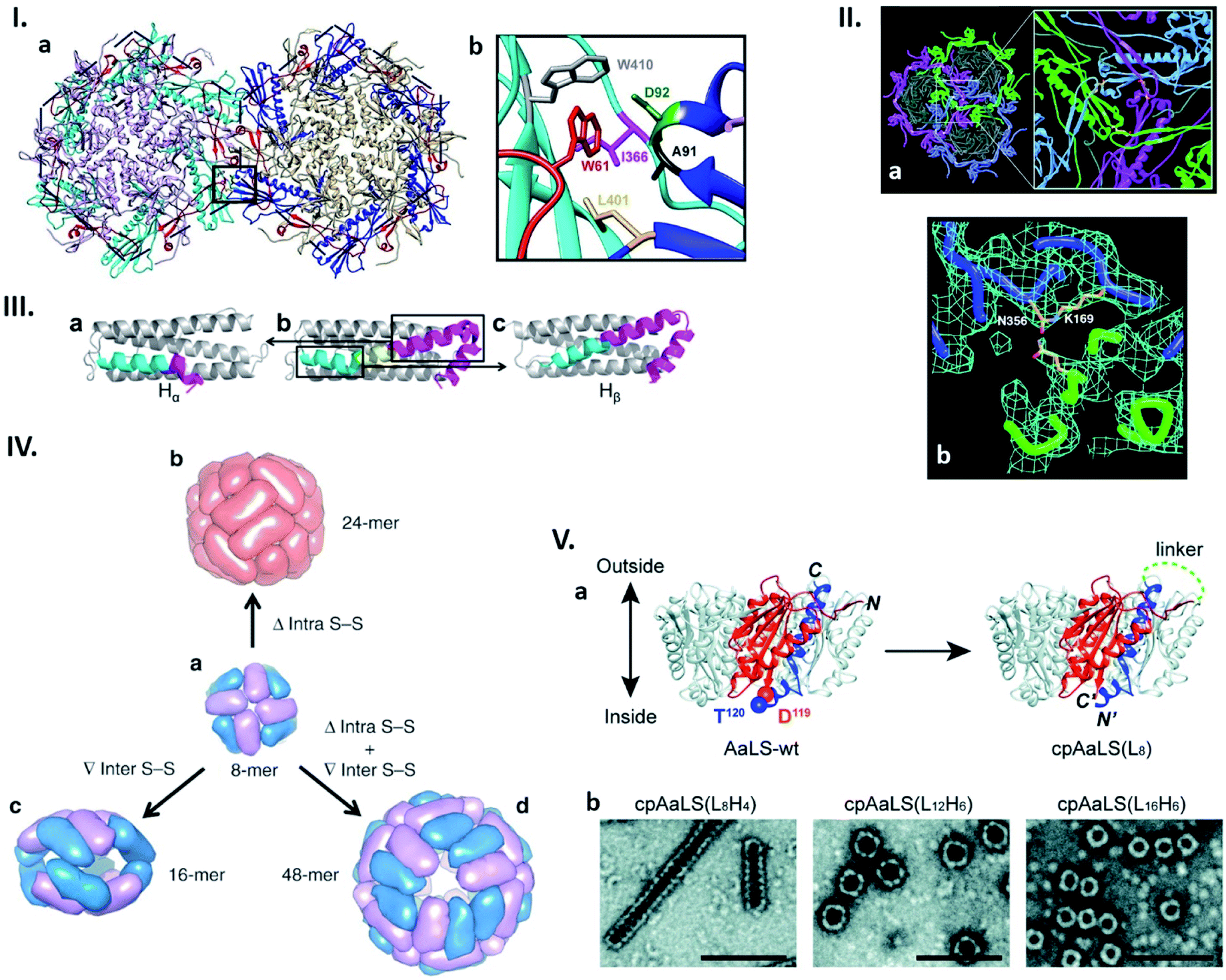 | ||
| Fig. 2 Connectability in natural and re-engineered protein capsids. (I) Hydrophobic interactions stabilizing the bacteriophage P22 capsid. (a) Two neighbouring capsomers of P22. (b) Enlarged image of the region surrounded by the box in (a) shows the residues forming the hydrophobic peg and pocket. Highlighted residues were subjected to mutagenesis. Reproduced with permission from ref. 19; Copyright (2019) American Society for Microbiology. (II) Bacteriophage HK97 capsomer cross-linking. (a) Three neighbouring subunits are covalently linked by isopeptide bonds (highlighted in the enlarged view). (b) Electron density map of the isopeptide bond formed by the ε amino group of the Lys169 of one subunit and the γ carbon of the Asn356 amide group of the other subunit. Reproduced with permission from ref. 23; Copyright (2000) The American Association for the Advancement of Science. (III) Change in the structure of the native human heavy chain ferritin (HFtn) subunit after deletion of six amino acids residues. Native HFtn subunit (b) with highlighted deleted amino acids (yellow) and two parts of the helix, which are rearranged after deletion. When the C-terminal part of the helix (purple) moves toward the N-terminal part (cyan), a subunit termed Hα is produced (a). The opposite rearrangement generates a subunit termed Hβ (c). Reproduced with permission from ref. 25; Copyright (2016) American Chemical Society. (IV) Regulation of the ferritin quaternary states by introducing/deleting disulphide bonds. (a) Four of each subunit, Hα (blue) and Hβ (purple), assemble into 8-mer bowl-like proteins (NF-8). (b) Deletion of intra S–S bond results in the conversion of NF-8 into a 24-mer ferritin-like protein cage (red). (c) Insertion of inter S–S bonds led to the conversion of NF-8 into a 16-mer protein cage. (d) Deletion of the same intra S–S and insertion of the inter S–S bonds caused the conversion of NF-8 into a 48-mer protein cage. Reproduced with permission from ref. 26; Copyright (2019) Springer Nature. (V) Circular permutation of Aquifex aeolicus lumazine synthase (AaLS). (a) Structure of the wild type AaLS (AaLS-wt) pentamer (left). One monomer unit is coloured: residues 1–119, orange; residues 120–156, blue. The native termini in AaLS-wt are connected by peptide linker with different lengths ((LxHy), where x and y represent the number of total amino acids and histidines, respectively) and new termini are introduced between residues 119 and 120, resulting in circularly permuted AaLS (cpAaLS) (right). (b) The assemblies of cpAaLS variants possessing different linkers form structures depicted on negative stain TEM images (scale bar = 100 nm). Reproduced from ref. 30 with permission from The Royal Society of Chemistry. | ||
The wide variety of structures and functionalities demonstrated by nature has inspired researchers to develop useful nanodevices based on protein cages. There are numerous studies exemplifying their significant potential to serve as vaccines,11 drug delivery vehicles,12 nanoreactors,13,14 templates for synthesis of inorganic nanoparticles,15,16 and catalytic materials.17,18 In such applications, characteristics of naturally-existing protein cages need to be tailored for individual purposes. An obvious example is guest selectivity. While natural cages often encapsulate specific guest molecules, e.g. genomic DNA/RNA in the case of viral capsids, packaging of unrelated, foreign cargoes is required for most applications. Lumenal volume and surface charges may need to be adjusted for such guests. For delivery purposes, controlled assembly–disassembly of protein cages is advantageous for packaging and releasing cargo molecules at desired timings and locations. Given these diverse demands, there is considerable interest in development of new methodologies to engineer protein cages possessing desired properties.
A key functionality in protein cage design is “connectability”. Protein cages are constructed from individual protein subunits. In virus capsids these are called capsomers, a term we abuse here to refer to the basic building block units of protein cages in general. The capsomers self-assemble into cages through protein–protein interactions composed of highly organized non-covalent bonding networks. Rearrangement of such capsomer–capsomer interfaces to modulate their connectability leads to new morphologies, controlled stability, and triggered assembly/disassembly of protein cages. Additionally, engineering of protein cage surfaces can endow them with new connectability to self or partner molecules, enabling their arrangement in ordered macrostructures. In this review, we highlight the recent advances in engineering of intra- and inter-cage connectability towards custom-design of protein cage-based technologies.
2. Connectability of capsomer interface
In nature, protein cage assembly occurs through the large number of identical interactions between capsomer subunits. Each interface is predominantly composed of non-covalent interactions including hydrogen bonds, hydrophobic interactions and π–π stacking as well as electrostatic interactions. Since the stability and morphology of cage structures are established on highly organized networks of these interactions, their modification may result in a variety of cage structures with morphologies not observed in nature. In this section, we describe recent efforts to understand the theory underlying the hierarchical assembly of naturally-existing protein cages, to develop new nanoarchitectures by reengineering them, as well as to generate entirely artificial protein cages by connecting protein building blocks that naturally do not assemble into cage structures.2.1. Cage formation through protein–protein interactions
Due to their repetitive and cooperative interactions, a small number of amino acid changes in monomer subunits of cage-forming proteins can have a significant impact on the assembled structures. One such example was recently shown by Asija et al. with the bacteriophage P22 capsid (Fig. 2I).19 Single mutations in the residues that form a hydrophobic network in the capsomer–capsomer interface lead to destabilization of the assembly and/or morphological immaturity. Similarly, a recent cryo-EM single particle reconstruction study on T4 bacteriophage revealed that a mutation near the quasi-threefold axis of the hexameric subunits changes the angles between adjacent capsomers, converting the wild type prolate structure to isomeric assemblies.20 Such flexible and sensitive morphology is assumed to be a common feature across proteins sharing the same protein fold, Hong Kong 97 (HK97). This is shared with many other cage-forming proteins including capsids derived from herpes simplex virus type 1 (HSV-1), murine cytomegalovirus (MCMV), and bacterial nanocompartment encapsulins.21,22 Nature can further avoid undesired morphology changes by stabilizing the assembly through formation of a covalent isopeptide bond between a lysine of one coat protein and an asparagine of an adjacent subunit as observed in the mature capsid of Hong Kong 97 virus (Fig. 2II).23Naturally existing protein cages have been subjected to more intensive reengineering to modulate their assembled morphologies. Zhang et al. have shown this in human heavy chain ferritin (HFtn), a cage composed of 24 identical monomer units with octahedral symmetry. Here a peptide insertion and deletion on the key subunit interface results in cage that is a 16-mer assembly with a lower symmetry24 as well as 48-mer cage consisting of equal numbers of two different subunits derived from the same polypeptide (Fig. 2III),25 respectively. These different quaternary states including the wild type-like 24-mer can be further regulated by deletion of intra- or introduction of inter-subunit disulphide bonds (Fig. 2IV).26 Circular permutation, a modality to alter the connectivity of secondary structure elements in a protein,27 has also been employed to modulate the assembly states of shell proteins derived from the propanediol utilization (Pdu) compartment and lumazine synthase.28,29 In the former protein's case, the tile-forming hexameric subunit was converted to a pentamer that self-assembles into a dodecahedral cage,28 while the latter forms expanded spherical cages, compared to the wild type assembly, as well as tubular structures with morphologies dependent on the length of the peptide linker connecting the native termini (Fig. 2V).29,30 These redesign approaches, together with mutagenesis studies, highlight how protein–protein interactions are precisely organized in naturally-existing protein cages to achieve defined structures. Their manipulation presents a powerful strategy towards production of proteinaceous compartments with designed size and shape.
Structures of naturally existing protein cages reliant on highly sophisticated non-covalent interaction networks have inspired the construction of entirely artificial ones with morphologies defined in a rational manner. Recent advances in computational approaches have enabled production of proteins that spontaneously assemble into defined quaternary structures. Such de novo designed protein cages are achieved by docking of oligomeric proteins into a target symmetric architecture (Fig. 3Ia–c), followed by engineering protein–protein interfaces to drive self-assembly (Fig. 3Id and e).31 This symmetry-based approach has resulted in an impressive library of protein architectures including two-component cages with tetrahedral and icosahedral symmetries.31–35
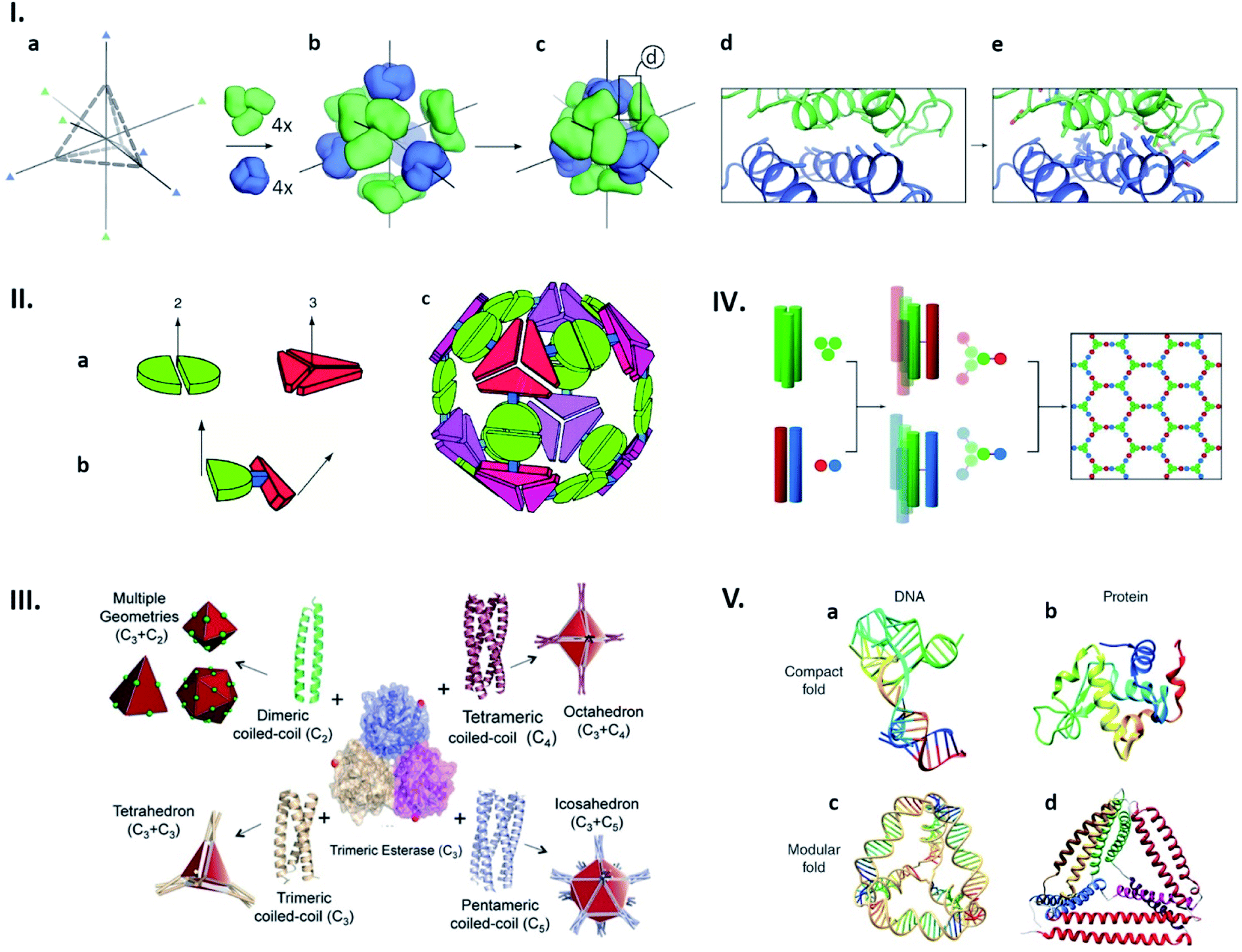 | ||
| Fig. 3 Strategies for artificial protein cage formation through protein–protein interactions. (I) Symmetry-based design and computationally re-engineered interfaces. After choosing initial architecture (here tetrahedral) with 3-fold symmetry axes (a) and arrangement of four copies each of two different trimeric proteins (green and blue) along symmetry axes (b and c), potential interfaces with multiple contacting amino-acids were remodelled using computational simulations (d and e). Reproduced with permission from ref. 31; Copyright (2014) Springer Nature. (II) Multimeric fusion proteins as building blocks. Dimeric and trimeric proteins, shown by the green and red shapes, respectively (a), are fused via semi-rigid linker (b), resulting in self-assembly into artificial cage (c). Reproduced with permission from ref. 36; Copyright (2001) National Academy of Sciences, U.S.A. (III) Coiled coils with different oligomerization states fused with trimeric esterase enable formation of cages with planned geometry. Red dot indicates esterase C-termini and a point of fusion. Reproduced with permission from ref. 42; Copyright (2017) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (IV) Self-assembled cage-like particles (SAGEs). Helixes from homotrimeric (green) and heterodimeric (red-blue) coiled coils are linked via disulphide bonds to form six-helix building blocks. When mixed, building blocks assemble into a hexagonal network. Reproduced with permission from ref. 45; Copyright (2013) American Association for the Advancement of Science. (V) Analogy between DNA and protein folds. (a and b) Native structures of a DNA aptamer (a) and a globular protein (b) can be formed by complex interactions between residues. (c and d) Modular folds (origami) of DNA (c) and polypeptide (d) are both stabilized by simple and defined pairing systems. Reproduced with permission from ref. 47; Copyright (2014) Wiley Periodicals, Inc. | ||
Cage-like structures can be constructed using protein building blocks that do not naturally form cages. This strategy was first devised by the Yeates group (Fig. 3II).36,37 In this concept, a protein which forms an oligomer is covalently fused to another protein which adapts a different quaternary state (Fig. 3IIa and b). The resulting fusion protein can self-assemble into symmetrical nanostructures (Fig. 3IIc). Since their first demonstration of a tetrahedral cage formation,36 this hybrid approach has been greatly expanded and diversified.38,39 Along the same lines, coiled-coil forming peptides afford ideal glues for connecting protein building blocks with designed symmetries.40,41 For example, a trimeric esterase equipped with three-, four- or five-helix coiled coils forms tetrahedral,42 octahedral,43 or icosahedral44 cages, respectively (Fig. 3II).42 Coiled-coil peptides also serve as the sole building blocks to construct self-assembled cage-like particles (SAGEs).45,46 SAGEs are composed of noncovalent heterodimeric and homotrimeric coiled coil bundles, where helices from different bundles are connected via disulphide bonds (Fig. 3IV).45 Furthermore, due to the well-defined pairing specificity, coiled coil peptides can be exploited to produce programmable macrostructures in a way somewhat analogous to the base-pairing complementarity seen in DNA origami (Fig. 3V).47–49 Together with considerations of symmetry, these approaches to connect protein building blocks using defined protein–protein interactions have greatly expanded the design flexibility of artificial protein cage construction.
2.2. Cage formation through metal coordination
In the design of artificial protein cages, a major concern is their controlled assembly and disassembly. Protein cages that can be disassembled into capsomer subunits by certain stimuli are particularly useful since they can liberate cargo molecules only at desired times and locations. This property may be highly desirable in a number of situations, delivery of cargo molecules to cells for instance. However, both naturally existing and artificially designed protein cages that assemble through protein–protein interaction networks are typically very stable and require harsh conditions, e.g. high concentration of chaotropic reagents, to induce their disassembly. To realise controlled (dis)assembly then, it is necessary to exploit alternative bonding chemistries for connecting capsomer subunits. In this regard, reversible metal coordination is one promising approach being utilised in artificial protein cage development.Metal-mediated assembly has been demonstrated in work where the native protein–protein interactions in a protein cage were transformed into engineered metal coordination sites. HFtn was used and two pairs of histidines were introduced at the C2 symmetrical interfaces, followed by elimination of nearby hydrophobic interactions and hydrogen bonds by mutagenesis.50 While wild type HFtn spontaneously assembles, the resulting variants were shown to assemble only in the presence of Cu(II) into the wild type-like cage structure that can be disassembled into the monomer units by addition of a chelating agent, ethylenediaminetetraacetic acid (EDTA). A similar replacement strategy has been employed in the above mentioned coiled-coil mediated cage formation. When the trimeric esterase was equipped with a coiled-coil motif designed to trimerize upon binding with divalent metal ions, the defined tetrahedral cage formation was observed by addition of Ni(II), Co(II), Cu(II), and Zn(II).51
We have shown a simple, distinct strategy for construction of artificial protein cages by connecting protein subunits via metal coordination (Fig. 4I).52–54 In this work, a toroidal shape protein, tryptophan RNA-binding attenuation protein (TRAP), was employed as the building block (Fig. 4Ia).52 TRAP mutants containing a cysteine residue around the rim of the torus were found to assemble into a uniform, hollow spherical structure by reaction with monovalent gold ions. Determination of the structure using cryo-EM revealed that the presence of Au(I) triggers TRAP rings to self-assemble into a protein cage consisting of 24 copies of the undecameric ring (Fig. 4Ib).52 Each ring is surrounded by five neighbouring rings and connected to each of them via two cross-linking thiol–Au(I)–thiol coordination bonds between opposing cysteines (Fig. 4Ic).52 Note that this Au(I)-mediated TRAP cage assembly can achieve a yield of >80%, remarkably higher than many other artificial protein cage formation efficiencies. The TRAP-cages show an extremely high stability under a variety of harsh conditions but can be easily disassembled into the capsomer units by addition of thiol- or phosphine-containing compounds including a major cellular reducing agent, glutathione. Such a disassembly characteristic may mark the TRAP-cage as suitable as a vehicle for cytosolic delivery of cargo molecules.
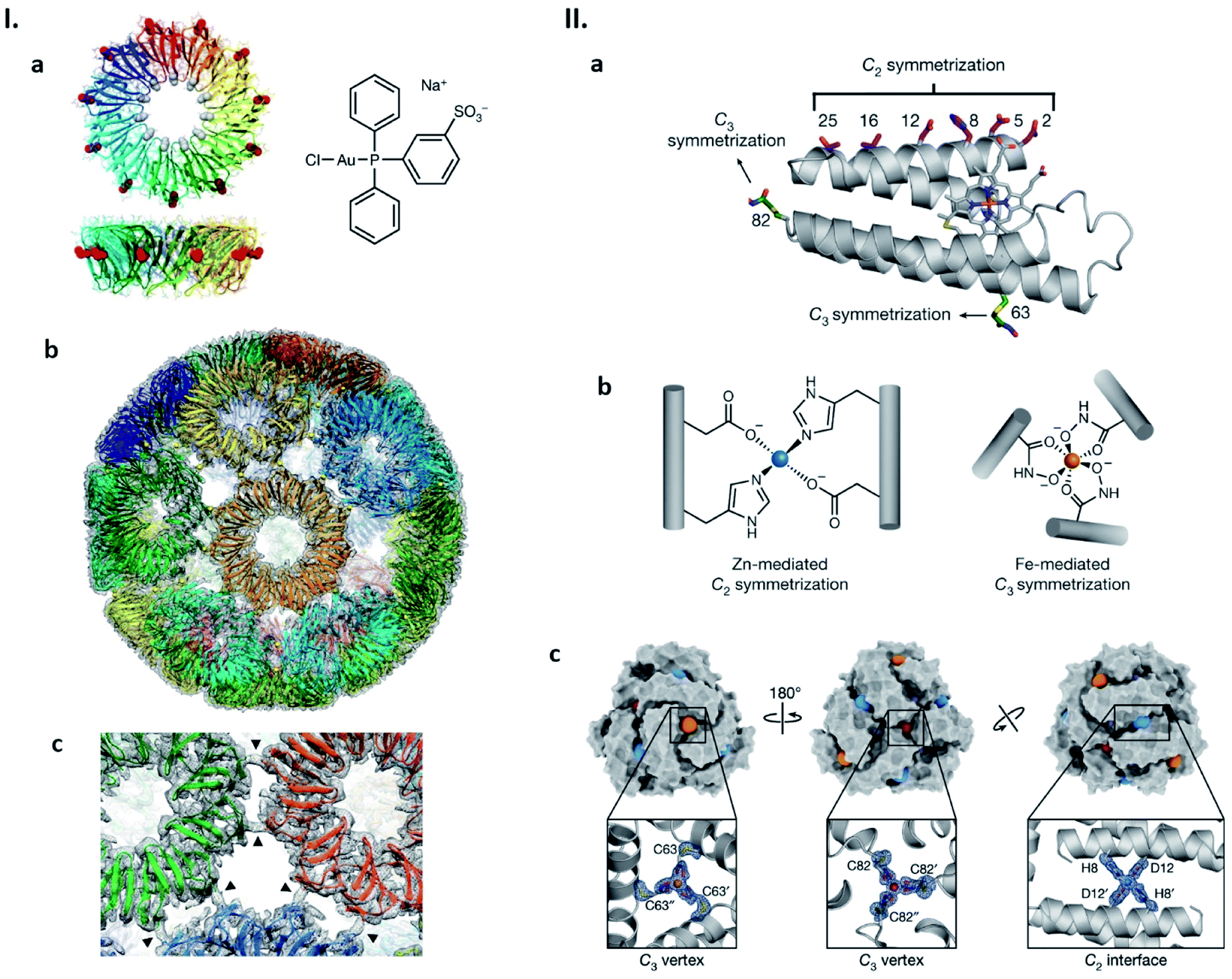 | ||
| Fig. 4 Strategies for artificial protein cage formation through metal coordination. (I) Au(I)-mediated TRAP-cage assembly. (a) TRAP ring structure in two orthogonal views with substituted Cys35 shown as red spheres (left), and the compound used as the Au(I) source (right). (b) Model of TRAP-cage overlapped with its electron density map. Gold atoms are shown as spheres. (c) Magnified image at the TRAP ring–ring interface. The arrowheads indicate density bridges connecting neighbouring rings. Reproduced with permission from ref. 52; Copyright (2019) Springer Nature. (II) Components and structure of bimetallic cages. (a) Structure of cytochrome cb562. Residues constituting metal-binding sites are shown as sticks. (b) Zn(II) and Fe(III)-binding motifs inducing protein dimerization and trimerization, respectively. (c) Structure of the dodecameric bimetallic cage with enlarged Zn(II) and Fe(III)-binding motifs. Fe(III) and Zn(II) ions are represented as orange/red and blue spheres, respectively. Reproduced with permission from ref. 55; Copyright (2020) Springer Nature. | ||
Appropriate ligand selection and its precise positioning while taking into account coordination chemistry are important considerations for achieving metal-mediated protein cage assemblies. In the case of the TRAP cysteine mutant, cage formation was observed with metal ions, Au(I) and Hg(II), possessing a preference for linear coordination with soft ligands. Little or no TRAP-cage assemblies were seen upon the addition of Au(III), Cu(I) and Zn(II). The Tezcan group has considered such aspects carefully and recently described cytochrome cb562 variants in which hydroxamate groups were introduced at appropriate positions via chemical methods and zinc-binding motifs by mutagenesis (Fig. 4IIa and b).55 Upon simultaneous addition of Fe(III) and Zn(II), the proteins assemble into dodecameric and hexameric cages. X-ray crystallography showed that the polyhedral cages have tightly tiled shells without the large pores that are often found in other artificial protein cages (Fig. 4IIc).55 Similar to the Au(I)-mediated TRAP-cage, disassembly of these bimetallic cages can be triggered by addition of compounds that remove or reduce the constituent metal ions.
3. Connectability of cage exterior
The building blocks of protein cages are DNA-encoded polypeptide chains displaying multiple functional groups on their side chains. This makes protein cages amenable to both chemical and genetic modifications. Exterior decoration using such methods can modulate protein cage connectability with other molecules. This allows their use as versatile platforms for medical- and nanotechnological-applications to be explored. While different approaches for such exterior decoration have been devised and summarised in several reviews,56,57 here we update and highlight recent efforts to functionalize protein cage surfaces with a particular focus on studies aiming at macromolecular display and lattice formation.3.1 Macromolecular display
Genetic fusion of peptides/proteins to cage-forming proteins is one of the most commonly used approaches for displaying targets on the exterior surface of cages. Due to the identical, multi-copy nature of the protein cage building blocks, this fusion approach typically leads to protein self-assemblies displaying a number of target polypeptides with high density. Since such polyvalent arrays are favourable for stimulating immunogenicity, protein cages have been extensively exploited as scaffolds to arrange antigens in vaccine development.11,58,59 As such, a computationally designed two-component protein cage has been genetically modified with envelope protein-derived antigens from respiratory syncytial virus (RSV) (Fig. 5I) and human immunodeficiency virus type 1 (HIV-1) (Fig. 5II) resulting in their decoration of the cage exterior.58,59 As expected, the protein cages displaying these viral antigens enhance in vitro immune responses, producing neutralizing antibodies. The fact that these two protein components do not assemble into the cage structure until mixed gives this system useful properties. For example, by mixing decorated and undecorated protein components prior to assembly, the density of displayed antigens can be controlled. Moreover, production and purification of individual protein components carrying antigen may allow them more easily to be correctly folded and purified prior to cage assembly.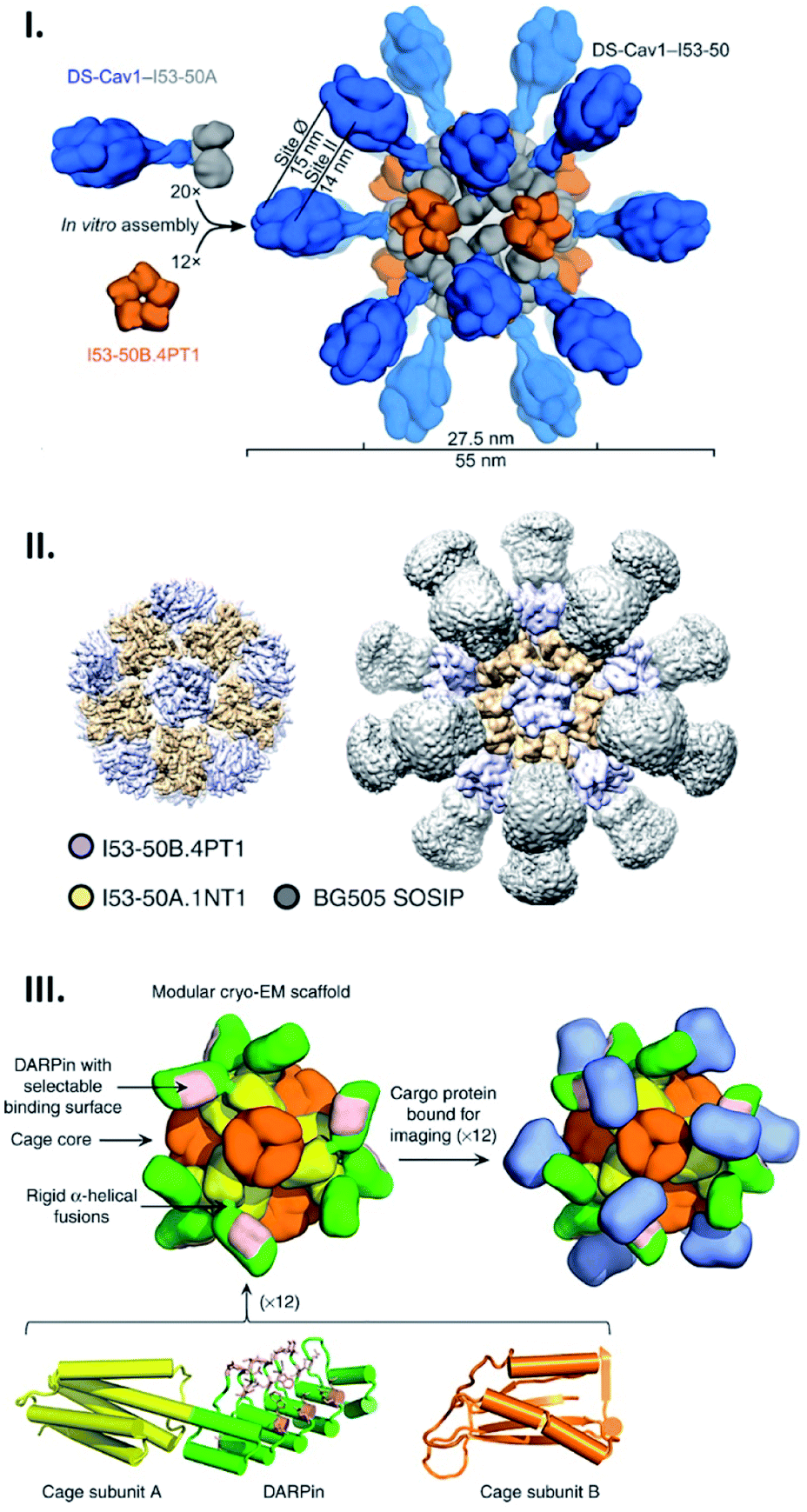 | ||
| Fig. 5 Artificial protein cages as a tool for macromolecular display. (I and II) Structures of two-component artificial cages genetically modified with envelope protein-derived antigens from RSV (DS-Cav1) (I) or HIV-1 (BG505 SOSIP) (II). The scaffold assembly consists of 20 trimeric (I53-50A) and 12 pentameric (I53-50B) building blocks, and antigens are fused with the trimeric subunits. Reproduced with permission from ref. 58 and 59; Copyright (2019) Elsevier and Springer Nature. (III) Structural model of protein scaffold for cryo-EM imaging. The artificial protein cage with tetrahedral symmetry is composed of 12 copies each of two protein subunits, A (yellow) and B (orange). DARPin (green) fused with subunit A via helical linker contains binding surface (pink) to capture cargo protein (blue) for cryo-EM imaging. Reproduced with permission from ref. 60; Copyright (2019) Springer Nature. | ||
Apart from their potential medical applications, protein cages have other possible uses: a cage has been recently demonstrated to serve as a scaffold in cryo-EM imaging to capture and structurally investigate an otherwise difficult-to-observe small protein.60 For this purpose, Liu et al. engineered an artificial 2-component protein cage displaying 12 copies of a designed ankyrin repeat protein (DARPin) targeting green fluorescent proteins (GFPs) using a genetic fusion strategy (Fig. 5III). This adaptor-conjugated nanoscaffold binds with GFPs, displaying them in an ordered manner, and at the same time results in a particle of much greater size than the isolated GFP. This facilitates imaging in cryo-EM followed by structure determination using single particle reconstruction. This technique resulted in the determination of the 3.8 Å structure of the 26 kDa GFP, something which would not have been achievable for the GFP alone.
3.2 Lattice formation
Protein cages can serve as building blocks that assemble into lattices. A common approach to form such structures utilizes electrostatic interactions, where protein cage surface charges are countered by complimentarily charged connectors to drive their assembly. Anionic protein cages, such as capsids from cowpea chlorotic mottle virus (CCMV) or ferritin from Pyrococcus furiosus (PfFtn), as well as polyvalent cations, e.g. dendrimers, have been exploited as building blocks and connectors in electrostatic-based lattice formation.61–65 As such, building on earlier work,63 we have recently described an ordered array based on ferritin from Thermotoga maritima (TmFtn) (Fig. 6I).66 These protein cages possessing a negatively charged exterior can assemble into superlattice structures when an appropriate size of positively charged gold nanoparticles (AuNPs) and ionic strength are provided.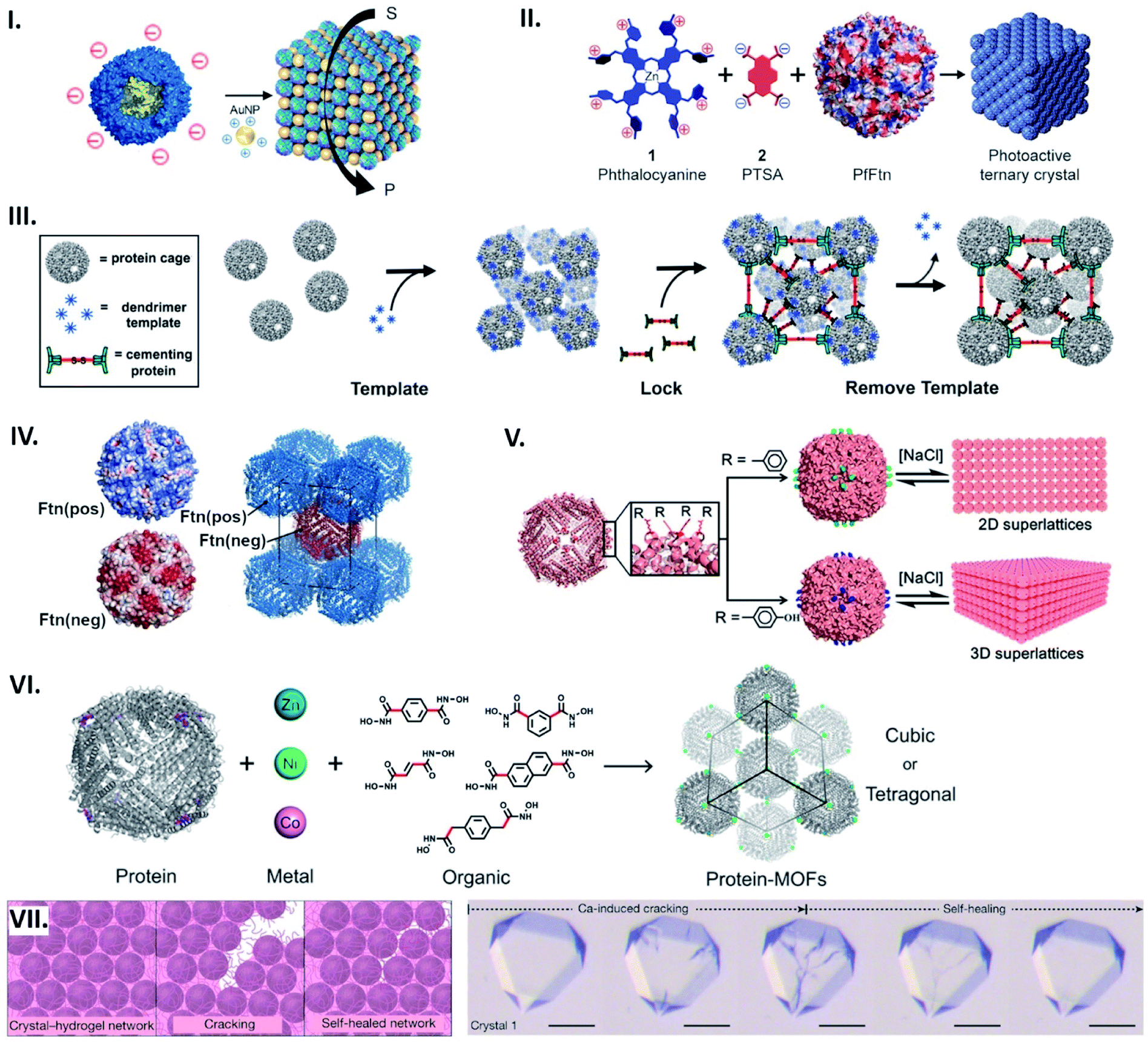 | ||
| Fig. 6 Assembling protein cages into lattices. (I) Common strategy for lattice assembly utilizing electrostatic interactions, where protein cage surface charges (e.g. TmFtn) are countered by complimentarily charged connectors (e.g. positively charged AuNPs) to drive their assembly. Reproduced with permission from ref. 66; Copyright (2019) American Chemical Society (DOI: 10.1021/acs.nanolett.9b01148, further permissions related to the material excerpted should be directed to the ACS). (II) An octacationic zinc phthalocyanine 1 and a tetraanionic pyrene 2 derivative used as a connector for construction of the ferritin lattice affords light-induced singlet oxygen production. Reproduced with permission from ref. 18; Copyright (2015) American Chemical Society (DOI: 10.1021/acsnano.5b07167, further permissions related to the material excerpted should be directed to the ACS). (III) Positively charged dendrimer serves as a template for lattice formation of P22 particles fused with negatively charged peptide. 3D structure is further locked with cementing protein, followed by template removal. Reproduced with permission from ref. 67; Copyright (2018) American Chemical Society. (IV) Engineered HFtn with positive (Ftn(pos)) and negative (Ftn(neg)) surface charge self-assemble into a tetragonal lattice. Reproduced with permission from ref. 69; Copyright (2016) American Chemical Society. (V) Lattice assembly utilizing aromatic stacking. Substitution to phenylalanine (top) or tyrosine (bottom) on the exterior surface of HFtn results in formation of a 2D array or 3D lattice, respectively. Reproduced with permission from ref. 70; Copyright (2018) American Chemical Society. (VI) Metal coordination-assisted lattice formation. Selection of suitable protein nodes equipped with metal-binding motifs, metal ions with preferred geometries, and dihydroxamate linkers with different shapes and lengths results in formation of cubic or tetragonal lattices. Reproduced with permission from ref. 71; Copyright (2017) American Chemical Society. (VII) Scheme of crack formation and self-healing of the Ca-HFtn crystal-hydrogel network (left). Light microscopy images of crystal–hydrogel hybrids, depicting the self-healing of Ca-induced cracking (scale bare = 100 μm) (right). Reproduced with permission from ref. 72; Copyright (2018) Springer Nature. | ||
In the lattice formation systems, guests packaged in protein cages as well as connectors impart superlattices with additional functionalities. Iron cores in ferritin cages, for instance, can function as a contrast agent for magnetic resonance imaging and superlattice formation of such ferritins with gold-nanoparticles has been shown to increase the relaxation rate.63 In another example, a cationic phthalocyanine derivative used as a connector for construction of the ferritin lattice affords light-induced singlet oxygen production (Fig. 6II).18 A lattice-like nanomaterial possessing glycolytic activity has been demonstrated by taking advantage of the salt-dependent assembly of TmFtn cages together with their negatively charged lumenal surfaces (Fig. 6I).66 This meant that they could be loaded with a positively charged lysozyme prior to being assembled into the lattice.
To promote electrostatic-based lattice assembly, additional net charges can be introduced on protein cage exteriors using genetic modifications. For example, bacteriophage P22 virus-like particles were equipped with a negatively charged peptide to form lattices upon addition of positively charged dendrimers.67 In this system, the resulting assemblies were further fixed with a cementing protein that binds to the P22 capsid exterior (Fig. 6III). In the same vein, four residues positioned on the exterior surface of DNA-binding protein from starved cells (Dps) were mutated with glutamates, resulting in highly ordered architectures assembled with zinc cations.68 By engineering with opposing surface charges, protein cages can act as both building blocks and connectors: Künzle et al. have computationally designed both positively and negatively supercharged variants of HFtn that self-assemble into crystalline superlattices (Fig. 6IV).69
Apart from electrostatic interactions, aromatic stacking can be utilised to build supramolecular architectures comprised of one type of building block. A single mutation at a position lining the C4 symmetric pores near the exterior surface of HFtn to phenylalanine resulted in a 2D array, while substitutions to tyrosine or tryptophan produced 3D lattices (Fig. 6V).70 Assembly and disassembly of such lattices can be controlled by ionic strength in an opposite manner to electrostatic-based systems.
Metal coordination presents a potent strategy for controlling the assembly geometry of protein cage-based lattices with design principles adopted from metal–organic frameworks (MOFs). This approach has been demonstrated through lattices prepared by connecting metal–ferritin nodes via organic crosslinkers.71 Incorporation of tripodal coordinating motifs to the C3 symmetric vertices of HFtn allowed binding of Zn(II), Ni(II) or Co(II) while leaving an empty metal orbital for dihydroxamate crosslinkers with varied length and shape. By selecting appropriate crosslinkers and coordination geometries, protein cage lattices with different symmetries and unit cell dimensions were obtained in a modular fashion (Fig. 6VI).
Metal-mediated protein cage superlattices can be further functionalized by incorporating other materials. A mutant of HFtn, K86Q, is known to form protein cage crystals via coordinated Ca ions.8 The Tezcan group recently integrated Ca-HFtn crystals with poly(acrylate-acrylamide) copolymer hydrogels.72 Because of the dynamic bonding interactions of the hydrogel network, a swelling–contraction behaviour of the resulting lattice can be controlled by ionic strength and pH. Furthermore, such reversible interactions between the lattice components enabled the protein cage crystals to tolerate fragmentation and self-heal (Fig. 6VII).
4. Conclusions and outlook
Engineered non-covalent interactions to modulate intra- and inter-cage “connectability” have produced a series of hollow proteinaceous particles as well as protein-cage-based nanomaterials with diverse morphologies and functionalities. Among many successful approaches, those exploiting a bonding chemistry not employed by naturally existing protein cages hold great promise for development of novel nanoarchitectures with characteristics not found in nature. In this regard, connecting proteins via metal ions has been suggested as a flexible and modular modality for construction of proteinaceous nanostructures in a target shape with the ability of triggered assembly and disassembly. This is due to the reversible bond formation and well-defined coordination chemistry of the metal linkages. While the metal-based approach will continue to be developed, it is perhaps worthy to explore other types of bonding chemistry, e.g. covalent bonds that are cleaved by desired stimuli, which would increase the library of design strategies and endow nanomaterials with new capabilities. In such approaches, a serious concern is poor production yield of target structures due to multiple assembly states or random protein aggregations, often observed in artificial protein cage formation.32,35,73 Appropriate selection of site-specific bond formation chemistry as well as of building blocks based on a precise assembly prediction will be important for obtaining monodisperse protein nanoassemblies with desired morphologies and functions.Conflicts of interest
JGH is named as an inventor on a patent application related to protein-cage assembly construction. He is also the founder of and holds equity in nCage Therapeutics LLC, which aims to commercialise protein cages for therapeutic applications.Acknowledgements
JGH and KM were funded by the National Science Centre (NCN, Poland) grant no. 2016/20/W/NZ1/00095 (Symfonia-4), and YA by the NCN grant 2018/31/D/NZ1/01102 (Sonata-14). YA is also grateful for a European Molecular Biology Organization (EMBO) Installation grant no. Z/IPZ/00315 and its agency the Ministry of Science and Higher Education (MNiSW, Poland).References
- L. Sun, X. Zhang, S. Gao, P. A. Rao, V. Padilla-Sanchez, Z. Chen, S. Sun, Y. Xiang, S. Subramaniam, V. B. Rao and M. G. Rossmann, Nat. Commun., 2015, 6, 7548 CrossRef PubMed.
- K. N. Parent, R. Khayat, L. H. Tu, M. M. Suhanovsky, J. R. Cortines, C. M. Teschke, J. E. Johnson and T. S. Baker, Structure, 2010, 18, 390–401 CrossRef CAS PubMed.
- P. D. Hempstead, S. J. Yewdall, A. R. Fernie, D. M. Lawson, P. J. Artymiuk, D. W. Rice, G. C. Ford and P. M. Harrison, J. Mol. Biol., 1997, 268, 424–448 CrossRef CAS PubMed.
- X. Zhang, W. Meining, M. Fischer, A. Bacher and R. Ladenstein, J. Mol. Biol., 2001, 306, 1099–1114 CrossRef CAS PubMed.
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277–290 CrossRef CAS PubMed.
- Viral Molecular Machines, ed. M. G. Rossmann and V. B. Rao, Springer, New York, 2012 Search PubMed.
- P. Pushko, P. Pumpens and E. Grens, Intervirology, 2013, 56, 141–165 CrossRef CAS PubMed.
- D. M. Lawson, P. J. Artymiuk, S. J. Yewdall, J. M. A. Smith, J. C. Livingstone, A. Treffry, A. Luzzago, S. Levi, P. Arosio, G. Cesareni, C. D. Thomas, W. V. Shaw and P. M. Harrison, Nature, 1991, 349, 541–544 CrossRef CAS PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Günther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CrossRef CAS PubMed.
- R. Ladenstein, M. Fischer and A. Bacher, FEBS J., 2013, 280, 2537–2563 CrossRef CAS PubMed.
- M. Neek, T. Il Kim and S. W. Wang, Nanomedicine, 2019, 15, 164–174 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS PubMed.
- Y. Azuma, D. L. V. Bader and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 860–863 CrossRef CAS PubMed.
- A. Liu, C. H. H. Traulsen and J. J. L. M. Cornelissen, ACS Catal., 2016, 6, 3084–3091 CrossRef CAS.
- B. Maity, S. Abe and T. Ueno, Nat. Commun., 2017, 8, 14820 CrossRef CAS PubMed.
- Z. Zhou, G. J. Bedwell, R. Li, S. Palchoudhury, P. E. Prevelige and A. Gupta, Langmuir, 2017, 33, 5925–5931 CrossRef CAS PubMed.
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige, B. Lee and T. Douglas, ACS Nano, 2018, 12, 942–953 CrossRef CAS PubMed.
- J. Mikkilä, E. Anaya-Plaza, V. Liljeström, J. R. Caston, T. Torres, A. De La Escosura and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef PubMed.
- K. Asija and C. M. Teschke, J. Virol., 2019, 93, e00727-19 CrossRef PubMed.
- Z. Chen, L. Sun, Z. Zhang, A. Fokine, V. Padilla-Sanchez, D. Hanein, W. Jiang, M. G. Rossmann and V. B. Rao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8184–E8193 CrossRef CAS PubMed.
- M. M. Suhanovsky and C. M. Teschke, Virology, 2015, 479–480, 487–497 CrossRef CAS PubMed.
- R. L. Duda and C. M. Teschke, Curr. Opin. Virol., 2019, 36, 9–16 CrossRef CAS PubMed.
- W. R. Wikoff, L. Liljas, R. L. Duda, H. Tsuruta, R. W. Hendrix and J. E. Johnson, Science, 2000, 289, 2129–2133 CrossRef CAS PubMed.
- S. Zhang, J. Zang, W. Wang, H. Chen, X. Zhang, F. Wang, H. Wang and G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 16064–16070 CrossRef CAS PubMed.
- S. Zhang, J. Zang, X. Zhang, H. Chen, B. Mikami and G. Zhao, ACS Nano, 2016, 10, 10382–10388 CrossRef CAS PubMed.
- J. Zang, H. Chen, X. Zhang, C. Zhang, J. Guo, M. Du and G. Zhao, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- Y. Yu and S. Lutz, Trends Biotechnol., 2011, 29, 18–25 CrossRef CAS PubMed.
- J. Jorda, D. J. Leibly, M. C. Thompson and T. O. Yeates, Chem. Commun., 2016, 52, 5041–5044 RSC.
- Y. Azuma, M. Herger and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 558–561 CrossRef CAS PubMed.
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- K. A. Cannon, R. U. Park, S. E. Boyken, U. Nattermann, S. Yi, D. Baker, N. P. King and T. O. Yeates, Protein Sci., 2019, 1–11 Search PubMed.
- J. B. Bale, S. Gonen, Y. Liu, W. Sheffler, D. Ellis, C. Thomas, D. Cascio, T. O. Yeates, T. Gonen, N. P. King and D. Baker, Science, 2016, 353, 389–394 CrossRef CAS PubMed.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. André, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- Y. Hsia, J. B. Bale, S. Gonen, D. Shi, W. Sheffler, K. K. Fong, U. Nattermann, C. Xu, P. S. Huang, R. Ravichandran, S. Yi, T. N. Davis, T. Gonen, N. P. King and D. Baker, Nature, 2016, 535, 136–139 CrossRef CAS PubMed.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS PubMed.
- Y. T. Lai, N. P. King and T. O. Yeates, Trends Cell Biol., 2012, 22, 653–661 CrossRef CAS PubMed.
- Y. T. Lai, E. Reading, G. L. Hura, K. L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065–1071 CrossRef CAS PubMed.
- K. A. Cannon, V. N. Nguyen, C. Morgan and T. O. Yeates, ACS Synth. Biol., 2020, 9, 517–524 CrossRef CAS PubMed.
- D. P. Patterson, A. M. Desai, M. M. B. Holl and E. N. G. Marsh, RSC Adv., 2011, 1, 1004–1012 RSC.
- D. P. Patterson, M. Su, T. M. Franzmann, A. Sciore, G. Skiniotis and E. N. G. Marsh, Protein Sci., 2014, 23, 190–199 CrossRef CAS PubMed.
- S. Badieyan, A. Sciore, J. D. Eschweiler, P. Koldewey, A. S. Cristie-David, B. T. Ruotolo, J. C. A. Bardwell, M. Su and E. N. G. Marsh, ChemBioChem, 2017, 18, 1888–1892 CrossRef CAS PubMed.
- A. Sciore, M. Su, P. Koldewey, J. D. Eschweiler, K. A. Diffley, B. M. Linhares, B. T. Ruotolo, J. C. A. Bardwell, G. Skiniotis and E. N. G. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8681–8686 CrossRef CAS PubMed.
- A. S. Cristie-David, J. Chen, D. B. Nowak, A. L. Bondy, K. Sun, S. I. Park, M. M. Banaszak Holl, M. Su and E. N. G. Marsh, J. Am. Chem. Soc., 2019, 141, 9207–9216 CrossRef PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- M. Mosayebi, D. K. Shoemark, J. M. Fletcher, R. B. Sessions, N. Linden, D. N. Woolfson and T. B. Liverpool, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9014–9019 CrossRef CAS PubMed.
- V. Kočar, S. Božič Abram, T. Doles, N. Bašić, H. Gradišar, T. Pisanski and R. Jerala, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 218–237 Search PubMed.
- A. Ljubetič, F. Lapenta, H. Gradišar, I. Drobnak, J. Aupič, Ž. Strmšek, D. Lainšček, I. Hafner-Bratkovič, A. Majerle, N. Krivec, M. Benčina, T. Pisanski, T. Ć. Veličković, A. Round, J. M. Carazo, R. Melero and R. Jerala, Nat. Biotechnol., 2017, 35, 1094–1101 CrossRef PubMed.
- F. Lapenta, J. Aupič, Ž. Strmšek and R. Jerala, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- D. J. E. Huard, K. M. Kane and F. Akif Tezcan, Nat. Chem. Biol., 2013, 9, 169–176 CrossRef CAS PubMed.
- A. S. Cristie-David and E. N. G. Marsh, Protein Sci., 2019, 28, 1620–1629 CAS.
- A. D. Malay, N. Miyazaki, A. Biela, S. Chakraborti, K. Majsterkiewicz, I. Stupka, C. S. Kaplan, A. Kowalczyk, B. M. A. G. Piette, G. K. A. Hochberg, D. Wu, T. P. Wrobel, A. Fineberg, M. S. Kushwah, M. Kelemen, P. Vavpetič, P. Pelicon, P. Kukura, J. L. P. Benesch, K. Iwasaki and J. G. Heddle, Nature, 2019, 569, 438–442 CrossRef CAS PubMed.
- A. D. Malay, J. G. Heddle, S. Tomita, K. Iwasaki, N. Miyazaki, K. Sumitomo, H. Yanagi, I. Yamashita and Y. Uraoka, Nano Lett., 2012, 12, 2056–2059 CrossRef CAS PubMed.
- M. Imamura, T. Uchihashi, T. Ando, A. Leifert, U. Simon, A. D. Malay and J. G. Heddle, Nano Lett., 2015, 15, 1331–1335 CrossRef CAS PubMed.
- E. Golub, R. H. Subramanian, J. Esselborn, R. G. Alberstein, J. B. Bailey, J. A. Chiong, X. Yan, T. Booth, T. S. Baker and F. A. Tezcan, Nature, 2020, 578, 172–176 CrossRef CAS PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- M. Künzle, M. Lach, M. Budiarta and T. Beck, ChemBioChem, 2019, 20, 1637–1641 CrossRef PubMed.
- P. J. M. Brouwer, A. Antanasijevic, Z. Berndsen, A. Yasmeen, B. Fiala, T. P. L. Bijl, I. Bontjer, J. B. Bale, W. Sheffler, J. D. Allen, A. Schorcht, J. A. Burger, M. Camacho, D. Ellis, C. A. Cottrell, A. J. Behrens, M. Catalano, I. del Moral-Sánchez, T. J. Ketas, C. LaBranche, M. J. van Gils, K. Sliepen, L. J. Stewart, M. Crispin, D. C. Montefiori, D. Baker, J. P. Moore, P. J. Klasse, A. B. Ward, N. P. King and R. W. Sanders, Nat. Commun., 2019, 10, 1–17 CrossRef CAS PubMed.
- J. Marcandalli, B. Fiala, S. Ols, M. Perotti, W. de van der Schueren, J. Snijder, E. Hodge, M. Benhaim, R. Ravichandran, L. Carter, W. Sheffler, L. Brunner, M. Lawrenz, P. Dubois, A. Lanzavecchia, F. Sallusto, K. K. Lee, D. Veesler, C. E. Correnti, L. J. Stewart, D. Baker, K. Loré, L. Perez and N. P. King, Cell, 2019, 176, 1420–1431.e17 CrossRef CAS PubMed.
- Y. Liu, D. T. Huynh and T. O. Yeates, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- M. A. Kostiainen, P. Hiekkataipale, J. Á. De La Torre, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem., 2011, 21, 2112–2117 RSC.
- M. A. Kostiainen, P. Ceci, M. Fornara, P. Hiekkataipale, O. Kasyutich, R. J. M. Nolte, J. J. L. M. Cornelissen, R. D. Desautels and J. Van Lierop, ACS Nano, 2011, 5, 6394–6402 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- V. Liljeström, J. Mikkilä and M. A. Kostiainen, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- V. Liljeström, J. Seitsonen and M. A. Kostiainen, ACS Nano, 2015, 9, 11278–11285 CrossRef PubMed.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stępień, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef CAS PubMed.
- K. McCoy, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2018, 12, 3541–3550 CrossRef CAS PubMed.
- H. Chen, K. Zhou, Y. Wang, J. Zang and G. Zhao, Chem. Commun., 2019, 55, 11299–11302 RSC.
- M. Künzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 CrossRef PubMed.
- K. Zhou, J. Zang, H. Chen, W. Wang, H. Wang and G. Zhao, ACS Nano, 2018, 12, 11323–11332 CrossRef CAS PubMed.
- J. B. Bailey, L. Zhang, J. A. Chiong, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 8160–8166 CrossRef CAS PubMed.
- L. Zhang, J. B. Bailey, R. H. Subramanian, A. Groisman and F. A. Tezcan, Nature, 2018, 557, 86–91 CrossRef CAS PubMed.
- Y. T. Lai, L. Jiang, W. Chen and T. O. Yeates, Protein Eng., Des. Sel., 2015, 28, 491–499 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |