
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneously formed and embedding-type ternary MoSe2/MoO2/nitrogen-doped carbon for fast and stable Na-ion storage†
Yuanxing
Yun
a,
Jie
Shao
 *ac,
Xuefang
Shang
b,
Wei
Wang
ad,
Weibo
Huang
a,
Qunting
Qu
*ad and
Honghe
Zheng
*ad
*ac,
Xuefang
Shang
b,
Wei
Wang
ad,
Weibo
Huang
a,
Qunting
Qu
*ad and
Honghe
Zheng
*ad
aCollege of Energy, Soochow Institute for Energy and Materials InnovationS, Soochow University, Suzhou, Jiangsu 215006, China. E-mail: qtqu@suda.edu.cn; hhzheng@suda.edu.cn; shaojie@suda.edu.cn
bKey Laboratory of Medical Molecular Probes, School of Basic Medical Sciences, Xinxiang Medical University, Jinsui Road 601, Xinxiang, Henan 453003, China
cCollege of Chemistry, Chemical Engineering and Material Science, Soochow University, Suzhou, Jiangsu 215006, China
dSuzhou Huaying New Energy Materials and Technology Co., Ltd., Suzhou, Jiangsu 215100, China
First published on 25th February 2020
Abstract
To obtain an electrode material that is capable of manifesting high Na-ion storage capacity during long-term cycling at a rapid discharge/charge rate, ternary heterophases MoSe2/MoO2/carbon are rationally designed and synthesized through a supermolecule-assisted strategy. Through using supermolecules that are constructed from MoO42− and polydopamine as the precursor and sulfonated polystyrene microspheres as the sacrificial template, the in situ formed ternary phases MoSe2/MoO2/carbon are fabricated into a hollow microspherical structure, which is assembled from ultrathin nanosheets with MoSe2 and MoO2 nanocrystallites strongly embedded in a nitrogen-doped carbon matrix. In the ternary phases, the MoSe2 phase contributes to a high Na-ion storage capacity by virtue of its layered crystalline structure with a wide interlayer space, while the surrounding MoO2 and porous nitrogen-doped carbon phases are conducive to rate behaviour and cycling stability of the ternary hybrids since both the two phases are beneficial for electronic transport and structural stability of MoSe2 during repeated sodiation/desodiation reaction. The as-prepared MoSe2/MoO2/carbon manifests excellent rate behaviour (a Na-ion storage capacity of 461 mA h g−1 at an extremely high current density of 70 A g−1) and outstanding cycle performance (610 mA h g−1 after 1000 cycles).
Introduction
Sodium-ion batteries (SIBs) have attracted more and more attention during the past decade due to the abundant reserves and low price of sodium-related sources. Despite the analogous electrochemical reaction process to that in lithium-ion batteries, SIBs suffer seriously from a poor rate behavior because of the sluggish sodiation/desodiation kinetics restricted by the large radius of Na+ and low intrinsic ionic/electronic conductivity of most anode materials.1–6 How can one obtain a SIB anode that can deliver a decent capacity at a rapid discharge/charge rate? The primary strategy is to design an anode material that could transport both Na-ions and electrons quickly. In terms of the former, it is desirable that the crystal lattice structure of active materials should provide convenient channels to facilitate the migration of Na-ions. In this regard, MoSe2, as a transition metal dichalcogenide with a typical 2D graphene-like layered structure and a large interlayer distance of ≈0.65 nm, stands out from various candidates for Na-ion storage.7–14 Based on an electrochemical conversion reaction from MoSe2 to NaSe2 and Mo, the theoretical capacity of MoSe2 reaches 422 mA h g−1, which is quite high as compared with those of other SIB anode materials.15–18 In spite of the superior Na-ion transport ability of MoSe2, its electronic conductivity is not good enough, thereby restricting its high rate behavior. So combining MoSe2 with various electronically conductive carbons has been proposed and demonstrated to be an effective strategy.19–22 MoSe2-covered N, P-doped carbon nanosheets manifest an improved charge transfer kinetics and retain a capacity of 216 mA h g−1 at a high current density of 15 A g−1.9 Hollow carbon nanosphere encapsulated MoSe2 nanosheets exhibit good rate behavior maintaining a capacity of 382 mA h g−1 at a current density of 10 A g−1.23 However, from the viewpoint of the structure–activity relationship, MoSe2-covered carbon nanosheets cannot provide fast electron transport pathways from the top, while the hollow carbon nanosphere encapsulated MoSe2 cannot ensure fast electron transport in the interior. Therefore, constructing an embedding-type structured MoSe2/carbon hybrid with ultrafine MoSe2 homogeneously distributed in a carbon matrix, which may offer a continuous charge transport pathway throughout the whole material, seems more effective in improving the rate behavior of MoSe2-based materials.Besides the rate behavior, MoSe2 is confronted with the issue of poor cycling stability when being used as the anode material of SIBs.8 On the one hand, MoSe2 as a typical two-dimensional material is most commonly fabricated into a nanosheet-like structure, which tends to restack or aggregate during repeated sodiation/desodiation, leading to gradual loss of active sites.24 On the other hand, the as-formed Na2Se after the first sodiation process could not be completely recovered to the MoSe2 phase and might be converted to Se element in the subsequent desodiation reaction.9,25–27 Thus the electrochemical mechanism of MoSe2 after the first cycle would be analogous to those of sulfur or selenium-based cathodes in Li–S, Li–Se or Na–Se batteries involving the conversion reactions of S to Li2S, Se to Li2Se, or Se to Na2Se, respectively. For these batteries, the formation of soluble polyselenide/polysulfide intermediates always leads to a severe shuttle effect and rapid capacity degradation during discharge/charge.28,29 Various porous carbons and transition metal oxides have been adopted for physical or chemical confinement to prevent polyselenide/polysulfide dissolution from sulfur or selenium-based cathodes and improve the cycling performance of these batteries.30–33 By analogy to these sulfur or selenium-based cathodes, the combination of MoSe2 with carbons or transition metal oxides is also expected to bring about improved cycling stability of MoSe2 for Na-ion storage.
With these backgrounds, this work proposes a supermolecule-assisted strategy to synthesize embedding-type structured ternary heterophases MoSe2/MoO2/carbon. Through the incomplete selenization of Mo-based oxides during pyrolysis of MoO42−-polydopamine (Mo-PDA) supermolecules, the ternary heterophases with MoSe2 and MoO2 nanoparticles uniformly embedded in a carbon matrix are obtained. Compared to the totally selenized binary phases MoSe2/carbon and unselenized binary phases MoO2/carbon, the ternary phases MoSe2/MoO2/carbon exhibit significantly improved Na-ion storage performance. By virtue of the superior Na-ion storage capability of MoSe2, good electronic conductivity of MoO2 and carbon, and the unique embedding-type structure in which the surrounding MoO2 and carbon phases can preserve the structural stability of MoSe2, the ternary heterophases exhibit excellent rate behavior (a Na-ion storage capacity of 461 mA h g−1 at an extremely high current density of 70 A g−1) and outstanding long-term cycling performance, maintaining a capacity of 610 mA h g−1 after 1000 cycles.
Experimental
Materials preparation
Sulfonated polystyrene (SPS) microspheres were prepared according to previous reports34,35 and adopted as the sacrificial template to synthesize hollow microspherical MoSe2/MoO2/carbon. Firstly, 30 mg of SPS microspheres were dispersed in a solution mixture containing 15 mg of ammonium molybdate and 15 mg of dopamine hydrochloride dissolved in a mixed solvent of ethanol and distilled water (2/1 volume ratio). The pH of the solution is adjusted to 9 using ammonium hydroxide. The solution mixture gradually turned dark red after several hours of stirring due to the formation of MoO42−-polydopamine (Mo-PDA) supermolecules on SPS. The SPS@Mo-PDA solid was collected from the solution through centrifugation, and then washed successively with distilled water and ethanol, and at last dried at 60 °C overnight. To prepare hollow microspherical MoSe2/MoO2/carbon ternary phases, the obtained core–shell SPS@Mo-PDA was pyrolyzed and selenized in a tube furnace with a flow of high-quality Ar gas. Se powder was used as the Se source. SPS@Mo-PDA and Se powder were separately placed at both ends of the same ceramic boat, in which Se powder was located at the upstream. The ceramic boat is kept open with no covering on it. The mass of Se powder was around three times that of SPS@Mo-PDA, and the temperature of the tube furnace was raised to 500 °C with a rate of 2 °C min−1 and maintained at this temperature for 3 h. Totally selenized MoSe2/carbon binary phases were prepared through a similar procedure except that the heating rate was set at 1 °C min−1 and the temperature was maintained at 500 °C for 4 h. In addition, another empty ceramic boat was covered in an inverted position on the ceramic boat containing SPS@Mo–PDA and Se powder to ensure complete selenization. The preparation procedure of unselenized MoO2/carbon binary phases was similar to that of MoSe2/MoO2/carbon except that Se powder was absent.Electrochemical testing
The working electrode laminate was prepared by spreading the electrode slurry onto copper foil using a coating machine. The slurry was obtained by ultrasonically dispersing a mixture of active materials (MoSe2/MoO2/NC, MoO2/NC or MoSe2/NC), conductive agent (super-P Li), and binder (sodium alginate) in distilled water. The mass ratio of active material, conductive agent, and binder in the mixture is 7/2/1. The working electrode laminate was cut into small disks with a diameter of 13 mm using a punching machine. The average loading of active substances on each electrode disk is 1.0 mg cm−2. CR-2032 type coin cells were assembled in a glovebox filled with high-purity argon using a sodium disk as the counter electrode, 1.0 M NaClO4 dissolved in propylene carbonate as the electrolyte, and a glass fiber filter (Whatman) as the separator. The coin cells were galvanostatically discharged and charged at various current densities in the potential range of 0.01–3.0 V using a LAND CT2001A charge/discharge tester. The specific capacity was calculated based on the total mass of the ternary or binary composites, namely, the mass of NC was also taken into account. The cyclic voltammetric (CV) data were collected at a scan rate of 0.1 mV s−1 using a CHI 660C electrochemical workstation.Results and discussion
Supermolecules that are composed of inorganic nodes/clusters uniformly dispersed in organic ligands at the molecular level can be used as the precursors to synthesize some hybrids of metal oxides and carbon. Because of the unique structure of supermolecules, the obtained metal oxide nanoparticles could be homogeneously and strongly embedded in or wrapped by the organic ligand-derived carbon matrix, which is favorable for the electronic transport throughout the whole electrode. In this work, a kind of supermolecule, MoO42−-polydopamine complex (denoted as Mo-PDA), is used as the precursor to synthesize ternary heterophases MoSe2/MoO2/carbon. To obtain a hierarchical hollow microspherical structure, sulfonated polystyrene (SPS) microspheres are adopted as the sacrificial template.34,35 A schematic is shown in Fig. 1 to illustrate the formation of MoSe2/MoO2/carbon. First, SPS microspheres are dispersed in a solution mixture containing ammonium molybdate and dopamine hydrochloride, and the pH of the solution is adjusted to 9 using ammonium hydroxide. The self-polymerization of dopamine preferentially occurs on the surface of SPS microspheres due to the sulfonated and active surface of SPS. Meanwhile, the catechol groups of dopamine possess a strong complexing ability towards MoO42− during its self-polymerization,36–38 and thus Mo-PDA supermolecules are formed on SPS. The obtained core–shell SPS@Mo-PDA is then pyrolyzed and selenized in a tube furnace with a flow of high-quality Ar gas. During pyrolysis, the inner SPS core could decompose completely and meanwhile the Mo-PDA shell could be in situ converted into MoSe2/MoO2 and N-doped carbon (NC), forming a hollow microspherical structure with MoSe2/MoO2 nanoparticles uniformly embedded in the NC matrix. The N-doping of carbon originates from the fact that PDA is a N-containing precursor. The incomplete selenization of the Mo-PDA derived Mo oxides generates the ternary heterophases MoSe2/MoO2/NC. Through slightly changing the reaction parameters, totally selenized binary phases MoSe2/NC can be obtained (Fig. S1†). Unselenized binary phases MoO2/NC (Fig. S2†) are prepared through the same procedure but in the absence of Se powder. | ||
| Fig. 1 Schematic illustration of the synthesis procedure of hollow microspherical MoSe2/MoO2/carbon ternary phases. | ||
Scanning electron microscopy (SEM) images of the intermediate SPS@Mo–PDA and end product MoSe2/MoO2/NC are shown in Fig. 2. The morphology of SPS@Mo-PDA (Fig. 2a) exhibits the same microspherical structure to that of the original SPS template. Compared to the smooth surface of pure SPS (Fig. S3†), nanosheet-like Mo-PDA is obviously grown on the surface of microspheres. After pyrolysis and selenization, the microspherical morphology is perfectly maintained, and the 2D nanosheet-like structure becomes more distinct due to the formation of crystalline-state MoSe2/MoO2/NC (Fig. 2b–d). The MoSe2/MoO2/NC microspheres possess a homogeneous diameter of around 600 nm. No extra particles or uneven aggregates are observed, suggesting that Mo-PDA supermolecules preferentially grow on the surface of SPS, which guarantees the homogeneous microspherical morphology of the product. In addition, a hollow interior is supposed to be formed in these microspheres since some holes as indicated by the red arrows in Fig. 2d are distinguishable.
 | ||
| Fig. 2 SEM images of (a) the intermediate SPS@Mo-PDA and (b–d) end product MoSe2/MoO2/NC. | ||
The hollow interior of MoSe2/MoO2/NC microspheres is also clearly observed through transmission electron microscopy (TEM). As shown in Fig. 3a, the black edges are in sharp contrast to the grey central areas, and the walls of the hollow microspheres are composed of ultrathin nanosheets. From the high-resolution TEM images (Fig. 3b and S4†), the lattice fringes with interplanar spacings of 0.28 and 0.65 nm are easily found, which can be indexed to the (100) and (002) crystalline planes of MoSe2, respectively. The lattice fringes with an interplanar spacing of 0.20 nm correspond to the (200) crystalline planes of MoO2. Besides MoSe2 and MoO2 crystallites, some porous and undefined regions, which should be amorphous carbon, are distributed in the hybrid. The dimensions of MoSe2 and MoO2 crystallites are observed to be less than 5 nm, so the structure of the ternary hybrid is described as ultrasmall MoSe2 and MoO2 crystallites embedded in a carbon matrix, which is the typical structure of supermolecule-derived materials. During the preparation of the ternary MoSe2/MoO2/NC hybrid, the PDA-derived carbon plays the role of restricting the growth of MoSe2 and MoO2, thereby giving rise to the formation of ultrasmall MoSe2 and MoO2 crystallites. Moreover, the in situ formation of N-doped carbon and MoSe2/MoO2 through the use of the Mo-PDA precursor could generate a strong interfacial interaction among the neighbouring phases, which is beneficial for Li-ion and electron transfer across the interface.
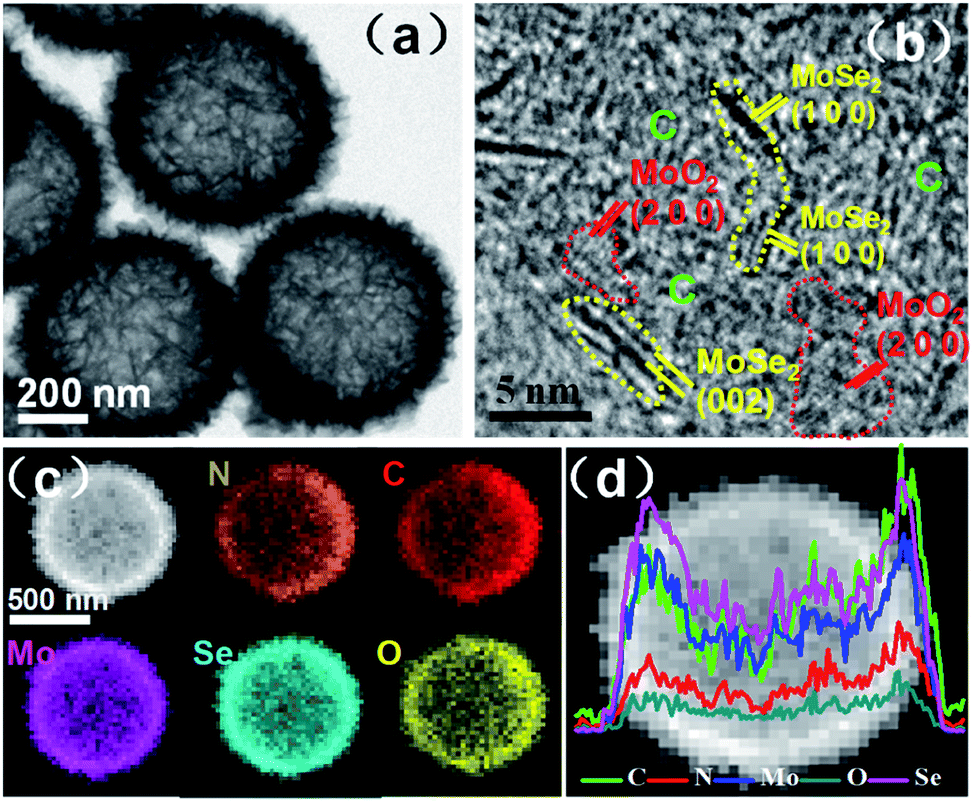 | ||
| Fig. 3 (a) TEM and (b) high-resolution TEM images, (c) EDS elemental mapping images and (d) EDS linear scan profiles of hollow microspherical MoSe2/MoO2/NC ternary phases. | ||
The elemental mapping images (Fig. 3c) of the hybrid obtained through energy dispersive X-ray spectroscopy (EDS) confirm the coexistence of Mo, Se, O, C, and N elements, which are homogeneously distributed in the wall of hollow microspheres. The linear scan profiles of all the elements along the diameter of one typical microsphere (Fig. 3d) exhibit double peaks on both sides with the lowest intensity in central areas, which further demonstrate the hollow microspherical structure of the ternary hybrid. The Brunauer–Emmett–Teller (BET) surface area of MoSe2/MoO2/NC determined by N2 adsorption/desorption isotherms (Fig. S5†) is 190 m2 g−1. Such a high surface area should originate from the porous carbon and hollow microspherical structure of the hybrid phase as observed from TEM images.
The crystalline phases of the product were further investigated using X-ray diffraction (XRD). As shown in Fig. 4a, the diffraction peaks can be well indexed to the hybrid phases of hexagonal 2H-MoSe2 (JCPDS no. 29-0914) and monoclinic MoO2 (JPCDS no. 32-0671).39,40 Besides the typical peaks characteristic of MoSe2, several weak diffraction peaks at 26.0°, 36.8° and 53.5° are well consistent with standard peaks of MoO2, further validating the presence of MoO2 in the composite. Notably, all the XRD peaks of MoSe2/MoO2/NC are quite weak or broad, which is ascribed to the ultrasmall size of MoSe2 and MoO2 nanocrystallites. There are no obvious peaks characteristic of graphitic carbon, suggesting that the carbon derived from the organic ligands of Mo-PDA is mainly amorphous. Raman spectroscopy was used to further ascertain the presence of carbon and also verify the hybrid crystalline phases of MoSe2/MoO2/NC. As shown in Fig. 4b, the two characteristic peaks of carbon, D-band and G-band, appear at 1353 and 1580 cm−1, respectively. Besides, the two peaks at around 237 and 287 cm−1 correspond to the out-of-plane A1g and in-plane E12g modes of MoSe2,41 while other peaks at 492, 573 and 745 cm−1 are characteristic of MoO2.42
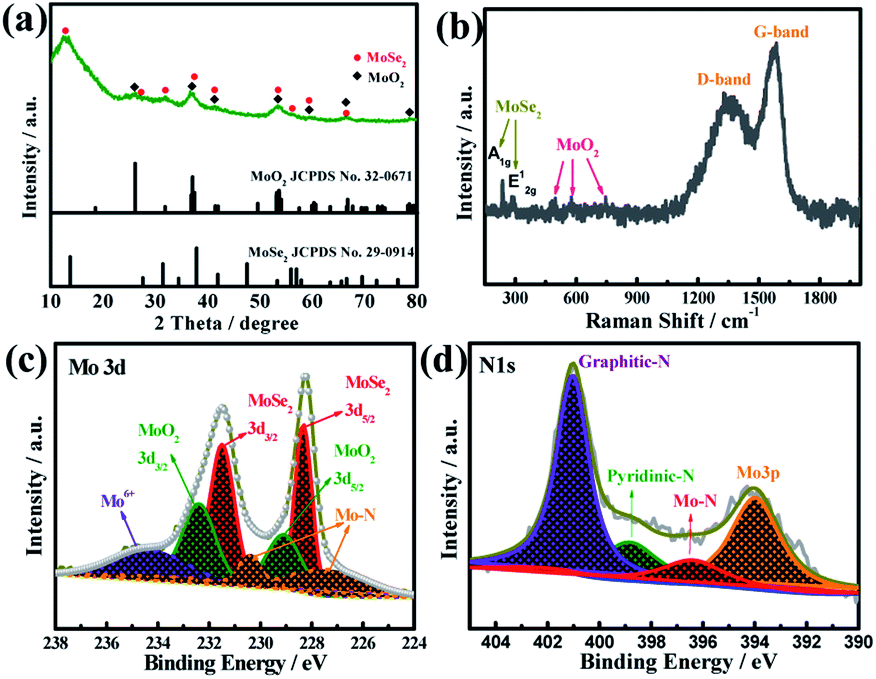 | ||
| Fig. 4 (a) XRD patterns and (b) Raman spectra of MoSe2/MoO2/NC, and (c and d) the deconvoluted XPS spectra in the core-level regions of Mo 3d and N 1s. | ||
X-ray photoelectron spectroscopy (XPS) was used to investigate the surface electronic state of each of the elements in the ternary phases MoSe2/MoO2/carbon. The survey spectrum reveals the existence of Mo, Se, O, C, and N elements (Fig. S6†). The deconvoluted spectra of the Mo 3d core level are shown in Fig. 4c. The doublet peaks at the binding energies of 231.5 and 228.3 eV can be attributed to the Mo 3d3/2 and Mo 3d5/2 spin-orbits of Mo4+ in the environment of MoSe2, while the neighboring doublet peaks at slightly higher binding energies of 232.4 eV and 229.2 eV signify the presence of MoO2.13,42,43 The one broad peak at a binding energy of around 234 eV usually originates from the slight oxidation of Mo4+ to Mo6+ when the sample is exposed to air.13 The deconvoluted satellite peaks at lower binding energies of 230.4 and 227.5 eV can be indexed to the Mo–N bond, the formation of which results from the incorporation of nitrogen into the hybrids through the use of a N-containing supermolecule precursor. The XPS spectrum of the Se 3d core level is split into two peaks at 54.9 and 53.8 eV, corresponding well with the Se 3d3/2 and Se 3d5/2 spin-orbits of Se2− (Fig. S7†).42 The deconvoluted XPS spectra of the C 1s core level (Fig. S8†) exhibit one dominant peak at 284.6 eV, which is characteristic of carbon materials. The deconvoluted XPS spectra in the core level region of N 1s are shown in Fig. 4d. The two binding energies at 398.8 and 401.1 eV can be identified as pyridinic-N and graphitic-N species located at the six-member ring of the carbon lattice. One additional peak at 396.5 eV can be attributed to the Mo–N bond.
To gain insight into the differences between the electronic states of Mo element in MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC, XPS spectra of binary MoO2/NC and MoSe2/NC in the core-level region of Mo 3d were also measured and are shown in Fig. S9 and S10,† respectively. In the case of binary MoO2/NC, the binding energies of 230.0 and 232.9 eV can be assigned to Mo4+ species, while the binding energies of 231.8 and 235.0 eV signify the presence of Mo6+. The relatively high content of Mo6+ in the surface of MoO2/NC could be caused by its oxidation in air since monoclinic MoO2 is a metastable phase. Besides, one extra weak peak of Mo 3d located at 228.9 eV is correlated with the Mo–N bond. In the case of binary MoSe2/NC, the doublet peaks at the binding energies of 229.2 and 232.3 eV can be attributed to the Mo 3d5/2 and Mo 3d3/2 spin-orbits of Mo4+ in the environment of MoSe2. The oxidation of Mo4+ by air is not obvious for MoSe2/NC because there are no obvious peaks at higher binding energy. Moreover, there are no deconvoluted peaks characteristic of the Mo–N bond. Therefore, it is speculated that the presence of Mo6+ in the ternary MoSe2/MoO2/NC should originate from the oxidation of the MoO2 phase by air, while the Mo–N bond is most probably formed between MoO2 and nitrogen-doped carbon phases.
All the TEM, EDS, XRD, Raman, and XPS results demonstrate that the obtained ternary hybrid MoSe2/MoO2/NC is composed of MoSe2, MoO2, and nitrogen-doped carbon. To quantitatively analyze the content of each phase in the ternary hybrid MoSe2/MoO2/NC, elemental analysis and inductively coupled plasma optical emission spectroscopy were used. The mass percentages of all the elements are listed in Table S1.† Through calculation, the mass fraction of MoSe2, MoO2, and nitrogen-doped carbon in the hybrid is around 39.4, 26.2, and 34.4 wt%, respectively.
The Na-ion storage performance of the as-prepared ternary phases MoSe2/MoO2/NC was investigated using a sodium disk as the counter electrode and compared with that of binary phases MoSe2/NC and MoO2/NC. Cyclic voltammetry (CV) was employed to study the redox reaction behaviour of the electrode materials and the CV curves obtained at a scan rate of 0.1 mV s−1 are shown in Fig. 5a, S11 and S12.† In the case of ternary phases MoSe2/MoO2/NC, during the first negative scan, there are three reduction peaks at around 1.1, 0.6, and 0.2 V, which are similar to those of MoSe2-based electrodes reported in the literature.9,21,25,27 The first reduction peak at 1.1 V is usually considered to be the intercalation of Na-ions into the MoSe2 lattice accompanied by the formation of the NaxMoSe2 phase. The other two reduction peaks at 0.6 and 0.2 V correspond to the solid-state interphase (SEI) film formation via the reductive decomposition of electrolyte and the conversion reaction from NaxMoSe2 to Mo/Na2Se, respectively.44 The following positive scan corresponds to the desodiation process, during which the main oxidation peak at 1.8 V and the shoulder-like peak at 2.2 V are in good agreement with the reported oxidation peaks of MoSe2-based electrodes in SIBs.21,25 The reaction mechanisms of these oxidation peaks are still controversial. Some researchers claimed that the desodiation process should be accompanied by the reversible conversion from metallic Mo back to MoSe2.17,23,24 Also, there are some reports proposing that the desodiation process is related to the oxidation of Na2Se rather than metallic Mo since Se instead of MoSe2 is obviously formed after the desodiation process.9,25,26 Through systematically studying the ex situ Raman spectra of MoSe2-based electrodes at different charge/discharge potentials, Yang et al.9 directly prove that Se is formed at the end of the first charge. Therefore, it is proposed that the Na-ion storage mechanism of MoSe2 after the first cycle most probably involves the reversible conversion reaction from Se to Na2Se.
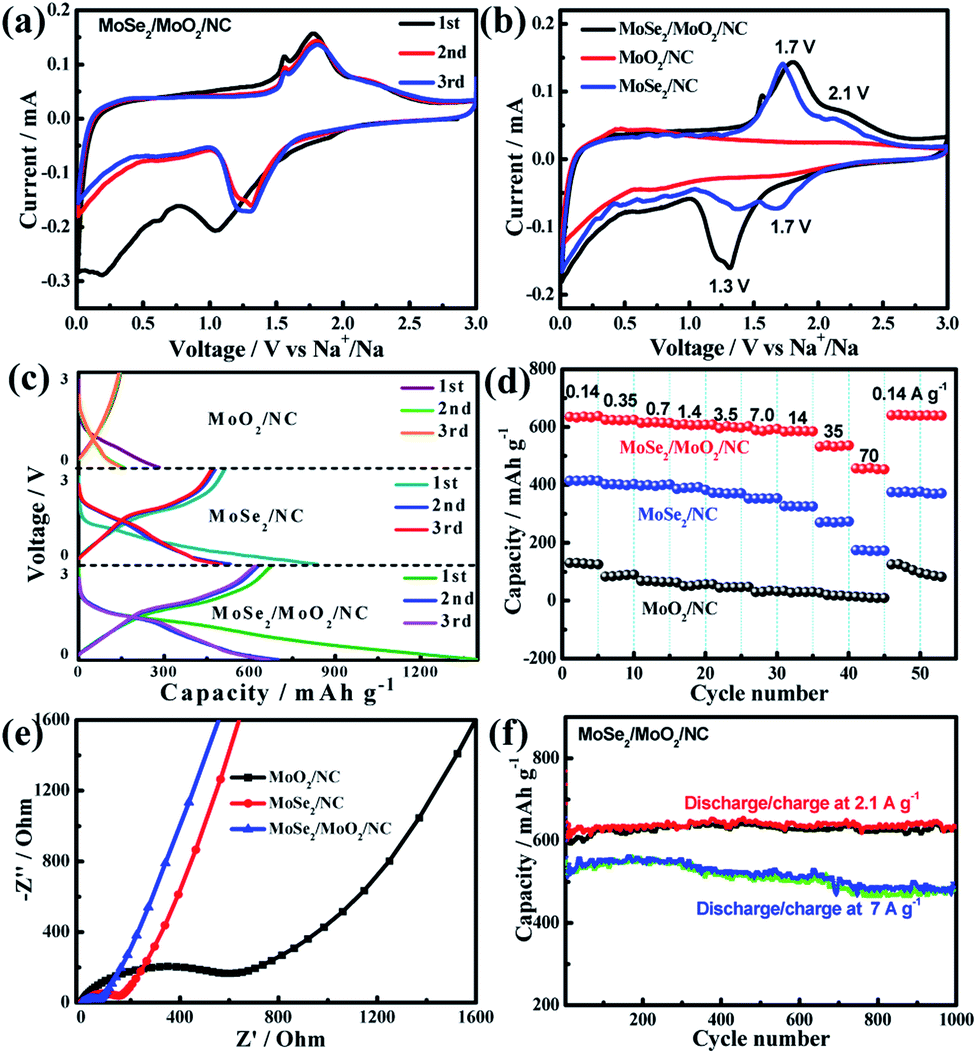 | ||
| Fig. 5 (a) CV curves of MoSe2/MoO2/NC during the first three scans at a scan rate of 0.1 mV s−1, (b) the CV curve comparison of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC electrodes in the second scan, (c) the first three discharge/charge curves at a current density of 0.14 A g−1, (d) the capacity evolution at various charge current rates ranging from 0.14 to 70 A g−1, (e) Nyquist plots of the three hybrid electrodes after five discharge/charge cycles, and (f) long-term cycling performance of the MoSe2/MoO2/NC electrode. | ||
The CV curves of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC electrodes in the second cycle are compared in Fig. 5b. The two couples of redox peaks at 1.7/2.1 V and 1.3/1.7 V observed for binary phases MoSe2/NC are typical of MoSe2-based electrodes in SIBs. Abouimrane et al.45 previously studied the Na-ion storage behaviour of a Se electrode and observed two discharge plateaus at 1.9 and 1.5 V in the sodiation process, so they proposed a multi-phase conversion reaction of Se, where intermediate phase polyselenides may be formed. The two reduction peaks of MoSe2/NC in the second cycle appear at similar potentials of 1.7 and 1.3 V, which actually should correspond to two potential plateaus in the discharge curves, further suggesting that the Na-ion storage mechanism of MoSe2 should be analogous to that of Se. So, intermediate phase polyselenides could also be formed during the cycling of MoSe2-based electrodes.42 The dissolution and shuttling of polyselenides may lead to a rapid capacity decline of MoSe2-based electrodes. In the case of binary phases MoO2/NC, no significant redox peaks are observed due to the poor Na-ion storage ability of MoO2. Through comparing the CV curves of MoSe2/MoO2/NC and MoSe2/NC, the main differences lie in that the first reduction peak of MoSe2/NC at 1.7 V is not clearly defined for MoSe2/MoO2/NC, but the reduction peak intensity of MoSe2/MoO2/NC at a lower potential of 1.3 V is much stronger. So it is assumed that such in situ formed ternary phases MoSe2/MoO2/NC could preclude the formation of intermediate phase polyselenides, which will be beneficial for the structural stabilization of electrodes during charge/discharge cycling.
Out of interest, the Li-ion storage behaviour of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC is also investigated through CV (Fig. S13–S15†). In terms of MoO2/NC, the two couples of sharp redox peaks at 1.3/1.4 V and 1.6/1.7 V correspond to the phase conversion of MoO2 upon lithiation/delithiation.46 The CV curves of MoSe2/NC exhibit one couple of reversible redox peaks at 1.8/2.2 V, which are very similar to those of Se-based electrodes in Li–Se batteries.28,30,47 So the Li-ion storage mechanism of MoSe2 after the first cycle is acknowledged to be based on the reversible conversion reaction of Se to Li2Se.48 By analogy to the Li-ion storage of MoSe2, it is most likely that MoSe2 would exhibit a similar conversion reaction of Se to Na2Se. In the case of ternary phases MoSe2/MoO2/NC, the redox peaks of both MoO2 and MoSe2 are clearly identified, which is another piece of evidence proving the co-existence of MoSe2 and MoO2 in the hybrid.
The first three discharge/charge curves of the as-prepared hybrids at a current density of 0.14 A g−1 are shown in Fig. 5c. The reversible (charge) capacity of MoO2/NC is quite low (around 124 mA h g−1), and this is because the crystalline structure of MoO2 is not favorable for Na-ion storage. The discharge curves of MoSe2/NC exhibit a sloping potential plateau at around 1.5 V, corresponding to the two reduction peaks at around 1.7 and 1.3 V in CV curves. The reversible capacity of MoSe2/NC in the second cycle is around 413 mA h g−1, close to the theoretical capacity of MoSe2. By comparison with MoSe2/NC, MoSe2/MoO2/NC manifests a similar potential change tendency during discharge/charge, while its capacity is considerably enhanced. The reversible capacity of MoSe2/MoO2/NC at a current density of 0.14 A g−1 in the second cycle is around 634 mA h g−1, which is significantly higher than that of carbon nanosphere encapsulated MoSe2 (498 mA h g−1 at 0.1 A g−1),25 MoSe2-covered N, P-doped carbon nanosheets (454 mA h g−1 at 0.5 A g−1),9 and MoO2 nanocluster decorated MoSe2/graphene (422 mA h g−1 at 0.1 A g−1).42 To calculate the theoretical capacity of the ternary MoSe2/MoO2/NC phases, the theoretical capacity and mass fraction of each phase should be considered. The theoretical capacity of MoSe2 for Na-ion storage is 422 mA h g−1. However, it is difficult to determine the theoretical capacities of the other two phases MoO2 and NC because their crystalline structures are unfavorable for Na-ion storage and their Na-ion storage capacity should mainly be capacitive. So here the practical capacity of MoO2/NC (124 mA h g−1) is assumed as the theoretical capacity of the other two components (MoO2/NC). Taking the mass fractions of MoSe2 and MoO2/NC (39.4% and 60.6%, respectively) into consideration, the theoretical capacity of the ternary phases is calculated to be 241 mA h g−1. The considerably enhanced capacity of MoSe2/MoO2/NC as compared to the theoretical value suggests a strong synergistic effect of the ternary phases for Na-ion storage.49 Firstly, the numerous pores in MoSe2/MoO2/NC as observed from TEM images are capable of accommodating Na-ions. Especially, the PDA-derived N-doped porous carbon can provide more active sites for Na-ion storage than undoped carbon. Secondly, the unique structure of MoSe2/MoO2/NC with ultrasmall MoSe2/MoO2 nanocrystallites uniformly embedded in the carbon matrix not only increases the electroactive sites but also favours electronic transport, thereby giving rise to a high utilization efficiency of active materials. Thirdly, the polymer/gel-like film formed on the electrode surface could reversibly store/release Na-ions, which generates some extra capacity for the MoSe2/MoO2/NC electrode. Lastly, besides the Na-ions stored in the bulk phase of MoSe2, some Na-ions may be stored in the pores or interstices of the ternary phases MoSe2/MoO2/NC. Pak and Stucky et al.50 reported that nanoscale pore engineering of MoO2 enables the formation of a metallic Li-rich phase between the Li-ion intercalated MoO2 phase, so its practical Li-storage capacity is considerably higher than the theoretical value. Similarly, it is also very probable that a metallic Na-rich phase may be formed in pores or interstices during the sodiation of MoSe2/MoO2/NC. All these factors contribute to the high capacity of the ternary phases MoSe2/MoO2/NC.
The rate behaviour of the as-prepared hybrids was tested by discharging the electrodes at a current density of 0.14 A g−1 and charging them at various current rates ranging from 0.14 to 70 A g−1. As shown in Fig. 5d, the MoSe2/MoO2/NC electrode exhibits an average capacity of 611, 594, 534, and 461 mA h g−1 at current densities of 1.4, 7.0, 35, and 70 A g−1, respectively. Once the current density is returned to 0.14 A g−1, the capacity rapidly recovers to 640 mA h g−1. The maximum current density used for the rate behaviour evaluation of MoSe2-based electrodes in previous reports is usually up to 10 A g−1. The high capacity of MoSe2/MoO2/NC at extremely high current densities of 35 and 70 A g−1 suggests its excellent high-rate capability, which undoubtedly should be related to the unique structure of ternary phases. In the MoSe2/MoO2/NC hybrid, ultrasmall MoSe2 and MoO2 nanocrystallites are strongly embedded into or surrounded by carbon, which can offer omnibearing electronic transport pathways through the whole electrode. Compared to the MoSe2/carbon nanocomposites with carbon coated onto or encapsulated into MoSe2 active materials, where the carbon can not provide effective conductive pathways from the bottom or top, the supermolecule-derived embedding-type MoSe2/MoO2/carbon nanocomposites possess unique advantages in electron transport. The nitrogen-doping of carbon can further increase its electronic conductivity, benefit its affinity to electrolyte, and also increase its electroactive sites for Na-ion storage. Furthermore, in the ternary MoSe2/MoO2/NC nanocomposites, the presence of the Mo–N bond as revealed by the XPS results could lead to an enhanced interfacial interaction between MoSe2/MoO2 and N-doped carbon because some nitrogen may be simultaneously coordinated to Mo and C. As a result, an intimate contact between MoSe2/MoO2 and N-doped carbon is achieved, which will facilitate the transport of electrons and Na-ions across the interface of heterophases. The good electronic conductivity of MoO2 resulting from the existence of delocalized electrons in its conduction band also facilitates the electronic transport of the ternary MoSe2/MoO2/NC electrode.46,51–53 In addition, the porous structure of carbon and ultrasmall nanocrystallite size of MoSe2/MoO2 can facilitate the access of electrolyte to the active material and shorten the diffusion distance of Na-ions in the electrode bulk. As a result, not only the charge-transfer reactions on the electrode/electrolyte interfaces but also the diffusion of Na-ions in the solid phase will be favourable, which bring about the excellent rate behaviour of MoSe2/MoO2/NC. By contrast, the binary phases MoO2/NC exhibit extremely low capacity at high current rates due to the low Na-ion storage capability of MoO2. The reversible capacities of MoSe2/NC at various current rates are obviously lower than those of MoSe2/MoO2/NC, suggesting the synergistic effect of ternary phases for improving the Na-ion storage capacity and rate performance.
To gain insight into the electrode reaction dynamics of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC, electrochemical impedance spectroscopy (EIS) was used. The Nyquist plots of the three electrodes after five complete sodiation/desodiation cycles are shown in Fig. 5e. The diameters of the depressed semicircles at medium-to-high frequency reflect the charge-transfer resistance (Rct) occurring on the electrode/electrolyte interface, while the inclined lines in the low frequency zone are ascribed to Warburg impedance, which is associated with solid-state Na-ion diffusion in the electrode bulk.44 The Rct value of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC is 67, 154, and 627 Ω, respectively. As we know, the charge-transfer reaction dynamics of the electrode is related to the transport of both electrons and Na-ions. Even though MoO2 should have better electronic conductivity, its Na-ion transport ability should be very poor during the electrochemical reactions as reflected by its very low Na-ion storage capacity. The much lower Rct of MoSe2/NC as compared to that of MoO2/NC suggests that the Na-ion transport ability of the electrode is also of great importance for a decreased Rct. Therefore, a synergistic effect of MoSe2 and MoO2 gives rise to the lowest Rct of ternary MoSe2/MoO2/NC, suggesting a rapid charge transfer reaction on the electrode/electrolyte interface. As estimated from the slope of the inclined lines at low frequency, the Na-ion diffusion resistance in the MoO2/NC electrode should be significantly higher than that in the other two electrodes. The different rate behaviour of MoSe2/MoO2/NC, MoSe2/NC, and MoO2/NC can be perfectly explained by their different charge transfer and Na-ion diffusion resistances. Specifically, the lower the resistances are, the better the rate behaviour is.
The long-term cycling performance of the MoSe2/MoO2/NC electrode is shown in Fig. 5f. The reversible capacity of MoSe2/MoO2/NC discharged/charged at 2.1 A g−1 is stabilized at around 610 mA h g−1 after the initial several formation cycles, and manifests negligible degradation during the subsequent 1000 cycles. At a higher discharge/charge rate of 7 A g−1, the reversible capacity of the 1000th cycle is around 480 mA h g−1, equalling to a capacity retention of around 91%. The electrochemical performance of some previously reported MoSe2-based anodes for Na-ion batteries is summarized in Table S2.† For instance, MoSe2-covered N, P-doped carbon nanosheets exhibit an initial Na-ion storage capacity of 378 mA h g−1 and a capacity retention of 87% after 1000 cycles at 0.5 A g−1;9 encapsulation-type structured MoSe2@hollow carbon nanospheres preserve a capacity of 501 mA h g−1 after 1000 cycles at 1 A g−1.23 These comparative data suggest the outstanding cycling stability of MoSe2/MoO2/NC for Na-ion storage, which undoubtedly is related to its unique structure and ternary phase composition. The pores and voids in the hierarchical structured MoSe2/MoO2/NC hollow microspheres assembled from ultrathin nanosheets work as the buffering areas to alleviate the volume change of MoSe2 upon sodiation/desodiation, enabling the electrode structure to be stable. TEM images of the MoSe2/MoO2/NC electrode after 1000 cycles (Fig. 6) show that not only the hollow microspherical structure but also the overall dimensions of these microspheres are well maintained, validating the structural stability of MoSe2/MoO2/NC during repeated sodiation/desodiation reaction. Also, the embedding-type structure of the in situ formed ternary phases MoSe2/MoO2/NC renders a good contact among the three heterophases, and the surrounding MoO2 and NC phases can restrict the aggregation of MoSe2 active materials during long-term cycling. In addition, by analogy to the sulphur-based cathodes in Li–S batteries, where both porous MoO2 and nitrogen-doped carbon have been combined with sulphur to prevent polysulfide dissolution and improve the cycling performance,32,42 the presence of MoO2 and nitrogen-doped carbon in the ternary phases MoSe2/MoO2/NC is supposed to play the same role of trapping polyselenides generated by the sodiation/desodiation reaction of MoSe2. Correspondingly, the cycling performance of MoSe2/MoO2/NC for Na-ion storage is significantly improved through the rational construction of embedding-type structured ternary phases.
 | ||
| Fig. 6 TEM images of MoSe2/MoO2/NC after 1000 sodiation/desodiation cycles. | ||
Conclusions
Strongly coupled ternary heterophases MoSe2/MoO2/NC are rationally designed and synthesized through a supermolecule-assisted strategy. The supermolecule-derived ternary phases are composed of MoSe2 and MoO2 nanocrystallites uniformly embedded in a nitrogen-doped carbon matrix. Compared to the binary phases MoSe2/NC and MoO2/NC, the ternary phases exhibit a strong synergistic effect for improving the Na-ion storage performance. The large interlayer space of MoSe2 favours Na-ion transport and storage. The surrounding electronically conductive MoO2 nanocrystallites and porous nitrogen-doped carbon not only provide fast electronic transport pathways through the whole electrode, but also play a role in preventing aggregation, alleviating volume change and trapping polyselenides engendered by the sodiation/desodiation reaction of MoSe2. As a result, the ternary phases manifest excellent rate behaviour and outstanding long cycle life. Such a supermolecule-assisted protocol for the in situ formation of ternary heterophases would provide a new strategy for the designed synthesis of superior nanocomposite electrodes for various applications in energy storage and conversion systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC no. 21301124 and 21875156) and The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJD150004).Notes and references
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- H. Zhang, I. Hasa and S. Passerini, Adv. Energy Mater., 2018, 8, 1702582 CrossRef.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- H. Han, X. Chen, J. Qian, F. Zhong, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Nanoscale, 2019, 11, 21999–22005 RSC.
- D. Cao, W. Kang, S. Wang, Y. Wang, K. Sun, L. Yang, X. Zhou, D. Sun and Y. Cao, J. Mater. Chem. A, 2019, 7, 8268–8276 RSC.
- Y. Wang, W. Kang, D. Cao, M. Zhang, Z. Kang, Z. Xiao, R. Wang and D. Sun, J. Mater. Chem. A, 2018, 6, 4776–4782 RSC.
- A. Eftekhari, Appl. Mater. Today, 2017, 8, 1–17 CrossRef.
- D. Xie, X. Xia, Y. Zhong, Y. Wang, D. Wang, X. Wang and J. Tu, Adv. Energy Mater., 2017, 7, 1601804 CrossRef.
- F. Niu, J. Yang, N. Wang, D. Zhang, W. Fan, J. Yang and Y. Qian, Adv. Funct. Mater., 2017, 27, 1700522 CrossRef.
- J. Ge, L. Fan, J. Wang, Q. Zhang, Z. Liu, E. Zhang, Q. Liu, X. Yu and B. Lu, Adv. Energy Mater., 2018, 8, 1801477 CrossRef.
- L. Zeng, B. Kang, F. Luo, Y. Fang, C. Zheng, J. Liu, R. Liu, Q. Chen, M. Wei and Q. Qian, Chem.–Eur. J., 2019, 25, 13411–13421 CrossRef CAS PubMed.
- J. Chen, A. Pan, Y. Wang, X. Cao, W. Zhang, X. Kong, Q. Su, J. Lin, G. Cao and S. Liang, Energy Storage Mater., 2019, 21, 97–106 CrossRef.
- J. Zhang, M. Wu, T. Liu, W. Kang and J. Xu, J. Mater. Chem. A, 2017, 5, 24859–24866 RSC.
- J. Zhang, W. Kang, M. Jiang, Y. You, Y. Cao, T.-W. Ng, D. Y. W. Yu, C.-S. Lee and J. Xu, Nanoscale, 2017, 9, 1484–1490 RSC.
- J. Gao, Y. Li, L. Shi, J. Li and G. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 20635–20642 CrossRef CAS PubMed.
- C. Zhao, H. Song, Q. Zhuang, Q. Ma, J. Liang, H. Peng, C. Mao, Z. Zhang and G. Li, Inorg. Chem. Front., 2018, 5, 1026–1032 RSC.
- H. Wang, X. Lan, D. Jiang, Y. Zhang, H. Zhong, Z. Zhang and Y. Jiang, J. Power Sources, 2015, 283, 187–194 CrossRef CAS.
- W. Kang, Y. Wang, D. Cao, Z. Kang and D. Sun, J. Alloys Compd., 2018, 743, 410–418 CrossRef CAS.
- G. D. Park, J. H. Kim, S.-K. Park and Y. C. Kang, ACS Appl. Mater. Interfaces, 2017, 9, 10673–10683 CrossRef CAS PubMed.
- X. Zhao, W. Cai, Y. Yang, X. Song, Z. Neale, H.-E. Wang, J. Sui and G. Cao, Nano Energy, 2018, 47, 224–234 CrossRef CAS.
- H. Liu, B. Liu, H. Guo, M. Liang, Y. Zhang, T. Borjigin, X. Yang, L. Wang and X. Sun, Nano Energy, 2018, 51, 639–648 CrossRef CAS.
- Y. Wang, Y. Wang, W. Kang, D. Cao, C. Li, D. Cao, Z. Kang, D. Sun, R. Wang and Y. Cao, Adv. Sci., 2019, 6, 1801222 CrossRef PubMed.
- H. Liu, H. Guo, B. Liu, M. Liang, Z. Lv, K. R. Adair and X. Sun, Adv. Funct. Mater., 2018, 28, 1707480 CrossRef.
- Y. C. Tang, Z. B. Zhao, Y. W. Wang, Y. F. Dong, Y. Liu, X. Z. Wang and J. S. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 32324–32332 CrossRef CAS PubMed.
- P. Ge, H. Hou, C. E. Banks, C. W. Foster, S. Li, Y. Zhang, J. He, C. Zhang and X. Ji, Energy Storage Mater., 2018, 12, 310–323 CrossRef.
- D. Xie, W. Tang, Y. Wang, X. Xia, Y. Zhong, D. Zhou, D. Wang, X. Wang and J. Tu, Nano Res., 2016, 9, 1618–1629 CrossRef CAS.
- Y. N. Ko, S. H. Choi, S. B. Park and Y. C. Kang, Nanoscale, 2014, 6, 10511–10515 RSC.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed.
- Y. Cui, X. Zhou, W. Guo, Y. Liu, T. Li, Y. Fu and L. Zhu, Batteries Supercaps, 2019, 2, 784–791 CrossRef CAS.
- C. Luo, J. Wang, L. Suo, J. Mao, X. Fan and C. Wang, J. Mater. Chem. A, 2015, 3, 555–561 RSC.
- L. Zeng, X. Chen, R. Liu, L. Lin, C. Zheng, L. Xu, F. Luo, Q. Qian, Q. Chen and M. Wei, J. Mater. Chem. A, 2017, 5, 22997–23005 RSC.
- Q. Qu, T. Gao, H. Zheng, Y. Wang, X. Li, X. Li, J. Chen, Y. Han, J. Shao and H. Zheng, Adv. Mater. Interfaces, 2015, 2, 1500048 CrossRef.
- T. Gao, J. Shao, X. Li, G. Zhu, Q. Lu, Y. Han, Q. Qu and H. Zheng, Chem. Commun., 2015, 51, 12459–12462 RSC.
- Z. Wan, J. Shao, J. Yun, H. Zheng, T. Gao, M. Shen, Q. Qu and H. Zheng, Small, 2014, 10, 4975–4981 CrossRef CAS PubMed.
- X. Li, X. Zheng, J. Shao, T. Gao, Q. Shi and Q. Qu, Chem.–Eur. J., 2016, 22, 376–381 CrossRef CAS PubMed.
- C. Wang, L. Sun, F. Zhang, X. Wang, Q. Sun, Y. Cheng and L. Wang, Small, 2017, 13, 1701246 CrossRef PubMed.
- D. Sun, Y. Tang, D. Ye, J. Yan, H. Zhou and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 5254–5262 CrossRef CAS PubMed.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, Z. Yao, F. Yang, S. Lin, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Mater., 2017, 29, 1700748 CrossRef PubMed.
- Y. Yun, J. Shao, Y. Chen, Z. Cao, Q. Qu and H. Zheng, ACS Appl. Nano Mater., 2019, 2, 1883–1889 CrossRef CAS.
- Z. Luo, J. Zhou, L. Wang, G. Fang, A. Pan and S. Liang, J. Mater. Chem. A, 2016, 4, 15302–15308 RSC.
- X. Zhao, H.-E. Wang, Y. Yang, Z. G. Neale, R. C. Massé, J. Cao, W. Cai, J. Sui and G. Cao, Energy Storage Mater., 2018, 12, 241–251 CrossRef.
- X. Zhao, J. Sui, F. Li, H. Fang, H. Wang, J. Li, W. Cai and G. Cao, Nanoscale, 2016, 8, 17902–17910 RSC.
- Z.-T. Shi, W. Kang, J. Xu, L.-L. Sun, C. Wu, L. Wang, Y.-Q. Yu, D. Y. W. Yu, W. Zhang and C.-S. Lee, Small, 2015, 11, 5667–5674 CrossRef CAS PubMed.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- Y. Yun, Z. Shi, J. Shao, Q. Qu, Y. Gao, Z. Chen, Y. Chen and H. Zheng, ChemNanoMat, 2018, 4, 1247–1253 CrossRef CAS.
- Y. J. Hong, K. C. Roh and Y. Chan Kang, J. Mater. Chem. A, 2018, 6, 4152–4160 RSC.
- M. Yousaf, Y. Wang, Y. Chen, Z. Wang, W. Aftab, A. Mahmood, W. Wang, S. Guo and R. P. S. Han, ACS Appl. Mater. Interfaces, 2018, 10, 14622–14631 CrossRef CAS PubMed.
- Y. Shi, C. Hua, B. Li, X. Fang, C. Yao, Y. Zhang, Y.-S. Hu, Z. Wang, L. Chen, D. Zhao and G. D. Stucky, Adv. Funct. Mater., 2013, 23, 1832–1838 CrossRef CAS.
- J. K. Shon, H. S. Lee, G. O. Park, J. Yoon, E. Park, G. S. Park, S. S. Kong, M. Jin, J.-M. Choi, H. Chang, S. Doo, J. M. Kim, W.-S. Yoon, C. Pak, H. Kim and G. D. Stucky, Nat. Commun., 2016, 7, 11049 CrossRef CAS PubMed.
- B. Hu, L. Mai, W. Chen and F. Yang, ACS Nano, 2009, 3, 478–482 CrossRef CAS PubMed.
- C. Jian, Q. Cai, W. Hong, J. Li and W. Liu, Small, 2018, 14, 1703798 CrossRef PubMed.
- X. Chen, G. Liu, W. Zheng, W. Feng, W. Cao, W. Hu and P. Hu, Adv. Funct. Mater., 2016, 26, 8537–8544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S15 and Tables S1 and S2. See DOI: 10.1039/c9na00815b |
| This journal is © The Royal Society of Chemistry 2020 |