
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the charge state of an Au nanocluster on the oxidized/reduced rutile TiO2 (110) surface using non-contact atomic force microscopy and Kelvin probe force microscopy
Yuuki
Adachi
 a,
Huan Fei
Wen
b,
Quanzhen
Zhang
a,
Masato
Miyazaki
a,
Yasuhiro
Sugawara
a and
Yan Jun
Li
*a
a,
Huan Fei
Wen
b,
Quanzhen
Zhang
a,
Masato
Miyazaki
a,
Yasuhiro
Sugawara
a and
Yan Jun
Li
*a
aDepartment of Applied Physics, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: liyanjun@ap.eng.osaka-u.ac.jp
bKey Laboratory of Instrumentation Science and Dynamic Measurement, School of Instrument and Electronics, North University of China, Taiyuan, Shanxi 030051, P. R. China
First published on 25th March 2020
Abstract
The charge state of Au nanoclusters on oxidized/reduced rutile TiO2 (110) surfaces were investigated by a combination of non-contact atomic force microscopy and Kelvin probe force microscopy at 78 K under ultra-high vacuum. We found that the Au nanoclusters supported on oxidized/reduced surfaces had a relatively positive/negative charge state, respectively, compared with the substrate. In addition, the distance dependence of LCPD verified the contrast observed in the KPFM images. The physical background of charge transfer observation can be explained by the model of charge attachment/detachment from multiple oxygen vacancies/adatoms surrounding Au nanoclusters. These results suggest that the electronic properties of the Au nanoclusters are dramatically influenced by the condition of the support used.
Introduction
Since the pioneering work of Haruta and Valden,1,2 a number of studies have been devoted to the Au/TiO2 surface and significant progress has been made. The size of Au nanoclusters crucially affects the catalytic activity of this system. In particular, Au nanoclusters having sizes between 2 nm and 6 nm dramatically promote the catalytic reactions.1–9 These findings demonstrate that the supported clusters, in general, may have unusual properties on this surface. Therefore, many researchers have been fascinated by this topic and tremendous progress has been made to investigate this catalytic activity. Particularly, the presence of these small nanoclusters is believed to play an important role; several studies have shown that the charge state of the Au nanoclusters can also dramatically alter the catalytic reaction.10–16 For example, the presence of cationic clusters was assumed to lower the reaction barrier between CO and molecular oxygen.14 In addition, several works have shown that the Au nanocluster charge state is strongly influenced by the choice of the oxide support in this system.10,15 Specifically, this means that the charge state of the Au nanoclusters can be assumed to be highly sensitive to the nature of the chemical environment of TiO2.15,16 Particularly, in the case of a reduced surface, previous experimental work involving XPS under UHV provided evidence for the electron transfer to the adsorbed Au nanoclusters.12 However, this method averages the characteristics of the macroscopic region and is difficult to understand at the microscopic scale. Thus, there is a lack of experimental research to individually detect the local charge redistribution of the Au nanoclusters supported on oxidized/reduced TiO2 surfaces at the microscopic scale. Scanning tunneling microscopy (STM) has been widely used to study the Au nanoclusters on these surfaces at the atomic scale.2,4–6,15,16 However, STM involves tunneling currents into the conducting substrate, which can easily result in unintended switching of the charge state during the measurements. Recently, atomic force microscopy (AFM), as a viable alternative, has been used to provide atomic-scale images of surfaces, characterize atoms and molecules,17–19 and manipulate the nanoclusters.20,21 In contrast to STM, AFM can avoid the unintended charge rearrangement due to its force modulation mechanism.17 Moreover, Kelvin probe force microscopy (KPFM) due to its atomic scale precision allows us to directly detect the different charge states within the atomic scale using the local contact potential difference (LCPD).22–24 Hence, in this work, AFM and KPFM were used to detect the different charge states of the Au nanoclusters on rutile TiO2 (110) surfaces depending on oxidation. We found that the Au nanoclusters supported on the oxidized surface were relatively positively charged, while the Au nanoclusters supported on the reduced surface were negatively charged compared to the substrate. For our charge state studies, the distance dependence of the LCPD values was recorded on top of the Au nanocluster and substrate, confirming the contrast observed in the oxidized/reduced surfaces of exactly different signs in KPFM measurements.Experimental details
The experiments were carried out using low-temperature ultra-high vacuum (UHV) atomic force microscopy (AFM). One common difficulty in this experiment is the pre-request of the r-TiO2 (110) surface, which can easily react with water and an unknown gas. Because the water molecules are known to produce a high coverage hydroxyl TiO2 (110) surface, which has significantly different properties compared with the reduced TiO2 (110) surface, clean reduced TiO2 (110) is necessary for comparison with the oxidized surface.25,26 Therefore, in this work, the Au nanocluster was deposited on oxidized/reduced TiO2 (110) at 300 K under ultra-high vacuum (<1.0 × 10−11 torr) to avoid the reaction with water molecules and contamination. The reduced TiO2 (110) sample was prepared by Ar+ sputtering and annealing at 900 K for several cycles before depositing the Au nanoclusters. The oxidized surface was prepared by exposing the sample to oxygen at room temperature for ∼0.5 L before depositing the Au nanoclusters. The observation chamber was also kept at an ultra-high vacuum condition (<1.0 × 10−11 torr) to keep the surface clean during the entire experimental run. The deflection of the cantilever was measured using the optical beam deflection method. The AFM unit was kept at liquid nitrogen temperature (78 K). The AFM measurements were performed in frequency-modulation (FM) detection mode. The cantilever was oscillated at its resonance frequency with a constant oscillation amplitude. We used an iridium (Ir)-coated Si cantilever (Nano sensors SD-T10L100, f0 = 800 kHz, A = 500–1000 pm). The Ir tips provided more stable AFM imaging compared to a bare Si tip. The tip was initially annealed to 600 K and then cleaned by Ar+ sputtering to remove any contamination before performing the experiments. The atom tracking method was used to compensate the thermal drift between the tip and surface during the measurements. KPFM measurements were recorded in the frequency-modulation mode. An AC bias voltage (VAC) at the frequency fAC and a DC bias voltage (VDC) were applied to the sample. VDC was adjusted to compensate the fAC component of the electrostatic force, providing the CPD value (VCPD). For details, please refer to our previous works.28,30Results and discussion
First, the surfaces were proven to be reduced/oxidized TiO2 (110) surfaces via atomically resolved AFM images. This is important to exclude the possibility of performing experiments on high coverage hydroxyl TiO2 (110), which would strongly alter the properties of the Au nanoclusters.25,26Fig. 1(a) and (b) show large area AFM images of the Au/reduced-TiO2 (110) surface and Au/oxidized-TiO2 (110) surface containing a number of Au nanoclusters on the terrace. The maximum diameter of the Au nanoclusters from these two images was determined to be smaller than 6 nm, which was in the range responsible for high catalytic activity.1–6 These findings are in agreement with previous STM studies, which show that Au nanoclusters nucleate on top of Ov or oxygen adatom (Oad).15,16Fig. 1(c) and (d) show atomically resolved AFM images measured on top of these surfaces. As shown by Fig. 1(c) and (d), several bright rows and dark rows are alternately aligned. The distance between these two rows is 0.65 nm. In addition, we can see several small dark spots and bright spots on top of the bright row. In particular, in Fig. 1(d), we can see several large bright spots on top of the dark row. The rutile TiO2 (110) surface contains two-fold coordinated protruding O atoms and five-fold coordinated Ti atoms, which are alternately aligned.4 Practical sample preparation under UHV leads to the creation of point defects such as Ov and a few double-OH defects. The oxygen molecules are known to dissociate and adsorb as Oad on top of these five-fold coordinated Ti atoms.28,29 Thus, the bright row and dark row observed in Fig. 1(c) and (d) correspond to the O row and Ti row. Moreover, the small dark spots and bright spots are the point defects such as Ov and double-OH on these surfaces. Additionally, the large bright spots observed in Fig. 1(d) are Oad.28,29 In the case of NC-AFM imaging on these surfaces, it is well known that the image contrast strongly depends on the chemical construction of the tip apex.27 In a recent work, three common contrasts (neutral, hole, and protrusion types) were observed on rutile TiO2 (110) surfaces; especially, the image obtained by the neutral mode tip is believed to give a contrast resembling the true geometrical structure and provides an easy way to identify the atomic species adsorbed on this surface.27 Therefore, the tips used in Fig. 1(c) and (d) are believed to correspond to a neutral mode because the contrast of atomically resolved images qualitatively resembles the true geometric structure. Here, it should be noted that the neutral mode tip was deliberately prepared by weakly poking the tip apex on these surfaces.29 The values for the coverage of Ov and OH defects (Fig. 1(c)) are 7.7% ML and 0.5% ML, respectively, as deduced from the extended AFM data, which also confirmed that the predominant species was Ov. As shown in Fig. 1(d), the values for the coverage of Ov and OH defects as well as Oad are 2.0% ML, 0.5% ML and 8.5% ML, respectively, as deduced from the AFM data, which confirms that the predominant species is Oad. From these results, we ambiguously conclude that our work is performed on reduced/oxidized TiO2 (110) surfaces, which are significantly covered by Ov or Oad, respectively. | ||
| Fig. 1 (a and b) AFM images of Au nanoclusters adsorbed on the reduced/oxidized-rutile TiO2 (110)-(1 × 1) surfaces. Numerous small Au nanoclusters were homogeneously distributed on the terraces. (c and d) Atomically resolved AFM images of reduced/oxidized-rutile TiO2 (110)-(1 × 1) surfaces. O and Ti atom rows on TiO2 (110) were observed as bright and dark by a tip, respectively, in the neutral mode.27 Imaging parameters: constant Δf mode, VS = 0 V, (a) 125 × 130 nm2; (b) 6.0 × 9.5 nm2; (c) 125 × 130 nm2; (d) 6.0 × 9.5 nm2. | ||
Next, the charge state of the Au nanoclusters was investigated by using high-resolution KPFM. Fig. 2 shows the topography images and LCPD images measured using AFM and KPFM at identical areas. As shown in Fig. 2(a)–(d), numerous contrast patterns of the Au nanoclusters on oxidized/reduced TiO2 (110) surfaces are obtained by AFM and KPFM. In order to comprehensively analyse these images, we studied the line scans on top the overall Au nanoclusters for each image. The corresponding line scans are depicted under each image. From the topography images (Fig. 2(a) and (b)), we can see that the oxidized/reduced-TiO2 (110) surfaces contain a number of Au nanoclusters on their terrace. The diameter of the Au nanoclusters is about 2–3 nm, which is in the range of high catalysis, as discussed above. Focusing on the LCPD images (Fig. 2(c) and (d)), we found that the overall contrast patterns of LCPD obtained on top of the Au nanoclusters in each image were relatively smaller/larger compared with that for the substrate. From Fig. 2(c) and (d), the smallest/largest LCPD value is found to be 0.75 V/0.83 V and 0.53 V/0.73 V on top of the Au nanoclusters, respectively. The origin of the overall difference of LCPD on top of the Au nanocluster is considered to be the difference in local charge redistribution at the Au nanoclusters.23,25 Given that the LCPD change is induced by this charge redistribution between the surface and Au nanoclusters, the Au nanoclusters are present in a relatively positive/negative charge state on these surfaces. We expect that this systematic trend of different charge states of the Au nanoclusters on reduced/oxidized TiO2 (110) surfaces is induced by the accumulation of more/less electrons in the Au nanoclusters on reduced/oxidized TiO2 (110) surfaces.
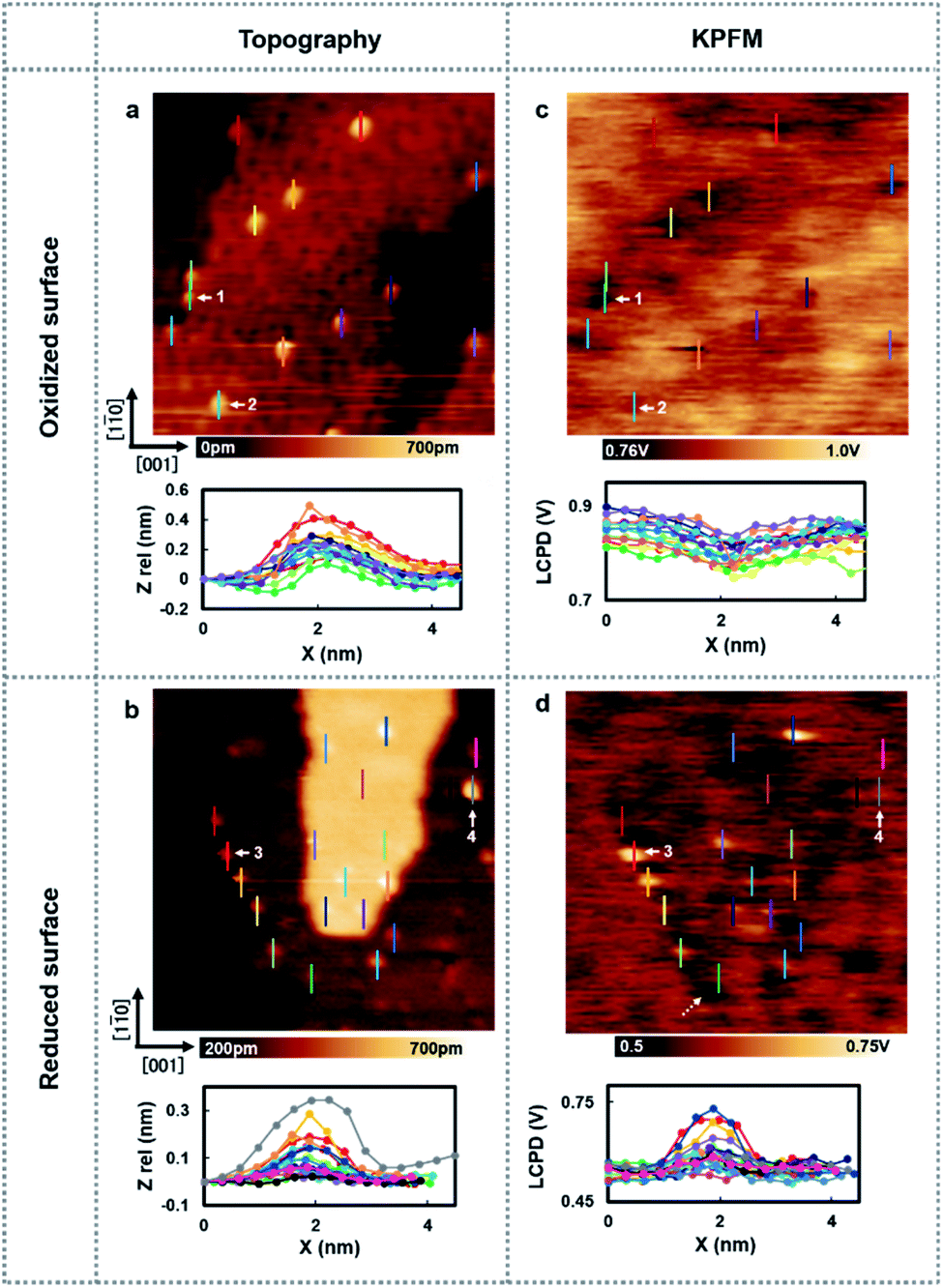 | ||
| Fig. 2 (a and b) AFM images of the Au nanoclusters adsorbed on the oxidized/reduced-rutile TiO2 (110)-(1 × 1) surfaces. Numerous small Au nanoclusters are homogeneously distributed on the terraces. Imaging size, both 35 × 35 nm2. (c and d) LCPD images of the Au nanoclusters adsorbed on the oxidized/reduced-rutile TiO2 (110)-(1 × 1) surfaces obtained at an identical area with (a) and (b). Imaging size, both 35 × 35 nm2. Inset: AFM or LCPD line profiles measured above several types of adsorbed Au nanoclusters as indicated by different colors. | ||
To establish a direct link between our experiments and physical background, we have illustrated a schematic model of these charge state differences induced by different oxidation surface states at the atomic scale. As shown in Fig. 3(a), because the electronegatively Oad tends to withdraw the electrons from the Au nanocluster, the Au nanocluster becomes positively charged.28,29 In contrast, Ov tends to supply excess electrons to the Au nanocluster, due to which the Au nanocluster becomes negatively charged (Fig. 3(b)). Here, we note that this measured charge redistribution should depend on both its size and the amount of vacancies attached to the Au nanocluster.31 In general, it cannot be said that the size of LCPD is always quantitatively proportional to the amount of charge.22–24 Nevertheless, in Fig. 2(a)–(d) we can find that some of the Au nanoclusters show relatively large absolute LCPD even though the size is small, as can be seen for the Au nanoclusters numbered 1 and 3 inside the images. On the other hand, some of the Au nanoclusters show relatively small absolute LCPD even though the size is large, as can be seen for the Au nanoclusters numbered 2 and 4 inside the images. This is probably because the charge state of the nanocluster depends not only on the size of the gold cluster but also on the shape and number of the surrounding defects and adsorbed oxygen. A comprehensive study of the size and shape as well as the existence of defects in the Au nanoclusters might be challenging because it will be necessary to discuss every condition properly at the atomic scale. Notably, there is also a macroscopic LCPD contrast on the background, as shown by the white dotted arrow in Fig. 2(d). The possible origin of this contrast is considered to be a step structure, which we previously observed on this surface.30 However, all of the LCPD line scans shown in Fig. 2 were measured exactly on top of the Au nanoclusters and overall showed smaller/larger values only on top of the Au nanoclusters compared to the surrounding substrate. Therefore, we conclude that the main contribution to the LCPD contrast observed on top of the Au nanoclusters significantly reflects the different charge states of the Au nanoclusters.
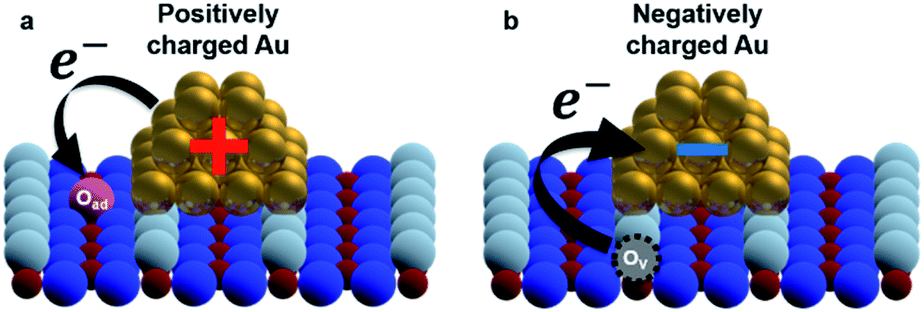 | ||
| Fig. 3 (a and b) Schematic model of an Au nanocluster on oxidized/reduced rutile TiO2(110)-(1 × 1) surfaces. O2c (white balls): two-fold coordinated bridge oxygen row; O3c (blue balls): in plane three-fold coordinated oxygen; Ti5c (red balls): rows of five-fold coordinated Ti atoms; OV (black dotted circles): oxygen vacancy; Au (gold balls): Au atoms; Oad (pink ball): oxygen adatom. | ||
To gain more insights into the strong contrast difference observed in the LCPD images, the system was further investigated by performing KPFM measurements as a function of the tip sample distance, ensuring that no changes in the tip or deformation of the Au nanoclusters could occur.22,25,28,30 The experiment was conducted as follows: the initial positions of the Au nanoclusters on oxide/reduced TiO2 surfaces were confirmed by AFM imaging. After AFM imaging, the tip was brought above the Au nanocluster and KPFM feedback was turned on. The tip was made to vertically approach the top of the Au nanocluster simultaneously collecting the value of LCPD. We performed these experiments in every redox condition of an Au nanocluster and the substrate. It should also be mentioned here that the spectroscopy data were acquired using ∼10 s for high sensitivity. As we can see in Fig. 4(a) and (b), on the oxidized/reduced surfaces, the LCPD of an Au nanocluster is always smaller/larger than that of substrate TiO2 at a range of measured distances. The LCPD value measured on top of the Au nanocluster starts to deviate from that of substrate TiO2 at around −0.8 nm in both cases with monotonic decrease/increase on the oxidized/reduced surfaces. These observations can be explained by the averaging effect, wherein the Au nanocluster area that contributes to the LCPD becomes larger as compared with the TiO2 surface due to the decrease in tip height.22 As the tip moves closely towards the Au nanocluster, the effect of the surrounding TiO2 became smaller, which explains the deviation of LCPD at around −0.8 nm. These systematic trends agree with the observations from Fig. 2(c) and (d); the LCPD values of the Au nanoclusters are overall smaller/larger than those for reduced/oxidized TiO2 (110) surfaces. In the case of reduced and oxidized surfaces, a previously performed theoretical work indicates that the charge transfer happens from the Au nanocluster to Ov or from oxygen molecules to the Au nanocluster based on matching electronic potentials between the cluster and the support.10 In particular, in the case of the reduced surface, a previous experimental work using XPS under UHV macroscopically shows the evidence of electron transfer to the adsorbed Au nanocluster.12 Indeed, the LCPD contrast and LCPD shift shown in Fig. 2(c), (d), 4(a) and (b) suggest that the charge redistribution occurring at the interface of metal/oxide results in Au nanoclusters being negatively charged in reduced conditions and positively charged in oxidized conditions. We believe that the charge redistribution initially creates an intrinsic dipole between the metal/oxide interface.22 Thus, the main contribution of the observed increase or decrease in LCPD may originate from the surface dipole, which is mainly modulated by the local charge state of the Au nanoclusters on the surface. The above-mentioned experimental results give a fundamental insight into our understanding of how reducible supports influence the Au nanocluster catalytic chemistry. Moreover, the donation or withdrawal of charge from the Au nanocluster builds up a dramatic activity difference on the metal/oxide interface.
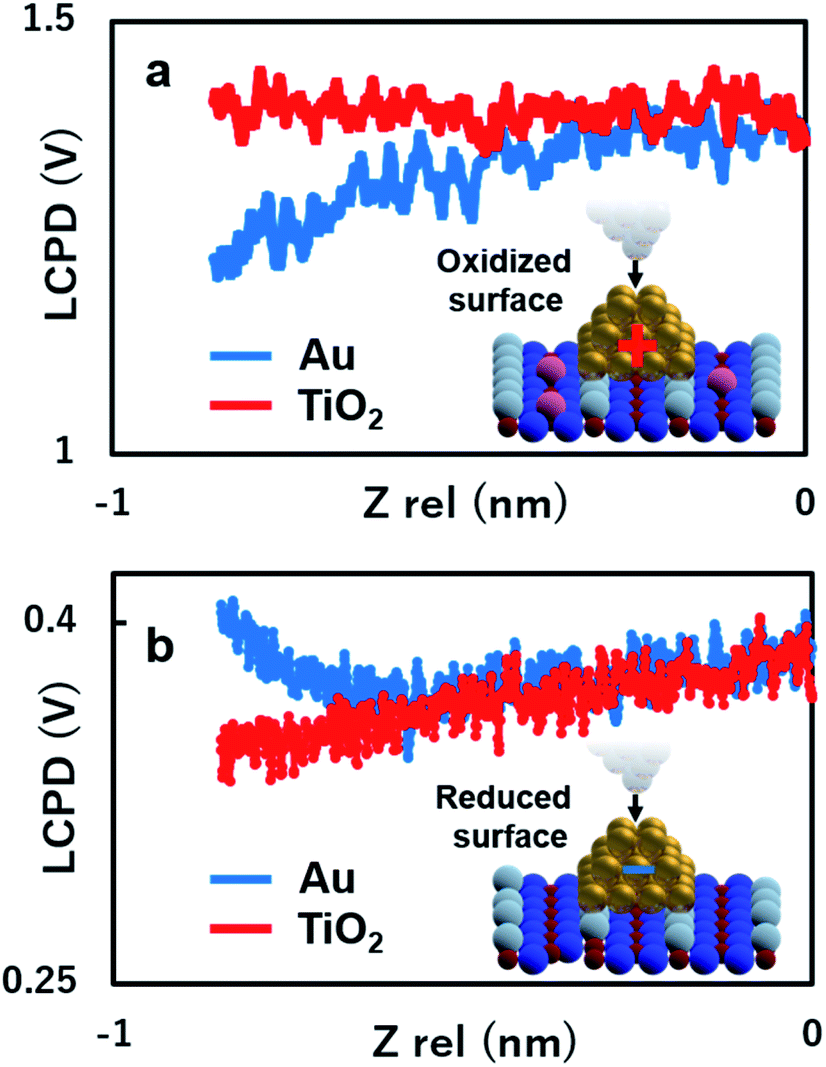 | ||
| Fig. 4 (a and b) Distance dependence of KPFM measured on top of the Au nanoclusters on oxidized/reduced-rutile TiO2 (110)-(1 × 1) surfaces. The blue spectrum shows the LCPD value obtained on top the Au cluster and red spectrum shows the LCPD value obtained on top of the TiO2 substrate. | ||
Conclusions
In conclusion, a direct observation of the charge state of Au nanoclusters was achieved on a rutile TiO2 (110) surface. We successfully demonstrated the different charge states using the AFM and KPFM techniques. A proposed model of charge transfer from Ov and Oad was established to explain the different charge states of the Au nanocluster on oxidized/reduced TiO2 (110) surfaces. These experimental results show excellent agreement with previously reported theoretical results, which involve charge attachment/detachment from multiple oxygen vacancies/adatoms surrounding the Au nanocluster. Our results give additional insights into understanding the atomic scale catalysis of an Au nanocluster on TiO2 (110) surfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (JSPS) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) by MEXT/JSPS KAKENHI Grant Number (16H06327, 16H06504 and 17H01061) and Osaka University's International Joint Research Promotion Program (J171013014, J171013007, J181013006, J191013005, J191013014, Ja19990011).References
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- X. Tong, L. Benz, P. Kemper, H. Metiu, M. T. Bowers and S. K. Buratto, J. Am. Chem. Soc., 2005, 127, 13516–13518 CrossRef CAS PubMed.
- X. Lai and D. W. Goodman, J. Mol. Catal. A: Chem., 2000, 162, 33–50 CrossRef CAS.
- L. Li, Y. Gao, H. Li, Y. Zhao, Y. Pei, Z. Chen and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 19336–19346 CrossRef CAS PubMed.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Am. Chem. Soc., 2004, 126, 5682–5683 CrossRef CAS PubMed.
- Y. G. Wang, Y. Yoon, V. A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240–2241 CrossRef CAS PubMed.
- Z. Jiang, W. Zhang, L. Jin, X. Yang, F. Xu, J. Zhu and W. Huang, J. Phys. Chem. C, 2007, 111, 12434–12439 CrossRef CAS.
- J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672–2673 CrossRef CAS PubMed.
- J. G. Wang and B. Hammer, Phys. Rev. Lett., 2006, 97, 136107 CrossRef CAS PubMed.
- D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard and F. Besenbacher, Science, 2007, 315, 1692–1696 CrossRef CAS PubMed.
- E. Wahlström, N. Lopez, R. Schaub, P. Thostrup, A. Rønnau, C. Africh and F. Besenbacher, Phys. Rev. Lett., 2003, 90, 026101 CrossRef PubMed.
- F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949 CrossRef CAS.
- H. Mönig, S. Amirjalayer, A. Timmer, Z. Hu, L. Liu, O. D. Arado, M. Cnudde, C. A. Strassert, W. Ji, M. Rohlfing and H. Fuchs, Nat. Nanotechnol., 2018, 7, 958–964 Search PubMed.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- T. Hynninen, G. Cabailh, A. S. Foster and C. Barth, Sci. Rep., 2013, 3, 1270 CrossRef PubMed.
- Y. Sugimoto, A. Yurtsever, N. Hirayama, M. Abe and S. Morita, Nat. Commun., 2014, 5, 4360 CrossRef CAS PubMed.
- L. Gross, F. Mohn, P. Liljeroth, J. Repp, F. J. Giessibl and G. Meyer, Science, 2009, 324, 1428–1431 CrossRef CAS PubMed.
- S. Akira, P. Cui and O. Hiroshi, J. Phys. Chem. B, 2006, 110, 13453–13457 CrossRef PubMed.
- W. Melitz, S. Jian, C. K. Andrew and L. Sangyeob, Surf. Sci. Rep., 2011, 66, 1–27 CrossRef CAS.
- H. Jing Chung, A. Yurtsever, Y. Sugimoto, M. Abe and S. Morita, Appl. Phys. Lett., 2011, 99, 123102 CrossRef.
- X. Tong, L. Benz, S. Chrétien, H. Metiu, M. T. Bowers and S. K. Buratto, J. Phys. Chem. C, 2010, 114, 3987–3990 CrossRef CAS.
- A. Yurtsever, D. Fernández-Torre, C. González, P. Jelínek, P. Pou, Y. Sugimoto and S. Morita, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125416 CrossRef.
- Q. Z. Zhang, Y. J. Li, H. F. Wen, Y. Adachi, M. Miyazaki, Y. Sugawara, R. Xu, H. Z. Cheng, J. Brndiar, L. Kantorovich and I. Štich, J. Am. Chem. Soc., 2018, 140, 15668–15674 CrossRef CAS PubMed.
- Y. Adachi, H. F. Wen, Q. Z. Zhang, M. Miyazaki, Y. Sugawara, H. Q. Sang, J. Brndiar, L. Kantorovich, I. Štich and Y. J. Li, ACS Nano, 2019, 13, 6917–6924 CrossRef CAS PubMed.
- M. Miyazaki, H. F. Wen, Q. Zhang, Y. Adachi, J. Brndiar, I. Štich, Y. J. Li and Y. Sugawara, Beilstein J. Nanotechnol., 2019, 10, 1228–1236 CrossRef CAS PubMed.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud and S. Fabris, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |