
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial activity of graphene oxide quantum dots: impacts of chemical reduction†
Mauricio D.
Rojas-Andrade
 a,
Tuan Anh
Nguyen
a,
William P.
Mistler
a,
Juan
Armas
a,
Jia En
Lu
a,
Graham
Roseman
a,
William R.
Hollingsworth
a,
Forrest
Nichols
a,
Glenn L.
Millhauser
a,
Alexander
Ayzner
a,
Chad
Saltikov
*b and
Shaowei
Chen
*a
a,
Tuan Anh
Nguyen
a,
William P.
Mistler
a,
Juan
Armas
a,
Jia En
Lu
a,
Graham
Roseman
a,
William R.
Hollingsworth
a,
Forrest
Nichols
a,
Glenn L.
Millhauser
a,
Alexander
Ayzner
a,
Chad
Saltikov
*b and
Shaowei
Chen
*a
aDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: shaowei@ucsc.edu
bDepartment of Microbiology and Environmental Toxicology, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: saltikov@ucsc.edu
First published on 20th January 2020
Abstract
Design and engineering of graphene-based functional nanomaterials for effective antimicrobial applications has been attracting extensive interest. In the present study, graphene oxide quantum dots (GOQDs) were prepared by chemical exfoliation of carbon fibers and exhibited apparent antimicrobial activity. Transmission electron microscopic measurements showed that the lateral length ranged from a few tens to a few hundred nanometers. Upon reduction by sodium borohydride, whereas the UV-vis absorption profile remained largely unchanged, steady-state photoluminescence measurements exhibited a marked blue-shift and increase in intensity of the emission, due to (partial) removal of phenanthroline-like structural defects within the carbon skeletons. Consistent results were obtained in Raman and time-resolved photoluminescence measurements. Interestingly, the samples exhibited apparent, but clearly different, antimicrobial activity against Staphylococcus epidermidis cells. In the dark and under photoirradiation (400 nm), the as-produced GOQDs exhibited markedly higher cytotoxicity than the chemically reduced counterparts, likely because of (i) effective removal by NaBH4 reduction of redox-active phenanthroline-like moieties that interacted with the electron-transport chain of the bacterial cells, and (ii) diminished production of hydroxyl radicals that were potent bactericidal agents after chemical reduction as a result of increased conjugation within the carbon skeletons.
Introduction
Graphene and its derivatives have gained significant attention due to their unique physical, chemical and mechanical properties that may be exploited for diverse applications. Among these, significant research efforts have been devoted to the efficacy of graphene derivatives for antimicrobial applications.1,2 Despite substantial progress, the exact mechanism of the graphene cytotoxicity has remained under active debate,3 where oxidative stress induction,4,5 protein dysfunction,6 membrane damage,7 and transcriptional arrest8 have been proposed to account for the antimicrobial activity. For instance, in a previous study,9 Biswas et al. synthesized graphene oxide nanosheets (GONS, 3 nm in length) and demonstrated their cytotoxicity towards bacterial cells, which was accounted for by the generation of reactive oxygen species (ROS) that damaged the bacterial cell membranes. Similar results were observed by Sun et al.10 who reported the antibacterial activity of 5 nm GONS in the presence of low concentrations of H2O2, and concluded that GONS catalyzed the decomposition of H2O2 into OH˙ radicals, affording the antimicrobial activity. Interestingly, such cytotoxicity may be enhanced by deliberate photoirradiation. For instance, Kuo et al.11 employed two-photon excitation to excite 7 nm GONS and damage bacterial cells. In addition, they characterized the type of ROS being generated, and found that both triplet oxygen (1O2) and superoxide anions (O2˙−) were formed upon photoexcitation. In another study,12 Perreault et al. examined the impacts of GONS size on the cytotoxicity, and argued that the mechanism of cytotoxicity varied with the GONS dimensions, where large GONS acted by wrapping around cells and starving them of their nutrients, while small GONS by causing oxidative stress that was proposed to arise from the intrinsic defects such as edges of sp2 domains and dangling bonds. This was concluded as X-ray photoelectron spectroscopy (XPS) studies did not show a significant variation of the oxygenated groups among the different sizes, but Raman measurements showed a marked increase of the intensity ratio of the D and G bands. The roles of graphene structural defect in the mechanism of cytotoxicity is further illustrated by Akhavan et al.13 who observed decreasing cytotoxicity of graphene oxide nanowalls after hydrazine reduction. This was ascribed to a significantly different composition of oxygen functional groups after chemical reduction, as manifested in XPS and Raman measurements.The observed dependence of cytotoxicity on the defect structure of graphene nanostructures is therefore both highly interesting and extremely significant, requiring a systematic investigation. In a recent study,14 oxygen functional groups were found to play a significant role in the phototoxicity of GOQDs, as a marked decrease in ROS production and bactericidal effects were observed when oxygen functional groups, particularly ketonic/carboxylic acid groups, were removed by derivatization. Although these results are promising, a thorough characterization of the defect structure of GOQDs is lacking, preventing a firm establishment of the mechanism of carbon nanostructure cytotoxicity.
By utilizing graphene oxide quantum dots (GOQDs, with lateral size 10–20 nm), the nature of structural defects can be more appropriately correlated with both the cytotoxicity and phototoxicity, and extended to other carbon nanomaterials. In this study, GOQDs were synthesized and the structures were varied readily by NaBH4 reduction. The corresponding cytotoxicity and phototoxicity towards bacterial cells were then characterized and compared. Experimentally, results from XPS, Raman spectroscopy, and electron paramagnetic resonance (EPR) measurements were obtained to quantify the defect structure, which was then correlated to the antibacterial activities both in the dark and under visible-light irradiation. Reductive removal of oxygen functional groups was found to lower both the cytotoxicity and phototoxicity of the GOQDs, likely due to a decrease of defect sites with oxidative activity and an increase of conjugation to hydroxyl groups rendering them inactive to photo-induced homolytic cleavage. These results suggest that the structural defects of carbon nanomaterials are directly correlated with their cytotoxicity and phototoxicity, providing a foundation for the rational design of low-cost, potent antimicrobial reagents.
Experimental section
Sample preparation
The synthesis of GOQDs has been described in detail previously.15,16 In brief, 1 g of commercially available carbon pitch fiber was added to a 500 mL, two-neck, round-bottom flask along with 60 mL of sulfuric acid and 40 mL of nitric acid. The mixture was first sonicated for 4 h, then refluxed at 125 °C for 16 h, where the appearance of a light-brown color signified the formation of GOQDs. After the reaction was completed, the solution was neutralized by sodium hydroxide, and dialyzed (MW 2000) for 72 h in Nanopure water with frequent changes of water. The purified GOQDs were concentrated by rotary evaporation, and dried in a vacuum chamber.To synthesize reduced GOQDs (rGOQDs), 100 mg of the as-prepared GOQDs were first dissolved in 5 mL of Nanopure water, producing a final concentration of 20 mg mL−1. Separately, 1 g of NaBH4 was dissolved in 1 mL of water, then added all at once into the GOQD solution under magnetic stirring at 1000 rpm. The reaction was allowed to stir for 5 min, then immediately dialyzed for 24 h with frequent changes of water, and the resulting solution was dried by rotary evaporation to afford powders of rGOQDs.
Characterization
Sample morphologies were examined with a Phillips CM300 transmission electron microscope (TEM) operated at 300 kV. Atomic force microscopy (AFM) topographic measurements were performed with a Molecular Imaging PicoLE SPM instrument. UV-vis absorption spectra were acquired with an Agilent Cary 60 UV-vis spectrometer using a 1 cm quartz cuvette, and steady-state photoluminescence measurements were performed with a PTI fluorospectrometer. FTIR measurements were performed on a PerkinElmer FTIR spectrometer with samples deposited onto a ZnSe disk. Raman spectra were acquired with a DeltaNu Advantage 532 Raman system powered by a 532 nm laser. XPS measurements were carried out with a PHI 5400/XPS instrument equipped with an Al Kα source operated at 350 W and 10−9 torr. 1H and 1H–13C HSQC (heteronuclear single quantum coherence) NMR measurements were carried out with a Varian Unity 500 MHz NMR spectrometer using saturated GOQD or rGOQD solutions in D2O.Electron paramagnetic resonance
EPR measurements were performed at room temperature using a Bruker EMX EPR spectrometer operating at an X-band frequency of approximately 9.4 GHz, and a Bruker ER 4122SHQE resonator. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was used as the spin trap in all experiments. The solutions consisted of DMPO (100 mM in water), along with GOQD or rGOQD (1 mg mL−1). A 50 μL aliquot of the solutions was loaded into a capillary tube. This capillary tube was then inserted into a quartz EPR tube (Wilmad) with a 4 mm outer diameter, and loaded into the EPR cavity resonator. All samples were analyzed two minutes after reagents were mixed and signals averaged over four scans. For photoirradiation experiments, the capillary tubes containing the DMPO solutions were suspended 5 cm above a 100 W LED array (Hongke Lighting) with a 395–400 nm emission band, and irradiated for 1 min before the acquisition of the scans. Spectra were recorded at the microwave power of 20.17 mW, modulation amplitude of 1 G, modulation frequency of 100 kHz, spectral window of 75 G, and resolution of 512 points.Time-resolved photoluminescence measurements
Time-correlated single photon counting (TCSPC) was carried out on a home-built apparatus. The excitation source was a pulsed Super K EXTREME (NKT Photonics) supercontinuum laser coupled to a Super K SELECT (NKT Photonics) acousto-optic filter and external RF driver (NKT Photonics) to select the wavelength of the excitation pulse. Measurements were carried out at a 78 MHz pulse repetition rate with 14.2 μW (400 nm) power, as measured near the sample. Both excitation and emission beams were horizontally polarized by mounted Glan-Thompson polarizers (Thorlabs). Emission light was collimated and refocused by a set of achromatic doublets (Thorlabs). Long pass filters were used to minimize the influence of the reflected excitation beam. Emission wavelengths were selected by an Acton Spectra Pro SP-2300 monochromator (Princeton Instruments), on which two detectors were mounted for steady-state and time-resolved measurements. An air cooled PIXIS 100 CCD (Princeton Instruments) was used to record the steady-state spectra. A hybrid PMT with minimal after-pulsing (Becker and Hickl) was used to record the time-resolved fluorescence decay. An SPC-130 photon counting module (Becker and Hickl) coupled to a Simple-Tau 130 table-top TCSPC system was used for photon counting. Emitted photons were collected for 5 s, and each measurement was repeated 50 times prior to averaging and subsequent analysis.Photocatalytic degradation of methylene blue
The photocatalytic degradation of methylene blue (MB) by GOQD and rGOQD was monitored by UV-vis spectroscopy in a 1 cm quartz cuvette. Experimentally, a 26 μM solution of MB was prepared in water. Then, 0.5 mL aliquots of this MB solution were added to 0.5 mL of either GOQD or rGOQD solutions at 0.1 mg mL−1, or pure water. These solutions were vortexed briefly and allowed to rest undisturbed for 15 min. An initial spectrum of these solutions was taken, then the cuvette was irradiated with a 395–400 nm LED held 10 cm away in 1 min intervals for a total of 15 min. Upon completion of each irradiation interval, the solution was mixed thoroughly by pipetting up and down, before a new spectrum was acquired. Similar measurements were also carried out in the presence of 10 mM mannitol as a hydroxyl radical scavenger.(Photo)toxicity assays
Staphylococcus epidermidis (ATCC 12228) cells were obtained by first spreading frozen liquid cultures (20% glycerol, −72 °C) on Luria broth (LB) agar plates and incubating them at 37 °C overnight. From these plates, one individual colony was selected and used to inoculate 3 mL of sterile liquid LB and incubated at 37 °C for 18 h under constant shaking at 250 rpm. Upon completion of this growth period, a 1 mL aliquot of this culture was removed and centrifuged at 5000 rpm for 5 min. The supernatant was discarded, and the pellet re-suspended in 1 mL sterile Nanopure water. Enough of the resuspended cell suspension was added to 5 mL of sterile Nanopure water to bring the optical density (OD) at 600 nm to 0.100. A 100 μL aliquot of this suspension was added to 1.5 mL centrifuge tubes containing 900 μL of GOQD or rGOQD solutions, both at 1 mg mL−1 concentration, or 900 μL of Nanopore water as a control. A 100 μL aliquot was removed after 5 min and spread onto a fresh LB agar plate using silica beads. All plates were incubated for 18 h at 37 °C, after which individual colonies were counted.For phototoxicity evaluation, a similar treatment was utilized, but the centrifuge tubes with the final resuspension were held 10 cm above a 100 W LED array (Hongke Lighting) with a 395–400 nm emission band, and irradiated for 3 min before plating.
Intracellular reactive oxygen species quantification
The production of ROS was quantified by using CellROX® green dye. Staphylococcus epidermidis cells were cultured for 18 h in LB as described above, and 500 μL aliquots removed and centrifuged at 5000 rpm for 5 min. The cell pellets were then resuspended in 500 μL of water, 1% H2O2, or 1 mg mL−1 solutions of GOQDs or rGOQDs and incubated at 37 °C for 5 min. After this incubation period, cell suspensions were centrifuged again and the pellets resuspended in 5 μM CellROX solutions in water. Cells were stained for 15 min in the dark, then centrifuged and resuspended in 500 μL of pure water. A 1 μL aliquot of these suspensions was dropped onto 1% agar pads (1 cm × 1 cm) and a 1.5 microscope coverslip (0.17 mm thickness) was placed on top. Fluorescence microscopy images were obtained on a Leica DM 4000B microscope fitted with a 63× water immersion lens. Samples were excited at 470 ± 40 nm and fluorescence emission collected at 525 ± 50 nm. Fluorescence intensity from 3 separate images for each sample were utilized for statistical analysis.Results and discussion
As described previously,15,16 GOQDs were prepared by acid exfoliation of the submicrometer-sized sp2 domains in carbon pitch fibers. Fig. 1A depicts a representative TEM image of the resulting GOQDs, which show a lateral size of 10–20 nm. The GOQDs also show clearly defined lattice fringes in high-resolution TEM measurements (Fig. 1B), where the interplanar distance of 0.263 nm is consistent with the d spacing of graphene (1120) planes.17 AFM topographic study (Fig. 1C) shows good dispersion of the GOQDs without apparent agglomeration, mostly due to the abundant oxygenated moieties on the GOQD surface, and from the line scan, the height of the GOQDs was found to range from 0.5 to 2 nm, corresponding to 1 to 4 graphene layers (Fig. 1D). No apparent variation was observed of the GOQD morphology after NaBH4 reduction (rGOQD, not shown). Indeed, very consistent Raman spectra were obtained for GOQDs and rGOQD as well (Fig. S1†), where well-defined D and G bands can be identified at 1368 and 1593 cm−1 for GOQD, and 1370 cm−1 and 1594 cm−1 for rGOQD. This slight red-shift has been observed for graphene oxide nanostructures after chemical reduction, and ascribed to an increase of the number of small sp2 domains.18 Furthermore, the ratio of the D to G band intensity also showed a small decrease from 2.02 for GOQDs to 1.96 for rGOQDs, indicating that somewhat less defective structures were produced after chemical reduction within the carbon skeleton.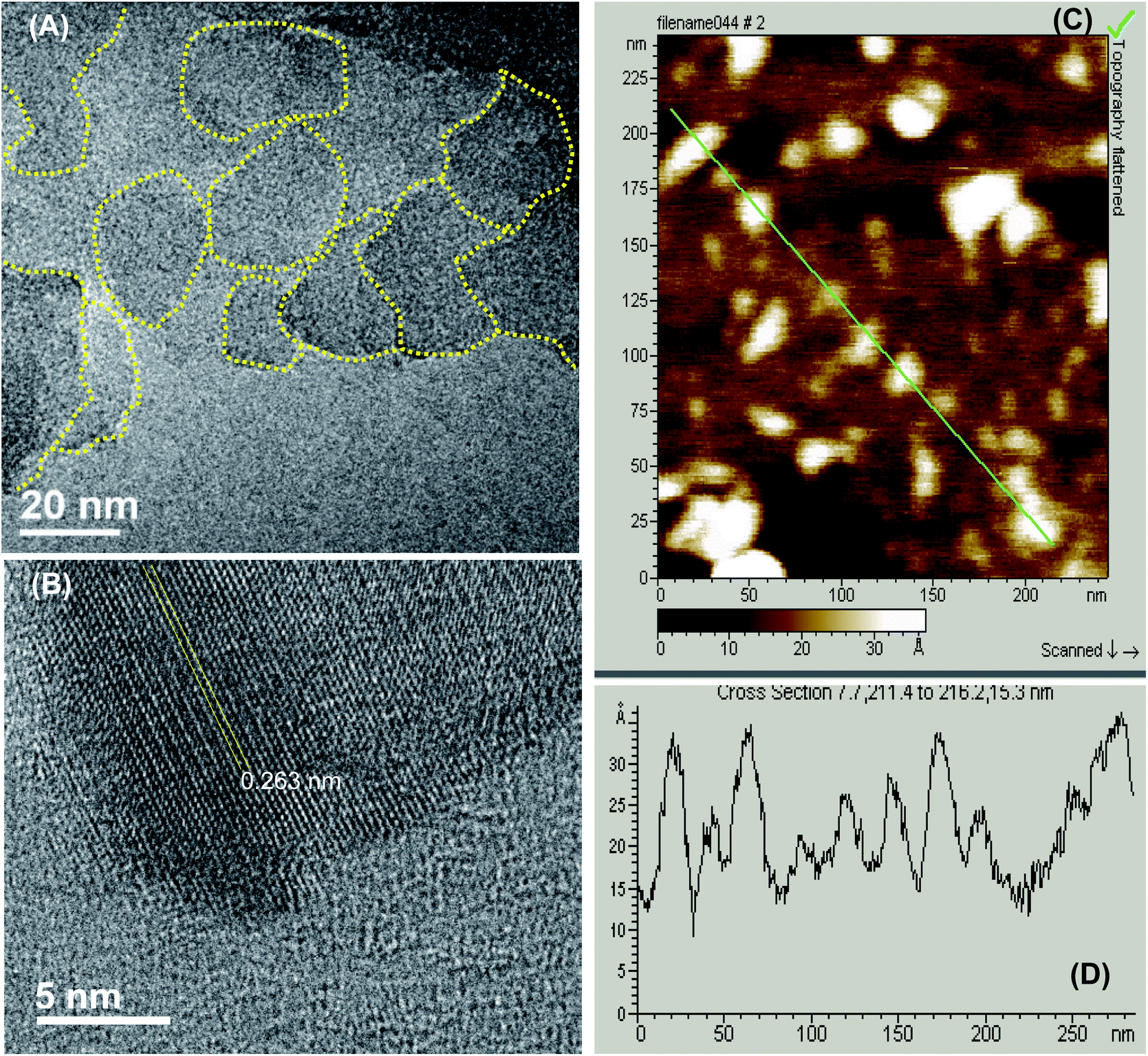 | ||
| Fig. 1 Representative TEM micrograph of GOQDs at different magnifications, with a scale bar of (A) 100 nm and (B) 5 nm. (C) AFM topograph of GOQDs, with the height profile of the line scan shown in (D). | ||
The optical properties of GOQDs and rGOQDs were then examined by UV-vis and photoluminescence measurements. From the UV-vis absorption spectra in Fig. 2A, one can see that GOQDs exhibited a largely exponential decay profile with a characteristic peak at 230 nm, and three broad shoulders centered around 290, 360 and 460 nm (marked by asterisks). The peaks at 230 and 360 nm peaks are ascribed respectively to the π → π* transitions of small and large sp2 carbon domains confined by edge and sp3 carbon defects, while the peak at 460 nm to n → π* transitions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties.19–21 Upon NaBH4 reduction, however, a slight redshift of the 230 nm band and disappearance of the 455 nm shoulder is observed, along with the emergence of a well-defined peak centered around 280 nm, corresponding to the π → π* transitions of larger sp2 carbon domains consisting of 2 or 3 phenyl rings.20 From the corresponding Tauc's plots (not shown) the optical bandgaps were estimated to be ca. 3.50 eV for GOQD and 3.15 eV for rGOQD, both of which are in agreement with values typically reported for nanosized graphene structures.22–24
O moieties.19–21 Upon NaBH4 reduction, however, a slight redshift of the 230 nm band and disappearance of the 455 nm shoulder is observed, along with the emergence of a well-defined peak centered around 280 nm, corresponding to the π → π* transitions of larger sp2 carbon domains consisting of 2 or 3 phenyl rings.20 From the corresponding Tauc's plots (not shown) the optical bandgaps were estimated to be ca. 3.50 eV for GOQD and 3.15 eV for rGOQD, both of which are in agreement with values typically reported for nanosized graphene structures.22–24
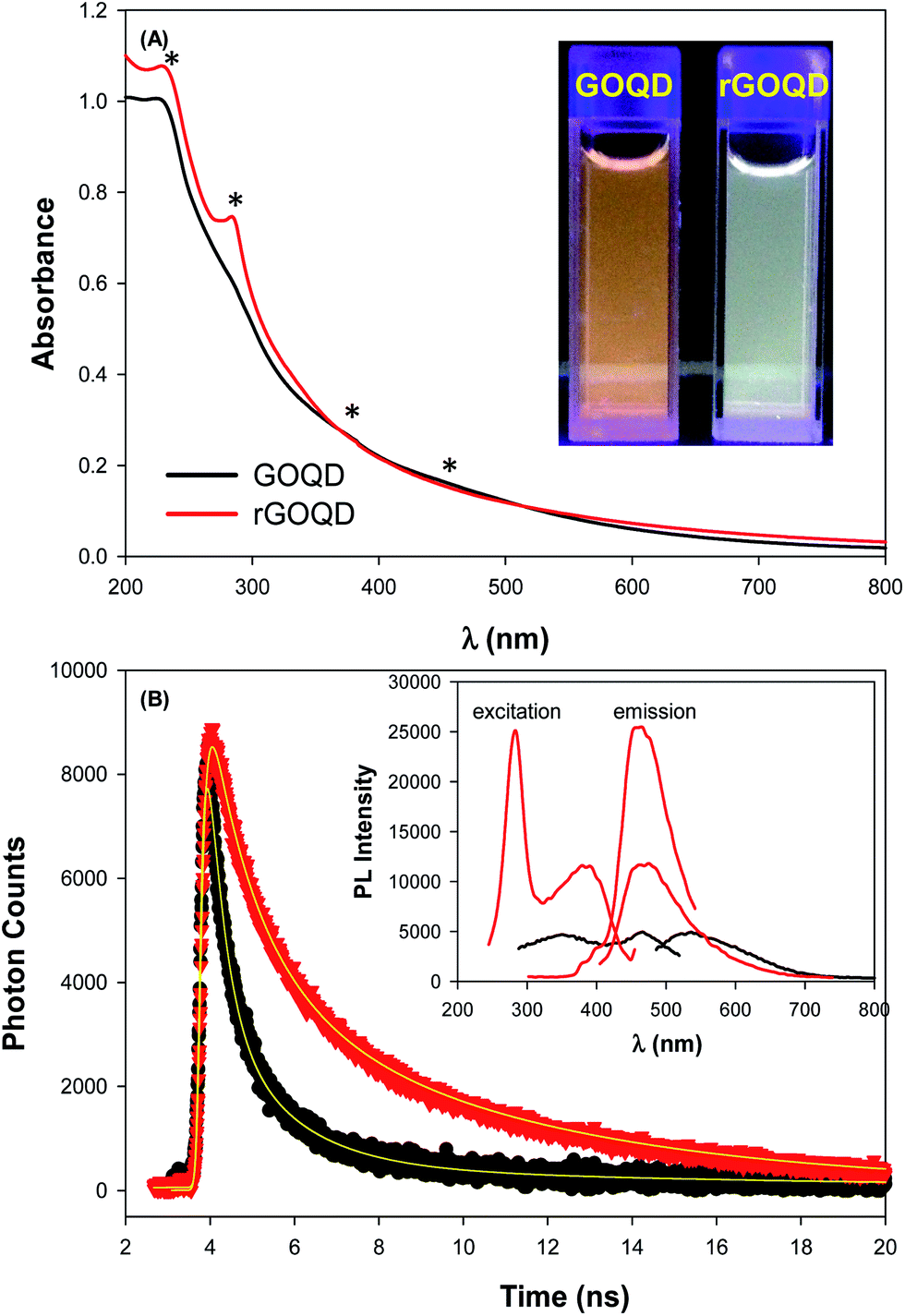 | ||
| Fig. 2 (A) UV-vis and (B) time-resolved photoluminescence spectra of GOQD and rGOQD in water. Inset to (A) is a photograph of both solutions under 365 nm photoirradiation, and inset to (B) is the corresponding steady-state photoluminescence spectra. Symbols are experimental data and solid curves are exponential fits. | ||
When excited at the respective absorption peak wavelength positions, apparent photoluminescence emissions emerged for both samples. Fig. 2B inset shows the steady-state photoluminescence measurements of the two samples. One can see that GOQDs displayed two excitation peaks (λex) centered at 350 and 465 nm, very close to the absorption peak positions observed in UV-vis measurements, and a corresponding emission peak (λem) at 535 nm. Upon NaBH4 chemical reduction, however, four key differences were observed: (i) a complete loss of the excitation band centered at 455 nm; (ii) the appearance of a new excitation band centered at 280 nm; (iii) a 35 nm redshift of the 350 nm excitation band; and finally (iv) a 64 nm blueshift of the 535 nm emission band to 460 nm. Photographs of the sample solutions under 365 nm photoirradiation were included in the inset to Fig. 2A, where the color appearance was clearly different, yellow-green for GOQD and blue-green for rGOQDs solutions. Furthermore, one can see that the photoluminescence intensity more than doubled after NaBH4 reduction, which probably arose from the prolonged lifetime of the GOQD emission, as manifested in time-resolved photoluminescence measurements (Fig. 2B). One can see that both emission decay profiles (at λex = 400 nm) can be well fitted by a single exponential equation, y = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−t/τ, from which the lifetime (τ) of the photoluminescence emission was estimated to be 1.05 ns for GOQDs and 1.73 ns for rGOQDs. The prolonged photoluminescence lifetime observed with rGOQDs is again in agreement with the removal of GOQD trap states (defects) upon NaBH4 reduction.25 This is likely due to the breaking of sp2-domain conjugation to carbonyl species located at the periphery and edge of the structures, where the 465 nm/530 nm emissions most likely originated from ketone groups on edge sites26 and the 280 nm and 350 nm excitation bands from small and large sp3-confined sp2-domains, respectively.27
e−t/τ, from which the lifetime (τ) of the photoluminescence emission was estimated to be 1.05 ns for GOQDs and 1.73 ns for rGOQDs. The prolonged photoluminescence lifetime observed with rGOQDs is again in agreement with the removal of GOQD trap states (defects) upon NaBH4 reduction.25 This is likely due to the breaking of sp2-domain conjugation to carbonyl species located at the periphery and edge of the structures, where the 465 nm/530 nm emissions most likely originated from ketone groups on edge sites26 and the 280 nm and 350 nm excitation bands from small and large sp3-confined sp2-domains, respectively.27
The change in surface functional groups before and after NaBH4 reduction was further examined by FTIR spectroscopic measurements. The resulting spectra (Fig. S2†) for GOQD and rGOQD clearly demonstrate the characteristic vibrations of oxidized graphene, where the vibrational bands at ca. 1710, 1600, 1430, 1340, and 1260 cm−1 can be assigned to the carbonyl, vinyl, carboxyl, phenolic, and hydroxy/epoxy species, respectively.15,16 Notably, after chemical reduction, the intensity of the carbonyl and hydroxy/epoxy vibrations, relative to that of CC, decreased by ca. 25%, suggesting an effective removal of these oxygen functional groups from the GOQD surfaces. Additionally, the CC stretching vibration red-shifts from 1610 cm−1 to 1598 cm−1, which can be ascribed to the removal of electron-withdrawing groups in the vicinity of sp2 domains, in agreement with the observed decrease in peak intensity for carbonyl and hydroxy/epoxy species.28 Consistent results were obtained in 1H NMR measurements where signals for the aromatic protons were found to increase markedly (Fig. S3†).
Further structural characterization was carried out by XPS measurements. Fig. 3A shows the survey spectra of the two samples, where C 1s and O 1s electrons can be clearly identified at the binding energy of ca. 285 and 531 eV, respectively; and after NaBH4 reduction, the peak intensity of the O 1s electrons relative to that of C 1s decreased by more than 10% (no other elements can be identified, indicative of high purity of the samples). This suggests partial removal of oxygenated species from the GOQD surface by NaBH4 reduction. The high-resolution scans of the C 1s electrons are depicted in Fig. 3B and C, respectively. One can see that deconvolution yields four major components for each sample. These can be identified at 283.8, 284.2, 285.4, and 287.6 eV for the as-produced GOQDs (Fig. 3B), which may be assigned to the carbons in CC, C–O, CO and O–CO, respectively; for comparison, similar peaks are resolved for rGOQDs with the corresponding binding energies at 284.0, 284.7, 285.8, and 287.7 eV (Fig. 3C).15,16 The blue-shift in the binding energy is likely a result of decreased number of electron-withdrawing groups, such as ketones and hydroxyl groups, in contact with sp2 carbons.29 This is confirmed by the change in carbon composition quantified by the integrated peak areas, as summarized in Table 1. One can see a major loss of carbonyl species after NaBH4 reduction, from 19.2% for GOQDs to 8.3% for rGOQDs, as well a decrease in C–O bonds from 29.7% in GOQD to 25.1% in rGOQD. The fraction of CC bonds increases, however, from 36.2% for GOQDs to 44.1% for rGOQDs, suggesting an increase of the fraction of sp2 carbon after NaBH4 reduction.
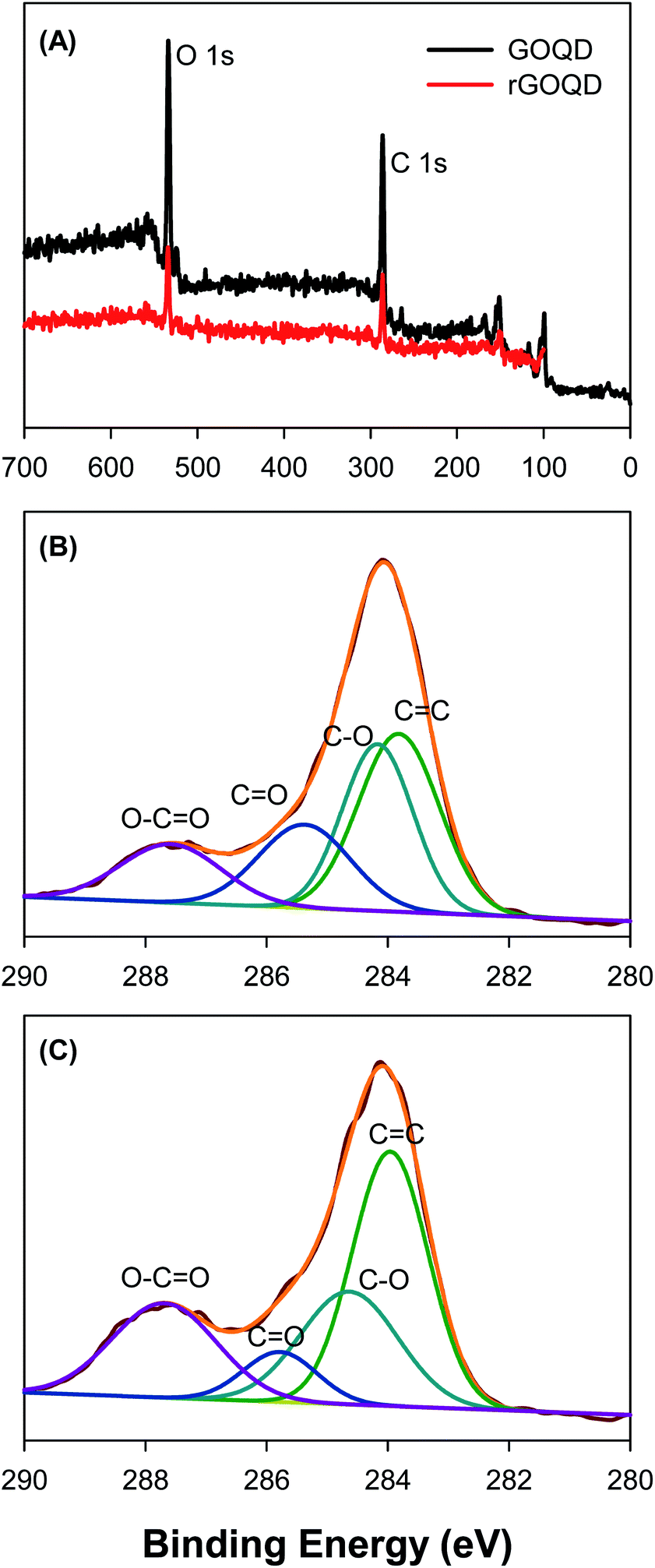 | ||
| Fig. 3 (A) XPS survey spectra of GOQD and rGOQD. High-resolution C 1s spectra for (B) GOQD and (C) rGOQD with the corresponding peak assignments. In panels (B) and (C), black curves are experimental data and colored curves are deconvolution fits. | ||
| CC |
C–O | CO |
O–CO |
|||||
|---|---|---|---|---|---|---|---|---|
| (eV) | (%) | (eV) | (%) | (eV) | (%) | (eV) | (%) | |
| GOQD | 283.8 | 36.2 | 284.2 | 29.7 | 285.4 | 19.2 | 287.6 | 14.9 |
| rGOQD | 284.0 | 44.1 | 284.7 | 25.1 | 285.8 | 8.3 | 287.7 | 22.5 |
The notable change in surface functional group composition was utilized to gain further insights into the structural correlation between graphene nanostructures and their (photo)toxicity. Staphylococcus epidermidis was chosen as an illustrating model bacterial species, due to its particularly relevant medical applicability in the pressing issues of antibiotic resistance and nosocomial infections.30,31 To accomplish this, a 1 mL aliquot of an overnight cell culture was first washed and resuspended in a 1 mg mL−1 GOQD or rGOQD solution. Then, a 100 μL aliquot was taken after 5 min and spread on fresh LB agar plates with silica beads. These plates were subsequently incubated at 37 °C for 18 h and viability assessed by the colony-counting method. The change in viability is shown in Fig. 4A in terms of % colony-forming units (CFU) which is normalized to the CFUs of the control (water). Fig. 4B displays the photographs of the plates after incubation with these cell suspensions. A substantial loss in viability can be seen for the bacteria incubated with GOQDs, whereas no obvious change in the number of colonies was observed for those with rGOQDs. This result is interesting in that NaBH4 reduction of GOQDs resulted in an almost complete loss of cytotoxicity towards bacterial cells. Note that a decreasing cytotoxicity of graphene oxide nanowalls was also observed after hydrazine reduction.13 The reason for this may lie in the chemical functional groups that exist in the GOQD structure before reduction. Tian et al.32 synthesized carbon nanoparticles by refluxing natural gas soot in strong acids to yield highly luminescent carbon nanoparticles. They further characterized the resulting carbon nanostructures by cyclic voltammetry and observed two redox peaks which were assigned to phenanthroquinone-like moieties. As the synthetic procedure for the preparation of GOQDs in the present study was similar, it is reasonable to expect these species were also formed on the GOQD surface, as manifested in XPS measurements (Fig. 3 and Table 1). Phenanthroquinone derivatives have been shown to induce cellular toxicity due to its redox activity, and are therefore likely to contribute to the observed cytotoxicity of GOQDs.33 The proposed mechanism of this moiety's toxicity toward bacteria involves the oxidation of vital electron-transport chain (ETC) redox mediators, namely, electron carriers such as NADH, FADH2, redox centers of proteins in complex I–IV, and membrane-embedded redox shuttles cytochrome c as well as various quinone derivatives.3Staphylococci species are known to utilize menaquinone34,35 as a redox shuttle, and this is the most likely target of this oxidative toxicity for several reasons: (i) they are a freely soluble species found in the membrane allowing relatively unimpeded contact with exogenous species, as compared to redox centers embedded in proteins and redox-active molecules in the cytoplasm which necessitate translocation through the plasma membrane; (ii) they are structurally similar to GOQDs which provides a vital driving forces for their interaction, likely through π–π stacking; and (iii) they have a relatively more negative reduction potential than other quinone species,36 making them highly likely reductants of phenanthroquinone moieties on GOQDs.
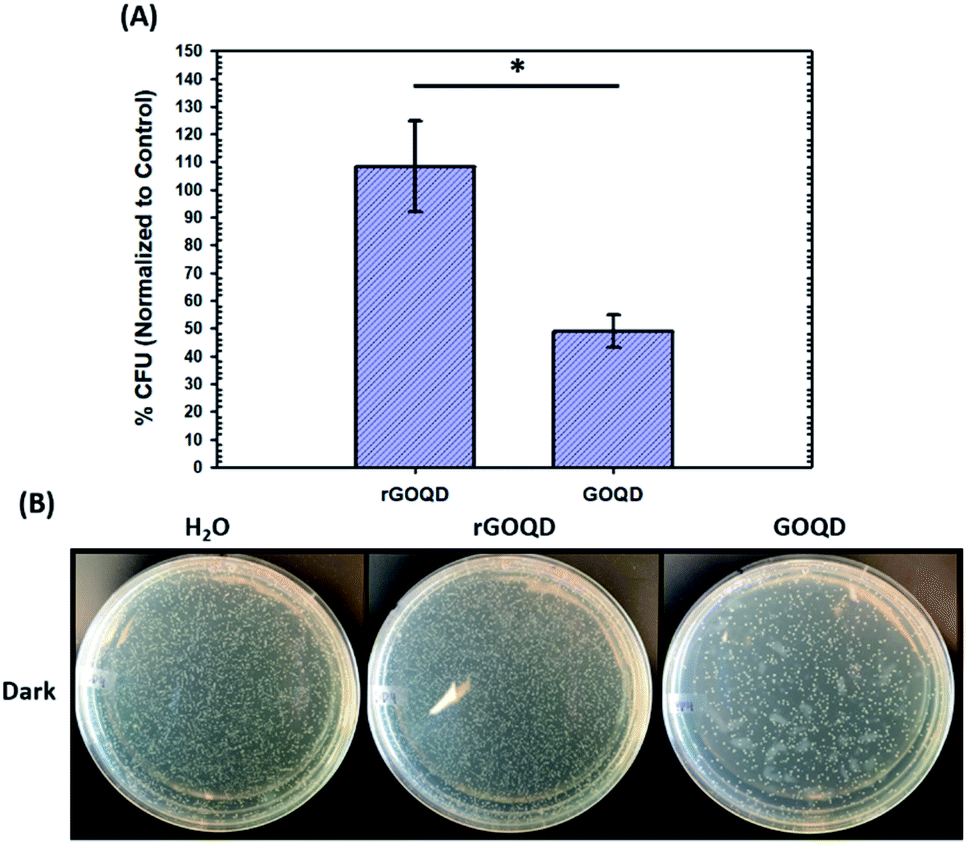 | ||
| Fig. 4 Cytotoxicity of GOQDs and rGOQDs towards Staphylococcus epidermidis. (A) Comparison of relative change in viability after incubation of cell suspensions in GOQD or rGOQD solutions for 5 min in the dark. (B) Photographs of LB agar plates after incubation with cell suspensions in the dark. Statistical significance was evaluated by two-tailed t-tests where *p < 0.05. | ||
Upon oxidation of reduced menaquinone by phenanthroquinone moieties, respiratory arrest and subsequent collapse of the proton motive force ensue as illustrated in Fig. S4.† Without a constant flow of electrons to oxygen molecules, incomplete reduction of oxygen to H2O2 becomes more prevalent which creates an abundance of hydroxyl radicals through Fenton reactions and the Haber–Weiss cycle, resulting in oxidative damage to cellular components. Additionally, disruption of electron flow also collapses the proton gradient as these electron-transfer reactions are coupled to proton translocation across the membrane. This creates a change in membrane potential, which subsequently inactivates ATP synthase, resulting in a lack of ATP production and, consequently, a depletion of a cell's energy source. As a heightened level of ROS is expected under these circumstances, the intracellular ROS concentration was measured in cells exposed to GOQD and rGOQD solutions. Micrographs of cells incubated with CellROX green after a 5 min exposure to GOQD and rGOQD solutions are shown in Fig. 5A, and average fluorescence intensities are summarized in Fig. 5B. A clear increase in the fluorescence intensity is observed in cells incubated with GOQD solutions, whereas no significant change in fluorescence was observed in the presence of rGOQD solutions. The oxidative nature of GOQDs was further investigated in vitro via Ellman's assay (Fig. S5†) using reduced glutathione as a model intracellular thiol. This colorimetric assay demonstrated the enhanced propensity for thiol oxidation that GOQDs have over rGOQD, in agreement with the CellROX assay results and the proposed cytotoxicity model. Cellular death due to oxidation of electron-transport species is therefore presented as the most reasonable mechanism of action for highly oxidative graphitic structures such as the ones presented in this study, and can reasonably be extended to other carbon nanostructures.
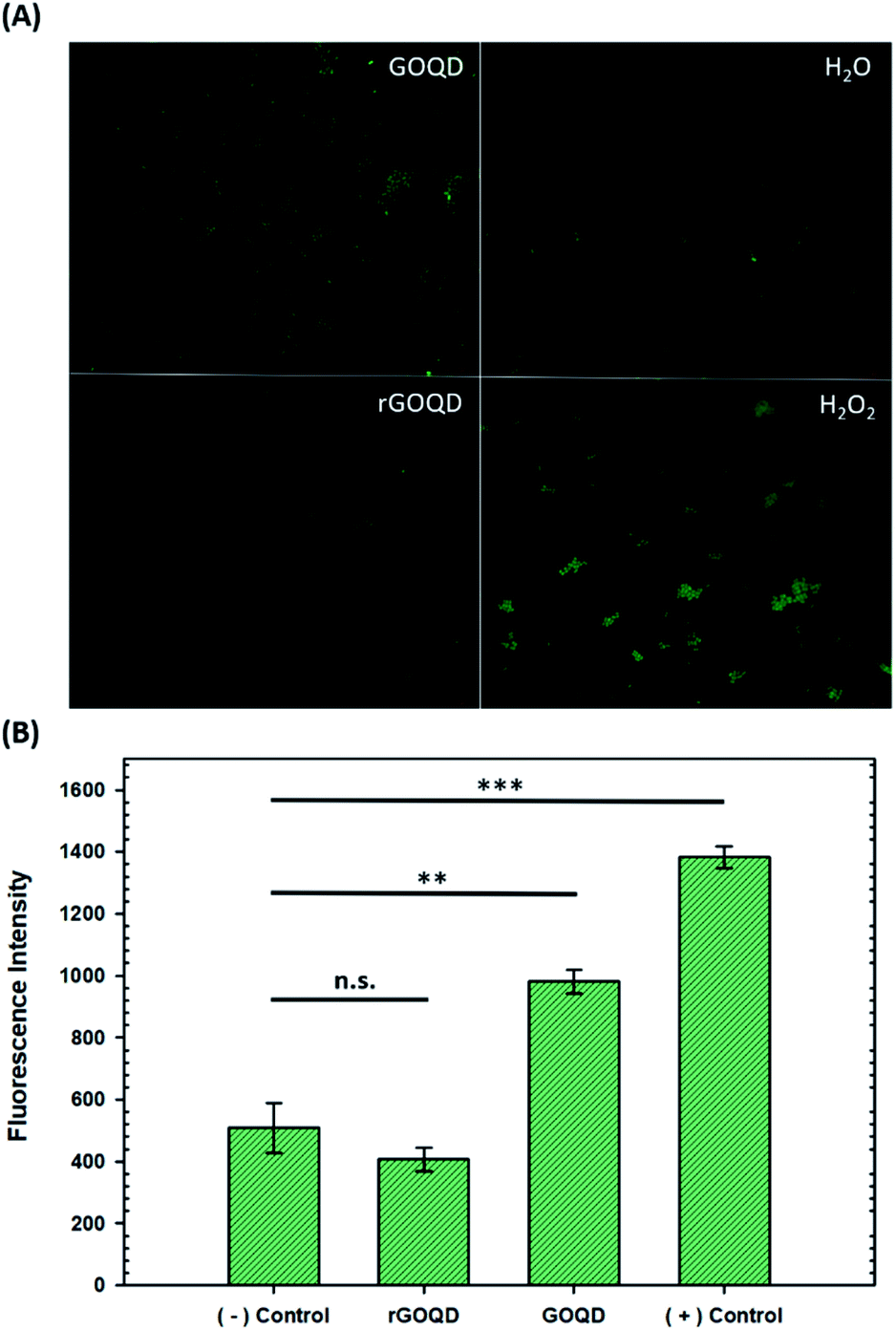 | ||
| Fig. 5 Evaluation of intracellular reactive oxygen species generation. (A) Representative micrographs of Staphylococcus epidermidis cells stained with CellROX green after a 5 min incubation with GOQD, rGOQD, water (− control), and 1% H2O2 (+ control). (B) Bar charts summarizing average fluorescence intensity of cells after exposure. Statistical significance is evaluated by two-tailed t-tests where p > 0.05 is defined as not significant (n.s.), *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
As graphene nanostructures have frequently been presented as photocatalytic agents with the potential to inactivate bacteria through phototoxic mechanisms, this activity was also evaluated for these structures.37–41 GOQD and rGOQD solutions were incubated with bacterial cell suspensions in a similar fashion as cytotoxicity assays and exposed to 400 nm light for a total of 3 min. The change in viability after this exposure is summarized in Fig. 6A, and photographs of the resulting plates are shown in Fig. 6B. A noticeable loss (30%) of viability is observed for cells irradiated in the presence of GOQD whereas cells irradiated in the presence of rGOQD solution actually observed an increase in viability relative to the control. Note that the GOQD solutions present an additional 21% loss of viability as compared to incubation for the same amount of time in the dark (Fig. S6†), which can be attributed to the phototoxic effects of GOQD structures. Compared to the water control which demonstrated a 30% loss in viability, rGOQD solutions demonstrate a remarkable preservation of viability which can be attributed to the antioxidant nature of rGOQD surface functional groups. As a significant loss of carbonyl and hydroxy/epoxy groups is observed after NaBH4 reduction (vide ante), these species are determined not to be the radical scavengers responsible for this antioxidant activity.42 Alternatively, CC double bonds,43 especially those that comprise aromatic systems,44 are known to undergo hydroxyl addition in the presence of hydroxyl radicals. Notably, both the 1H NMR and XPS results presented above indicate that rGOQDs have a significantly greater vinyl and aromatic composition than GOQDs, indicating hydroxyl radical addition across CC double bonds is more likely the mechanism of antioxidant activity.45
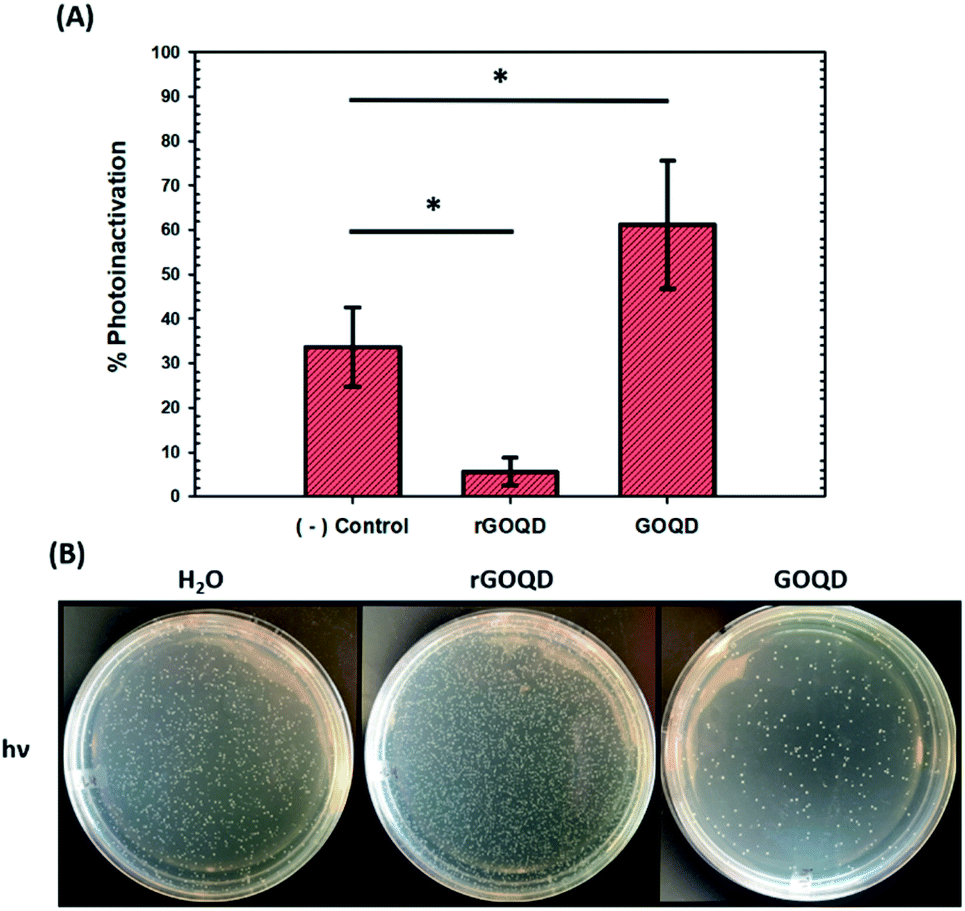 | ||
| Fig. 6 Phototoxicity of GOQDs and rGOQDs towards Staphylococcus epidermidis. (A) Change in viability after irradiation of cell suspensions in GOQD, and rGOQD solutions for 3 min by 400 nm LED. (B) Photographs of LB agar plates after incubation with cell suspensions after irradiation. Statistical significance was evaluated by two-tailed t-tests where *p < 0.05. | ||
EPR measurements were then carried out to characterize the formation of radical species upon photoirradiation with DMPO as the spin trap. The spectra of the GOQD and rGOQD solutions acquired without photoirradiation are shown in Fig. 7A. A consistent profile was observed in the absence of DMPO (Fig. S7†). Interestingly, both samples display a signal (#) centered at 3369 G with a corresponding g value of 2.0034 ± 0.0005, in contrast to pure water. This measured g value lies between those of carbon-centered radicals (g ≈ 2.0023) and oxygen-centered radicals (g > 2.004), and is characteristic of carbon-centered radicals in the proximity of oxygen functionalities.46,47 As both GOQD and rGOQD structures have a significant oxygen content, this assignment is justified. More specifically, this signal likely originates from phenalenyl-like radical species, due to the inherent stability of such species towards oxidation in air48 and to σ-dimerization49 that allowed this signal to persist under ambient conditions for extended periods of time.50 Although GOQD and rGOQD solutions are at the same concentration, GOQDs demonstrate more than two-fold stronger signal, indicating that a significant number of these species were removed by NaBH4 reduction.
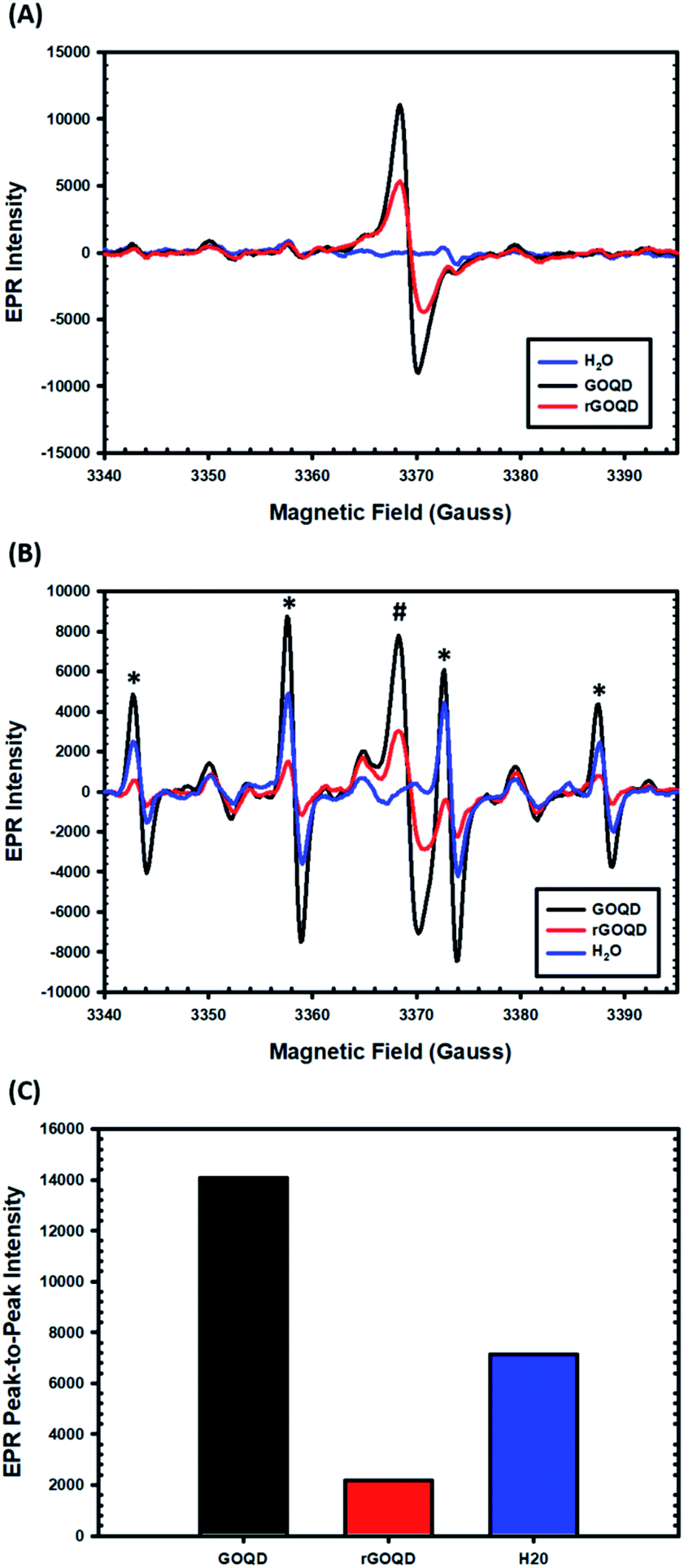 | ||
| Fig. 7 EPR spectra of GOQD and rGOQD solutions (1 mg mL−1 in water) in the presence of DMPO that are collected (A) in the dark and (B) after photoirradiation by 400 nm light for 1 min. In panel (B), the # and * symbols denote signals from intrinsic phenalenyl species and DMPO/hydroxyl radical adducts, respectively. Panel (C) shows the corresponding peak-to-peak intensities for hydroxyl adduct after photoirradiation in (B). | ||
These spectra changed significantly upon photoirradiation by 400 nm light, however, as shown in Fig. 7B. Four distinct new peaks (marked with asterisks) emerge with an intensity ratio of 1:2:2:1 and hyperfine couplings of aN = aH = 14.9 G, characteristic of hydroxyl radical adducts to DMPO.51,52 The peak-to-peak intensities of the signals centered at 3358 G are compared in Fig. 7C, demonstrating a notable, seven-fold increase in hydroxyl adduct formation in the presence of GOQDs, whereas water alone displayed an almost four-fold increase, as compared to rGOQDs, in agreement with bacterial viability changes under light irradiation. As there is an abundance of hydroxy groups on the GOQD and rGOQDs surfaces, hydroxyl radical formation from homolytic cleavage of these groups upon photo excitation is likely the source of the hydroxyl radical adducts observed in the EPR measurements. The main difference between GOQDs and rGOQD is the composition of vinyl carbons, and as C–O bonds have a much lower bond dissociation energy for alkyl carbons (322 kJ mol−1) than on vinyl (364 kJ mol−1) and especially aromatic (431 kJ mol−1) carbons, the expanded conjugation after reduction is proposed to be the underpinning reason for the significant difference in their hydroxyl radical formation under light excitation. Indeed, such an argument is supported by results from 1H–13C HSQC measurements (Fig. S8†), which shows that after NaBH4 reduction, hydroxy carbons exhibit significant correlations to vinylic protons, suggesting they are in electronic conjugation. This is also supported by theoretical bond dissociation energies of various peroxy-radical species,53 where it was found that conjugation to sites of oxygen reduction significantly lowered the R–O–O· bond strength and afforded more effective antioxidant structures.
The role of hydroxyl radicals in the observed phototoxicity and EPR measurements was further examined through the photocatalytic degradation of MB in the presence of GOQD and rGOQD. A typical absorption spectrum of MB in water after successive 1 min photoirradiation is shown in Fig. S9A,† and the change in absorption at 665 nm over time was plotted as ln(A/Ao) vs. time to extract the pseudo first-order rate constant (kdeg), as shown in Fig. S9B.† A striking difference in kdeg values were found, where in the presence of GOQDs, MB was degraded nearly twice as fast as in the presence of rGOQDs, in accordance with the aforementioned EPR and viability results. Upon addition of mannitol, a hydroxyl radical-specific scavenger,54,55 a significant decline in degradation rate was observed, as illustrated in Fig. S9B.† A comparison of kdeg values in the presence and absence of mannitol is given in Fig. S9C.† It is clear from the substantial difference in kdeg that hydroxyl radical species are indeed involved in the degradation of MB facilitated by GOQDs and rGOQDs. The overall contribution of hydroxyl radical species is given by the ratio of kdeg in the presence and absence of mannitol (kdeg/kdeg,o), which is 0.31, 0.73, and 0.55 for GOQD, rGOQD, and MB alone, respectively. A substantial decrease (69%) in kdeg was observed for GOQDs but only a relatively minor one (27%) observed for rGOQDs. This indicates that hydroxyl radical formation plays a fundamental role in the photooxidation of organic molecules by GOQD structures and suggests highly oxidized, defect-rich structures facilitate their formation.
This study presents a valuable in-depth analysis of both the structural changes graphene oxide quantum dots undergo upon NaBH4 reduction, and its effect on cytotoxicity and phototoxicity towards medically relevant model bacterium. The insights obtained from these results provide critical groundwork for understanding the antimicrobial mechanisms of carbon nanomaterials. Further investigation utilizing both different reducing agents and carbon nanostructures should provide conclusive evidence on the role of various functional groups in the reactivity of carbon nanomaterials under both ambient conditions and under light irradiation. This will be pursued in future studies.
Conclusions
In this study, chemical reduction of GOQDs by NaBH4 produces an astonishing change in both their physical properties and (photo)cytotoxicity. Results from a range of spectroscopic measurements suggest the rather effective removal of structural defects in the carbon matrix, which mostly converted oxygenated carbon moieties into CC double bonds. The change in surface functionality resulted in a marked diminishment in both cytotoxicity and phototoxicity. The decrease in cytotoxicity was proposed to originate from removal of oxidative phenanthraquinone-like moieties that act to disrupt bacterial electron-transport chain propagation and generate reactive oxygen species. The complete loss of phototoxicity was found to result from diminished formation of hydroxyl radicals as a result of the newly formed conjugation to hydroxyl groups that made homolytic cleavage unfavorable, as confirmed by the significant decrease in the activity of photocatalytic degradation of organic dyes. Taken together, these results provide substantial evidence for the structural basis of toxicity and phototoxicity of graphene-based nanostructures. Such fundamental insights may be exploited for the design and engineering of graphene derivatives for antimicrobial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Alfred Freeberg and Vanessa Ventura for assistance in data acquisition. This work was supported in part by the National Science Foundation (CHE-1710408 and CBET-1848841). TEM and XPS studies were carried out at the National Center for Electron Microscopy and Molecular Foundry, Lawrence Berkeley National Laboratory, as part of a user project.References
- A. Lukowiak, A. Kedziora and W. Strek, Adv. Colloid Interface Sci., 2016, 236, 101–112 CrossRef CAS PubMed.
- A. Soroush, D. Rice, M. S. Rahaman and F. Perreault, Carbon Nanostructures, 2016, pp. 287–322 Search PubMed.
- M. D. Rojas-Andrade, G. Chata, D. Rouholiman, J. L. Liu, C. Saltikov and S. W. Chen, Nanoscale, 2017, 9, 994–1006 RSC.
- S. Gurunathan, J. W. Han, A. A. Dayem, V. Eppakayala and J. H. Kim, Int. J. Nanomed., 2012, 7, 5901–5914 CrossRef CAS PubMed.
- J. H. Li, G. Wang, H. Q. Zhu, M. Zhang, X. H. Zheng, Z. F. Di, X. Y. Liu and X. Wang, Sci. Rep., 2014, 4, 4359 CrossRef PubMed.
- C. Santhosh, P. Kollu, S. Doshi, M. Sharma, D. Bahadur, M. T. Vanchinathan, P. Saravanan, B. S. Kim and A. N. Grace, RSC Adv., 2014, 4, 28300–28308 RSC.
- S. B. Liu, M. Hu, T. H. Zeng, R. Wu, R. R. Jiang, J. Wei, L. Wang, J. Kong and Y. Chen, Langmuir, 2012, 28, 12364–12372 CrossRef CAS PubMed.
- H. L. Ren, C. Wang, J. L. Zhang, X. J. Zhou, D. F. Xu, J. Zheng, S. W. Guo and J. Y. Zhang, ACS Nano, 2010, 4, 7169–7174 CrossRef CAS PubMed.
- A. Biswas, P. Khandelwal, R. Das, G. Salunke, A. Alam, S. Ghorai, S. Chattopadhyay and P. Poddar, J. Mater. Chem. B, 2017, 5, 785–796 RSC.
- H. J. Sun, N. Gao, K. Dong, J. S. Ren and X. G. Qu, ACS Nano, 2014, 8, 6202–6210 CrossRef CAS PubMed.
- W. S. Kuo, C. Y. Chang, H. H. Chen, C. L. L. Hsu, J. Y. Wang, H. F. Kao, L. C. S. Chou, Y. C. Chen, S. J. Chen, W. T. Chang, S. W. Tseng, P. C. Wu and Y. C. Pu, ACS Appl. Mater. Interfaces, 2016, 8, 30467–30474 CrossRef CAS PubMed.
- F. Perreault, A. F. de Faria, S. Nejati and M. Elimelech, ACS Nano, 2015, 9, 7226–7236 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- Y. Zhou, H. J. Sun, F. M. Wang, J. S. Ren and X. G. Qu, Chem. Commun., 2017, 53, 10588–10591 RSC.
- G. Q. He, Y. Song, K. Liu, A. Walter, S. Chen and S. W. Chen, ACS Catal., 2013, 3, 831–838 CrossRef CAS.
- Y. Song and S. W. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 14050–14060 CrossRef CAS PubMed.
- W. K. Zhang, Y. Q. Liu, X. R. Meng, T. Ding, Y. Q. Xu, H. Xu, Y. R. Ren, B. Y. Liu, J. J. Huang, J. H. Yang and X. M. Fang, Phys. Chem. Chem. Phys., 2015, 17, 22361–22366 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- K. P. Loh, Q. L. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- J. Z. Shang, L. Ma, J. W. Li, W. Ai, T. Yu and G. G. Gurzadyan, Sci. Rep., 2012, 2, 792 CrossRef PubMed.
- K. Rahimi, A. Yazdani and M. Ahmadirad, Mater. Des., 2018, 140, 222–230 CrossRef CAS.
- D. Bhatnagar, S. Singh, S. Yadav, A. Kumar and I. Kaur, Mater. Res. Express, 2017, 4, 015101 CrossRef.
- H. B. Yang, Y. Q. Dong, X. Z. Wang, S. Y. Khoo, B. Liu and C. M. Li, Sol. Energy Mater. Sol. Cells, 2013, 117, 214–218 CrossRef CAS.
- L. Tian, Y. Song, X. J. Chang and S. W. Chen, Scr. Mater., 2010, 62, 883–886 CrossRef CAS.
- L. Wang, S. J. Zhu, H. Y. Wang, S. N. Qu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, H. L. Xu, W. Han, B. Yang and H. B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- F. Liu, M. H. Jang, H. D. Ha, J. H. Kim, Y. H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS PubMed.
- P. G. Ren, D. X. Yan, X. Ji, T. Chen and Z. M. Li, Nanotechnology, 2011, 22, 055705 CrossRef PubMed.
- Y. Yamada, H. Yasuda, K. Murota, M. Nakamura, T. Sodesawa and S. Sato, J. Mater. Sci., 2013, 48, 8171–8198 CrossRef CAS.
- J. Y. H. Lee, I. R. Monk, A. G. da Silva, T. Seemann, K. Y. L. Chua, A. Kearns, R. Hill, N. Woodford, M. D. Bartels, B. Strommenger, F. Laurent, M. Dodemont, A. Deplano, R. Patel, A. R. Larsen, T. M. Korman, T. P. Stinear and B. P. Howden, Nat. Microbiol., 2018, 3, 1175–1185 CrossRef CAS PubMed.
- W. Ziebuhr, S. Hennig, M. Eckart, H. Kranzler, C. Batzilla and S. Kozitskaya, Int. J. Antimicrob. Agents, 2006, 28, S14–S20 CrossRef CAS PubMed.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. J. Chang and S. W. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- C. E. Rodriguez, M. Shinyashiki, J. Froines, R. C. Yu, J. M. Fukuto and A. K. Cho, Toxicology, 2004, 201, 185–196 CrossRef CAS PubMed.
- P. J. McNamara and R. A. Proctor, Int. J. Antimicrob. Agents, 2000, 14, 117–122 CrossRef CAS PubMed.
- M. D. Collins and D. Jones, Microbiol. Rev., 1981, 45, 316–354 CrossRef CAS PubMed.
- S. Kishi, K. Saito, Y. Kato and H. Ishikita, Photosynth. Res., 2017, 134, 193–200 CrossRef CAS PubMed.
- L. Guardia, S. Villar-Rodil, J. I. Paredes, R. Rozada, A. Martinez-Alonso and J. M. D. Tascon, Carbon, 2012, 50, 1014–1024 CrossRef CAS.
- D. Chen, H. Zhang, Y. Liu and J. H. Li, Energy Environ. Sci., 2013, 6, 1362–1387 RSC.
- J. C. Ge, M. H. Lan, W. M. Liu, Q. Y. Jia, L. Guo, B. J. Zhou, X. M. Meng, G. L. Niu and P. F. Wang, Sci. China Mater., 2016, 59, 12–19 CrossRef CAS.
- J. Y. Zhang, X. M. Lu, D. D. Tang, S. H. Wu, X. D. Hou, J. W. Liu and P. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 40808–40814 CrossRef CAS PubMed.
- R. Yin, T. Agrawal, U. Khan, G. K. Gupta, V. Rai, Y. Y. Huang and M. R. Hamblin, Nanomedicine, 2015, 10, 2379–2404 CrossRef CAS PubMed.
- Y. M. Wang, W. H. Kong, L. F. Wang, J. Z. Zhang, Y. Li, X. G. Liu and Y. Li, Phys. Chem. Chem. Phys., 2019, 21, 1336–1343 RSC.
- A. C. Lloyd, K. R. Darnall, A. M. Winer and J. N. Pitts, J. Phys. Chem., 1976, 80, 789–794 CrossRef CAS.
- T. P. Troy, M. Nakajima, N. Chalyavi, K. Nauta, S. H. Kable and T. W. Schmidt, J. Phys. Chem. A, 2012, 116, 7906–7915 CrossRef CAS PubMed.
- Y. Qiu, Z. Y. Wang, A. C. E. Owens, I. Kulaots, Y. T. Chen, A. B. Kane and R. H. Hurt, Nanoscale, 2014, 6, 11744–11755 RSC.
- U. Green, Y. Shenberger, Z. Aizenshtat, H. Cohen and S. Ruthstein, J. Visualized Exp., 2014,(86), e51548 Search PubMed.
- F. Tampieri, S. Silvestrini, R. Ricco, M. Maggini and A. Barbon, J. Mater. Chem. C, 2014, 2, 8105–8112 RSC.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- L. Beer, S. K. Mandal, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, Cryst. Growth Des., 2007, 7, 802–809 CrossRef CAS.
- X. L. Hou, J. L. Li, S. C. Drew, B. Tang, L. Sun and X. G. Wang, J. Phys. Chem. C, 2013, 117, 6788–6793 CrossRef CAS.
- E. Finkelstein, G. M. Rosen and E. J. Rauckman, Arch. Biochem. Biophys., 1980, 200, 1–16 CrossRef CAS PubMed.
- C. Mottley, H. D. Connor and R. P. Mason, Biochem. Biophys. Res. Commun., 1986, 141, 622–628 CrossRef CAS PubMed.
- J. S. Wright, H. Shadnia and L. L. Chepelev, J. Comput. Chem., 2009, 30, 1016–1026 CrossRef CAS PubMed.
- S. Goldstein and G. Czapski, Int. J. Radiat. Biol., 1984, 46, 725–729 CAS.
- J. M. Desesso, A. R. Scialli and G. C. Goeringer, Teratology, 1994, 49, 248–259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: 10.1039/c9na00698b |
| This journal is © The Royal Society of Chemistry 2020 |