
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid conjugated polymer/magnetic nanoparticle composite nanofibers through cooperative non-covalent interactions†
Lingyao
Meng
,
Brad W.
Watson
II
and
Yang
Qin
 *
*
Department of Chemistry & Chemical Biology, University of New Mexico, MSC03 2060, 1 UNM, Albuquerque, New Mexico 87131, USA. E-mail: yangqin@unm.edu
First published on 28th April 2020
Abstract
Hybrid organic–inorganic composites possessing both electronic and magnetic properties are promising materials for a wide range of applications. Controlled and ordered arrangement of the organic and inorganic components is key for synergistic cooperation toward desired functions. In this work, we report the self-assemblies of core–shell composite nanofibers from conjugated block copolymers and magnetic nanoparticles through the cooperation of orthogonal non-covalent interactions. We show that well-defined core–shell conjugated polymer nanofibers can be obtained through solvent induced self-assembly and polymer crystallization, while hydroxy and pyridine functional groups located at the shell of nanofibers can immobilize magnetic nanoparticles via hydrogen bonding and coordination interactions. These precisely arranged nanostructures possess electronic properties intrinsic to the polymers and are simultaneously responsive to external magnetic fields. We applied these composite nanofibers in organic solar cells and found that these non-covalent interactions led to controlled thin film morphologies containing uniformly dispersed nanoparticles, although high loadings of these inorganic components negatively impact device performance. Our methodology is general and can be utilized to control the spatial distribution of functionalized organic/inorganic building blocks, and the magnetic responsiveness and optoelectronic activities of these nanostructures may lead to new opportunities in energy and electronic applications.
1. Introduction
Hybrid organic–inorganic (O–I) materials constitute a remarkable and growing field that has led to a wide variety of applications owing to their structural diversity and emerging properties via synergistic cooperation between both phases.1–3 The idea of combining two distinct components to yield improved properties is not new and dates back to ancient ages as exemplified by the Maya blue pigments that are physical mixtures of plant dyes and clays. The nature of modern “O–I hybrid materials” has shifted from simple physical mixtures to hybrid structures containing multiple components arranged in space at the nanometer scale and/or molecular level. The collective properties from the resulting materials are thus highly dependent on the spatial arrangement and interactions among the different constituents. One of the most extensively studied O–I hybrid material systems is based on organic polymers and inorganic nanoparticles (NPs). With rational materials design and precise interfacial engineering, the resulting hybrid materials exhibit superior physical, electronic, magnetic, catalytic, and biological properties that are better than or not found in the individual constituents.4–11Since the discovery of metallic conductivity in doped polyacetylene that led to the Nobel Prize in 2000,12–14 conjugated polymers (CPs) have evolved into an enormous field and been considered to revolutionize the next generation of light-weight and flexible electronics.15–19 Numerous research directions have been established and extensively pursued in CPs, one of which is of particular interest in the field of O–I hybrid materials, i.e., incorporation of inorganic magnetic nanoparticles (NPs) into organic CPs, resulting in hybrid materials simultaneously possessing electronic/electric and magnetic properties. Commonly, CPs based on polypyrrole, polyaniline, and polythiophene backbones have been blended with magnetic NPs composed of pure metals, e.g., cobalt, or metal oxides such as magnetite (Fe3O4) and maghemite (γ-Fe2O3), and the resulting O–I hybrid materials have found applications in energy storage, magneto-optoelectronics, sensors, memory devices, catalysis, magnetic resonance imaging, and cancer therapy.20–31
As an extensively studied application of CPs, organic solar cells (OSCs) have been considered as low-cost, light-weight, and renewable alternative energy sources.32,33 With comprehensive structural designs and device engineering, efficiencies of CPs based OSCs have been steadily improved to over 15% during the past three decades.34–41 One interesting but less pursued direction is the incorporation of magnetic NPs into the active layers of OSCs, in which the interactions between photo-generated excitons within the organic phase and magnetism of the NPs can potentially enhance device performances. The power conversion efficiencies (PCEs) of OSCs employing blends of regio-regular poly(3-hexylthiophene) (P3HT) and phenyl-C61-butyric acid methyl ester (PCBM) doped with an appropriate amount of Fe3O4 NPs were shown to significantly increase over those without NP incorporation under otherwise identical experimental conditions. Such improvement in performance has been attributed to enhanced light absorption caused by NP surface plasmon effects,42 improved charge separation efficiencies due to a large coercive internal electric field from aligned NPs,43 and the generation of long-lived triplet excitons through increased spin–orbit coupling effects from the NPs.44,45 In all the above studies, Fe3O4 NPs were simply blended into the P3HT/PCBM active layers without specific morphological or O–I interfacial control, and it was found that high loadings of NPs beyond a few weight percent led to detrimental effects on both materials morphology and device performance. It is thus highly desirable to construct P3HT/PCBM/NP hybrid films with controlled morphology and interfaces in order to more accurately study the effects of NPs on electronic processes and to further improve device performance and stability.
P3HT is the most widely studied CP in OSCs, and is one of the few semi-crystalline CPs that can self-assemble into well-defined nanofiber (NF) structures with uniform widths of 15–25 nm and lengths up to several μm.46–49 Due to this unique crystallization property, P3HT has been utilized as homopolymers and telechelic polymers, as well as been incorporated into block copolymer (BCPs), for combination with various types of organic and inorganic nanoobjects towards hierarchically ordered complex structures.50–76 In this paper, we describe the construction of P3HT/Fe3O4 NP core–shell composite NFs through the cooperation of orthogonal non-covalent interactions including BCP self-assembly, P3HT crystallization, ligand–metal coordination, and hydrogen bonding interactions. The resulting hybrid nanostructures display electronic properties intrinsic to P3HT and enhanced magnetic responsiveness over those from simple blends of the two components. OSCs were fabricated using these hybrid composites that led to controlled and stable morphologies but no significant improvement on device performance.
2. Experimental
2.1 Materials and general methods
All reagents and solvents were used as received from Sigma Aldrich, Alfa Aesar or TCI America unless otherwise noted. Anhydrous tetrahydrofuran (THF) was distilled over sodium using benzophenone as an indicator and was collected in flame-dried, air-free storage flasks. All NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer and referenced internally to the residual solvent signals. Size exclusion chromatography was performed on a Waters 1515 system equipped with a 2414 refractive index detector and a 2707 auto-sampler. The mobile phase was chloroform with 0.5% (v/v) triethylamine passing through two styragel columns (Polymer Laboratories, 5 μm Mix-C) at a flow of 1 mL min−1, kept in a column heater at 35 °C. SEC results were calibrated by external polystyrene standards (Varian). Ultraviolet-visible (UV-Vis) absorption spectra were recorded on a Shimadzu UV-2401 PX spectrometer over a range of 300–900 nm using quartz cuvettes. Infrared spectra were generated by using a Bruker Alpha-P spectrometer, using a powder sample in ATR mode. Fluorescence emission spectra were measured using a Varian Cary Eclipse fluorimeter. X-ray diffraction (XRD) patterns were recorded using a Rigaku SmartLab diffractometer. The FT-IR spectrum was obtained using a Thermo Nicolet 380 FTIR spectrometer. Transmission electron microscopy (TEM) images and selected area electron diffraction (SAED) patterns were obtained on a JEOL-2010 TEM microscope operating at 200 kV. Samples were prepared by drop casting diluted sample solutions onto carbon coated copper grids (Ted Pella).2.2 Solar cell fabrication and measurement
All the ITO (China Shenzhen Southern Glass Display Ltd., Shenzhen, China, 8 ohm sq−1) glass substrates were cleaned with detergent, DI water, acetone and isopropanol with ultrasonication for 15 min each, and then treated with UV Ozone (Novascan PSD series) for 1 hour. The substrates were then transferred to a nitrogen filled glove box and a thin layer of MoO3 (10 nm) was thermally deposited using an Angstrom Engineering Amod deposition system. Next, a blend solution comprising hybrid NFs and PC61BM was spin coated at room temperature at 500 RPM for 30 s on top of the MoO3 surface. 100 nm Al electrodes were further deposited by thermal evaporation through a shadow mask, followed by annealing at 150 for 15 min. J–V characteristics of OSCs were obtained at room temperature by using a Keithley 2400 source-measuring unit under a simulated AM1.5G spectrum (100 mW cm−2) generated by using a Xe arc-lamp based solar simulator (Newport 67005 150-W) in a nitrogen filled glove box.2.3 Synthetic procedures
BCP1 and BCP2 were synthesized according to previously reported procedures.77–82The synthesis of citric acid coated IONP-CA was conducted according to previously published procedures.85 120 mg IONP-OA were dispersed in 15 mL of a 50/50 mixture of dichlorobenzene and N,N-dimethylformamide. Next, 0.1 g citric acid was added, and the mixture was stirred at 100 °C for 24 hours. It was later allowed to cool down to room temperature. The NPs were then precipitated by adding ethyl ether, and then separated via centrifugation, followed by washing with ethyl ether three times.
2.4 Preparation of hybrid nanofibers
Both P3HT and BCP NFs were fabricated through a mixed solvent approach. Typical procedures were as follows: 5 mg polymer was first dissolved in 0.4 mL chlorobenzene, before 0.1 mL acetone was added slowly with stirring. The mixture was further stirred at room temperature for 9 hours. Next, IONPs were added into the as-formed polymer NF solution with predetermined polymer/IONP weight ratios and stirred for 1 hour. The resulting hybrid NF solutions were directly used for solar cell fabrication and diluted 100 times using solvent mixtures of chlorobenzene and acetone (4/1, vol/vol) and drop-cast onto carbon coated grids for TEM analyses.3. Results and discussion
3.1 Synthesis of polymer nanofibers and IONPs
Chemical structures of the two conjugated block copolymers involved in this study, BCP1 and BCP2, are shown in Scheme 1, which were synthesized according to our previously reported procedures.86 The hydroxy groups in BCP1 were easily obtained through quantitative desilylation reactions of a polymer precursor and can be used as a facile synthetic handle for various functionalities, such as the pyridine moieties in BCP2 that possess high polarity and the ability to coordinate to metal-containing nanoparticles.86 The molecular weights of BCP1 and BCP2 are estimated by size-exclusion chromatography (SEC) to be ca. 32.9 kDa and ca. 46.6 kDa, respectively. The non-functionalized versus functionalized block length ratio (m/n, Scheme 1) in both polymers is estimated from SEC to be ca. 4.5 to 1. Through NMR analyses, the shorter functionalized block contains a statistical mixture of 3-hexylthiophene units and functionalized thiophene units in a ca. 5/4 ratio, which leads to an overall functional group concentration of ca. 8% in both polymers. For comparison, a P3HT homo-polymer having a molecular weight of ca. 37.9 kDa has also been prepared. Nanofibers (NFs) of these homo- and block co-polymers were obtained through a mixed-solvent approach by dissolving ca. 5 mg of the polymers in 0.4 mL of chlorobenzene, a good solvent for both P3HT and the functionalized blocks, followed by the addition of 0.1 mL acetone, a poor solvent for P3HT but a good solvent for the hydroxy and pyridine moieties in BCP1 and BCP2, respectively. The chlorobenzene/acetone ratio of 4/1 was optimized previously to afford the most well-defined NFs with sufficient solution stability.79–82 After stirring the mixtures for ca. 9 h, UV-vis absorption spectra (Fig. S1, ESI†) of the diluted solutions of all three polymers show clear structured profiles having λmax values at 514, 552 and 603 nm, indicating the formation of ordered aggregates.87,88 These mixture solutions were then drop cast onto carbon coated grids, and the corresponding transmission electron microscopy (TEM) images are shown in Fig. 1A–C. P3HT forms NFs with uniform widths of ca. 14.9 ± 1.7 nm and lengths up to a few μm. BCP1, on the other hand, forms NFs with similar average widths of ca. 15.2 ± 1.8 nm but with a large distribution of fiber lengths. We are not certain about the exact mechanisms behind this observation, and speculate that it is the relatively strong hydrogen bonding interactions between the hydroxy groups in BCP1 and acetone, which limits the formation of large polymer crystallites, i.e., long fibers. Based on the same argument, the pyridine moieties in BCP2 do not form hydrogen bonds with acetone but possess stronger dipole–dipole interactions with acetone than those from pure P3HT, which leads to the formation of NFs with intermediate lengths and widths of ca. 14.5 ± 1.5 nm. To be noted, the mixed-solvent approach does lead to less uniform and sometimes ill-defined P3HT NFs than those obtained from the so-called whisker method using a single marginal solvent,46–49 but it allows for much higher polymer concentrations (e.g., 10 mg mL−1 in our case vs. less than 1 mg mL−1 in commonly applied whisker methods) for device relevant applications. Such a mixed-solvent approach also allows for the possibility to control nanostructure morphology by fine-tuning the polymer–solvent interactions through functional group and non-solvent variations.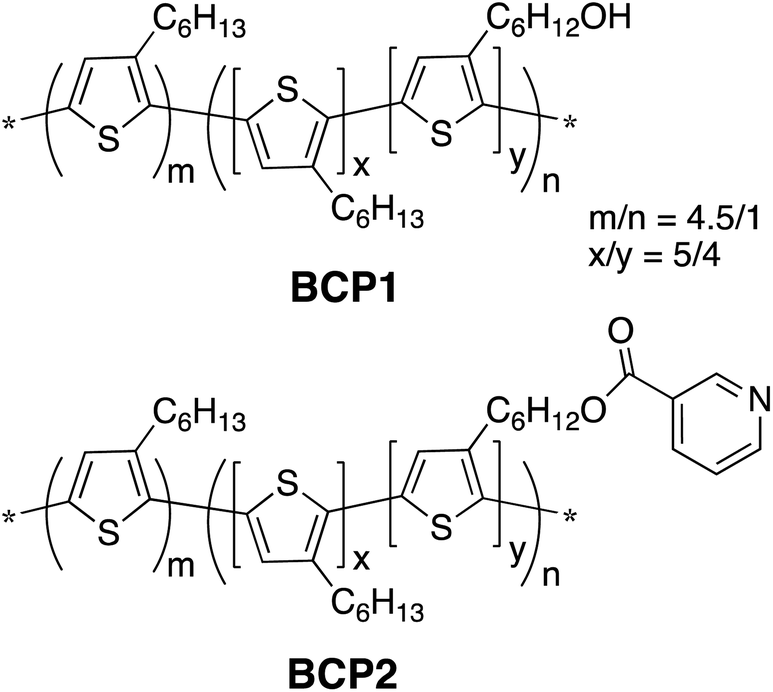 | ||
| Scheme 1 Chemical Structures of BCP1 and BCP2. | ||
 | ||
| Fig. 1 Transmission electron microscopy (TEM) images of (A) P3HT NFs; (B) BCP1 NFs; (C) BCP2 NFs; (D) IONP-OA; (E) IONP-L-OA; and (F) IONP-CA. Inserts: histograms of corresponding NF widths and nanoparticle diameters sampled from 100 individual objects. | ||
Iron oxide nanoparticles (IONPs) were prepared through thermal decomposition of Fe(acac)3 at high temperatures in the presence of oleic acid and oleylamine (OA) as surface stabilizing ligands. By varying the relative amount of iron precursors while keeping other reaction conditions constant, we obtained IONPs in two different sizes, namely IONP-OA and IONP-L-OA having diameters of 7.7 ± 0.9 nm and 20.3 ± 3.2 nm, respectively. Fig. 1D and E show the TEM images of these two IONPs, which are well dispersed without significant aggregation due to the long aliphatic chains of capping OA ligands. The selected area electron diffraction (SAED) and powder X-ray diffraction (PXRD) patterns of IONP-OA are shown in Fig. S2 (ESI†), which confirm that the IONPs as prepared are in Fe3O4 magnetite phase. We also performed ligand exchange reactions on IONP-OA with citric acid, and the TEM image of the resulting IONP-CA from acetone solutions is shown in Fig. 1F. IONP-CA appears smaller than IONP-OA with an average diameter of 5.0 ± 0.9 nm, which is understandable considering the much shorter citric acid capping ligand in IONP-CA. Significant aggregation is also observed for IONP-CA, likely caused by the strong hydrogen bonding interactions among the surface carboxylic ligands.
3.2 Self-assembly and magnetic responses of hybrid nanofibers
The self-assembly of polymer NFs and IONPs was conducted by adding IONPs to the pre-formed polymer NF solutions with a polymer/IONP weight ratio of ca. 2/1. The solutions were then stirred at room temperature for 1 h before being diluted 100 times with chlorobenzene/acetone solvent mixtures (4/1, v/v) for TEM analyses. Representative TEM images of these hybrid nanostructures are assembled in Fig. 2 and Table S1† summarizes the average numbers of nanoparticles associated with one polymer NF by sampling about 50 individual NFs.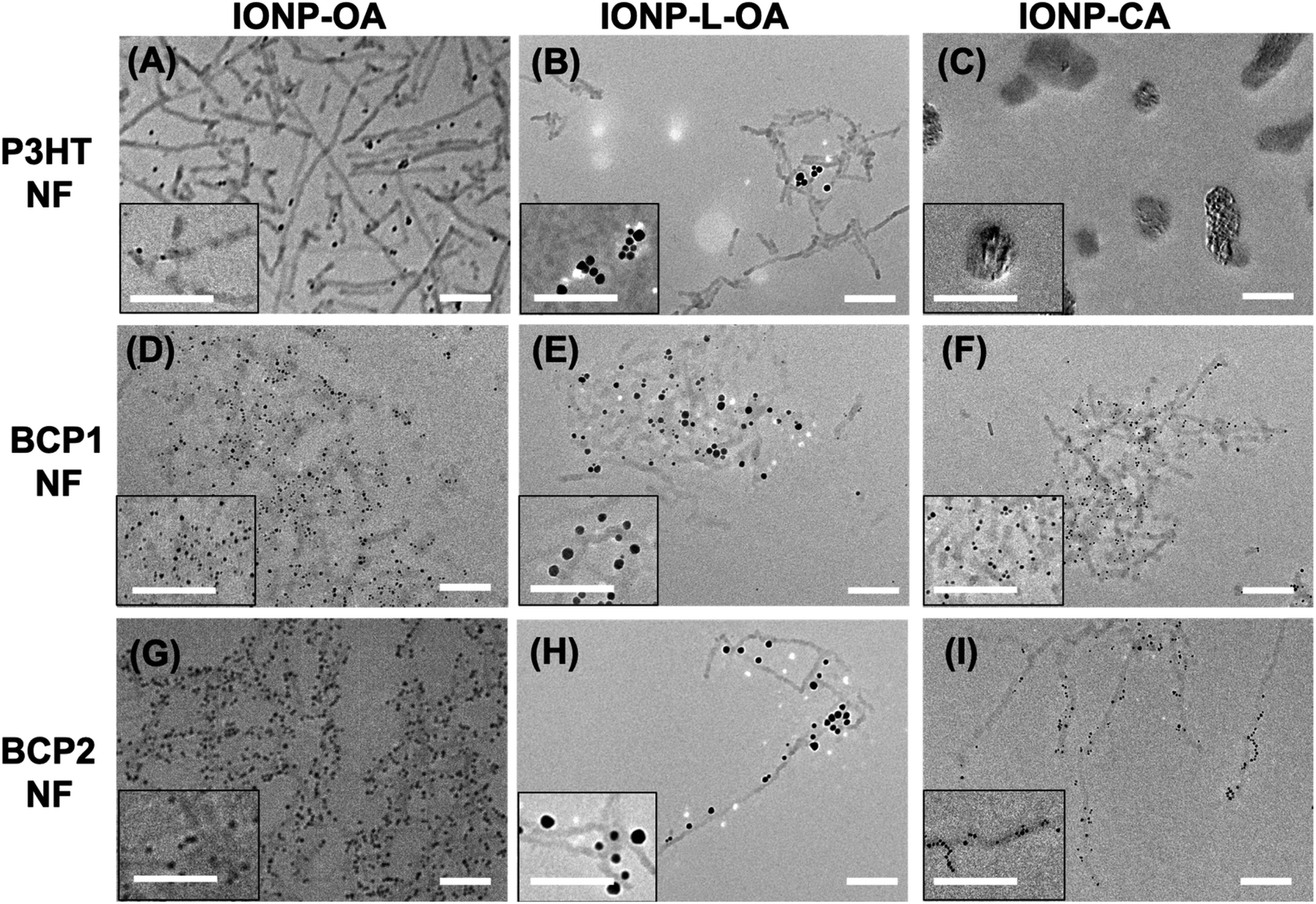 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of nanostructures from mixtures of P3HT NFs and (A) IONP-OA, (B) IONP-L-OA, and (C) IONP-CA; BCP1 NFs and (D) IONP-OA, (E) IONP-L-OA, and (F) IONP-CA; and BCP2 NFs and (G) IONP-OA, (H) IONP-L-OA, and (I) IONP-CA. The mixture solutions used for TEM analyses contain polymer NFs and IONPs at a ca. 2/1 weight ratio and polymer concentrations at ca. 0.1 mg mL−1. Scale bars in all: 200 nm. | ||
For P3HT NFs, IONP-OA and IONP-L-OA seem to well disperse within the networks of polymer NFs but without apparent association between them, as shown in Fig. 2A and B, respectively. These observations are expected since P3HT NFs have no specific, except hydrophobic, interactions with IONPs coated with long aliphatic chains. Thus, the organic and inorganic components can disperse well among each other without showing significant association. In the case of IONP-CA, irregularly shaped aggregates of a few hundred nanometers in size are observed in Fig. 2C. These aggregates appear to contain both the nanoparticles and polymers, but discrete P3HT NFs are no longer observed. IONP-CAs are known to self-aggregate (Fig. 1F), which is caused by the strong hydrogen bonding interactions among surface carboxylic groups, the hydrophilicity of which also make these nanoparticles incompatible with hydrophobic P3HT NFs. Thus, the appearances of large aggregates composed of both components are somewhat surprising, and we are currently investigating such formation mechanisms.
In the cases of BCP1 and BCP2 NFs, similar behaviors were observed with all three IONPs as shown in Fig. 2D through 2I. IONP-OAs are well dispersed within the networks of both BCP1 and BCP2 NFs, and most of the nanoparticles are found to closely associate and align along both sides of the NFs. The difference is that the density of IONP-OAs is found to be higher along BCP2 NFs, with less free, unattached nanoparticles, than for BCP1 hybrid NFs. Similarly, IONP-L-OAs are well dispersed and associated with both BCP1 and BCP2 NFs, with stronger attachment and less free particles observed for the latter. Interestingly, IONP-CAs no longer self-aggregate and are found to align with both BCP1 and BCP2 NFs. We rationalize the observations as the following. The hydroxy groups in BCP1 can form hydrogen-bonding interactions, in addition to hydrophobic interactions from the polymer main-chain, with the OA ligands on the surfaces of IONP-OAs and IONP-L-OAs. Such additional hydrogen-bonding interactions lead to closer association of IONPs with BCP1 NFs than with P3HT NFs. The apparent stronger attachment of IONP-OAs and IONP-L-OAs to BCP2 NFs is likely caused by stronger interactions between the pyridine moieties on BCP2 and IONPs. Besides hydrophobic and hydrogen-bonding interactions, pyridine groups can also coordinate to the surfaces of nanoparticles and partially replace the original ligands. To study such effects, we precipitated a well-dissolved BCP2/IONP-OA (2/1, wt/wt) solution in chlorobenzene into methanol and washed the precipitate extensively with methanol in order to remove any free OA ligands. The remaining powder could be attracted to a nearby permanent magnet, confirming the presence of IONPs, but was found to be insoluble in any solvent. This can be explained by cross-linking of polymer chains with IONPs as the cross-linkers, through pyridine coordination interactions. We also performed infrared (IR) spectroscopy on the precipitated BCP2/IONP powder as well as on BCP2 and IONP-OA individually, and the spectra are shown in Fig. S3 (ESI†). The signals at ca. 1710 cm−1 and between 1400 and 1600 cm−1, characteristic of pyridine moieties are clearly observed in both BCP2 and BCP2/IONP-OA precipitates, while the signals at ca. 1631 cm−1, 1561 cm−1, and 1454 cm−1, characteristic of OA ligands diminish in the spectrum of BCP2/IONP-OA precipitate, suggesting the replacement of the original ligands. As for IONP-CA, the carboxylic surface ligands can form hydrogen-bonding interactions with the hydroxy and pyridine groups in BCP1 and BCP2, respectively, leading to the observed NF attachment without significant self-aggregation.
It is well-known that ferromagnetic Fe3O4 can become superparamagnetic when it displays single magnetic domains as nanoparticles with sizes below 20 nm; and these nanoparticles can respond and self-assemble to external magnetic fields.89–91 We thus tested the magnetic responsiveness of the polymer/IONP composite NFs by placing a permanent magnetic cube (Neodymium Magnet N42, Applied Magnetics, ca. 100 Gauss at surface) next to the hybrid NF solutions. Photos were taken at the beginning and at the time when most of the solutes were attracted to the side of the magnet and the solutions became clear, the durations of which were also recorded. The results are summarized in Fig. 3 and corresponding videos are provided in the ESI.† Since all solutions are in identical vials and contain the same concentrations of polymers and NPs, (10 mg mL−1 and 5 mg mL−1, respectively), the different times during which the solutions become clear can be used to compare relative association strengths between different polymer NFs and IONPs. For IONP-OA and IONP-L-OA, similar trends are observed for the three polymer NFs. BCP2 NFs display the fastest clearing times of 30 seconds with IONP-OA and 4 minutes with IONP-L-OA, while the respective times for P3HT NFs are 4 and 14 minutes. This is consistent with TEM observations and the conclusion that BCP2 NFs form the strongest interactions with the OA coated NPs. For both P3HT and BCP2 NFs, it took longer for the IONP-L-OA composite solutions to clear out. This is likely because that the same amount of IONPs by weight was used in all cases and the much larger particles lead to significantly smaller number of particles relative to the number of NFs, so that the relative attractive forces experienced by the NFs are weaker in the cases of larger IONPs. Surprisingly, it took much longer (ca. 2.5 hours) for the solutions of BCP1 NFs complexed with both IONP-OA and IONP-L-OA nanoparticles to be cleared out. Although TEM images have suggested that the nanoparticles are more strongly attached to BCP1 NFs than to P3HT NFs, the NFs of BCP1 are much shorter, i.e., the number of BCP1 NFs is much higher than that of P3HT NFs under the same concentrations, leading to a smaller amount of NPs attached per NF and possibly bare NFs for BCP1. Thus, the BCP1 composite NFs may experience less attractive force from the magnet, and thus it took longer for the solutions to clear out. For hydrophilic IONP-CA, no clearing out events could be observed for P3HT NF solutions. This confirms the lack of interactions between P3HT and IONP-CA, and the seeming co-aggregates observed in the TEM image (Fig. 2C) are likely a result from the solvent evaporation process during the TEM sample preparation. For both BCP1 and BCP2 composite NFs with IONP-CA, the solutions were cleared out in 4 and 10 minutes respectively. The faster time for BCP1 suggests stronger interactions between the –OH groups and nanoparticle citric acid ligands.
 | ||
| Fig. 3 Photographs of solutions of composite NFs next to a permanent magnetic cube at the start time and the times when solutions became mostly clear. Durations for such processes to take place are shown above arrows (s: second; m: minute; h: hour). | ||
We also tested magnetic responsiveness of well-dissolved solutions of polymers and IONP-OA nanoparticles at the same weight ratios and concentrations in chlorobenzene as those in hybrid NF solutions. The photographs of these experiments are included in Fig. S4 (ESI†). In contrary to composite NFs, the well-dissolved solutions did not show clearing out events but displacement of solutions from the far side to the near side of the magnetic cube. The height differences between these edges are ca. 2.6 mm, 3.2 mm, 3.5 mm, and 4.0 mm respectively for solutions of pure IONP-OA, P3HT/IONP-OA, BCP1/IONP-OA, and BCP2/IONP-OA. We believe the height differences are caused by solute concentration differences or gradients between the near and far sides to the magnet; the higher the concentration differences the larger the height differences. Given that all solutions contain the same amount of IONP-OA and polymers, the larger height differences observed for solutions containing polymers than that for the pure nanoparticle solution confirm the existence of interactions and associations between the two components. It is also understood that the P3HT/IONP-OA solution displays the smallest height difference among the three polymer mixture solutions due to the relatively weak hydrophobic interactions, while the BCP2/IONP-OA system shows the largest height difference caused by the stronger hydrogen bonding and coordination interactions.
3.3 Organic solar cells and active layer morphology
We next applied our self-assembled hybrid NFs in organic solar cells (OSCs) in combination with the commonly used electron acceptor phenyl-C61-butyric acid methyl ester (PCBM). We used IONP-OA as the superparamagnetic particles in our studies since they are compatible and show varied interactions with all three polymer NFs, and their sizes are more uniform than those of IONP-L-OA and comparable with those applied in previous literature reports.42–45 As for the polymers, we chose P3HT and BCP2 NFs for direct comparison since devices employing BCP1 and PCBM under standard conditions showed very poor performance and are thus less suitable to conduct comparative studies on the effects of IONP incorporation. We first studied the device performance of P3HT NFs with various amounts of IONP-OA, using optimized conditions for P3HT/PCBM devices (i.e., P3HT NF/PCBM, 1/1, wt/wt, thermal annealed at 150 °C for 10 min), and the results are summarized in Table 1. Previous reports all concluded that by the addition of a few weight percent of Fe3O4 nanoparticles, the P3HT/PCBM device efficiencies were improved by up to 50%.42–45 In our case, devices employing P3HT NFs and PCBM gave a power conversion efficiency (PCE) of ca. 3.31 ± 0.29%, which is characteristic of this materials combination and comparable with the above mentioned reports. However, with just 1 wt% of IONP-OA added, the device PCE drops to ca. 2.67 ± 0.25%, as a result of slight decreases in all performance parameters, i.e., short circuit current (JSC), open circuit voltage (VOC), and fill factor (FF). With the additions of 5 and 50 wt% of IONP-OA, device performance further decreases to nearly non-functional cells for the latter. The same trend was observed for devices based on BCP2 NFs. Without the addition of IONP-OA, the devices using BCP2 NFs and PCBM perform somewhat worse than those using P3HT NFs, giving an average PCE of ca. 1.86 ± 0.27%. With the addition of just 1 wt% of IONP-OA, the device PCE is reduced by ca. 65% to 0.65 ± 0.10%, with the reduction in JSC as a major contributor. We thus did not attempt to further increase the amount of IONP-OA for these devices.| IONPb (wt%) | J SC (mA cm−2) | V OC (V) | FFe (%) | PCEf (%) | |
|---|---|---|---|---|---|
| a All devices are based on the following geometries: ITO/MoO3 (10 nm)/active layer (100 nm)/Al (100 nm). Active layers are obtained by spin-coating from chlorobenzene/acetone (4/1, v/v) of polymer NFs (10 mg mL−1) and PCBM (10 mg mL−1) with varied amounts of IONP-OA. Performance parameters are calculated from at least five individual cells. b Weight percentage relative to the polymer. c Short circuit current density. d Open circuit voltage. e Fill factor. f Power conversion efficiency. | |||||
| P3HT NF | 0 | 11.97 ± 1.74 | 0.54 ± 0.02 | 52 ± 6 | 3.31 ± 0.29 |
| 1 | 10.93 ± 1.79 | 0.51 ± 0.01 | 48 ± 4 | 2.67 ± 0.25 | |
| 5 | 5.59 ± 0.69 | 0.40 ± 0.01 | 51 ± 2 | 1.16 ± 0.20 | |
| 50 | 2.02 ± 0.36 | 0.17 ± 0.03 | 37 ± 3 | 0.12 ± 0.01 | |
| BCP2 NF | 0 | 6.93 ± 0.96 | 0.59 ± 0.01 | 46 ± 7 | 1.86 ± 0.27 |
| 1 | 3.73 ± 0.60 | 0.56 ± 0.02 | 31 ± 1 | 0.65 ± 0.10 | |
The TEM images of active layers of OSC devices employing P3HT and BCP2 NFs with 0 and 1 wt% IONP-OA are shown in Fig. 4. Without IONP-OA, both the P3HT and BCP2 active layers show polymer NFs and bulk heterojunction (BHJ) morphologies with domain sizes on the order of tens of nanometers. With the addition of IONP-OA, no large phase separation could be observed and the nanoparticles are well dispersed in both films. So the detrimental effect from IONP-OA addition on the device performance is unlikely to be caused by significant morphological changes induced by the inorganic nanoparticles. In order to probe the microscopic packing structures of the polymers, we performed X-ray diffraction (XRD) experiments on BCP2 NF thin films with PCBM, IONP-OA, and both, and the results are shown in Fig. S5 (ESI†). All films show (100), (200) and (300) peaks from polymer lamellar stacking motifs with similar intensities. The thin films containing PCBM, with or without IONP-OA, display slightly enhanced peaks around 20° (2θ), which correspond to π–π stacking among P3HT main-chains. As a result, the device performance drop is unlikely to result from IONP induced disruption on polymer crystalline structures. Based on the above observations, we suspect that, due to the specific interactions between polymer NFs and IONP-OA, the insulating inorganic nanoparticles are strongly associated with the polymer NFs and located in between polymer NFs and PCBM in the solid state, as suggested by TEM images, which can potentially act as a barrier for charge separation and thus decrease device performance. Confirmation of such a hypothesis will require more thorough and sophisticated photophysical studies that are currently under way and will be reported in future accounts.
 | ||
| Fig. 4 TEM images of device active layers employing (A) P3HT NF/PCBM; (B) P3HT NF/PCBM/IONP-OA; (C) BCP2 NF/PCBM; and (D) BCP2 NF/PCBM/IONP-OA. Scale bars in all: 200 nm. | ||
4. Conclusions
In summary, we have established a facile methodology to fabricate well-defined core–shell hybrid nanofibers from organic conjugated polymers and inorganic magnetic nanoparticles. The self-assembly processes employ several orthogonal non-covalent interactions including polymer crystallization, block copolymer self-assembly, hydrogen bonding, and coordination interactions. The resulting hybrid NFs display electronic properties from the polymer and magnetic responsiveness from the nanoparticles. Organic solar cells were fabricated using these hybrid nanofiber systems, but it was found that addition of iron oxide nanoparticles detrimentally affected device performance, which is likely caused by their insulating nature that limits charge transfer efficiency. Our methodology is general and can be applied to a wide range of conjugated polymers and inorganic nanoparticles, where controlled morphologies on the nanometer scales are necessary.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Science Foundation (DMR-1453083 and CHE-1904659) for financial support and research reported in this publication was partially supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103451.References
- P. Gómez-Romero and C. Sanchez, Functional Hybrid Materials. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511 RSC.
- L. Merhari, Hybrid Nanocomposites for Nanotechnology, Springer, 2009 Search PubMed.
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC.
- S. K. Kumar, N. Jouault, B. C. Benicewicz and T. Neely, Macromolecules, 2013, 46, 3199 CrossRef CAS.
- K. Miller, L. Wang, B. C. Benicewicz and A. W. Decho, Chem. Soc. Rev., 2015, 44, 7787 RSC.
- S. Kumar, B. C. Benicewicz, R. Vaia and K. Winey, Macromolecules, 2017, 50, 714 CrossRef CAS.
- S. H. Mir, L. A. Nagahara, T. Thundat, P. Mokarian-Tabari, H. Furukawa and A. Khosla, J. Electrochem. Soc., 2018, 165, B3137 CrossRef CAS.
- H. Jin, J. Li, J. Iocozzia, X. Zeng, P.-C. Wei, C. Yang, N. Li, Z. Liu, H. He, T. Zhu, J. Wang, Z. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15206 CrossRef CAS PubMed.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046 CrossRef CAS PubMed.
- H. Kim, S. So, A. Ribbe, Y. Liu, W. Hu, V. V. Duzhko, R. C. Hayward and T. Emrick, Chem. Commun., 2019, 55, 1833 RSC.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574 CrossRef PubMed.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581 CrossRef CAS PubMed.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591 CrossRef CAS PubMed.
- T. A. Skotheim and J. R. Reynolds, Handbook of Conducting Polymers, CRC Press, Boca Raton, 3rd edn, 2007, vol. 2, volume set Search PubMed.
- W. R. Salaneck, K. Seki, A. Kahn and J.-J. Pireaus, Conjugated Polymer and Molecular Interfaces – Science and Technology for Photonic and Optoelectronic Applications, Marcel Dekker, Inc., New York, Basel, 2002 Search PubMed.
- M. Leclerc and J.-F. Morin, Design and Synthesis of Conjugated Polymers, Wiley-VCH Verlag Gmbh & Co. KGaA, Weinheim, 2010 Search PubMed.
- B. Liu, Conjugated Polymers for Biological and Biomedical Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018 Search PubMed.
- M. Knaapila, Conjugated Polymers and Oligomers: Structural and Soft Matter Aspects (Materials and Energy), World Scientific Publishing, Singapore, 2018 Search PubMed.
- S. C. Wuang, K. G. Neoh, E.-T. Kang, D. W. Pack and D. E. Leckband, J. Mater. Chem., 2007, 17, 3354 RSC.
- K. R. Reddy, K. P. Lee and A. I. Gopalan, Colloids Surf., A, 2008, 320, 49 CrossRef CAS.
- H. M. Xiao, W. D. Zhang, M. X. Wan and S. Y. Fu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4446 CrossRef CAS.
- L. Kong, X. Lu, E. Jin, S. Jiang, X. Bian, W. Zhang and C. Wang, J. Solid State Chem., 2009, 182, 2081 CrossRef CAS.
- L. Li, C. Xiang, X. Liang and B. Hao, Synth. Met., 2010, 160, 28 CrossRef CAS.
- J. Azadmanjiri, P. Hojati-Talemi, G. Simon, K. Suzuki and C. Selomulya, Polym. Eng. Sci., 2011, 51, 247 CrossRef CAS.
- Y. Xie, X. Hong, X. Wang, J. Zhao, Y. Gao, Y. Ling, S. Yan, L. Shi and K. Zhang, Synth. Met., 2012, 162, 1643 CrossRef CAS.
- K. G. Alves, C. A. Andrade, S. L. Campello, R. E. de Souza and C. P. de Melo, J. Appl. Polym. Sci., 2013, 128, 3170 CrossRef CAS.
- M. Baghayeri, E. N. Zare and M. M. Lakouraj, Biosens. Bioelectron., 2014, 55, 259 CrossRef CAS PubMed.
- S. Varshney, A. Ohlan, V. Jain, V. Dutta and S. Dhawan, Mater. Chem. Phys., 2014, 143, 806 CrossRef CAS.
- Z. Xu, M. Gao, L. Yu, L. Lu, X. Xu and Y. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 17823 CrossRef CAS PubMed.
- H. Wang, N. Ma, Z. Yan, L. Deng, J. He, Y. Hou, Y. Jiang and G. Yu, Nanoscale, 2015, 7, 7189 RSC.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821 CrossRef CAS PubMed.
- O. Inganäs, Adv. Mater., 2018, 30, 1800388 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159 CrossRef CAS PubMed.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094 CrossRef CAS PubMed.
- B. Fan, Z. Zeng, W. Zhong, L. Ying, D. Zhang, M. Li, F. Peng, N. Li, F. Huang and Y. Cao, ACS Energy Lett., 2019, 4, 2466 CrossRef CAS.
- X. Xu, K. Feng, Z. Bi, W. Ma, G. Zhang and Q. Peng, Adv. Mater., 2019, 31, 1901872 CrossRef PubMed.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746 CrossRef CAS.
- Q. An, X. Ma, J. Gao and F. Zhang, Sci. Bull., 2019, 64, 504 CrossRef CAS.
- N. Zheng, K. Mahmood, W. Zhong, F. Liu, P. Zhu, Z. Wang, B. Xie, Z. Chen, K. Zhang, L. Ying, F. Huang and Y. Cao, Nano Energy, 2019, 58, 724 CrossRef CAS.
- Z. Çaldıran, M. Biber, Ö. Metin and Ş. Aydoğan, Optik, 2017, 142, 134 CrossRef.
- K. Wang, C. Yi, C. Liu, X. Hu, S. Chuang and X. Gong, Sci. Rep., 2015, 5, 9265 CrossRef CAS PubMed.
- W. Zhang, Y. Xu, H. Wang, C. Xu and S. Yang, Sol. Energy Mater. Sol. Cells, 2011, 95, 2880 CrossRef CAS.
- D. M. González, V. Körstgens, Y. Yao, L. Song, G. Santoro, S. V. Roth and P. Müller-Buschbaum, Adv. Energy Mater., 2015, 5, 1401770 CrossRef.
- K. J. Ihn, J. Moulton and P. Smith, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 735 CrossRef CAS.
- J. Liu, M. Arif, J. Zou, S. I. Khondaker and L. Zhai, Macromolecules, 2009, 42, 9390 CrossRef CAS.
- W. D. Oosterbaan, V. Vrindts, S. Berson, S. Guillerez, O. Douheret, B. Ruttens, J. D'Haen, P. Adriaensens, J. Manca, L. Lusten and D. Vanderzande, J. Mater. Chem., 2009, 19, 5424 RSC.
- J. D. Roehling, I. Arslan and A. J. Moulé, J. Mater. Chem., 2012, 22, 2498 RSC.
- J. Liu, J. Zou and L. Zhai, Macromol. Rapid Commun., 2009, 30, 1387 CrossRef CAS PubMed.
- B. K. Sarker, J. Liu, L. Zhai and S. I. Khondaker, ACS Appl. Mater. Interfaces, 2011, 3, 1180 CrossRef CAS PubMed.
- J. Liu, J. Moo-Young, M. McInnis, M. A. Pasquinelli and L. Zhai, Macromolecules, 2014, 47, 705 CrossRef CAS.
- S. Pan, L. He, J. Peng, F. Qiu and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 8686 CrossRef CAS PubMed.
- W. I. Park, D.-H. Kim, J. Jung, S. W. Hong, Z. Lin and M. Byun, Adv. Mater. Technol., 2019, 4, 1800554 CrossRef.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390 CrossRef CAS PubMed.
- A. C. Kamps, M. Fryd and S.-J. Park, ACS Nano, 2012, 6, 2844 CrossRef CAS PubMed.
- M. H. M. Cativo, D. K. Kim, R. A. Riggleman, K. G. Yager, S. S. Nonnenmann, H. Chao, D. A. Bonnell, C. T. Black, C. R. Kagan and S.-J. Park, ACS Nano, 2014, 8, 12755 CrossRef CAS PubMed.
- S. Oh, M. Yang, J. Bouffard, S. Hong and S.-J. Park, ACS Appl. Mater. Interfaces, 2017, 9, 12865 CrossRef CAS PubMed.
- B. A. G. Hammer, F. A. Bokel, R. C. Hayward and T. Emrick, Chem. Mater., 2011, 23, 4250 CrossRef CAS.
- F. A. Bokel, P. K. Sudeep, E. Pentzer, T. Emrick and R. C. Hayward, Macromolecules, 2011, 44, 1768 CrossRef CAS.
- E. Pentzer, F. A. Bokel, R. C. Hayward and T. Emrick, Adv. Mater., 2012, 24, 2254 CrossRef CAS PubMed.
- L. Bu, E. Pentzer, F. A. Bokel, T. Emrick and R. C. Hayward, ACS Nano, 2012, 6, 10924 CrossRef CAS PubMed.
- S. Chantarak, F. Liu, T. Emrick and T. P. Russell, Macromol. Chem. Phys., 2014, 215, 1647 CrossRef CAS.
- B. A. G. Hammer, M. A. Reyes-Martinez, F. A. Bokel, F. Liu, T. P. Russell, R. C. Hayward, A. L. Briseno and T. Emrick, J. Mater. Chem. C, 2014, 2, 9674 RSC.
- H. J. Kim, M. Skinner, H. Yu, J. H. Oh, A. L. Briseno, T. Emrick, B. J. Kim and R. C. Hayward, Nano Lett., 2015, 15, 5689 CrossRef CAS PubMed.
- D. E. Acevedo-Cartagena, J. Zhu, E. Trabanino, E. Pentzer, T. Emrick, S. S. Nonnenmann, A. L. Briseno and R. C. Hayward, ACS Macro Lett., 2015, 4, 483 CrossRef CAS.
- D. E. Acevedo-Cartagena, J. Zhu, M. Kocun, S. S. Nonnenmann and R. C. Hayward, Macromolecules, 2019, 52, 7756 CrossRef CAS.
- X. Yang, J. Ge, M. He, Z. Ye, X. Liu, J. Peng and F. Qiu, Macromolecules, 2016, 49, 287 CrossRef CAS.
- M. Zhu, S. Pan, Y. Wang, P. Tang, F. Qiu, Z. Lin and J. Peng, Angew. Chem., Int. Ed., 2018, 57, 8644 CrossRef CAS PubMed.
- H. Cui, X. Chen, Y. Wang, D. Wei, F. Qiu and F. Peng, Soft Matter, 2018, 14, 5906 RSC.
- M. He, L. Zhao, J. Wang, W. Han, Y. Yang, F. Qiu and Z. Lin, ACS Nano, 2010, 4, 3241 CrossRef CAS PubMed.
- L. Zhao, X. Pang, R. Adhikary, J. W. Petrich, M. Jeffries-El and Z. Lin, Adv. Mater., 2011, 23, 2844 CrossRef CAS PubMed.
- L. Zhao and Z. Lin, Adv. Mater., 2012, 24, 4353 CrossRef CAS PubMed.
- L. Zhao, X. Pang, R. Adhikary, J. W. Petrich and Z. Lin, Angew. Chem., Int. Ed., 2011, 50, 3958 CrossRef CAS PubMed.
- M. He, F. Qiu and Z. Lin, J. Phys. Chem. Lett., 2013, 4, 1788 CrossRef CAS PubMed.
- J. Jung, Y. J. Yoon, M. He and Z. Lin, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1641 CrossRef CAS.
- F. Li, J. Yang and Y. Qin, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3339 CrossRef CAS.
- F. Li, K. G. Yager, N. M. Dawson, J. Yang, K. J. Malloy and Y. Qin, Macromolecules, 2013, 46, 9021 CrossRef CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Chem. Mater., 2014, 26, 3747 CrossRef CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Polym. Chem., 2015, 6, 721 RSC.
- F. Li, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Polymer, 2015, 76, 220 CrossRef CAS.
- B. W. Watson II, L. Meng, C. Fetrow and Y. Qin, Polymers, 2016, 8, 408 CrossRef PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS PubMed.
- D. Wilson and M. Langell, Appl. Surf. Sci., 2014, 303, 6 CrossRef CAS.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158 CrossRef CAS PubMed.
- B. W. Watson, L. Meng, C. Fetrow and Y. Qin, Polymers, 2016, 8, 408 CrossRef PubMed.
- F. C. Spano, Chem. Phys., 2006, 325, 22 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429 CrossRef CAS PubMed.
- D. L. Huber, Small, 2005, 1, 482 CrossRef CAS PubMed.
- S. Kralj and D. Makovec, ACS Nano, 2015, 9, 9700 CrossRef CAS PubMed.
- G. Singh, H. Chan, T. Udayabhaskararao, E. Gelman, D. Peddis, A. Baskin, G. Leitus, P. Král and R. Klajn, Faraday Discuss., 2015, 181, 403 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption spectra of polymer nanofiber solutions, electron and X-ray diffraction patterns of IONP-OA, infrared spectra, photographs of solution magnetic responsiveness experiments, X-ray diffraction profiles of solar cell active layers, videos showing magnetic attraction of hybrid nanofiber solutions. See DOI: 10.1039/d0na00191k |
| This journal is © The Royal Society of Chemistry 2020 |