
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic framework templated Pd/CeO2@N-doped carbon for low-temperature CO oxidation†
Weidong
Fan‡
 a,
Dongyuan
Liu‡
a,
Xia
Wang
a,
Xiuping
Liu
b,
Dongwei
Cao
a,
Lili
Fan
*a,
Zhaodi
Huang
a,
Wenyue
Guo
a and
Daofeng
Sun
a
a,
Dongyuan
Liu‡
a,
Xia
Wang
a,
Xiuping
Liu
b,
Dongwei
Cao
a,
Lili
Fan
*a,
Zhaodi
Huang
a,
Wenyue
Guo
a and
Daofeng
Sun
a
aSchool of Materials Science and Engineering, College of Science, China University of Petroleum (East China), Qingdao, Shandong 266580, China. E-mail: lilifan@upc.edu.cn
bCollege of Materials Science and Engineering, Linyi University, Linyi, Shandong 276000, China
First published on 31st December 2019
Abstract
A new Pd/Ce based metal–organic framework is designed and synthesized as a self-sacrificial template for fabrication of an efficient catalyst for CO oxidation. The catalyst obtained by thermal annealing at 700 °C (Pd/CeO2@NC-700) is composed of N-doped carbon with embedded Pd and CeO2 nanoparticles, which are highly dispersed and closely connected in the N-doped carbon; the high Pd loading (33.7 wt%) and the coupling between Pd and the CeO2 phase synergistically boost the CO oxidation performance. The Pd/CeO2@NC-700 catalyst exhibits a 100% conversion temperature of 89 °C and excellent long-term stability. By combining structural characterization with density functional theory calculations, two possible CO oxidation pathways of TPB and TOP are revealed, in which the adsorbed O2 directly dissociates to O* atoms and activates CO* molecules. The transfer of O* between Pd and Ce (TPB) or Pd and Pd (TOP) facilitates the formation of 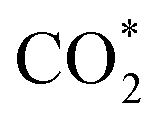 intermediates and finally results in the production of CO2. This work provides a new insight into the development of novel efficient catalysts for CO oxidation based on metal–organic frameworks.
intermediates and finally results in the production of CO2. This work provides a new insight into the development of novel efficient catalysts for CO oxidation based on metal–organic frameworks.
Introduction
The carbon monoxide (CO) oxidation reaction is indisputably important for mitigating pollution, which has attracted great attention in the aspect of the living environment and industrial research.1–3 Hence, development of highly efficient catalysts that can enhance this reaction is crucial.4–6 As one of the most commonly used reducible oxides, CeO2 has been studied extensively for CO oxidation catalysis due to its high oxygen storage capacity and redox properties. Meanwhile, CeO2 nanomaterials are also active carriers that can interact with catalytic components, such as precious metals (such as Pd, Pt, etc.), to enhance their stability and reactivity. Pd/CeO2 composites not only have the activity of Pd and CeO2, but also promote O2 adsorption through synergistic interactions between Pd and CeO2 (ref. 7) and can therefore be expected to be an efficient catalyst for CO oxidation.8–10Recently, progress has been made in Pd/CeO2 systems with different structures, and several preparation methods have been reported, such as wet impregnation, coprecipitation, and photoassisted deposition.11–13 Though it is generally acknowledged that a higher Pd content contributes to stronger catalytic activity, Pd/CeO2 materials fabricated in most of these cases possess a low content of Pd (0.081–11 wt%) as the agglomeration of Pd nanoparticles (NPs) might become more serious with the increase of the Pd content.14–18 Furthermore, to the best of our knowledge, there is still no effective method to acquire Pd/CeO2 materials with a high content and dispersion of Pd NPs, which greatly limits their activity. Thus, development of new synthesis approaches that can achieve a high content and uniform dispersion of Pd still represents a major challenge.
Metal–organic frameworks (MOFs), a new type of porous material that is constructed by self-assembly of inorganic metal centers and organic ligands,19,20 have recently attracted tremendous attention as self-sacrificial templates to derive promising metal (M)/metal oxide (MOx) composites,21,22 owing to the following unique advantages: (1) the metal centers of MOFs are periodically distributed throughout the whole framework and separated by organic ligands, which can effectively impede further agglomeration of metal centers and ensure the highly uniform distribution of NPs. (2) The type of atoms that coordinate with metal centers can be controlled by choosing appropriate ligands. There is a great chance that the metal centers connected to oxygen (O) atoms are transformed into MOx NPs, while the metal centers bonding with nitrogen (N) atoms are turned into M NPs.23 (3) Due to the high metal content in MOF structures, more Pd can be retained in the final products. (4) The carbon matrix derived from organic ligands usually exhibits a partly graphitized structure and can be doped with N atoms, which would facilitate the synergistic interactions between M and MOx NPs, ensuring the delivery of their catalytic activities and good stability. Hence, MOFs are promising precursors for the preparation of Pd/CeO2-based catalysts. However, to the best of our knowledge, targeted design and synthesis of proper MOF materials for deriving new Pd/CeO2 catalytic systems has not yet been reported.
Herein, we present a new Pd/Ce-MOF assembled from Pd and Ce ions and 2,2′-bipyridine-4,4′-dicarboxylic acid (4,4′-H2bpydc) as a sacrificial template for synthesizing a novel Pd/CeO2 catalytic system for CO oxidation. This new Pd/CeO2 catalytic system obtained by thermal annealing in a hydrogen/argon mixture atmosphere at 700 °C (Pd/CeO2-NC-700) is composed of Pd and CeO2 NPs embedded in N-doped carbon and possesses a high Pd loading of 33.7 wt%, and it exhibits excellent catalytic activity and stability for CO oxidation (T100 = 89 °C, 100% conversion temperature). By combining structural characterization with density functional theory (DFT) calculations, two possible CO oxidation pathways are revealed: the TPB and TOP pathways, which provide an in-depth theoretical understanding of the catalytic process. Thus, this work provides a new insight into the development of novel efficient catalysts for CO oxidation based on MOFs.
Experimental
Materials and reagents
All reagents were purchased from commercial suppliers and used without further purification. 4,4′-H2bpydc, palladium acetate and cerium(III) nitrate hexahydrate were purchased from Energy (China). Acetonitrile and 1,4-dioxane were supplied by Tianjin Fuyu Fine Chemical Co. Ltd.Synthesis of Pd/Ce-MOF
Pd/Ce-MOF was prepared by the solvothermal reaction. A mixture of 4,4′-H2bpydc (0.1 mmol, 0.0244 g), Pd(OAc)2 (0.1 mmol, 0.0225 g), and Ce(NO3)3·6H2O (0.433 mmol, 0.1880 g) was ultrasonically dissolved in a solution of acetonitrile (CH3CN) and H2O (5.5 mL, v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Then, the solution was transferred into a 10 mL vial, which was heated at 90 °C for 72 h. After cooling down to room temperature, orange orthorhombic prismatic crystals were obtained with a yield of 85% based on Pd. The solid product was washed thoroughly with a solution of CH3CN and H2O (v/v = 1:1) and dried in air at room temperature.
:1). Then, the solution was transferred into a 10 mL vial, which was heated at 90 °C for 72 h. After cooling down to room temperature, orange orthorhombic prismatic crystals were obtained with a yield of 85% based on Pd. The solid product was washed thoroughly with a solution of CH3CN and H2O (v/v = 1:1) and dried in air at room temperature.
Synthesis of Pd/CeO2@NC catalysts
Thermal annealing of the as-synthesized Pd/Ce-MOF was conducted at 400, 500, 600, 700, and 800 °C in a hydrogen/argon mixture atmosphere (H2, 5 vol%) for 6 hours. The obtained samples were denoted as Pd/CeO2@NC-X (where X represents the heating temperature).Catalytic CO oxidation
The CO oxidation activities of Pd/CeO2@NC-X were measured in a plug flow reactor using 50 mg of sieved (20–40 mesh) catalysts in a reactant atmosphere of 1 vol% CO/20 vol% O2/79 vol% N2 (from Deyang Gas Co., Jinan, 99.997% purity) with a flow rate of 67 mL min−1, corresponding to a space velocity of 80000 mL h−1 gcat−1. Prior to the measurement, the catalysts were treated with synthetic air (21 vol% O2/79 vol% N2) at 300 °C for 30 min. After this, the reactor was cooled down to room temperature under a flow of pure N2 gas. The catalytic tests were carried out in the reactant atmosphere by increasing the temperature of catalysts from room temperature to 300 °C (5 °C min−1). The outlet gas compositions of CO and CO2 were monitored online by nondispersive IR spectroscopy (Gasboard 3500, Wuhan Sifang Company, China).
Characterization
Infrared (IR) spectroscopy spectra were collected on a Nicolet 330 FTIR spectrometer within the 4000–500 cm−1 region. Elemental analyses (C, H, and N) were conducted on a PerkinElmer 240 elemental analyzer. Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA under a N2 flow by heating from room temperature to 900 °C (10 °C min−1). Powder X-ray diffraction (PXRD) measurements were carried out on an analytical X-Pert pro diffractometer with Cu-Kα radiation (γ = 1.5478 Å). Surface area and pore size distributions of the catalysts were determined using nitrogen adsorption–desorption isotherms obtained on a Micromeritics ASAP 2020 surface area analyzer at 77 K. The Pd contents were determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corp.). Scanning electron microscopy (SEM) images were obtained using a JEOL JSM-7600 field-emission SEM with an accelerating voltage of 5 kV. TEM images were collected on a FEI Tecnai F20 microscope operating at 200 kV. X-ray photoelectron spectroscopy (XPS) analysis was performed on an Axis Ultra XPS spectrometer (Kratos, U.K.) with 225 W of Al Kα radiation. The Raman spectrum was collected via a HORIBA Evolution Raman microscope using 532 nm incident wavelength.Models and methods of DFT
All calculations were performed using the projector augmented wave method (PAW)24 as implemented in the Vienna ab initio simulation package (VASP).25 Considering the exchange correlation interactions in the Kohn–Sham equations, the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) functionals was applied.26 We chose a cutoff energy for the plane wave basis set of 400 eV. Brillouin zone integration was performed using a 2 × 2 × 1 Gamma grid for each surface.For the calculations of Pd modified CeO2(111), we used a (3 × 3) supercell periodic slab with two O–Ce–O triple-layers. The bottom O–Ce–O triple-layer was fixed and only the top layer was relaxed when performing the geometric optimization.
The vacuum layer was set to 15 Å to avoid the interactions between replicated models. In order to determine the transition state (TS) and activation energies, the climbing-image nudged elastic band (CI-NEB)27 method was applied. Besides, the vibrational mode was used to verify the accuracy of the transition states.
The isolated CO and O2 molecules in the gas phase were optimized in a vacuum cube.
The adsorption energies were calculated using the following relationship:
| ΔEads = Eads/sub − Eads − Esub |
where Evac/sub, EO2 and Esub denote the total energy of the substrate with an oxygen vacancy, an oxygen molecule in the gas phase and the stoichiometric substrate, respectively.
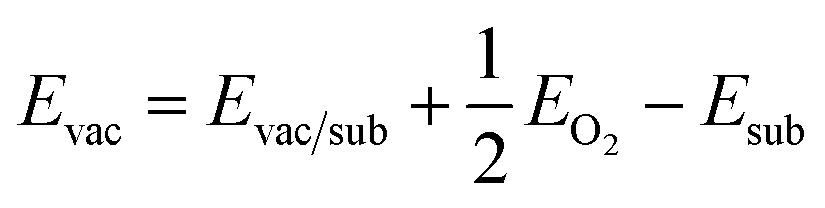
Results and discussion
The structure of Pd/Ce-MOF was resolved by single crystal X-ray diffraction (Fig. S1, Tables S1 and S2†), which revealed that the Pd and Ce centers are interconnected by 4,4′-bpydc2− molecules to form one-dimensional chains that are constructed into a three-dimensional framework by intermolecular force. The powder X-ray diffraction (PXRD) pattern (Fig. S2†) and Fourier transform infrared (FTIR) spectrum (Fig. S3†) indicate the successful synthesis of Pd/Ce-MOF with high crystallinity and phase purity. From thermogravimetric analysis (TGA), we observe that the Pd/Ce-MOF structure can remain stable up to 330 °C. The following weight loss at higher temperature is due to the collapse of the MOF structure and the decomposition of the organic ligands (Fig. S4†). Based on these results, an appropriate range of thermal annealing temperature from 400 to 800 °C was selected. Under a hydrogen/argon mixture atmosphere (H2, 5 vol%), the Pd/CeMOF crystals were transformed into Pd/CeO2@NC-X (where X represents the heating temperature) catalysts (Scheme 1).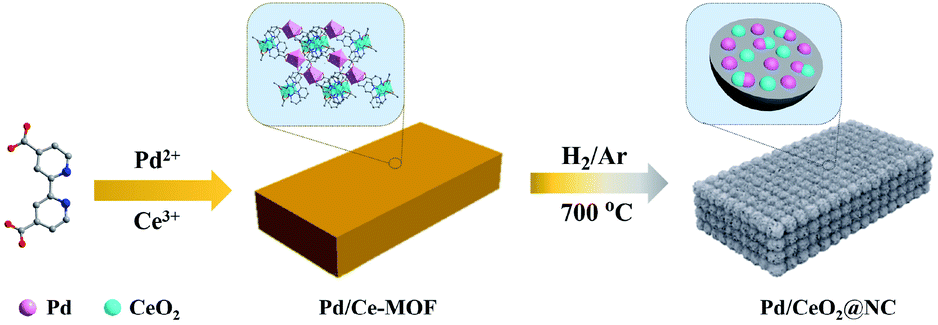 | ||
| Scheme 1 Schematic illustration of the preparation of the Pd/CeO2@NC-700 catalyst. | ||
As revealed in the SEM image in Fig. 1a, the as-synthesized Pd/Ce-MOF crystals present irregular bulk morphology. The inset optical picture also confirms the good crystallinity of the MOF crystal. After the annealing treatment at 700 °C in a hydrogen/argon atmosphere, the sample is denoted as Pd/CeO2@NC-700 and retains the original MOF crystal shape well (Fig. 1b). More structural details of Pd/CeO2@NC-700 are shown in SEM images with higher magnification (Fig. 1c–e). It can be observed that these blocks are composed of abundant uniform sphere-like nanostructures with diameters of 70–100 nm. By TEM characterization, these sphere-like nanostructures are confirmed to exist throughout the whole bulk (Fig. 1f). The high-resolution TEM image (Fig. 1g) and selected area electron diffraction pattern (Fig. 1h) verify that the sphere-like nanostructure consists of Pd and CeO2 NPs as well as carbon species derived from organic ligands. As shown, the lattice fringes of about 0.196 and 0.221 nm agree well with the (200) and (111) crystal planes of Pd metal, while the lattice fringe with an interplanar distance of 0.310 nm can be ascribed to the (111) plane of CeO2. The Pd and CeO2 phase are highly dispersed in Pd/CeO2@NC-700 and simultaneously closely connected with each other as shown by the red circles marked in Fig. 1g, which has been proved by DFT calculation to be important in facilitating the catalytic activity for CO oxidation. The elemental mapping images of Pd/CeO2@NC-700 further confirm the uniform distribution of Pd, Ce, C, N, and O inherited from the MOF precursor (Fig. 1i–n).
 | ||
| Fig. 1 (a) SEM image of the Pd/Ce-MOF (inset: optical photomicrograph of a Pd/Ce-MOF crystal); (b–e) SEM images, (f) TEM image, (g) HRTEM image and (h) selected area electron diffraction pattern of Pd/CeO2@NC-700; (i–n) elemental mapping images of Pd, Ce, C, N, and O distributions in Pd/CeO2@NC-700. | ||
TEM characterization of Pd/CeO2@NC-600 and Pd/CeO2@NC-800 was also performed for comparison and the TEM images are presented in Fig. S5.† Similar to Pd/CeO2@NC-700, the particles of Pd and CeO2 in Pd/CeO2@NC-600 are closely connected and their lattice fringes can be clearly recognized as Pd (111) and CeO2 (111). For the Pd/CeO2@NC-800 sample, the particles of Pd and CeO2 are still closely connected, but no clear lattice fringes of CeO2 can be observed, indicating its amorphous structure at higher temperature. Furthermore, as illustrated in the marked red circle in Fig. S5b,† some of the lattice fringes of Pd cannot be distinguished. This indicates that the transformation of ceria from the crystalline to amorphous phase affects the long-range order of the Pd crystal as well, which can be further proved by the XRD and XPS results.
Fig. 2a shows the PXRD patterns of the Pd/CeO2@NC-X catalysts obtained from 400 to 800 °C. The peaks present at 2θ values of 40.1°, 46.7°, and 68.1° in the pattern of Pd/CeO2@NC-400 correspond to the (111), (200) and (220) crystal planes of Pd metal (PDF no. 46-1043). There is no CeO2 phase formed at the lower annealing temperature of 400 °C. When the temperature reaches 500 °C, a series of new diffraction peaks appear due to the formation of the CeO2 phase. The peaks appearing at 2θ values of 28.6°, 33.1°, 47.5°, 56.3°, 59.1°, and 69.4° are associated with the (111), (200), (220), (311), (222), and (400) crystal planes of CeO2 (PDF no. 34-0394). From 500 to 700 °C, the intensity of the diffraction peaks gradually increases with the temperature, and the full width at half maximum (FWHM) gradually decreases, indicating the higher crystallinity of Pd and the CeO2 phase at a higher temperature. No extra peaks of impurities are found, suggesting a complete conversion of the Pd/Ce-MOF to Pd/CeO2@NC-X materials. When the annealing temperature is as high as 800 °C, the crystallinity of the product decreases remarkably and only large and broad peaks of Pd can be clearly distinguished, which indicates that the crystalline ceria was transformed to amorphous ceria at higher temperature. It should be mentioned that the larger Pd nanoparticle sizes of Pd/CeO2@NC-600 (22.8 nm) and 700 (41.2 nm) calculated from XRD data are due to the inevitable formation of a few larger Pd particles at 600 and 700 °C. As for Pd/CeO2@NC-800, the XRD-based Pd size is 3.9 nm and no similar phenomenon is observed. The impediment to the further growth of Pd particles can be attributed to the above discussed transformation of CeO2 from the crystalline to amorphous phase. Thus, for Pd/CeO2@NC-700, the presence of both the crystalline Pd and CeO2 phase and the largest surface area confirmed using N2 adsorption isotherms (Fig. S6†) are believed to contribute to its high catalytic activity for CO oxidation.
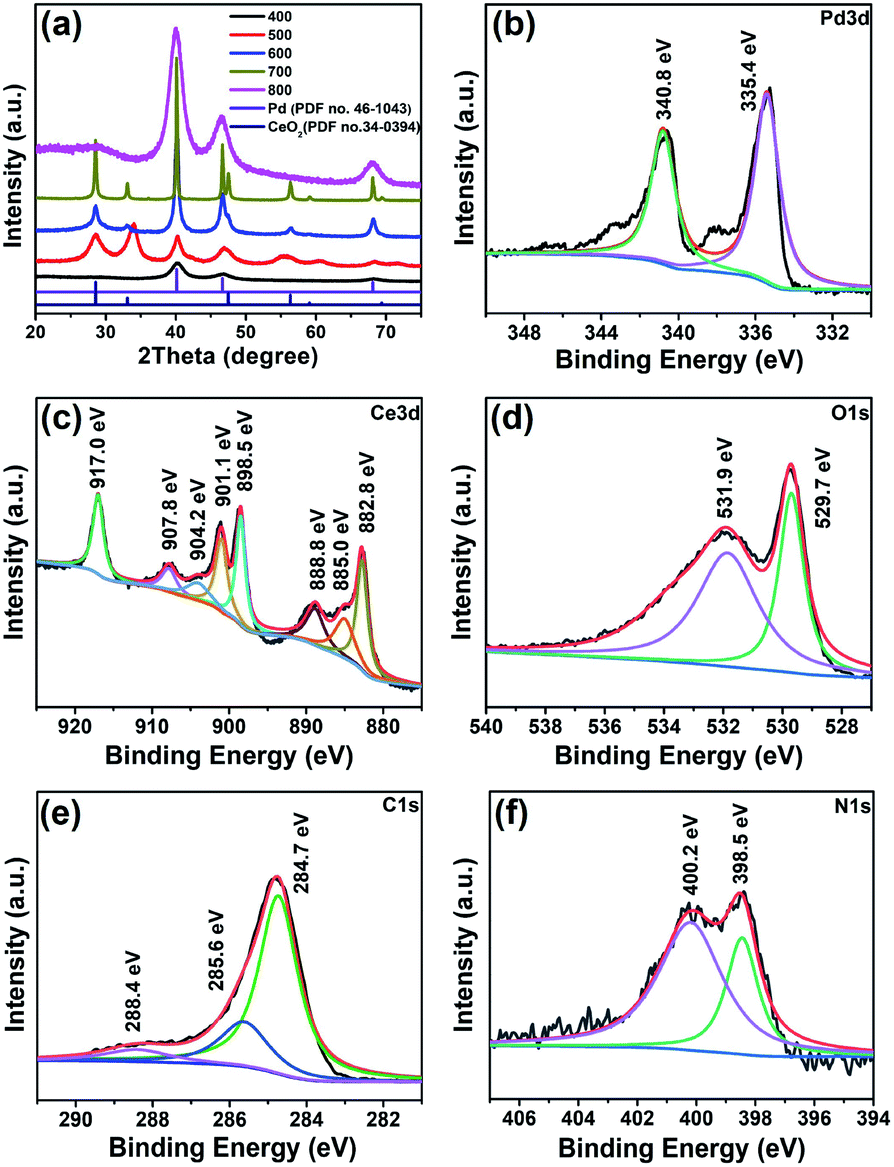 | ||
| Fig. 2 (a) PXRD patterns of the Pd/CeO2@NC-X catalysts obtained at different temperatures and XPS spectra of (b) Pd 3d, (c) Ce 3d, (d) O 1s, (e) C 1s and (f) N 1s for Pd/CeO2@NC-700. | ||
The Pd/CeO2@NC-700 sample was also investigated by X-ray photoelectron spectroscopy (XPS) to probe the alterations of the surface electronic structure (Fig. 2b–f). The presence of elemental Ce, Pd, C, N, and O was verified. The Pd 3d XPS spectrum (Fig. 2b) demonstrates that the Pd phase in Pd/CeO2@NC-700 is zero-valent with binding energies of Pd 3d5/2 at 335.4 eV and Pd 3d3/2 at 340.8 eV, respectively, indicating complete reduction of the Pd2+ ions after annealing at 700 °C. The Ce 3d XPS spectrum (Fig. 2c) can be deconvoluted into three kinds of peaks such as those associated with Ce4+ (882.8 and 901.1 eV) and Ce3+ (885.0 and 904.2 eV) and satellite peaks (888.8, 898.5, 907.8 and 917.0 eV), which also indicates that the main valence state of Ce in Pd/CeO2@NC-700 is +4 according to the integral area ratio of the peaks. In the O 1s spectrum (Fig. 2d), the peaks at 529.7 and 531.9 eV are ascribed to the lattice oxygen of CeO2 and the surface adsorbed oxygen species, respectively. As for the C 1s spectrum (Fig. 2e), three peaks at 284.7, 285.6 and 288.4 eV are deconvoluted corresponding to C–C (sp2), C–N and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. In the N 1s XPS spectrum (Fig. 2f), two fitted peaks around 398.5 and 400.2 eV can be respectively assigned to pyridinic and pyrrolic N species. The presence of N doped carbon and its partially graphitized structure demonstrated by Raman characterization (Fig. S7†) are speculated to play an important role in maintaining better activity and stability of Pd/CeO2@NC-700 during the catalytic process.
O, respectively. In the N 1s XPS spectrum (Fig. 2f), two fitted peaks around 398.5 and 400.2 eV can be respectively assigned to pyridinic and pyrrolic N species. The presence of N doped carbon and its partially graphitized structure demonstrated by Raman characterization (Fig. S7†) are speculated to play an important role in maintaining better activity and stability of Pd/CeO2@NC-700 during the catalytic process.
The XPS measurements of Pd/CeO2@NC-600 and Pd/CeO2@NC-800 were also carried out for comparison. As illustrated in Fig. S8,† Pd/CeO2@NC-600 shows similar XPS spectra of Pd 3d, Ce 3d, C 1s and N 1s to those of Pd/CeO2@NC-700. However, a difference is observed between the spectra of O 1s. The peak assigned to lattice oxygen increases significantly from 600 to 700 °C, indicating the increase of the surface atomic ratio of lattice oxygen in CeO2, which is in accordance with the crystallinity enhancement of CeO2 shown in the XRD pattern of Pd/CeO2@NC-700. When the annealing temperature increases to 800 °C, the peaks associated with Ce4+ (882.6 and 901.0 eV) are greatly reduced, and the peaks originating from Ce3+ (885.4 and 904.1 eV) increase remarkably, suggesting the reduction of Ce4+ to Ce3+ in Pd/CeO2@NC-800. Furthermore, it is also observed that the peak of lattice oxygen in the O 1s spectrum of Pd/CeO2@NC-800 almost disappears, which reveals the transformation of crystalline ceria to amorphous ceria at higher temperature. This is the reason why Pd/CeO2@NC-800 shows no XRD diffraction peaks of CeO2, consistent with the TEM results.
The amount of Pd in Pd/CeO2@NC-700 is determined by ICPAES (Table S3†) and is calculated to be 33.7 wt%. This is higher than that of all the reported M/CeO2 (M = Pd, Pt or Au) composites (Table 1) to the best of our knowledge, which ensures the good catalytic activity of the Pd/CeO2@NC-700 catalyst. It also higher than the theoretical value of Pd (20.6 wt%) in the crystal structure of Pd/Ce-MOF (Table S3†). This can be attributed to the mass loss during the thermal annealing process.
| Materials | Content of the catalyst (mg) | T 100 (°C) | Flow rate (mL min−1) | Pd loading (wt%) | Activity (mol gM−1 h−1) | Ref. |
|---|---|---|---|---|---|---|
| Pd@CeO2 | 600 | 95 | 100 (1% CO/5% O2/N2) | 11.0 | 6.8 | 8 |
| h-Pd–CeO2 | 10 | 150 | 50 (1% CO/20% O2/Ar) | 2.0 | 63.9 | 9 |
| Pd@CeO2/Al2O3 | 200 | 110 | 50 (1% CO/20% O2/N2) | 11.0 | 30.4 | 10 |
| Pt@CeO2 | 8 | 135 | 30 (1% CO/20% O2/N2) | 8.7 | 19.2 | 33 |
| Au@CeO2 | 200 | 155 | 50 (1% CO/1.6% O2/He) | 1.0 | 11.2 | 34 |
| Pt–CeO2 | 12.5 | 140 | 50 (1% CO/20% O2/He) | 7.7 | 23.2 | 35 |
| Au/scCeO2 | 15 | — | 22.5(1% CO/20% O2/N2) | 3.7 | 19.8 | 36 |
| Pd/CeO2@NC-700 | 50 | 89 | 67(1% CO/20% O2/N2) | 33.7 | 31.9 | This work |
Due to the high Pd content, the Pd/CeO2@NC-700 catalyst is expected to show Pd-like catalytic activity for exhaust emission control. And therefore, its catalytic activity and stability for CO oxidation to CO2 were evaluated.28–30 The CO conversion was calculated according to the equation: CO conversion = COreaction/COinput.31,32 As shown in Fig. 3, the five samples of Pd/CeO2@NC-400, 500, 600, 700 and 800 exhibit a similar initial conversion of 20% at room temperature. With the increase of temperature, the conversion increases rapidly. The T100 (100% conversion temperature) of the five samples follows the trend of Pd/CeO2@NC-800 (137 °C) < Pd/CeO2@NC-400 (121 °C) < Pd/CeO2@NC-500 (114 °C) < Pd/CeO2@NC-600 (108 °C) < Pd/CeO2@NC-700 (89 °C) (Fig. 3a). Apparently, the annealing temperature has a big impact on their catalytic activities. This is due to the structural differences brought about by temperature, which has also been confirmed by the XRD results.37–40 The sample obtained at 700 °C exhibits much higher catalytic activity than its counterparts mainly owing to its higher crystallinity of Pd and CeO2, thus enhancing the catalytic activity for CO oxidation.41,42 Compared with other reported CeO2-based composites, the Pd/CeO2@NC-700 catalyst exhibits better activity with a lower T100 of only 89 °C (Table 1).
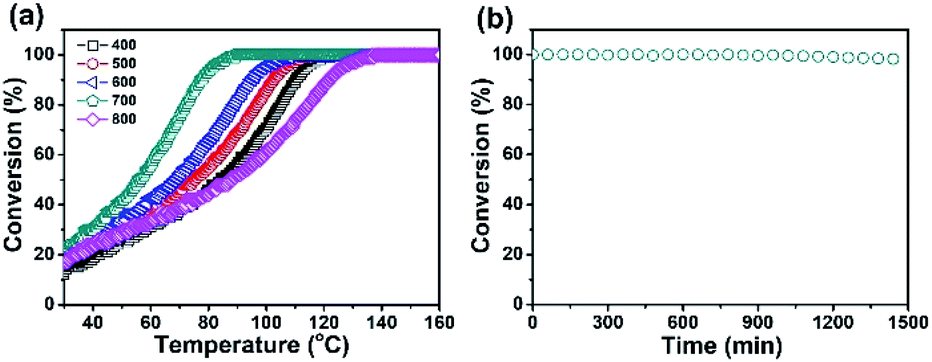 | ||
| Fig. 3 (a) CO conversion by using Pd/CeO2@NC-X catalysts and (b) catalytic stability of the Pd/CeO2@NC-700 catalyst in the CO oxidation reaction (t = 90 °C). | ||
Durability is another pivotal concern for evaluating a catalyst. Thus, the cycling test of Pd/CeO2@NC-700 was carried out to study its stability. As shown in Fig. 3b, the Pd/CeO2@NC-700 catalyst shows excellent catalytic stability at 90 °C for more than 24 h and no obvious reduction of the conversion of CO oxidation reaction is observed after this process. From the TEM images obtained after the stability test (Fig. S10†), it can be seen that the Pd/CeO2@NC-700 catalyst retains its original nanostructure, indicating its good catalytic performance under long-term cycling conditions.
It is apparent that the coupling of Pd and the CeO2 phase is crucial for synergistically boosting the CO oxidation performance of Pd/CeO2@NC-700. Thus, we also carried out systematic DFT calculations to provide an in-depth theoretical understanding of the catalytic process. Since the majority of Ce ions have a valency of +4 in the Pd/CeO2@NC-700 catalyst, the density of oxygen vacancies would be relatively low according to the charge-compensation mechanism.43 Furthermore, according to Fig. 3b, the Pd/CeO2@NC-700 catalyst shows excellent catalytic stability, which indicates that it is difficult to produce oxygen vacancies on its surface.44–46 In other words, the Pd/CeO2@NC-700 surface can barely activate the adsorbed CO by the Mars-van Krevelen (M-vK) mechanism,47 in which the lattice O atom directly interacts with CO to form CO2 leaving an oxygen vacancy.48 Additionally, the formation energy of the oxygen vacancy (2.308 eV) is higher than the dissociation energy of adsorbed O2 (1.745 eV). Therefore, we constructed the defect-free CeO2 (111) surface as the support of the catalyst.
The surface model is assembled with a Pd8 cluster placed on the stoichiometric CeO2 (111) surface. Before this, the free Pd8 cluster structure was optimized in a vacuum and the lowest energy of Pd8 was adopted. Then the study of the CO oxidation reaction was carried out on Pd8/CeO2 (111), which follows the well-accepted Langmuir–Hinshelwood mechanism that molecules must first be adsorbed on the ceria surface before undergoing oxidation. We found that a stable chemisorption configuration can be formed between the CO molecule and Pd8 cluster supported on CeO2. The lattice O atoms of ceria are not involved in CO oxidation as we have confirmed that the stable adsorption sites for O2 molecules are on the Pd8/CeO2 (111) surface. Hence, the adsorbed O2 on the surface can easily be activated and dissociated.
Due to the divergence in stable adsorption sites on the Pd8/CeO2 (111) surface, we explored two alternative reaction pathways for CO oxidation (Fig. 4), which do not follow the M-vK mechanism according to the above discussion. It is worth mentioning that there is a competitive adsorption relationship between CO and O2, i.e. the CO and O2 molecules cannot be stably adsorbed on the same facet of the Pd8 cluster. The CO molecule prefers to adsorb on the Pd8 cluster and coordinate with two Pd atoms with adsorption energies of −1.13 and −2.04 eV in TPB (three phase boundary)49 and TOP (both CO and O2 molecules are adsorbed on top of the Pd8 cluster), respectively. The bond length of the adsorbed CO molecule is 1.18 Å in both the pathways, slightly longer than that of CO in the vacuum phase (1.14 Å), which is attributed to the charge transfer from C to the Pd8 cluster. These indicate that CO can interact with the Pd8 cluster by forming a chemical bond.
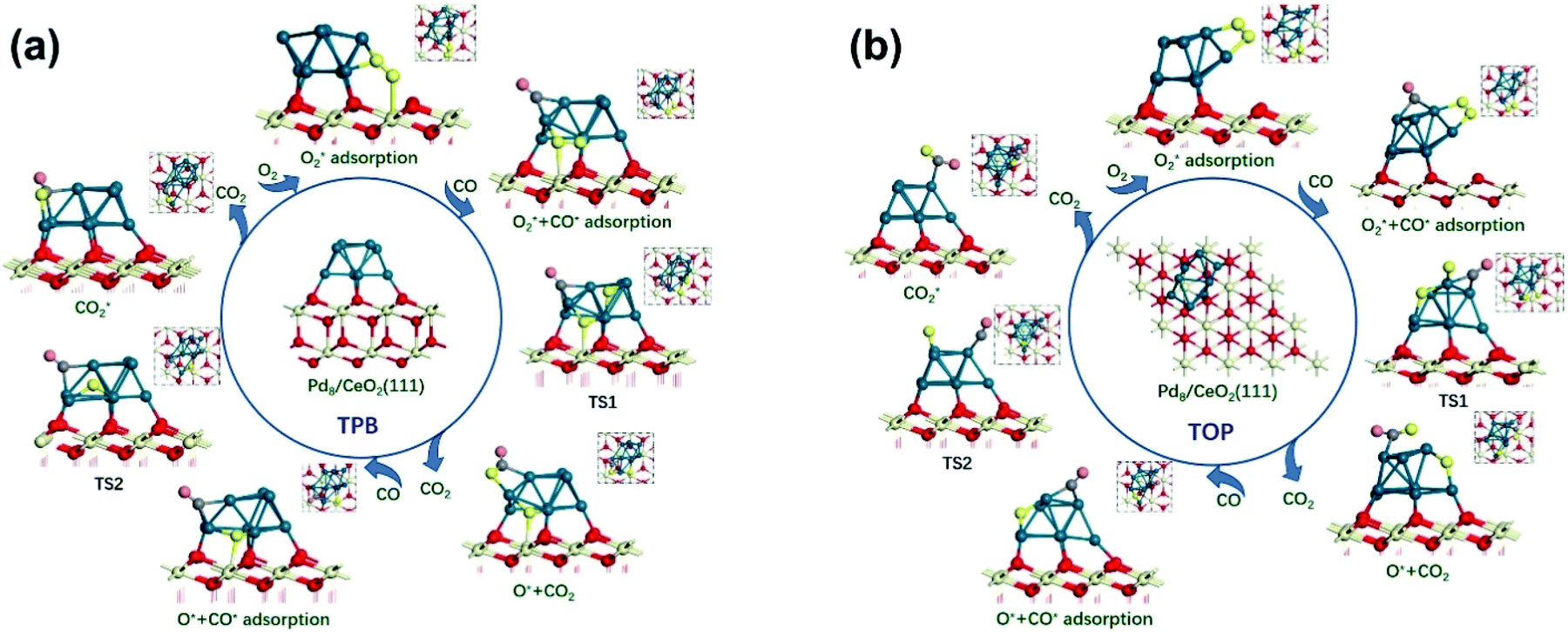 | ||
| Fig. 4 The reaction path for CO oxidation to CO2 on Pd/CeO2 (111) surfaces: (a) the TPB pathway and (b) the TOP pathway. Atom key: Pd (dark green), Ce (light yellow), C (gray), and O (red, yellow, and rose). | ||
The formation of the bent O–C–O type from CO and O* dissociated from O2 is an exothermic elementary step and requires energy barriers of 1.38 and 1.05 eV in the TPB and TOP pathway (Fig. 5), respectively. In the TPB pathway, the most energy-consuming step is the migration of O* from the Pd8 cluster to CO. It is important to note that the migration of O* is affected by the morphology of the Pd8 cluster. The topmost Pd8 layer consists of four Pd atoms before the CO oxidation reaction. The co-adsorption of CO and O2 affected the structure of the topmost Pd8 layer, leading to one Pd atom spontaneously interacting with a surface lattice O atom. Because of the two different molecular adsorption configurations of the two pathways, the impact of structural deformation on the reaction is also different. Therefore, the Pd8 cluster undergoes surface reconstruction while the O* diffuses on the surface, which entails different activation energy consumption in the two pathways. Once the bent O–C–O intermediate is formed, the valence electrons of C completely interact with O* and are no longer bonded to the Pd8 cluster, leading to the O–C–O intermediate spontaneously desorbing to the gas phase without an energy barrier.
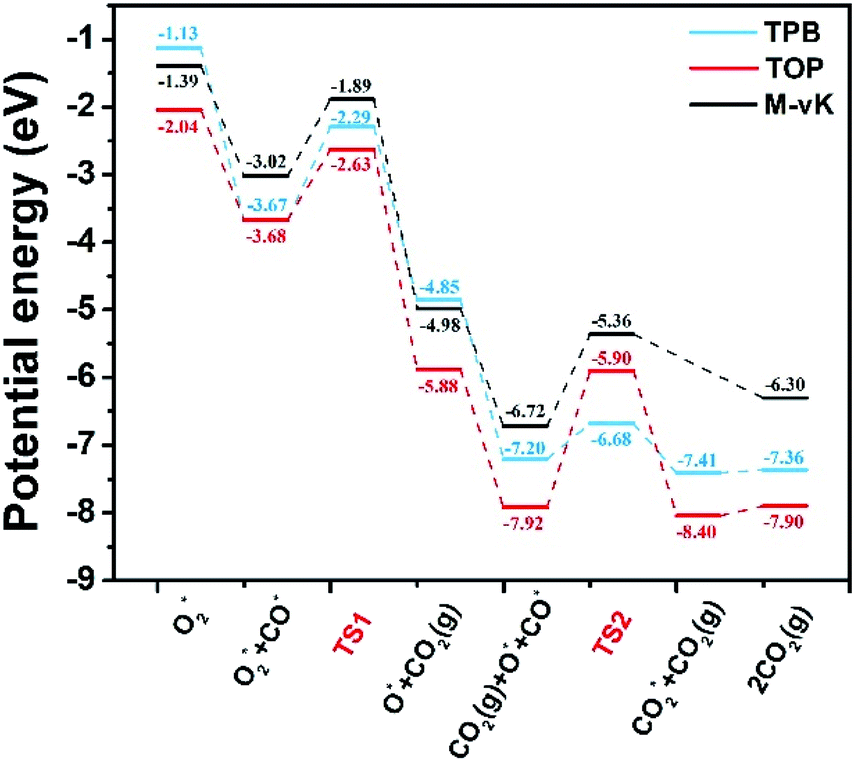 | ||
| Fig. 5 Potential energy diagrams for CO oxidation. The elementary reaction barriers are given in eV. M-vK represents the CO oxidation pathway on the reduced Pd8/CeO2 surface (the well accepted Mars-van Krevelen mechanism50). | ||
It can be determined that the most stable adsorption sites of subsequent CO bonding to the Pd8 cluster are affected by O* dissociated from O2. In the TPB pathway, the O* spontaneously forms an ionic bond with Ce4+, since the Ce4+ on the surface is unsaturated. However, if O* is adsorbed on the bridge sites of the Pd8 cluster, it would lead to a situation where the subsequent CO cannot be adsorbed on the adjacent facets of the Pd8 cluster.
Understandably, the activation energy of TS2 in the TPB pathway is 0.52 eV, which is significantly lower than that in the TOP and M-vK pathways since the dissociated O* can easily be activated in the TPB area. However, in the TOP pathway, the O–CO formation requires 2.02 eV to overcome the energy barrier which is significantly higher than that for the first CO2 production. This is attributed to the specific diffusion route of O*. Obviously, the most difficult step in the O* diffusion process is the rotation of the O–Pd bond, which is caused by the reconstruction of the Pd8 cluster. The O* atom has to climb the facet to combine with CO. A complete cycle of the CO oxidation reaction contains two subsequent CO adsorption and oxidation steps. However, by comparing the final states of the two pathways, it can be observed that there is a difference in the energy of the solid surface after the removal of CO2 molecules. This is because the Pd8 cluster undergoes surface reconstruction after the dissociated O* bonds with Ce4+ in the TPB pathway, and its configuration is slightly different from the initial state. Nevertheless, in the TOP pathway, the Pd8 cluster can always maintain the most stable configuration in which five Pd atoms strongly interact with lattice oxygen of ceria. The accompanying disadvantage is that this stable cluster configuration is difficult to relax, which hinders migration of dissociated O*.
Based on the DFT calculations, two possible CO oxidation pathways are determined on the Pd/CeO2@NC-700 surface. Compared with the M-vK mechanism, the adsorbed O2 directly dissociates to O* and activates a CO* molecule rather than a lattice O atom. The activation energy of the M-vK mechanism in two subsequent CO oxidation reactions is slightly higher than that of the TPB and TOP pathways because creating an oxygen vacancy in this surface is not energetically favorable. The transfer of O* between Pd and Ce (TPB) or Pd and Pd (TOP) facilitates the formation of 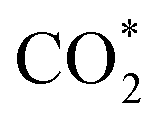 intermediates and finally results in the production of CO2.
intermediates and finally results in the production of CO2.
All the above results suggest that the as-synthesized Pd/CeO2@NC-700 material is a promising alternative catalyst for CO oxidation. There are three reasons for its excellent catalytic activity and stability: (1) the high content of Pd (33.7 wt%) inherited from the MOF structure accelerates the reaction dynamics; (2) the coupling of Pd and the CeO2 phase synergistically boosts the catalytic performance; (3) the N-doped and partly graphitized carbon matrix ensures the delivery of their catalytic activities and the good stability. These results give way to great expectations for future MOF-derived catalysts for efficient CO oxidation.
Conclusions
In summary, we have successfully designed and synthesized a series of Pd/CeO2@NC catalysts by one-step thermal annealing of a new Pd/Ce-MOF precursor, after which the Pd2+ and Ce3+ centers in Pd/Ce-MOF were completely transformed to Pd and CeO2 NPs, respectively. In the sample obtained at 700 °C (Pd/CeO2@NC-700), the Pd loading is as high as 33.7 wt%. The coupling of Pd and the CeO2 phase synergistically boosts the CO oxidation performance of Pd/CeO2@NC-700. The 100% conversion temperature of CO is as low as 89 °C with simultaneously excellent long-term stability. DFT calculations reveal two possible CO oxidation pathways: the TPB and TOP pathways. This confirms that the transfer of dissociated O* between Pd and Ce (TPB) or Pd and Pd (TOP) facilitates the formation of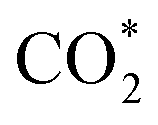 intermediates and finally results in the production of CO2. The results demonstrate that using MOFs as self-sacrificial templates provides a new insight into fabricating novel nanocatalysts for efficient CO oxidation.
intermediates and finally results in the production of CO2. The results demonstrate that using MOFs as self-sacrificial templates provides a new insight into fabricating novel nanocatalysts for efficient CO oxidation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Grant Nos. 21571187, 21601205, and 21875285), the Taishan Scholar Foundation (ts201511019), and the Fundamental Research Funds for the Central Universities (18CX02047A).Notes and references
- T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- H. Guo, Y. He, Y. Wang, L. Liu, X. Yang, S. Wang, Z. Huang and Q. Wei, J. Mater. Chem. A, 2013, 1, 7494–7499 RSC.
- W. Chen, T. Li, Q. Hu, C. Li, Y. Li and H. Guo, Catal. Commun., 2015, 64, 62–65 CrossRef CAS.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- E. Sasmaz, C. Wang, M. J. Lance and J. Lauterbach, J. Mater. Chem. A, 2017, 5, 12998–13008 RSC.
- Y. Dai, V. P. Kumar, C. Zhu, M. J. MacLachlan, K. J. Smith and M. O. Wolf, ACS Appl. Mater. Interfaces, 2018, 10, 477–487 CrossRef CAS PubMed.
- J. Ye, Y. Xia, D.-G. Cheng, F. Chen and X. Zhan, Int. J. Hydrogen Energy, 2019, 44, 17985–17994 CrossRef CAS.
- J.-Y. Luo, M. Meng, H. Xian, Y.-B. Tu, X.-G. Li and T. Ding, Catal. Lett., 2009, 133, 328–333 CrossRef CAS.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- G. Li, L. Li, D. Jiang, Y. Li and J. Shi, Nanoscale, 2015, 7, 5691–5698 RSC.
- L. Adijanto, D. A. Bennett, C. Chen, A. S. Yu, M. Cargnello, P. Fornasiero, R. J. Gorte and J. M. Vohs, Nano Lett., 2013, 13, 2252–2257 CrossRef CAS PubMed.
- N. L. Wieder, M. Cargnello, K. Bakhmutsky, T. Montini, P. Fornasiero and R. J. Gorte, J. Phys. Chem. C, 2011, 115, 915–919 CrossRef CAS.
- X. Wang, Y. Zhang, S. Song, X. Yang, Z. Wang, R. Jin and H. Zhang, Angew. Chem., Int. Ed., 2016, 55, 4542–4546 CrossRef CAS PubMed.
- X. Wang, D. Liu, J. Li, J. Zhen, F. Wang and H. Zhang, Chem. Sci., 2015, 6, 2877–2884 RSC.
- C. Du, Y. Guo, Y. Guo, X.-Q. Gong and G. Lu, J. Mater. Chem. A, 2015, 3, 23230–23239 RSC.
- M. Cargnello, N. L. Wieder, T. Montini, R. J. Gorte and P. Fornasiero, J. Am. Chem. Soc., 2010, 132, 1402–1409 CrossRef CAS PubMed.
- H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. D. Benedetto, F. DÏAcapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55, 6004–6007 CrossRef CAS PubMed.
- S. Zhang, C.-R. Chang, Z.-Q. Huang, J. Li, Z. Wu, Y. Ma, Z. Zhang, Y. Wang and Y. Qu, J. Am. Chem. Soc., 2016, 138, 2629–2637 CrossRef CAS PubMed.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H.-C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef CAS PubMed.
- G. Liu, Y. Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231–6234 CrossRef CAS PubMed.
- J. Chen, G. Xia, P. Jiang, Y. Yang, R. Li, R. Shi, J. Su and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13378–13383 CrossRef CAS PubMed.
- Y. Wang, S. Yuan, Z. Hu, T. Kundu, J. Zhang, S. B. Peh, Y. Cheng, J. Dong, D. Yuan, H.-C. Zhou and D. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 7118–7126 CrossRef CAS.
- Y. Qian, Q. Liu, E. Sarnello, C. Tang, M. Chng, J. Shui, T. Li, S. J. Pennycook, M. Han and D. Zhao, ACS Mater. Lett., 2019, 1, 37–43 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. JóNsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- W. Ji, Z. Xu, P. Liu, S. Zhang, W. Zhou, H. Li, T. Zhang, L. Li, X. Lu, J. Wu, W. Zhang and F. Huo, ACS Appl. Mater. Interfaces, 2017, 9, 15394–15398 CrossRef CAS PubMed.
- L.-W. Guo, P.-P. Du, X.-P. Fu, C. Ma, J. Zeng, R. Si, Y.-Y. Huang, C.-J. Jia, Y.-W. Zhang and C.-H. Yan, Nat. Commun., 2016, 7, 13481 CrossRef CAS PubMed.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- W.-W. Wang, W.-Z. Yu, P.-P. Du, H. Xu, Z. Jin, R. Si, C. Ma, S. Shi, C.-J. Jia and C.-H. Yan, ACS Catal., 2017, 7, 1313–1329 CrossRef CAS.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864–15872 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 RSC.
- H.-P. Zhou, H.-S. Wu, J. Shen, A.-X. Yin, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2010, 132, 4998–4999 CrossRef CAS PubMed.
- Z.-R. Tang, J. K. Edwards, J. K. Bartley, S. H. Taylor, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 249, 208–219 CrossRef CAS.
- W. Liu, A. F. Sarofim and M. Flytzani-stephanopoulos, Chem. Eng. Sci., 1994, 49, 4871–4888 CrossRef CAS.
- A. I. Boronin, E. M. Slavinskaya, I. G. Danilova, R. V. Gulyaev, Y. I. Amosov, P. A. Kuznetsov, I. A. Polukhina, S. V. Koscheev, V. I. Zaikovskii and A. S. Noskov, Catal. Today, 2009, 144, 201–211 CrossRef CAS.
- R. V. Gulyaev, A. I. Stadnichenko, E. M. Slavinskaya, A. S. Ivanova, S. V. Koscheev and A. I. Boronin, Appl. Catal., A, 2012, 439–440, 41–50 CrossRef CAS.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Starch, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- G. L. Markaryan, L. N. Ikryannikova, G. P. Muravieva, A. O. Turakulova, B. G. Kostyuk, E. V. Lunina, V. V. Lunin, E. Zhilinskaya and A. Aboukais, Colloids Surf., A, 1999, 151, 435–447 CrossRef CAS.
- C. Bozo, N. Guilhaume and J. M. Herrmann, J. Catal., 2001, 203, 393–406 CrossRef CAS.
- Y. Liu, L. Fan, Y. Cai, W. Zhang, B. Wang and B. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 23614–23623 CrossRef CAS PubMed.
- S. Gao, M. Cao, W. Li and R. Cao, J. Mater. Chem. A, 2014, 2, 12185–12193 RSC.
- S.-I. Yamamoto, H. Kinoshita, H. Hashimotob and Y. Nishina, Nanoscale, 2014, 6, 6501–6505 RSC.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- F. Chen, D. Liu, J. Zhang, P. Hu, X.-Q. Gong and G. Lu, Phys. Chem. Chem. Phys., 2012, 14, 16573–16580 RSC.
- G.-Q. Kong, S. Ou, C. Zou and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 19851–19857 CrossRef CAS PubMed.
- S. Wang, M. Zheng, M. Li, X. Wu and C. Xia, J. Mater. Chem. A, 2016, 4, 5745–5754 RSC.
- J.-X. Liu, Y. Su, I. A. W. Filot and E. J. M. Hensen, J. Am. Chem. Soc., 2018, 140, 4580–4587 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal data, PXRD patterns, FT-IR spectra, TGA curves, N2 adsorption–desorption isotherms, a Raman spectrum and TEM images. See DOI: 10.1039/c9na00744j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |