
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrodroplet photofuel cells to harvest high-density energy and dye degradation†
Siddharth
Thakur
 a,
Nayan Mani
Das†
b,
Sunny
Kumar
a,
Ashok Kumar
Dasmahapatra
ab and
Dipankar
Bandyopadhyay
*ab
a,
Nayan Mani
Das†
b,
Sunny
Kumar
a,
Ashok Kumar
Dasmahapatra
ab and
Dipankar
Bandyopadhyay
*ab
aDepartment of Chemical Engineering, Indian Institute of Technology Guwahati, Guwahati – 781039, India
bCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati – 781039, India. E-mail: dipban@iitg.ac.in
First published on 28th February 2020
Abstract
In this study, a membraneless photofuel cell, namely, μ-DropFC, was designed and developed to harvest chemical and solar energies simultaneously. The prototypes can also perform environmental remediation to demonstrate their multitasking potential as a sustainable hybrid device in a single embodiment. A hydrogen peroxide (H2O2) microdroplet at optimal pH and salt loading was utilized as a fuel integrated with Al as an anode and zinc phthalocyanine (ZnPC)-coated Cu as a cathode. The presence of n-type semiconductor ZnPC in between the electrolyte and metal enabled the formation of a photo-active Schottky junction suitable for power generation under light. Concurrently, the oxidation and reduction of H2O2 on the electrodes helped in the conversion of chemical energy into the electrical one in the same membraneless setup. The suspension of Au nanoparticles (Au NPs) in the droplet helped in enhancing the overall power density under photonic illumination through the effects of localized surface plasmon resonance (LSPR). Furthermore, the presence of photo-active n-type CdS NPs enabled the catalytic photo-degradation of dyes under light in the same embodiment. A 40 μL μ-DropFC could show a significantly high open circuit potential of ∼0.58 V along with a power density of 0.72 mW cm−2. Under the same condition, the integration of ten such μ-DropFCs could produce a power density of ∼7 mW cm−2 at an efficiency of 3.4%, showing the potential of the prototype for a very large scale integration (VLSI). The μ-DropFC could also degrade ∼85% of an industrial pollutant, rhodamine 6G, in 1 h while generating a power density of ∼0.6 mW cm−2. The performance parameters of μ-DropFCs were found to be either comparable or superior to the existing prototypes. In a way, the affordable, portable, membraneless, and high-performance μ-DropFC could harvest energy from multiple resources while engaging in environmental remediation.
1. Introduction
Infusing the efficacies of nanoscale science1,2 in microfluidic devices for the development of affordable and portable energy harvesters with a significantly high power density has attracted major research attention in the recent years. The inventions of state-of-the-art thermoelectric, triboelectric, piezo-acoustic, electrokinetic, or Marangoni energy harvesters are directed towards this end.3–9 Interestingly, the traditional renewable10 or non-renewable11 energy-harvesting technologies have also been undergoing a paradigm shift from the regime of macroscopic to the very large scale integration (e.g. VLSI) of micro or nanoscopic prototypes for enhanced efficiency, higher throughput, and escalated power density.12 Thus, it is not very surprising that a flurry of research activities has also been observed in the miniaturization along with process intensification13 of more widely employed commercial energy harvesters such as batteries,14,15 photovoltaic cells,16 or fuel cells17,18 for better efficiency.In particular, since the path-breaking invention by Sir William Grove in the 18th century19 to convert chemical energy into the electrical one, fuel cells (FCs) have shown remarkable progress20 despite numerous setbacks.21 For example, the utility of the FCs as isolated or distributed power resources has now been translated into a few megawatt plants for power supply.17 A large varieties of FCs composed of polymer electrolyte membranes, phosphoric acid, methanol or alkali have made appearance at different lengths and performance scales not only to power energy-intensive rockets but also to run miniaturized micro-transmitters or biomedical devices.22–25 Presently, the fuels utilized in the FCs are either hydrogen (H2), methanol (CH3OH), methane (CH4) or their different combinations.26 In particular, H2 has been the preferred fuel over others owing to its higher energy density and green chemistry to produce water during energy harvesting.26,27
However, as an alternative, hydrogen peroxide (H2O2) has lately been employed as an alternative fuel for FCs because of its high energy density and non-toxicity until a concentration of 20 M.28 Much like H2, peroxide is also a carbon-free and green energy source producing water and oxygen during energy harvesting.29 In short time, the peroxide FCs have evolved from simple oxidizers to standalone energy sources28,30 because (i) the existence of peroxide in the liquid state under ambient conditions makes it a better candidate for fuel storage than H2; (ii) although peroxide possesses a lower gravimetric energy density, it has a significantly higher volumetric energy density; and (iii) they have higher fuel efficiency suitable for the space or underwater applications.23,24 However, the peroxide FCs also face limitations associated with the production,31 preservation,32,33 supply, and storage28 of rather unstable H2O2 at industrial scale.
In the recent years, the utilization of membranes in FCs has been found to be slowly waning, which is paving the way to much simpler membraneless single-compartment FCs. The concept was first realized by Yamazaki et al.34 who employed H2O2 as both an electron acceptor and a fuel with an Ag cathode alongside using Au, Pt, Pd or Ni anodes. Subsequently, various cathodic (e.g. metals, inert materials, conducting dyes or their combinations)34,35 and anodic (e.g. Al, Ni, C, and noble metals)30,36,37 materials have been experimented for membraneless FCs over the years. In particular, ferrocyanides along with phthalocyanines and porphyrins38 (PCs) have been found to be excellent candidates for H2O2 reduction owing to their low-onset potentials and cost-effectiveness.30,36,39–41 Interestingly, an Al (anode)–H2O2 (fuel) combination is found to possess a significantly higher specific energy (∼17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W h kg−1),18,42 which is even higher than some of the state-of-the-art Li-ion batteries (∼12000 W h kg−1).43 However, the membraneless peroxide FCs also suffer from the limitations associated with the crossover of reactants and, thus, are suitable for only low-power intensive systems.
000 W h kg−1),18,42 which is even higher than some of the state-of-the-art Li-ion batteries (∼12000 W h kg−1).43 However, the membraneless peroxide FCs also suffer from the limitations associated with the crossover of reactants and, thus, are suitable for only low-power intensive systems.
In view of this background, we report the design and development of a peroxide microdroplet-based membraneless FC, namely, μ-DropFC, which is capable of converting the combined influences of the chemical and light energies into the electrical one under the same embodiment. In a sense, the droplet-based device is found to be an exception to most of the previous attempts, where film-based approaches were preferred.5,8 While the usage of the microdroplet architecture helps in gaining advantages from the confinement effects due to miniaturization, the photo-activity of the droplet helps in improving the open circuit voltage (ψoc) and current density (J) of the μ-DropFC.16
The photo-activity44,45 inside μ-DropFCs has been facilitated by the localized surface plasmon resonance (LSPR)45 of the suspended gold nanoparticles (Au NPs) under light sources. The presence of Au NPs also further enhances the rate of charge transfer between the electrodes by improving the electrical conductivity of the microdroplet.46 To ensure a lower economic footprint, the μ-DropFC is integrated with an Al-foil anode and a Cu–ZnPC cathode, which promotes an augmented catalytic breakdown of H2O2 under photonic excitation.47,48 Low-work-function Cu also facilitates a stronger adhesion with ZnPC to provide a strong electronic conductivity.49–52 Importantly, in such a simple embodiment, a single μ-DropFC can show a significantly high ψoc of ∼0.58 V and a power density of ∼0.72 mW cm−2 per unit mass of the fuel. Ten such μ-DropFCs show a power density of ∼7 mW cm−2 at an efficiency of 3.4%, which highlights the potential of such device for μ-VLSI and scale up.
Furthermore, to enhance the overall utility of the prototype, the μ-DropFC setup has been utilized in environmental remediation processes such as dye degradation, while simultaneously being involved in energy harvesting from the peroxide fuel and photonic excitations. For this purpose, we synthesized a nanocomposite of Au NPs embedded in a matrix of cadmium sulfide nanoparticles (CdS NPs) before suspending them in the peroxide fuel to breakdown the “stubborn” organic dyes under photonic excitation.53 As a model system, we show that the Au and CdS NPs with a powerful photocatalytic activity54 can break rhodamine 6G dye under light while demonstrating a significantly higher power density of ∼0.62 mW cm−2. In some way, the simple and affordable μ-DropFC emulate the state-of-the-art microbial FCs, which have been operated in tandem with dye degradation to simultaneously perform dual objectives.55,56
Concisely, the study showcases the versatility of a droplet8 or digital57 microfluidic device loaded with the efficacies of nanoscience for efficient energy harvesting from electrochemical and solar resources as well as performing environmental remediation.55,56,58
2. Experimental methods
2.1. Materials
The chemicals 50% (w/v) hydrogen peroxide solution (H2O2), cadmium nitrate (CdNO3), sodium sulfide (Na2S), hydrochloric acid (37% (w/w), HCl) and sodium hydroxide (NaOH) were purchased from Merck, India. The chemicals zinc phthalocyanine (ZnPC) and rhodamine 6G (Rh6G) were purchased from Sigma Aldrich, India. Copper wires and aluminum foil were purchased from a local vendor. De-ionized water was employed for cleaning, washing and the preparation of the solutions. Gold(III) chloride hydrate, sodium borohydride, trisodium citrate dihydrate, and sodium chloride were purchased from Sigma Aldrich, India. All the chemicals were of analytical grade and were utilized without further purification.2.2. Characterizations
The open circuit voltage (ψoc) was measured using a digital nanovoltmeter (SES Instruments). The Keithley 2640A sourcemeter in connection with the Kickstarter software was used for potential-amperometry (I–ψ) and chrono-amperometry (J–t) characterizations. The surface morphologies and lattice structures (SAED patterns) of the nanocomposites were detected using a field emission transmission electron microscope (FETEM, Jeol India). A field emission scanning electron microscope equipped with an energy-dispersive X-ray spectrometer (FESEM-EDX, Zeiss) was used to confirm the elemental composition of different materials. The material characterization of the nanocomposites and electrodes was carried out using a UV-Visible spectrophotometer (SHIMADZU UV-2600 model) and Raman spectrometer (Horiba). A Kelvin Probe Force Microscope (KPFM) was also employed to determine the work function of the electrode after cell operation.2.3. Methods
To prepare the composite of Au and CdS NPs, first, 20 mL of prepared Au NPs solution (20 nM) was added to a beaker, where 10 mL each of aqueous solution of 0.01 M CdNO3 and 0.01 M Na2S solution in 0.1 M NaOH were added simultaneously. The reaction temperature was maintained at 60 °C for 12 h while the solution was stirred continuously. The FETEM and UV-Vis spectra in Section I of the ESI† confirmed the formation of the composite of Au and CdS NPs. The sizes of the Au NPs in the composite were around 20 nm, while CdS NPs were around 2–5 nm. This was confirmed by the FETEM images, which have been shown in Fig. S1(a) and (b), Section I of the ESI.† The effect of the harsh peroxide solutions on the characteristic UV spectra of the nanocomposites is also depicted in Fig. S2(a) and (b), Section I of the ESI.† These plots establish the limited effect of the peroxide moieties on the prepared Au/CdS NPs.
| Sl. no. | Compositions |
|---|---|
| a Concentration of Au/CdS NP mixture was unknown since they were synthesized in situ, as discussed below. | |
| Fuel 1 | Mixture of 10 μL 0.3 M aqueous H2O2 with 10 μL of 0.1 M aqueous HCl and 10 μL of 1 M of aqueous NaCl |
| Fuels 2I–2V | (I) 10 μL of Fuel 1 with 10 μL of 20 nM aqueous Au NPs |
| (II) 10 μL of Fuel 1 with 20 μL of 20 nM aqueous Au NPs | |
| (III) 10 μL of Fuel 1 with 30 μL of 20 nM aqueous Au NPs | |
| (IV) 10 μL of Fuel 1 with 40 μL of 20 nM aqueous Au NPs | |
| (V) 10 μL of Fuel 1 with 50 μL of 20 nM aqueous Au NPs | |
| Fuels 3I–3Va | (I) 10 μL of Fuel 1 with 10 μL of aqueous Au/CdS NPs mixture |
| (II) 10 μL of Fuel 1 with 20 μL of aqueous Au/CdS NPs mixture | |
| (III) 10 μL of Fuel 1 with 30 μL of aqueous Au/CdS NPs mixture | |
| (IV) 10 μL of Fuel 1 with 40 μL of aqueous Au/CdS NPs mixture | |
| (V) 10 μL of Fuel 1 with 50 μL of aqueous Au/CdS NPs mixture | |
| Fuel 4 | 10 μL of Fuel 1 with 10 μL of 0.5 mg mL−1 aqueous rhodamine 6G |
| Fuel 5 | 10 μL of Fuel 1 with 10 μL of 0.5 mg mL−1 aqueous rhodamine 6G + and 10 μL of 20 nM aqueous Au NPs |
| Fuel 6a | 10 μL of Fuel 1 with 10 μL of 0.5 mg mL−1 aqueous rhodamine 6G + and 10 μL of aqueous Au/CdS NPs mixture |
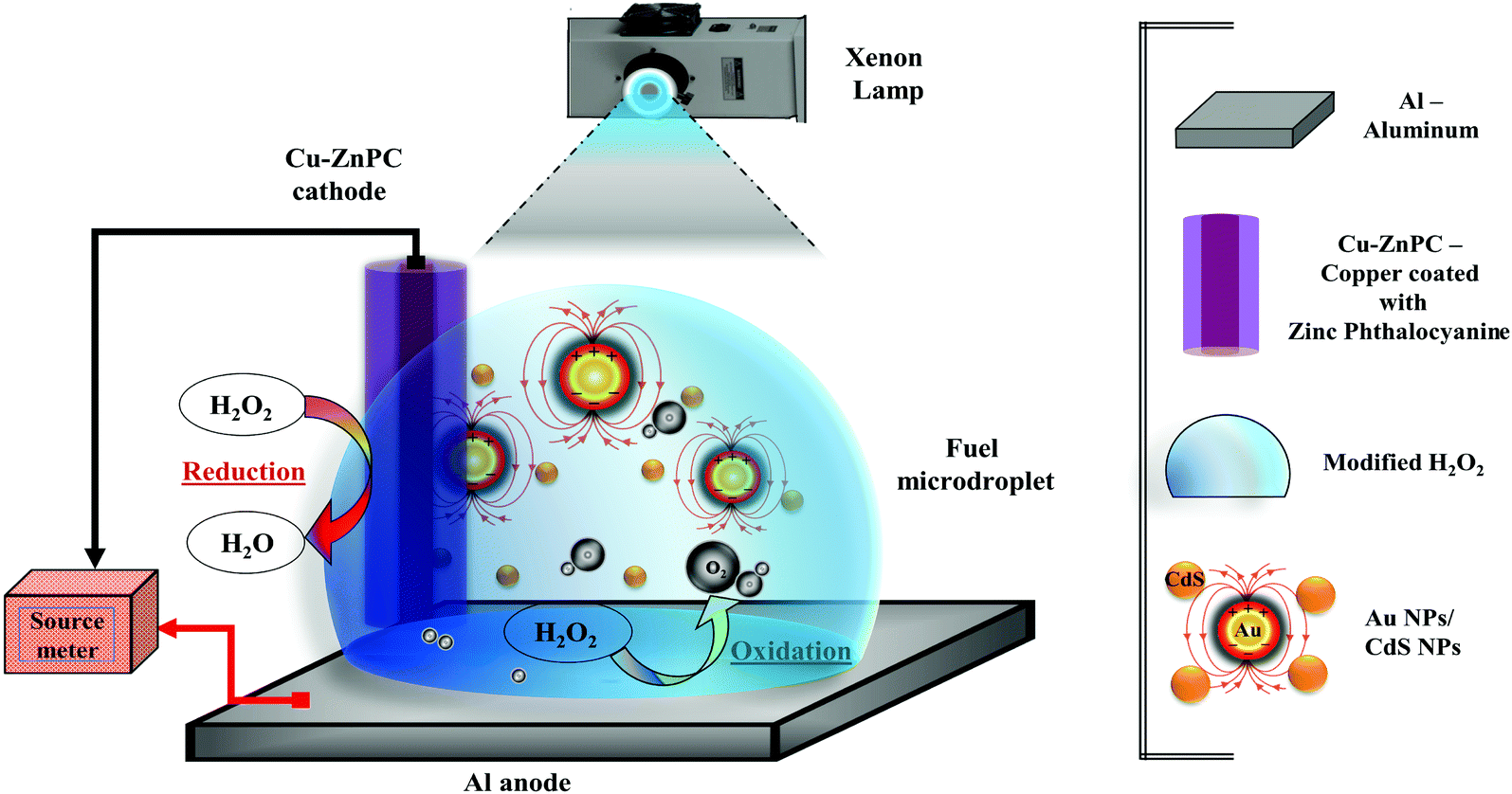 | ||
| Fig. 1 Schematic of the μ-DropFC setup comprising of a Cu–ZnPC cathode, an Al foil anode, a microdroplet with the fuel (0.3 M H2O2, 0.1 M HCl, 1 M NaCl), additives (Au/CdS NPs) and light source. The image also schematically shows the redox half-reactions at the cathode and anode, H2O2 → O2 + 2H+ + 2e− and H2O2 + 2H+ + 2e− → 2H2O, respectively. The legends aid in the identification of the different components and their placements. | ||
3. Results and discussion
3.1 Mechanistic details
Fig. 2 justifies the choices of different materials chosen for the proposed μ-DropFC. Subsequently, the energy band diagrams shown also uncover the working principle of the μ-DropFC, under different conditions.59Fig. 2(a) schematically depicts the basic state of the μ-DropFC before any reaction started or light illumination took place.59 The schematic diagram shows the Fermi levels (EF) of Cu, ZnPC, peroxide solution, and Al alongside showing the band-gap for the p-type semiconductor ZnPC with the conduction and valence bands at EC = −3.8 eV and EV = −5.8 eV.59,60 The presence of ZnPC ensured the formation of a Schottky junction between metallic Cu, p-type SC ZnPC, and electrolyte. Furthermore, the peroxide electrolyte was reactive at room temperature and the typical energy levels Dox (e.g. −5.3 eV)30,61 and DR (e.g. −6.4 eV)30,61 denote their oxidation and reduction potentials. | ||
| Fig. 2 Schematics of the energy band diagrams displaying the various stages of the operation of the μ-DropFC. (a) Initial condition, even before equilibration, when the contact has just been established between the components. (b) Equilibrium under the dark condition when the Fermi levels of different materials equilibrated before photonic illumination. (c) Effects after the illumination using the Xe lamp. (d) The situation when an anodic bias has been applied to the setup shown in (c). (e) Influence of the LSPR of the Au NPs under illumination. (f) Influence of the Au/CdS NPs. The images also depict the overviews of redox reactions occurring at the anode and cathode. | ||
It may be noted here that once the droplet was integrated with the electrodes, the setup started harvesting energy even under dark conditions and the energy band diagram after such equilibration is shown in Fig. 2(b). The electrolyte loaded with H2O2 underwent oxidation at the Al-foil-anode to release O2 gas in the atmosphere and produce H+ ions in the solution following the half-reaction, H2O2 → O2 + 2H+ + 2e−.36 The peroxide decomposition in the bulk under acidic and basic conditions is elucidated in detail in Section II of ESI.† The oxidation of the anode led to a progressive deposition of oxides at the Al-foil-anode when Al combined with hydroxyl ions of the droplet. This was confirmed by the Raman and KPFM characterization of the electrodes before and after the analysis, as shown in Fig. S4b, c, and S5 of Section III of ESI.†62 The reduction of H2O2 occurred at the ZnPC–Cu cathode, which eventually formed water, following the half-reaction, H2O2 + 2H+ + 2e− → 2H2O.36
Interestingly, the KPFM helped in identifying the work function of the Al anode before the reaction to be ∼4.577 eV, as depicted in Fig. 2(a). The schematic diagram in Fig. 2(b) shows that these redox reactions initiate a charge transport across the electrodes, which was the initiation of the conversion of chemical energy into electrical energy. Since the circuit was complete, the different components of the μ-DropFC strived to equilibrate their Fermi level. However, upon contact with the low-work-function Cu, an electron flow occurred from the metal to ZnPC via the formation of mixed states,51,52 which led to the balancing of the potentials at these two surfaces, as shown in Fig. 2(b). Further, during equilibration of the Fermi level, the conduction and valence bands of the p-type SC ZnPC underwent a band bending near the SC–electrolyte and metal–SC interfaces, as shown in Fig. 2(b).59,60
The band diagram under dark conditions also helped the charge transfer at the mixed-molecule ZnPC–Cu interface. Previous studies have shown that interfacial dipoles could be created at the metal–SC junctions, which influenced the Schottky barrier height.51,52 Especially when the SC layer is physisorbed on a metal surface, charge redistribution might take place across the junction to affect the interface dipoles. In such a scenario, to maintain the Pauli exclusion principle valid, the charges present around the interface are pushed back into the metal, leading to a slight reduction in its work-function. Importantly, the interactions between the ZnPC and Cu in the proposed μ-DropFC could be explained along the similar lines. The physisorbed ZnPC on Cu created interfacial states at the junction, and the ZnPC molecules were “pulled” towards the Cu core owing to the lower work function of the metal.52 This made the interaction between SC and metal surfaces result in a stronger bond, and subsequently, the charge transfer from Cu to the HOMO of ZnPC facilitated the reduction of peroxide into water near the SC–electrolyte interface.52
Fig. 2(c) shows the effect of the illumination of a Xe lamp on the μ-DropFC, wherein the photonic excitation pumps in more electrons into the system due to the electron transfer from valence to conduction bands of ZnPC molecules.60 Subsequently, the influence of anodic bias to the pattern of band bending of the p-type ZnPC is shown in Fig. 2(d) under the illumination of a Xe lamp. The image also suggests a redistribution of the Fermi levels with the application of the anodic bias. It can be seen that at the anode, the Fermi level went down due to the introduction of positive charges, which facilitated the reaction further as more electron transfer could take place from the oxidation reaction of H2O2. Subsequently, on the cathode side, the Fermi level increased due to injection of negative charges, which further increased the rate of reduction reaction. In a way, at the electrode interfaces, charges were either introduced (cathode) or removed (anode), which influenced the rate of reaction. This led to higher electron transfer from the metal to the p-type SC. Furthermore, since the inherent charge density over the p-type SC increased under light, this facilitated higher rate of charge transfer to the adjacent electrolyte due to the Fermi energy difference. The effect of the addition of the Au NPs to the μ-DropFC and the subsequent effect of LSPR are schematically shown in Fig. 2(e). The photonic excitation on the Au NPs promoted LSPR on the same, which, in turn, improved the photoactivity of the μ-DropFC. The presence of Au NPs also increased the electrical conductivity of the microdroplet, which improved the rate of electron transfer through the circuit.
Concisely, Fig. 2 helps in understanding that the usage of each and every component contributed to the increased performance of the μ-DropFC. Importantly, the μ-DropFC setup was found to be rather robust because the output characteristics did not alter much when the experiments were repeated on the same system. This was shown by reporting the small reduction in ψoc measured for the duration of a single operation cycle when the same set of electrodes were operated 3 times, as shown in Fig. S4a of Section III of the ESI.† However, the change in the ψoc values changed significantly after three cycles, owing to the electrode degradation. Again, the KPFM analysis revealed a significant change in the surface potentials upon exposure of the Al-foil-anode to peroxide in the presence of an anodic bias, as shown in Fig. S5 in Section III of the ESI.†
Fig. 2(f) depicts the variations in the band configurations upon the addition of n-type CdS NPs in the solution in the form of Au/CdS NPs. This facilitated the formation of a p–n junction when some of the n-type CdS NPs got adsorbed at the p-type ZnPC. Thus, in dark condition and in the presence of forward bias, the flow of electrons could take place from CdS to ZnPC. However, under light, the electronic transitions of both the ZnPC and CdS molecules changed the scenario. The transition in the n-type CdS facilitated its oxidation into CdSO4 in the presence of peroxide fuels63 while the electron in the conduction band of ZnPC after transition due to photonic excitation was transferred to CdS for further oxidation of the same. In this manner, the configuration ensured that the addition of CdS did not disturb the mainstream flow of electrons in the μ-DropFC. It may be noted here that the major purpose of addition of CdS NPs were to degrade the dyes under light. However, to ensure such activity, we did not compromise much with the power density of the μ-DropFC owing to the photoactivity of the CdS NPs, as shown in Fig. 2(f). It may also be noted here that the Au/CdS NPs were mostly suspended in the bulk rather than adsorbed on the electrodes, which influenced the kinetics of the electron flow rather than the thermodynamics. Furthermore, these nanoparticles increased the rate of decomposition of the peroxide fuel due to catalytic activity. The charge transfer in the electrolyte was also facilitated by the presence of Na+ and Cl− ions in the electrolyte.8
3.2 μ-DropFC characterization
The ESI Video 2† shows the performances of the μ-DropFCs under dark and illuminated conditions. Clearly, the setup generated a higher output under light following the mechanism mentioned previously. The electrical characterization of the μ-DropFCs was performed using a set of nanovoltmeter and sourcemeter. For this purpose, linear sweep voltammetry (LSV) was employed with a voltage scan from 0 V to the open circuit voltage (ψoc) of the system. The testing parameters were kept the same for all experiments reported unless mentioned otherwise. In what follows, we discuss the results associated with the μ-DropFCs under the light. The results thus obtained were shown as the P–ψ characteristics in Fig. 3. It may be noted here that all the P–ψ plots show a progressive increase in P with ψ until they reach a maximum value of Pmax before they decrease with a further increase in ψ to a zero value at ψoc. It may also be noted here that, initially, we optimized the loading of salt and acid in the native μ-DropFCs before we performed the experiments with the NPs. Fig. S3 in Section II of ESI† show the characteristics of a few native μ-DropFCs when the salt and acid/alkali loadings were varied. Fig. S3 in Section II of ESI† also shows that although the alkalinity of the solution improved the power density of the energy harvester, it reduced the life time and ψoc. In contrast, at an acidic pH 1, steady and continuous degradation of peroxide for a longer duration led to a better performing and long-lasting μ-DropFC. These experiments uncovered that Fuel 1 in Table 1, a mixture of 10 μL 0.3 M aqueous H2O2 with 10 μL of 0.1 M aqueous HCl and 10 μL of 1 M of aqueous NaCl was the near-optimal native system, which led to Pmax ∼ 0.35 mW cm−2 at ψoc = 0.38 V, as shown in plot (a) of Fig. 3.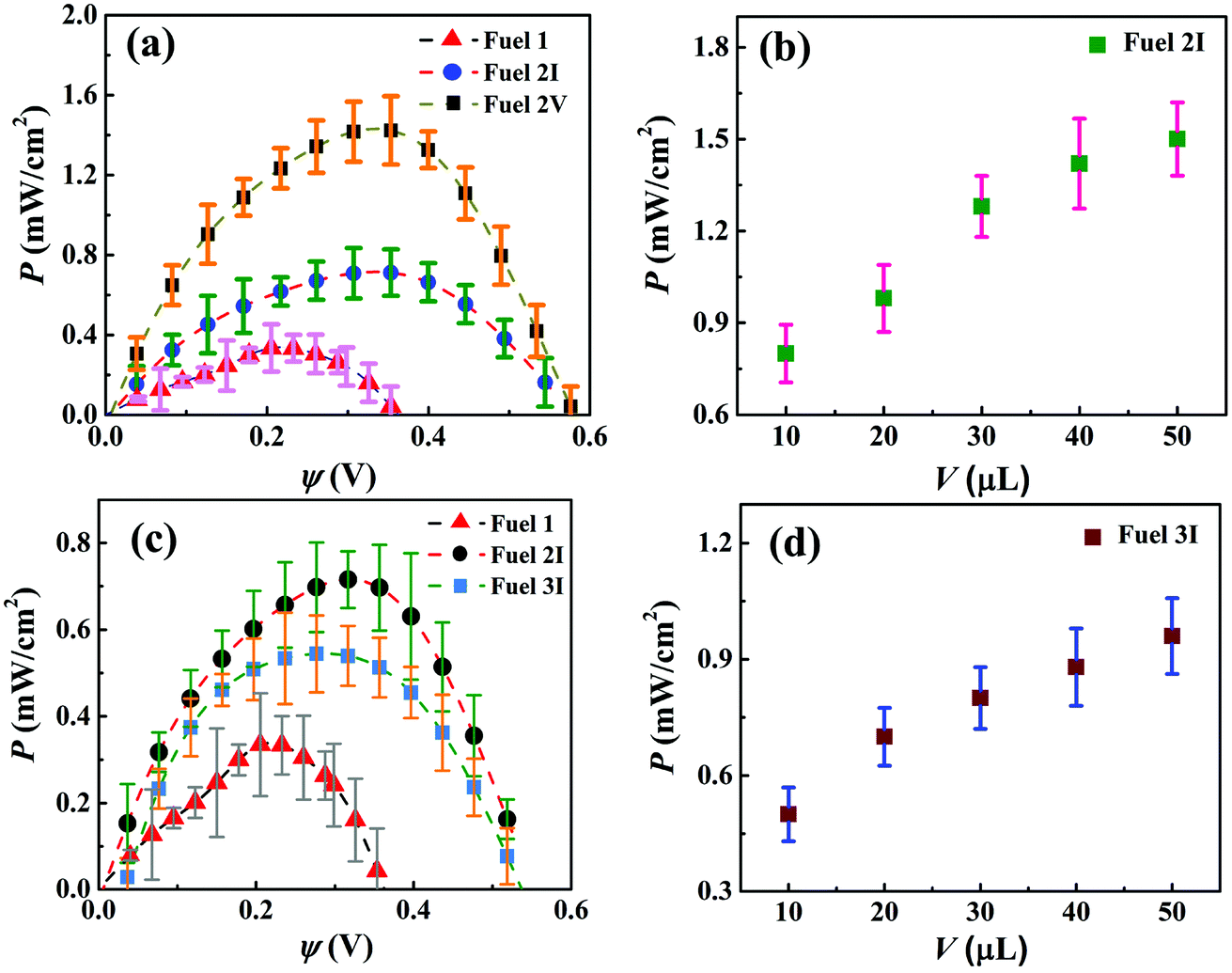 | ||
| Fig. 3 Power–potential (P–ψ) curves for the different μ-DropFCs containing different volumes of additives – Au NPs and Au/CdS NPs, respectively. (a) Performance curves indicating the variation in power density (P) with ψ of three different fuels: 1, 2I and 2V. (b) Effect of changing the volume (V) of the Au NPs suspension on the overall power generation (P). (c) Power–potential (P–ψ) curves for Fuels 1, 2I and 3I. (d) Effect of changing the volume (V) of the Au/CdS NPs suspension on the overall power generation, P. All the results were obtained under the application of a voltage sweep from 0 V to ψoc, and then by measuring the resulting output currents. Details of Fuels 1–6 are provided in Table 1. | ||
Interestingly, plot (a) also shows nearly a four-fold increase in Pmax when the native system was loaded with a suspension of Au NPs, named as Fuel 2V. The combined influence of the LSPR of the Au NPs under light and enhanced electrical conductivity of the system led to the improvement of the power harvesting parameters, Pmax ∼ 1.5 mW cm−2. Plot (b) shows the effect of variation of the volume of Au NPs, Fuel 2I–2V, on the P–ψ values of the μ-DropFCs. The plot suggests that although increasing the volume of the Au NPs led to higher P values, the increment saturated due to diffusional resistance at higher values. The Au NPs improved the output parameters because (i) the LSPR under light improved charge transfer between the electrodes, (ii) under acidic conditions Au NPs catalyzed the degradation of peroxide fuels, (iii) they also acted as electron relays for better charge transfer, and (iv) they reduced the electrical resistance of the composite fuel droplet marginally.46,56 Furthermore, the incorporation of the Au NPs or Au/CdS NPs primarily affected the reaction kinetics, as they were suspended in the bulk rather than adsorbed at the electrodes. With the increase in solution volume, the amount of NPs that participated in the charge transfer processes increased, thereby facilitating faster redox reactions at the electrodes, which resulted in higher output. Furthermore, with the increase in solution volume, the area occupied by the composite fuel droplet also increased marginally since the anode surface was partially hydrophilic. This led to an increase in the number of reaction sites available for the redox reaction to occur. Thus, with a progressive increase in the droplet volume, the output cell characteristics were enhanced under external radiation due to the various above-mentioned reasons. Importantly, with the increase in the droplet volume, the resistance to diffusion near the electrodes also decreased; however, a saturation limit was observed beyond the addition of a certain volume of NPs.
The performance of Fuel 3I–3V is summarized in plots (c) and (d). Plot (c) suggests that, in comparison to Fuel 1, they performed better showing Pmax ∼ 0.59 mW cm2; however, the performance was not as good as with Fuel 2. Again, plot (d) shows the increase in the performance characteristics before saturation with the loading of Au/CdS NPs. It may be noted here that the addition of Au/CdS NPs was intended towards their capability of photocatalytic degradation of dyes, which is to be discussed later. However, from the pure energy harvesting point of view, Fuel 2 with an optimal loading of Au NPs was found to be superlative, as summarized in Fig. 3.
Fig. 4(a) shows the J (current density)–t (time) plot, for Fuels 2I and 3I. The plots suggest that the solution containing only Au NPs displayed an initial higher current density of ∼5 mA cm−2 when compared with the solution containing Au/CdS NPs (∼3.5 mA cm−2), upon the application of a bias of 0.1 V. Thereafter, the decay during the initial ∼200 s could be due to the mismatch between the high rate of redox reactions occurring at the interface and the limited charge transfer at the cathode. In such a scenario, the μ-DropFCs with Au NPs showed enhancement due to the higher ionic conductivity, better charge-transfer rates due to the presence of charge mediators, and occurrence of faster chemical kinetics at the electrode interface due to the enhanced plasmonics.8,45,56 However, the plot also shows that the μ-DropFCs with Fuel 3I registered a sustained charge density of about 1.7 mA cm−2, which was higher than that of Fuel 2I even after a long period of time. The ratios of J values after 0 s and 1500 s were ∼0.3 (Fuel 2I) and ∼0.5 (Fuel 3I), which indicated the slower degradation of the Au/CdS NP system.
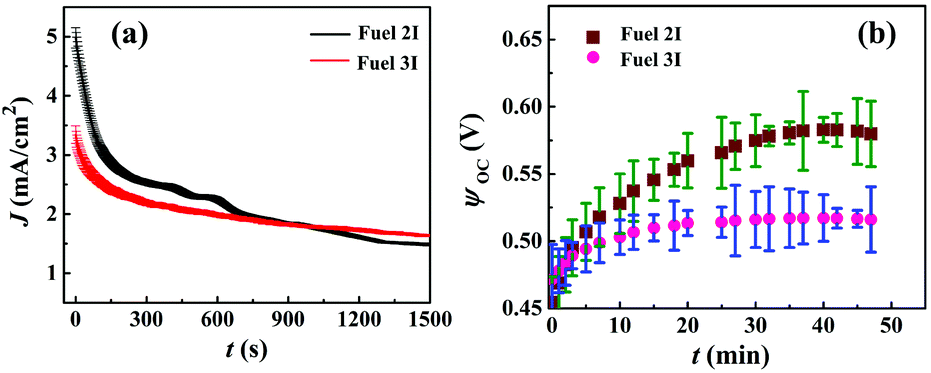 | ||
| Fig. 4 (a) Chronoamperometric current density (J) versus time (t) studies for different μ-DropFCs with the Fuels 2I and 3I at an applied voltage of 0.1 V. (b) The corresponding open-circuit potential (ψoc) with t for 50 min. To demonstrate long-term performance of μ-DropFCs, the chronoamperometry test was performed for different systems. | ||
Plot (b) in Fig. 4 highlights the difference in ψoc for Fuels 2I (0.58 V) and 3I (0.52 V). The plots suggest the generation of a high ψoc upon the addition of the photo-sensitive NPs under a longer duration of light illumination for ∼50 min. Herein, the ψoc values signify the overall driving force of the system, which is a measure of the thermodynamic potential difference between the electrodes.
It may be noted here that for the system containing additional additives in the electrolyte, ψoc is the difference between the chemical potential (μ) of the electrolyte and the potential of the conduction band of the photo-sensitive electrode (e.g. ZnPC in the present setup), ψoc = EC − μ.8,64 The external irradiation might cause intense localized electric field to develop on Au NPs, which effectively could reduce μ to, μ′ = μ − qV, where q represents the charge of the particle and V, the potential developed due to the field around the excited particle.8,64 Subsequently, an enhancement in ψoc could be expected in a μ-DropFC loaded with Au NPs under a prolonged light exposure, as shown in plot (b). In comparison, for the case of Au/CdS NPs, a lower ψoc value was observed, which could be attributed to the generation of lower value of potential difference between the electrodes. In the presence of light, the photo-excitation generated the electron–hole pair in CdS NPs in which the conduction band was at a lower potential value. This caused electron transfer from CdS NPs to oxidize the peroxide fuel. In the process, a fraction of the photonic energies was taken up by CdS NPs for peroxide degradation in the bulk rather than at the electrode, which contributed to a reduced ψoc.
3.3 Energy harvesting with dye degradation
As discussed previously, the μ-DropFC setup could also be utilized as a dye degradation system without much alteration of the components. For this purpose, Au/CdS NPs were involved to exploit their act as superior photo-catalysts under light. The energy harvesting performance involving P–ψ characteristics and J–t curves of such μ-DropFCs is summarized in Fig. S6 of Section IV in the ESI.† These plots concluded that while the addition of Au NPs in the μ-DropFC loaded with Rh6G (Fuel 5) provided an enhanced power density, the addition of Au/CdS NPs in the same (Fuel 6) led to the more efficient dye degradation. This could be because of the catalytic nature of Au/CdS NPs, which degraded some peroxide amount in the bulk of the droplet and thereby lowered the overall energy output. Importantly, Fig. S6 of Section IV in the ESI† also suggest that although the μ-DropFC loaded with Au/CdS NPs and Rh6G (Fuel 6) harvested energy at a lower density, it gave a sustained output of 1.25 mA cm−2, at an applied voltage of +0.1 V, for a longer period of time.Fig. 5(a) schematically shows the energy band diagrams displaying the various stages of the operation of the μ-DropFC when loaded with Fuels 4–6. In the presence of light, the Au NPs catalyzed the formation of OH radicals via the degradation of peroxide molecules,65 which formed one of the many reactive oxygen species (ROS) engaged in the degradation of the dye, as shown in eqn (1) and (2). The OH radicals were also produced due to the oxidation of water in the presence of holes generated by the n-type CdS NPs under photonic excitations, as shown in eqn (1). Subsequently, the dissolved oxygen accepted the electrons from the LSPR-prone Au NPs to form the superoxide radicals, which subsequently broke down the dye molecules, as shown in eqn (3) and (4). The mechanism for the degradation of dye is summarized as follows:56
| Au–CdS + hν → Au(e−)–CdS(h+), | (1) |
| Au(e−)–CdS(h+) + H2O → Au(e−)–CdS + ˙OH + H+, | (2) |
| Au(e−)–CdS + O2 → Au–CdS + O2−, | (3) |
| O2− + ˙OH + Rh6G → CO2 + H2O + organic products. | (4) |
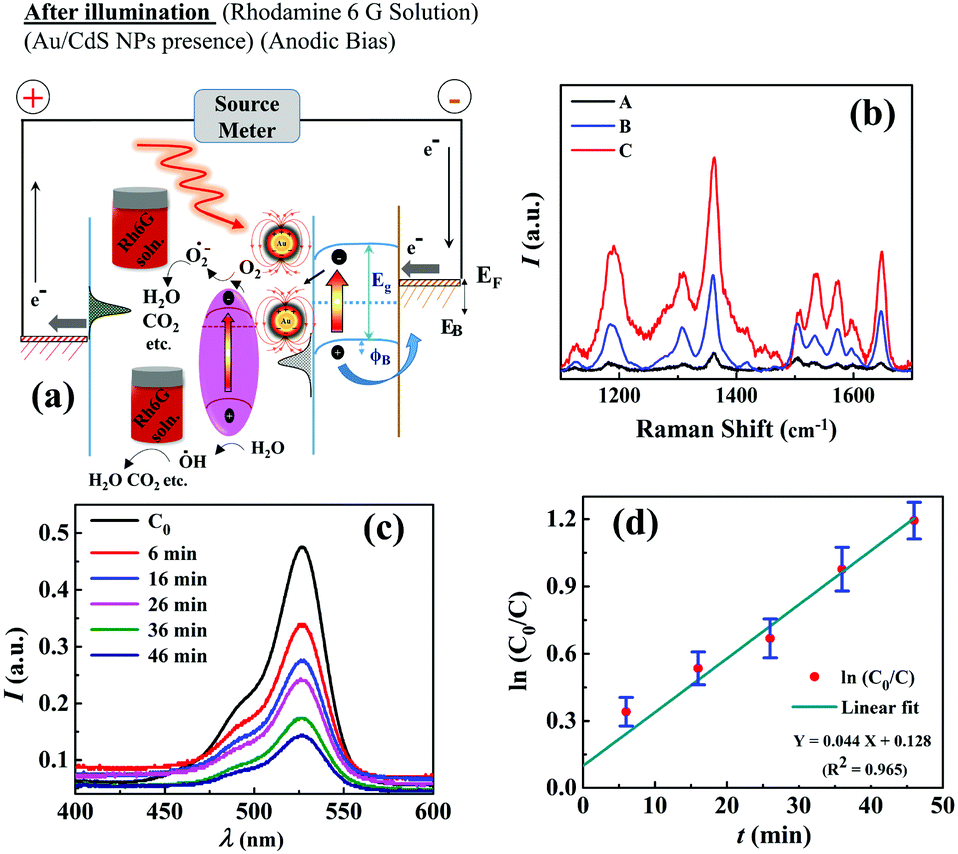 | ||
| Fig. 5 (a) Schematic of the energy band diagrams displaying the various stages of the operation of the μ-DropFC when loaded with the Fuels 4–6. (b) Plasmonic effect of Au NPs in the form of enhanced Raman spectra of Rh6G molecules. Spectrum A corresponds to a pure Rh6G solution in water (0.5 mg mL−1), spectrum B shows the same when 0.5 mg mL−1 of aqueous Rh6G solution was mixed with 0.3 M aqueous H2O2 loaded with Au/CdS NPs and spectrum C shows the same when only Au/CdS NPs were added to the Rh6G solution in the absence of peroxide. (c) UV-Visible plots at different time intervals for Fuel 6, measured from 6 min (t1) to 46 min (t5). (d) Variation in the normalized concentration of Rh6G (lnC0/Ct) with time, where the initial concentration is C0 and the concentration at time t is Ct. The kinetics was found to be similar to a pseudo-first-order reaction with a rate constant of k ∼ 0.044 min−1. | ||
The Au/CdS nanocomposites are known to have significantly better photocatalytic properties than bare CdS NPs. The increment in the degradation efficiency was because of the improved charge generation and transfer rates for CdS NPs in the presence of Au NPs.66 Importantly, the setup was shown to reduce ∼85% of Rh6G in 50 min along with harvesting energy at a decent rate. To confirm the LSPR effect of Au NPs, we performed the surface enhanced Raman spectroscopy (SERS) study, as summarized in Fig. 5(b). The plot shows that, upon irradiation with a 633 nm laser in the Rh6G solution loaded with either Au NPs or Au/CdS NPs, enhanced Raman peaks were observed. We also analyzed the rate of dye degradation of the μ-DropFCs employing UV-Visible spectroscopy. Fig. 5(c) shows the variation in the peak intensity of Rh6G with time for Fuel 6. The plot also shows that the intensity of peaks decreased with time to mark the degradation of the complex dye molecule inside the μ-DropFC. Image (d) shows the variation in the normalized concentration of Rh6G (lnC0/Ct) with time, where the initial concentration is C0 and the concentration at time t is Ct. The kinetics for this system was found to be a pseudo first order with a rate constant of k ∼ 0.044 min−1.53
3.4 Performance, VLSI, and efficiency
The μ-DropFC system showed enhanced output characteristics in the presence of additives like Au NPs and Au/CdS NPs. The additives not only played an important role in enhancing the power densities but also aided the transformation of the μ-DropFC setup to be realized as a dye degradation setup. In what follows, we have analyzed the role of diverse additives in promoting the output characteristics: (i) addition of salt NaCl led to an increase in the electrical conductivity, which further increased when salts like CdSO4 were formed during the operation due to the oxidation of CdS NPs; (ii) in the presence of high H+ loading at a lower pH value the controlled breakup of peroxide fuel in the presence of the Au NPs56,65 aided high-density-power generation for a longer period; (iii) since the cell operated under a light source, the incident radiation caused LSPR to create plasmonic hot-spots around the Au NPs; (iv) the Au NPs displayed electron relay effect under a large potential difference between the two electrodes to transfer charges at a higher rate. Table 2 provides an overall outlook on the performance of the different fuels utilized for the μ-DropFC. The results presented so far uncovered that the addition of Au NPs in Fuel 1 to generate Fuels 2I–2V helped in enhancing the Pmax, ψoc, and ψmax (the voltage corresponding to Pmax).| Fuels | ψ oc (V) | ψ max (V) | P max (mW cm−2) |
|---|---|---|---|
| Fuel 1 | 0.38 | 0.24 | 0.35 |
| Fuel 2I | 0.59 | 0.38 | 0.72 |
| Fuel 3I | 0.55 | 0.36 | 0.57 |
| Fuel 4 | 0.36 | 0.22 | 0.39 |
| Fuel 5 | 0.56 | 0.44 | 0.61 |
| Fuel 6 | 0.54 | 0.41 | 0.51 |
Furthermore, the addition of Au/CdS NPs (Fuels 3I–3V) did improve the overall characteristics of the system over native Fuel 1; however, the values were lower compared to Fuels 2I–2V. Importantly, the addition of Rh6G solution did significantly reduce the output values because it hardly contributed to the energy production attributes. To optimize the NPs additives in the fuel, we performed further experiments, which have been summarized in Section V of ESI.† Fig. S7† shows that the addition of ∼70 μL of Au NPs solution in Fuel 1 can be optimal for harvesting high-density energy for a longer duration. Thus, we chose this fuel to show a pilot-scale μ-VLSI potential of the proposed μ-DropFC setup in Fig. 6.
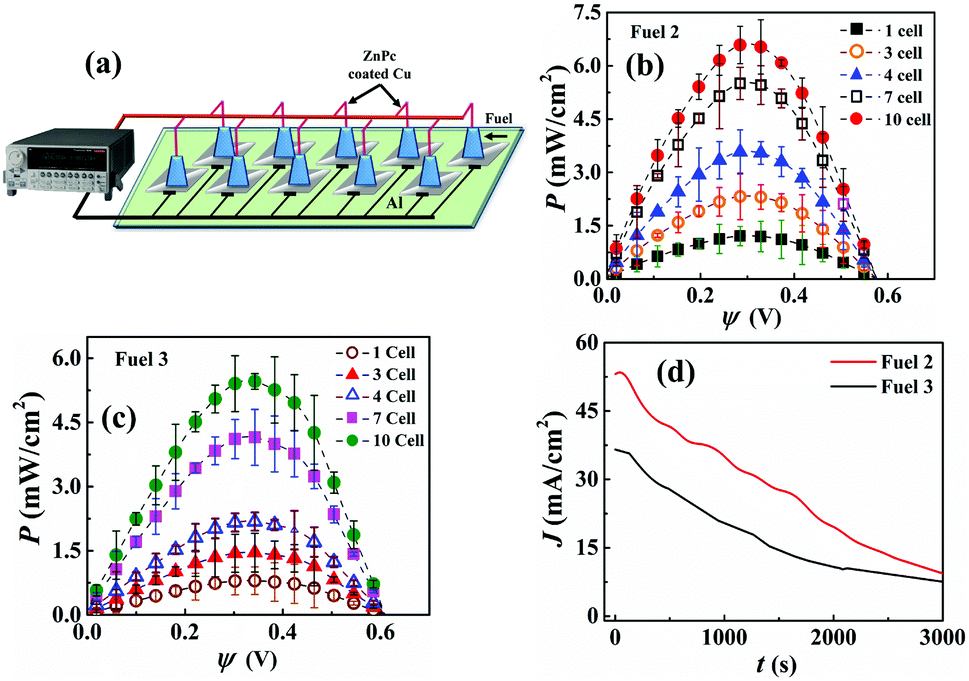 | ||
| Fig. 6 (a) Schematic of the μ-VLSI setup comprising of 10 μ-DropFCs connected in parallel with each other. Plot (b) and (c) P–ψ curves for the VLSI with the increasing number of μ-DropFCs. In plot (b) for each μ-DropFC, 70 μL of Au NPs was added to 10 μL of the native system, while in plot (c) 70 μL of Au/CdS NPs was added to 10 μL of the native system. Plot (d) I–t data for the VLSI setup of 10 μ-DropFCs consisting of the fuels mentioned for plots (b) and (c). | ||
Fig. 6(a) shows the pilot-scale μ-VLSI setup comprised of 10 μ-DropFCs connected in parallel with each other. Plots (b) and (c) show the corresponding P–ψ analysis for Fuels 2 and 3 with the exception that 70 μL Au NPs and Au/CdS NPs were added to prepare each μ-DropFC. Fig. 6(b) shows a 14-fold increase in Pmax to ∼7 mW cm−2 when the number of μ-DropFCs with Fuel 2 was increased from one to ten. It may also be noted that we achieved such power density for the maximum fuel mixture volume of 1 mL (100 μL per cell for 10 cells). Fig. 6(b) shows a 10-fold increase in Pmax to ∼5 mW cm−2 when the number of μ-DropFCs with Fuel 3 was increased from one to ten. Again, the usage of Au NPs led to a better performance than the Au/CdS NPs for the reasons discussed previously.
It may be noted herein that, initially for a single cell, the power density value reached ∼0.72 mW cm−2 when it contained 10 μL of Au NPs along with other constituents making up the total cell volume to be ∼40 μL. The power density thereafter increased to ∼1.8 mW cm−2 when the Au NPs volume increased to ∼80 μL. For the large-scale integration experiment, an optimal value of 70 μL of Au NPs was chosen, which increased the cell volume to ∼100 μL after adding all the other additives. Subsequently, different number of cells (3, 4, 7, and 10) were connected and characterized. Due to the parallel connection within the system, the current values added up and resulted in the overall enhancement of the output power values. This circuit design allowed us to harvest high power densities from the setup. There was also a change in the ψoc values observed; however, the change was not significant when compared with that of the single cell. As the number of cells increased, there was a gradual increase in the maximum power density of the configuration and upon employing linear sweep voltammetry technique, it was observed that the Pmax value was obtained near 0.36 V. Thereafter, the 10 cells that were connected in parallel to harvest a high power density value from the system eventually resulted in generating ∼7 mW cm−2. There was an overall loss in the output efficiency, which could be assigned to various factors, which include the ohmic resistances arising from different cell connections, non-uniform cathode coating (ZnPC) across different cells and time-sensitive system properties like peroxide decomposition.
Fig. 6(d) shows the chronoamperometry plot for the VLSI system with 10 cells, which confirmed that the overall current density for Fuel 2 was higher than Fuel 3. The tests were performed for 50 min to obtain a variation of J from 55 mA cm−2 to 10 mA cm−2 (35 mA cm−2 to 10 mA cm−2) for Fuel 2 (Fuel 3) with a potential of 0.1 V. The enhanced current density values could be attributed to the inherent electronic and optical properties of the proposed prototype, which substantially improved under the presence of light.
Table 3 shows a comparison of the different performance parameters of the proposed μ-DropFC with the others available in the literature. The values summarized in the table suggest that Pmax of μ-DropFCs are quite comparable if not better than the others referred with respect to the working volume of the fuel. In a way the proposed setup has its uniqueness in the development of a portable and affordable μ-DropFC for harvesting high density power from both chemical and light energies utilizing significantly low fuel volumes, which can also be simultaneously utilized for dye degradation.
| An | Ct | ψ oc (V) | H2O2 [M] | P max (mW cm−2) | V F (mL) | Ref. |
|---|---|---|---|---|---|---|
| a Not explicitly mentioned. An: anode; Ct: cathode; VF: volume of fuels utilized per cell; FePC: iron phthalocyanine; PB: Prussian blue. | ||||||
| Mg | PB | 2.3 | 0.5 | 4.9 | 2 | 18 |
| Ni | PB | 0.6 | 0.5 | 1.5 | 50 | 36 |
| Ni | [FeII(H2O)2]3[CoIII (CN)6]2 | 0.8 | 0.3 | 9.9 | —a | 37 |
| Ni | Pedot:PSS | 0.5 | 0.1 | 0.3 | —a | 41 |
| Ni | FePC | 0.5 | 0.3 | 0.01 | —a | 47 |
| Al | PB | 0.6 | 0.5 | 0.8 | 0.5 | 61 |
| Al | ZnPC–Cu | 0.6 | 0.3 | 1.4 – 1 cell, 7.1 – 10 cells | 0.07 – 1 cell, 0.1 – 10 cells | This work |
Table 4 shows the efficiency of the different μ-DropFCs reported in this work. The calculations of efficiencies for the different μ-DropFCs are summarized in Section VI of ESI.† The overall efficiency of the μ-DropFC was determined employing the following formula: εO = εT × εE × εPV, where εO denotes the overall efficiency of the system, εT is the thermodynamic efficiency of the system, εE is the electrochemical efficiency and εPV is the photovoltaic efficiency of the system.
| System | ψ oc | P max | ε PV | ε T | ε E | ε O (%) |
|---|---|---|---|---|---|---|
| a 70 μL of Fuel 2 or 3 was taken for the VLSI system. | ||||||
| Fuel 1 | 0.38 | 0.35 | 0.005 | 0.63 | 0.35 | 0.11 |
| Fuel 2I | 0.59 | 0.72 | 0.01 | 0.63 | 0.55 | 0.36 |
| Fuel 2V | 0.62 | 1.5 | 0.021 | 0.63 | 0.57 | 0.75 |
| Fuel 3I | 0.55 | 0.57 | 0.008 | 0.63 | 0.51 | 0.26 |
| Fuel 3V | 0.57 | 0.95 | 0.014 | 0.63 | 0.53 | 0.47 |
| Fuel 5 | 0.56 | 0.61 | 0.008 | 0.63 | 0.52 | 0.26 |
| Fuel 2a | 0.63 | 7.2 | 0.102 | 0.63 | 0.53 | 3.54 |
| Fuel 3a | 0.61 | 5.7 | 0.08 | 0.63 | 0.54 | 2.83 |
The data suggest that the efficiency of the Au NP-loaded μ-DropFCs was the best among the proposed systems. The output power density increased by ∼10-fold when 10 such units were integrated under the same amount of photonic illumination, which highlighted the scope for improving the overall efficiency of the setup through VLSI. The data reported in Table 4 shows the capacity of the proposed μ-DropFC for harvesting high density power in a very efficient manner, which can be improved further with the help of process intensification and VLSI in the future.
Conclusions
A peroxide microdroplet has been designed and developed into a high-performance fuel cell, namely, μ-DropFC, which is also capable of harvesting solar energy. For this purpose, a peroxide microdroplet at optimal pH and salt loading has been utilized as the native system integrated into an Al-anode and a ZnPC–Cu cathode. The n-type ZnPC enabled the formation of a photo-active Schottky junction at the electrolyte and metal interfaces, which is found to be suitable for power generation under light. Oxidation and reduction of H2O2 on the electrodes helped in the conversion of chemical energy into the electrical one in a membraneless setup. The use of suspended Au NPs enhanced the power density under photonic illumination through LSPR. The addition of n-type CdS NPs infuses the capacity of catalytic photo-degradation of dyes under light. The simple, low-cost, portable, and membraneless setup possesses significant power densities of ∼0.8 mW cm−2 per cell and 7 mW cm−2 at an efficiency of 3.5% when a group of 10 cells have been integrated. The single-cell setup requires significantly less working fluid volume and can give a very high power density at a voltage of ∼0.38 V. The single-cell setup also delivers a current density of ∼1.5 mA cm−2 at an applied voltage of 0.1 V for 30 min. Furthermore, the single-cell setup is capable of degrading ∼85% of rhodamine 6G dye in 1 h while simultaneously generating energy at a power density of ∼0.6 mW cm−2. A comparison with previous works shows the superiority of the proposed μ-DropFC for harvesting high density power, which can be improved further with the help of process intensification and VLSI in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MeitY grant no. 5(9)/2012-NANO, MHRD IMPRINT program, grant no. 8058, and DST SERB, Grant no. EMR/2016/001824, Government of India, for the financial aids. We thank the Department of Chemical Engineering, Centre for Nanotechnology and Central Instrumental Facility, IIT Guwahati for the facilities and support provided during the work.Notes and references
- K. S. Dhathathreyan, N. Rajalakshmi and R. Balaji, in Nanotechnology for Energy Sustainability, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, pp. 569–596 Search PubMed.
- F. Zhu, J. Kim, K.-C. Tsao, J. Zhang and H. Yang, Curr. Opin. Chem. Eng., 2015, 8, 89–97 CrossRef.
- Z. L. Wang and W. Wu, Angew. Chem., Int. Ed., 2012, 51, 11700–11721 CrossRef CAS PubMed.
- N. Wang, L. Han, H. He, N.-H. Park and K. Koumoto, Energy Environ. Sci., 2011, 4, 3676–3679 RSC.
- M. Bhattacharjee, V. Pasumarthi, J. Chaudhuri, A. K. Singh, H. Nemade and D. Bandyopadhyay, Nanoscale, 2016, 8, 6118–6128 RSC.
- S. S. Das, V. M. Pedireddi, A. Bandopadhyay, P. Saha and S. Chakraborty, 2019, arXiv:1907.09999.
- X. Xuan, Electrophoresis, 2019, 40, 2484–2513 CAS.
- M. Bhattacharjee, S. Timung, T. K. Mandal and D. Bandyopadhyay, Nanoscale Adv., 2019, 1, 1155–1164 RSC.
- L. Manjakkal, A. Vilouras and R. Dahiya, IEEE Sens. J., 2018, 18, 7779–7785 CAS.
- J. Chen, J.-H. Cui and L. Wang, IEEE Trans. Very Large Scale Integr. Syst., 2015, 23, 1931–1935 Search PubMed.
- R. Vooradi, S. B. Anne, A. K. Tula, M. R. Eden and R. Gani, BMC Chem. Eng., 2019, 1, 7–24 CrossRef.
- K. V. Selvan and M. S. Mohamed Ali, Renewable Sustainable Energy Rev., 2016, 54, 1035–1047 CrossRef.
- A. Kundu, J. H. Jang, J. H. Gil, C. R. Jung, H. R. Lee, S.-H. Kim, B. Ku and Y. S. Oh, J. Power Sources, 2007, 170, 67–78 CrossRef CAS.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- S. Mamidi, M. Kakunuri and C. S. Sharma, ECS Trans., 2018, 85, 21–27 CrossRef CAS.
- S. Thakur, S. Rarotra, M. Bhattacharjee, S. Mitra, G. Natu, T. K. Mandal, A. K. Dasmahapatra and D. Bandyopadhyay, Phys. Rev. Appl., 2018, 10, 064012–064125 CrossRef CAS.
- S. Hardman, A. Chandan and R. Steinberger-Wilckens, J. Power Sources, 2015, 287, 297–306 CrossRef CAS.
- S. A. Mousavi Shaegh, S. M. Mousavi Ehteshami, S. H. Chan, N.-T. Nguyen and S. N. Tan, RSC Adv., 2014, 4, 37284–37287 RSC.
- W. R. Grove, London, Edinburgh Dublin Philos. Mag. J. Sci., 1839, 14, 127–130 CrossRef.
- M. L. Perry and T. F. Fuller, J. Electrochem. Soc., 2002, 149, S59–S67 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- N. Luo, G. H. Miley, R. J. Gimlin, R. L. Burton, J. Rusek and F. Holcomb, J. Propul. Power, 2008, 24, 583–589 CrossRef CAS.
- R. German, in IEEE Proceedings OCEANS, 1989, vol. 3, pp. 843–848 Search PubMed.
- M. A. Modestino, D. Fernandez Rivas, S. M. H. Hashemi, J. G. E. Gardeniers and D. Psaltis, Energy Environ. Sci., 2016, 9, 3381–3391 RSC.
- L. Carrette, K. A. Friedrich and U. Stimming, Fuel Cells, 2001, 1, 5–39 CrossRef CAS.
- R. P. O'Hayre, S.-W. Cha, W. G. Colella and F. B. Prinz, Fuel Cell Fundamentals, John Wiley & Sons, 2016 Search PubMed.
- R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS.
- L. An, T. Zhao, X. Yan, X. Zhou and P. Tan, Sci. Bull., 2015, 60, 55–64 CrossRef CAS.
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334–7336 RSC.
- S. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. A. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS.
- M. Asadnia, S. M. Mousavi Ehteshami, S. H. Chan and M. E. Warkiani, RSC Adv., 2017, 7, 40755–40760 RSC.
- S. A. Mousavi Shaegh, N.-T. Nguyen, S. M. Mousavi Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225–8228 RSC.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- G. Lisak, T. Tamaki and T. Ogawa, Anal. Chem., 2017, 89, 3943–3951 CrossRef CAS PubMed.
- A. Kozawa, V. E. Zilionis and R. J. Brodd, J. Electrochem. Soc., 1970, 117, 1470–1474 CrossRef CAS.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813–819 CrossRef CAS.
- E. Miglbauer, P. J. Wójcik and E. D. Głowacki, Chem. Commun., 2018, 54, 11873–11876 RSC.
- A. E. Sanli and A. Aytaç, Int. J. Hydrogen Energy, 2011, 36, 869–875 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- M. Haruta, Gold Bull., 2004, 37, 27–36 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed.
- K. Mallick, M. J. Witcomb and M. S. Scurrell, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 797–801 CrossRef CAS.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 RSC.
- I. F. Bohrer, N. C. Colesniuc, J. Park, K. I. Schuller, A. C. Kummel and W. C. Trogler, J. Am. Chem. Soc., 2008, 130, 3712–3713 CrossRef PubMed.
- I.-H. Yeo and D. C. Johnson, J. Electroanal. Chem., 2001, 495, 110–119 CrossRef CAS.
- A. Maizelis, J. Electrochem. Energy Convers. Storage, 2019, 16, 041003–041009 CrossRef CAS.
- A. Ferretti, C. Baldacchini, A. Calzolari, R. Di Felice, A. Ruini, E. Molinari and M. G. Betti, Phys. Rev. Lett., 2007, 99, 046802–046805 CrossRef PubMed.
- B. Amin, S. Nazir and U. Schwingenschlögl, Sci. Rep., 2013, 3, 1705–1710 CrossRef.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- Y. Jiao, X.-M. Ma, F.-F. Wang, K. Wang, P.-P. Zhang, Z.-Z. Wang and L. Yang, Micro Nano Lett., 2012, 7, 631–633 CrossRef.
- S. Kalathil, J. Lee and M. H. Cho, Bioresour. Technol., 2012, 119, 22–27 CrossRef CAS PubMed.
- T. H. Han, M. M. Khan, S. Kalathil, J. Lee and M. H. Cho, Ind. Eng. Chem. Res., 2013, 52, 8174–8181 CrossRef CAS.
- G. Chen, X. Liu, S. Li, M. Dong and D. Jiang, Lab Chip, 2018, 18, 1026–1034 RSC.
- Y. Liu, L. Liu and F. Yang, RSC Adv., 2016, 6, 12068–12075 RSC.
- A. J. Nozik and R. Memming, J. Phys. Chem., 1996, 100, 13061–13078 CrossRef CAS.
- D. O. Grynko, O. M. Fedoryak, P. S. Smertenko, N. A. Ogurtsov, A. A. Pud, Y. V. Noskov and O. P. Dimitriev, J. Phys. D: Appl. Phys., 2013, 46, 495114–495122 CrossRef.
- S. M. Mousavi Ehteshami, M. Asadnia, S. N. Tan and S. H. Chan, J. Power Sources, 2016, 301, 392–395 CrossRef CAS.
- D. Tuschel, Spectroscopy, 2016, 31, 14–21 CAS.
- W. Lee, H. Kim, D.-R. Jung, J. Kim, C. Nahm, J. Lee, S. Kang, B. Lee and B. Park, Nanoscale Res. Lett., 2012, 7, 672–676 CrossRef PubMed.
- Y.-H. Su, Y.-F. Ke, S.-L. Cai and Q.-Y. Yao, Light: Sci. Appl., 2012, 1, e14–e18 CrossRef.
- W. He, Y.-T. Zhou, W. G. Wamer, X. Hu, X. Wu, Z. Zheng, M. D. Boudreau and J.-J. Yin, Biomaterials, 2013, 34, 765–773 CrossRef CAS PubMed.
- L. Wang, R. Li, J. Liu, J. Han and M. Huang, J. Mater. Sci., 2017, 52, 1847–1855 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Nanocomposites characterization, effect of pH on power generation in presence of additives, electrode characterization before and after analysis, cell characterization employing dye degradation moieties, optimal additive loading in VLSI, efficiency calculations and description of supplementary videos. See DOI: 10.1039/c9na00785g |
| ‡ Department of Physics, Sipajhar College, Sipajhar Darrang – 784145, India. |
| This journal is © The Royal Society of Chemistry 2020 |