
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the role of zinc dithiocarbamate complexes as single source precursors to ZnS nanomaterials
Husn-Ubayda
Islam
ab,
Anna
Roffey
a,
Nathan
Hollingsworth
a,
Wim
Bras
 bc,
Gopinathan
Sankar
a,
Nora H.
De Leeuw
d and
Graeme
Hogarth
*e
bc,
Gopinathan
Sankar
a,
Nora H.
De Leeuw
d and
Graeme
Hogarth
*e
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H OAJ, UK
bNetherlands Organisation for Scientific Research DUBBLE@ESRF, 38043 Grenoble, France
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
dSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
eDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 1DB, UK. E-mail: graeme.hogarth@kcl.ac.uk
First published on 9th January 2020
Abstract
Zinc sulfide is an important wide-band gap semi-conductor and dithiocarbamate complexes [Zn(S2CNR2)2] find widespread use as single-source precursors for the controlled synthesis of ZnS nanoparticulate modifications. Decomposition of [Zn(S2CNiBu2)2] in oleylamine gives high aspect ratio wurtzite nanowires, the average length of which was increased upon addition of thiuram disulfide to the decomposition mixture. To provide further insight into the decomposition process, X-ray absorption spectroscopy (XAS) of [Zn(S2CNMe2)2] was performed in the solid-state, in non-coordinating xylene and in oleylamine. In the solid-state, dimeric [Zn(S2CNMe2)2]2 was characterised in accord with the single crystal X-ray structure, while in xylene this breaks down into tetrahedral monomers. In situ XAS in oleylamine (RNH2) shows that the coordination sphere is further modified, amine binding to give five-coordinate [Zn(S2CNMe2)2(RNH2)]. This species is stable to ca. 70 °C, above which amine dissociates and at ca. 90 °C decomposition occurs to generate ZnS. The relatively low temperature onset of nanoparticle formation is associated with amine-exchange leading to the in situ formation of [Zn(S2CNMe2)(S2CNHR)] which has a low temperature decomposition pathway. Combining these observations with the previous work of others allows us to propose a detailed mechanistic scheme for the overall process.
Introduction
Zinc sulfide (ZnS) is a wide band-gap (3.7 eV) semiconductor that finds use in optoelectronic devices due to its unique photoluminescence and electroluminescence properties.1 Nano-dimensional ZnS is especially important as both its optical and electronic properties can be tailored by controlling the size and shape of the nanostructures and over the past decade a range of different nanoparticulate forms of ZnS have been prepared through judicious choice of synthetic methods and control over the synthetic parameters,2 however, many of these methods employ either toxic gases such as H2S and H2 and/or have to be carried out in an inert atmosphere. More synthetically appealing is the single source precursor (SSP) approach,3 especially when the precursors are air and moisture stable, since this allows control of nanoparticulate shape-size-structure without the need for elaborate experimental facilities. Dithiocarbamate complexes are easily prepared from readily available starting materials4 and consequently find use as SSPs towards a range of metal-sulfides.5–10 Zinc dithiocarbamates, [Zn(S2CNR2)2], are particularly attractive SSPs since they are air and moisture stable solids which are easily prepared from the requisite secondary amine in water.4 They have been used extensively in the production of ZnS either as pure nanomaterials,10–19 quaternary zinc-containing sulfide nanomaterials20 or as shells on quantum dots.21 In this contribution we detail our use of [Zn(S2CNMe2)2] (1) and [Zn(S2CNiBu2)2] (2) (Chart 1) as SSPs towards ultra-thin wurtzite nanowires and also probe mechanistic aspects of the precursor decomposition process by in situ X-ray absorption spectroscopy (XAS) studies of 1. By piecing together our in situ findings with previous NMR studies22,23 we have put together a plausible reaction mechanism which will allow others to better rationalise their own findings and logically plan synthetic routes to ZnS nanomaterials. | ||
| Chart 1 | ||
Results and discussion
(i) Synthesis
Dithiocarbamate complexes [Zn(S2CNMe2)2] (1) and [Zn(S2CNiBu2)2] (2) were prepared by standard methods. Decomposition of SSPs is normally carried out in a high boiling point solvent which can act both as a heat sink and capping agent, with long chain primary amines such as oleylamine being widely utilised.24 In order to ensure that our synthetic and in situ studies (see later) were carried out under similar conditions, we prepared ZnS from the decomposition of [Zn(S2CNiBu2)2] (2) in oleylamine, being chosen over 1 due to its greater solubility in this solvent. A 5 mM solution of 2 in oleylamine was heated to 230 °C and held for 1 h. Following this, the mixture was slowly cooled to room temperature and methanol added to precipitate the generated nanoparticles, which were isolated by centrifugation, washing being repeated three times and the material produced air dried. Solid samples had an off-white appearance and were characterised by powder X-ray diffraction (PXRD) as wurtzite (Fig. 1). Interestingly, the peak at 28.5° 2θ for the (002) lattice plane is more intense than in the reference pattern for bulk wurtzite (ICDD card no. 36-1450) which suggests preferential growth along the c axis.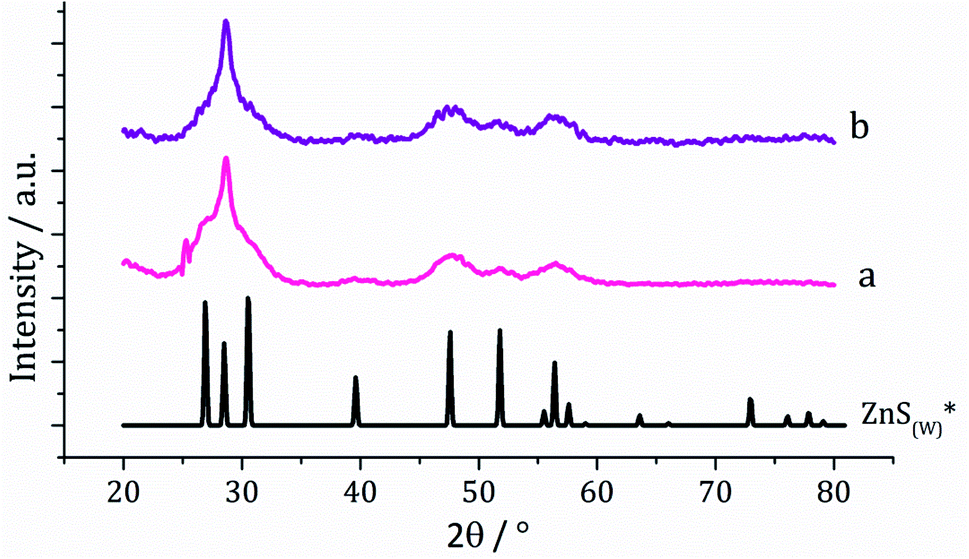 | ||
| Fig. 1 PXRD patterns using Cu Kα radiation for samples prepared from (a) 2 and (b) 2 with added 3, with reference pattern for bulk wurtzite (2H) (ICDD card no. 36-1450). | ||
Transmission Electron Microscopy (TEM) showed that high aspect ratio nanowires were produced (Fig. 2) with an average particle length of 34.3 nm (SD 26.2 nm) and width of 3.3 nm (SD 0.8 nm) and HRTEM the shows d-spacings of 3.17 Å, corresponding to the (002) lattice plane in wurtzite (ICDD card no. 36-1450), being consistent with PXRD data. These dimensions are similar to the nanowires generated upon heating [Zn(S2CNEt2)2] (3)11 or [Zn(S2CNBz2)2] (4)12 in oleylamine.
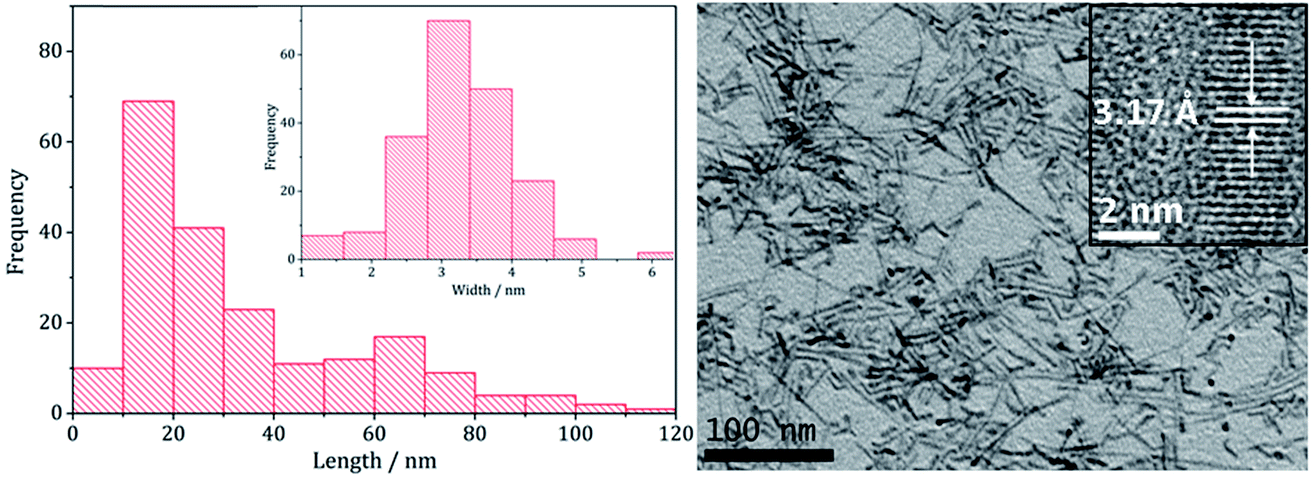 | ||
| Fig. 2 Particle length histogram with width histogram insert (left) and TEM image with HRTEM insert (right), of the sample prepared from 2. | ||
In recent work on the decomposition of [Fe(S2CNR2)3] we found that addition of thiuram disulfide, (R2NCS2)2, led to the formation of different phases of iron sulfides.25 As [Zn(S2CNR2)2] are known to add dithiocarbamate to generate the tris(dithiocarbamate) species, [Zn(S2CNR2)3]−,26 then potentially thiuram disulfide addition may slow down the decomposition process (oleylamine provides a reducing environment). Decomposition of 2 in the presence of 10 mM of the thiuram disulfide 3 (Chart 1) also afforded wurtzite nanowires, being of the same width (3.2 nm ± 0.8 nm) but slightly longer (45.0 nm ± 25.1 nm).
The formation of wurtzite nanowires upon decomposition of zinc dithiocarbamate complexes has previously been reported.11–14 Thus Lieber and co-workers prepared wires with a diameter of ca. 18 nm upon decomposition of [Zn(S2CNEt2)2] by a nanocluster-catalysed vapour-liquid-solid method,13 while Wang and co-workers prepared ultra-thin nanowires with diameters of ca. 3–5.5 nm from decomposition of the same precursor in oleylamine at 300 °C;11 even thinner nanowires (ca. 2 nm) have been prepared from [Zn(S2CNnBu2)2] in dodecylamine at 280 °C.14
(ii) X-ray absorption spectroscopy (XAS) studies of [Zn(S2CNMe2)2] (1)
For the XAS studies we chose [Zn(S2CNMe2)2] (1) as its simple structure enables easier interpretation of data, whilst still rendering relevant information on the class of SSP. We sequentially investigated the structure of 1 in the solid-state, the non-coordinating (high boiling solvent) xylene and oleylamine. The results of these studies are summarised in Fig. 3 and 4. | ||
| Fig. 3 XANES spectra of 1 as a solid (black), dissolved in xylene (red), and dissolved in oleylamine (blue). The relevance of peaks marked (A–C) is discussed in the text. | ||
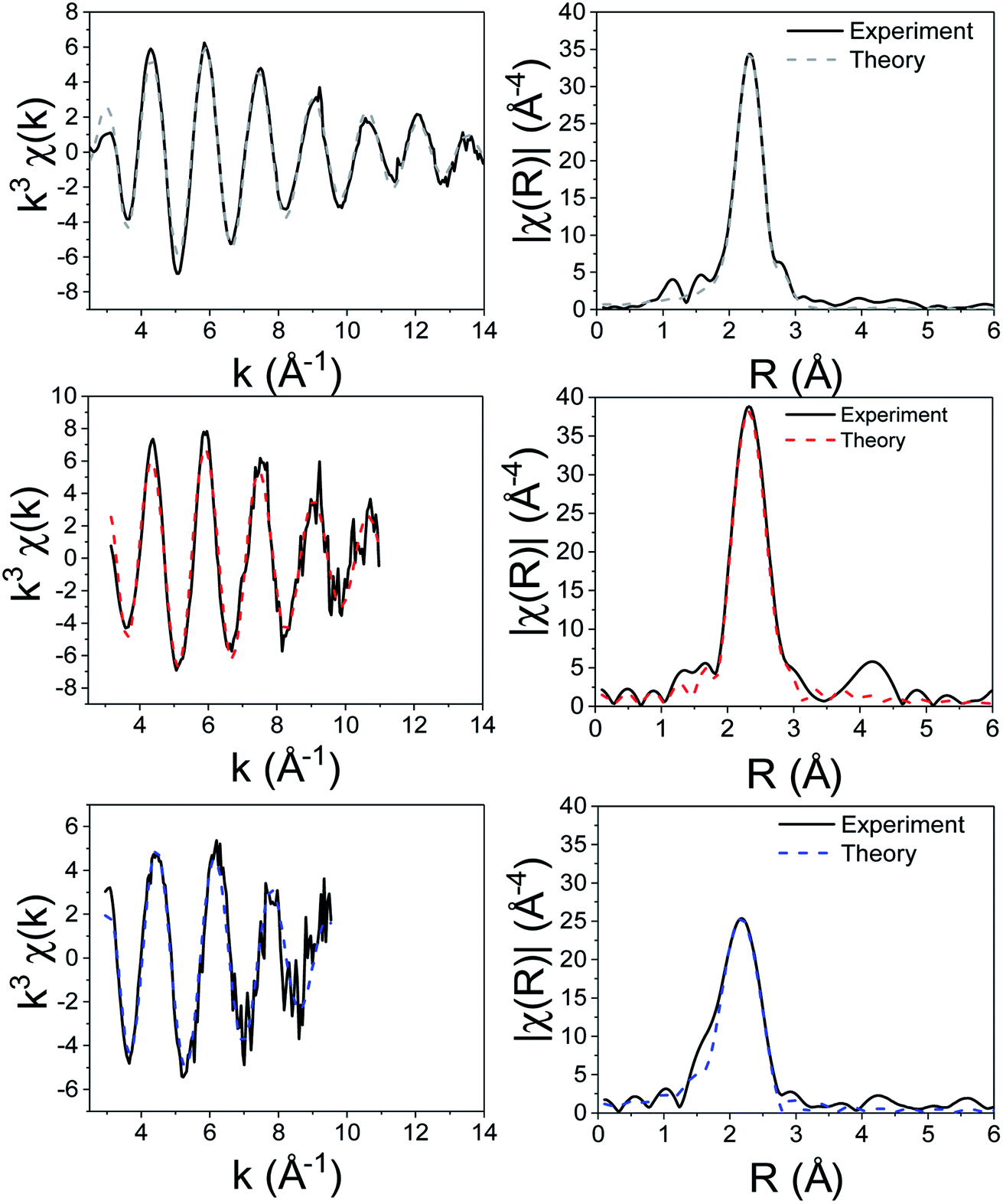 | ||
| Fig. 4 k 3-Weighted EXAFS and Fourier transformed (real space) data of 1 solid (top), in xylene (middle) and in oleylamine (bottom). | ||
We first investigated 1 in the solid-state. While in the gas phase it is monomeric with four equivalent Zn–S distances of 2.28 Å,27 a single crystal X-ray diffraction study shows it is dimeric in the solid-state containing two bridging dithiocarbamate ligands giving an overall boat-type conformation with non-degenerate Zn–S bond distances.28 The XANES spectrum exhibits a straight edge jump followed by a multi-featured region including white line intensity A, and two further peaks B and peak C. The EXAFS of 1 can be modelled accurately to the crystal structure. Thus there are two Zn–S distances at 2.32 Å arising from bonding of the zinc centre to the terminal bidentate dithiocarbamate ligand; XRD shows these bonds to be 2.31 and 2.33 Å. There are also two Zn–S distances at 2.38 and 2.41 Å resulting from the bridging ligands; found at 2.37 and 2.43 Å in the crystal structure.28 A fifth Zn–S distance is detected at 2.95 Å. This is a typical feature of dimeric zinc dithiocarbamates and arises in 1 at 3.03 Å.28
Spectra of 1 in xylene, while comparable to that in the solid-state with similar features after the edge position, also show some significant differences; specifically the ratio between A and B, and the shape of C differ. Indeed, the EXAFS can be best fit to a monomeric structure with two sets of Zn–S bond distances at 2.30 and 2.39 Å. The crystal structure of [Zn(S2CNBz2)2] shows a monomeric species containing two inequivalent Zn–S bond distances of 2.33 and 2.36 Å.29 A further two Zn–C interatomic distances are found at 2.73 Å, being similar to the Zn–C distance of 2.74 Å in the solid-state.
The XANES spectrum of 1 in oleylamine is quite distinct from 1 in the solid-state or in xylene; peaks B and C are absent suggesting significant changes in the local structure. The EXAFS was also modified with oleylamine, showing a Zn–N interatomic distance of 2.06 Å, similar to other Zn–N literature values,30,31 suggesting that oleylamine is bound to the metal centre to give five-coordinate [Zn(S2CNMe2)2(RNH2)] (1.RNH2). The structure of the five-coordinated complex shows a Debye–Waller factor that is far higher than values found for 1 in the solid-state or dissolved in xylene. This is likely due to the dynamic nature of the five-coordinated complex which can interchange between trigonal bipyramidal and square-based pyramid geometries. Table 1 summarises the results of EXAFS modelling studies.
| Ex situ XAS | ||||||
|---|---|---|---|---|---|---|
| Sample | Scatter | N | R XRD (Å) | R EXAFS (Å) | σ 2 (Å2) | F |
| 1 solid | S | 2 | 2.31–2.33 | 2.32 ± 0.02 | 0.010 | 20 |
| S | 1 | 2.37 | 2.38 ± 0.02 | 0.010 | ||
| S | 1 | 2.43 | 2.41 ± 0.02 | 0.010 | ||
| C | 1 | 2.77 | 2.74 ± 0.02 | 0.006 | ||
| S | 1 | 3.03 | 2.95 ± 0.03 | 0.015 | ||
| C | 1 | 3.02 | 3.00 ± 0.03 | 0.006 | ||
| 1 in xylene | S | 2 | — | 2.30 ± 0.03 | 0.007 | 25 |
| S | 2 | — | 2.39 ± 0.02 | 0.007 | ||
| C | 2 | — | 2.73 ± 0.02 | 0.008 | ||
| 1 in oleylamine | N | 1 | — | 2.06 ± 0.02 | 0.020 | 27 |
| S | 4 | — | 2.25 ± 0.03 | 0.020 | ||
These studies show that the structure of 1 (Scheme 1) and analogues are sensitive to the nature of surrounding coordination environment. Most importantly they show that under the standard decomposition conditions, which take place in a coordinating solvent such as oleylamine, then it is actually a species of the form [Zn(S2CNR2)2(RNH2)] which undergoes decomposition. Indeed, as early as the 1960s, the reversible 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation of 1 with a range of amines was reported by Saville and co-workers32,33 and subsequently the crystal structures of [Zn(S2CNEt2)2(NHiBu2)],31, [Zn(S2CNEt2)2(NHC5H10)]34 and related five-coordinate complexes16 have been reported. We have no evidence of a six-coordinate complex even in neat oleylamine and as far as we are aware, while six-coordinate adducts with aromatic diamines such as [Zn(S2CNR2)2(2,2′-bipy)] and [Zn(S2CNR2)2(1,10-phen)]35 are well-known, fully authenticated examples of six-coordinate [Zn(S2CNR2)2(amine)2] remain elusive.
:1 complexation of 1 with a range of amines was reported by Saville and co-workers32,33 and subsequently the crystal structures of [Zn(S2CNEt2)2(NHiBu2)],31, [Zn(S2CNEt2)2(NHC5H10)]34 and related five-coordinate complexes16 have been reported. We have no evidence of a six-coordinate complex even in neat oleylamine and as far as we are aware, while six-coordinate adducts with aromatic diamines such as [Zn(S2CNR2)2(2,2′-bipy)] and [Zn(S2CNR2)2(1,10-phen)]35 are well-known, fully authenticated examples of six-coordinate [Zn(S2CNR2)2(amine)2] remain elusive.
 | ||
| Scheme 1 Changes to 1 upon dissolution in xylene and oleylamine. | ||
(iii) In situ XAS studies of the decomposition of 1
XAS spectra for the decomposition of 1 in oleylamine are shown in Fig. 5. XANES (Fig. 5a) spectra shows that [Zn(S2CNMe2)2(RNH2)] (1.RNH2) is stable up to 71 °C. Above this temperature, the peak previously labelled B decreases significantly along with a slight reduction in C. The edge position remains remarkably constant as a result of the stable oxidation state, while a slight drop in white line intensity may indicate a decrease in overall coordination.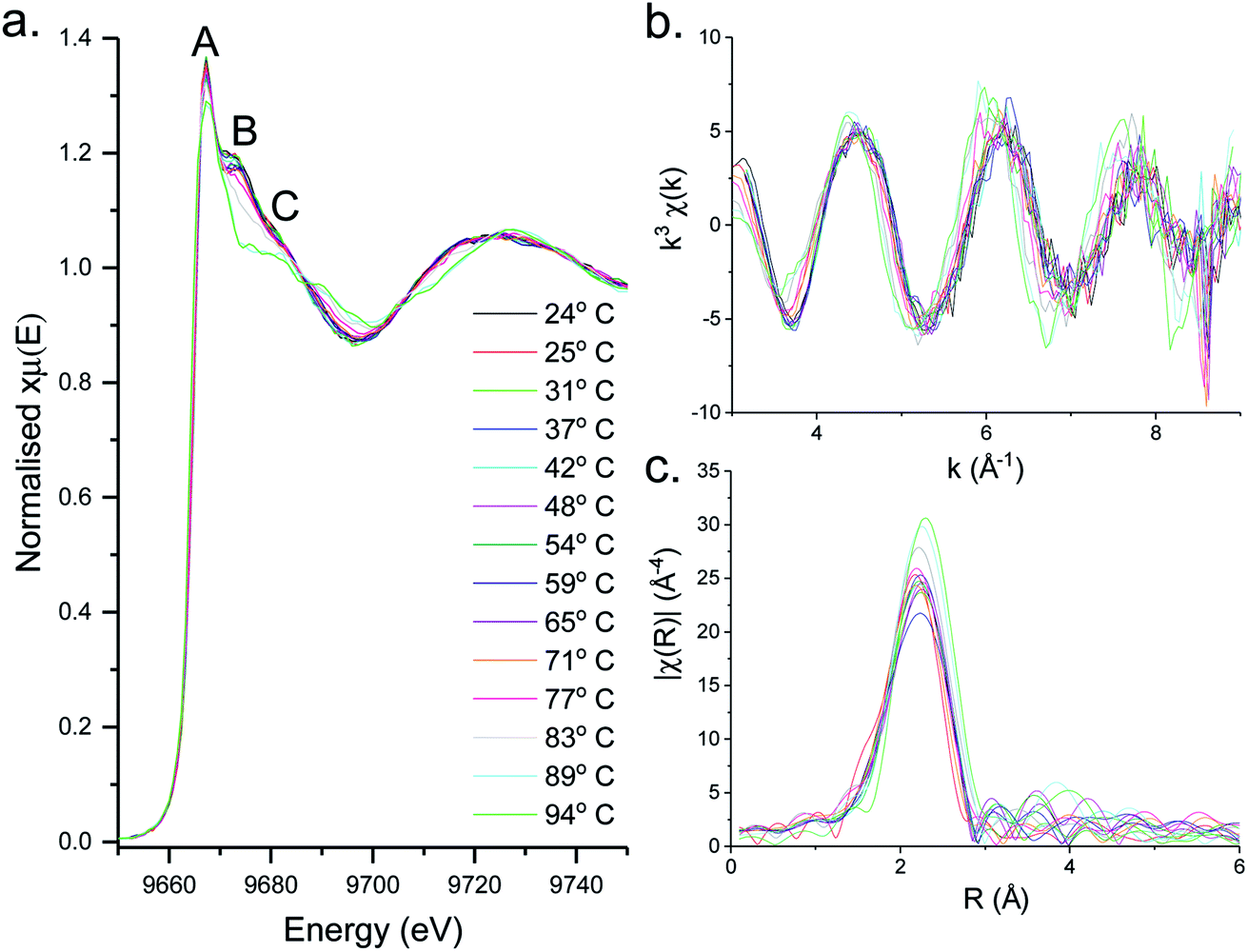 | ||
| Fig. 5 In situ (a) XANES spectra, (b) EXAFS and (c) FT of the solvothermal decomposition of 1 in oleylamine up to 94 °C at a ramp rate of 1 °C min−1. The relevance of the peaks marked (A–C) is discussed in the text. | ||
EXAFS and FT data (Fig. 6) also suggest changes above ca. 71 °C (Table 2). Thus data best fits to 0.84 bound oleylamine ligands with an interatomic distance of 2.06 Å, in addition to four Zn–S bonds at 2.29 Å, and the disorder is slightly reduced. At 77 and 83 °C there are only 0.73 and 0.63 Zn–N bond distances respectively at 2.06 Å, and four Zn–S bond distances at 2.32 Å. By 89 °C, while the four Zn–S distances remain at 2.32 Å, zinc–oleylamine interactions are no longer detected, suggesting that coordination of the amine is lost. A further lengthening of the four Zn–S bond distances to 2.33 Å occurs at 94 °C the Zn–S bond distance of 2.33 Å (±0.03 Å) being similar to those found in both wurtzite (2.334 Å) and sphalerite (2.347 Å).
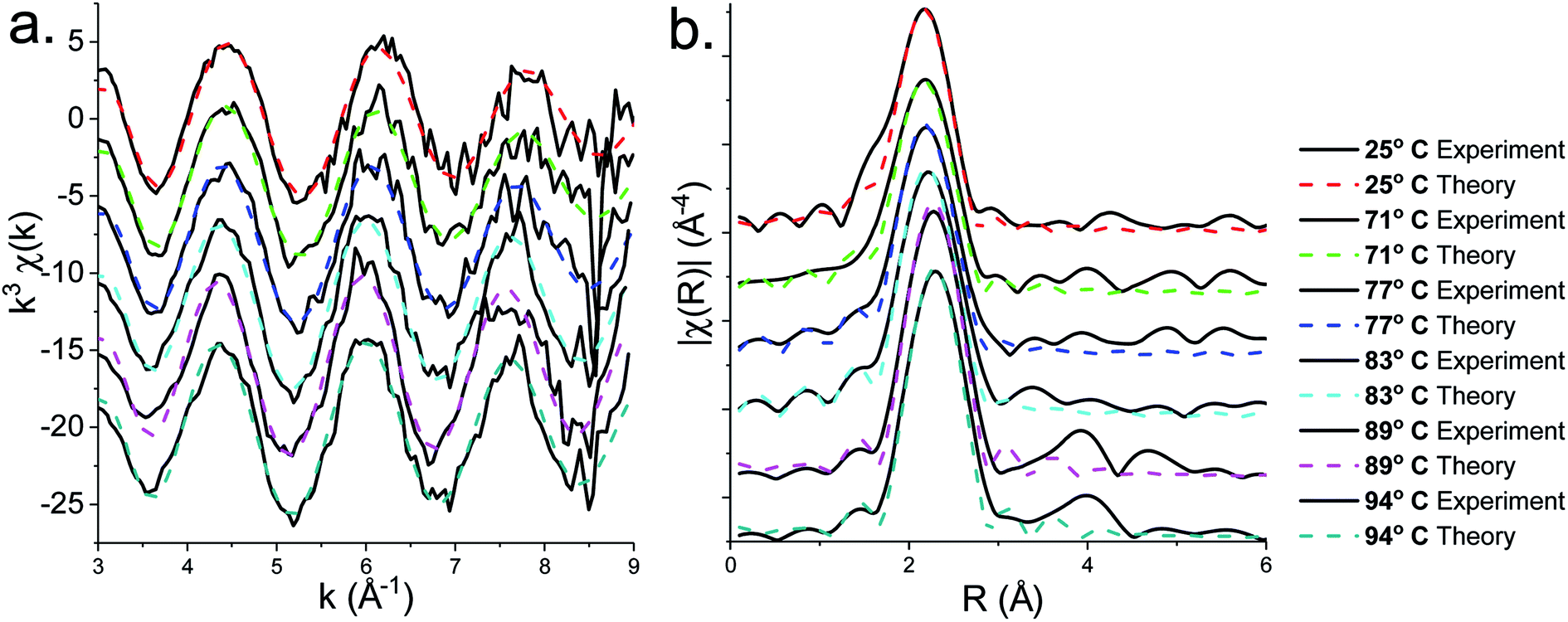 | ||
| Fig. 6 (a) EXAFS and (b) FT fits for the solvothermal decomposition of 1 in oleylamine at 25 °C, 71 °C, 77 °C, 83 °C, 89 °C and 94 °C. | ||
| Temperature (°C) | Scatter | N | R EXAFS (Å) | 2σ2 (Å2) | F |
|---|---|---|---|---|---|
| 71 | N | 0.84 | 2.06 ± 0.02 | 0.019 | 27 |
| S | 4 | 2.29 ± 0.03 | 0.019 | ||
| 77 | N | 0.73 | 2.06 ± 0.02 | 0.016 | 27 |
| S | 4 | 2.30 ± 0.03 | 0.016 | ||
| 83 | N | 0.63 | 2.06 ± 0.02 | 0.013 | 23 |
| S | 4 | 2.32 ± 0.03 | 0.013 | ||
| 89 | S | 4 | 2.32 ± 0.03 | 0.009 | 28 |
| 94 | S | 4 | 2.33 ± 0.03 | 0.009 | 26 |
Thus it appears that the decomposition takes place in two steps. Firstly, oleylamine dissociates from the zinc at ca. 70 °C, and following this decomposition occurs suddenly. In support of this low temperature transformation, Shen and co-workers have recently reported that decomposition of [Zn(S2CNEt2)2] in BuNH2 occurs at 60 °C, giving wurtzite nanosheets,18 although it is a slow process (ca. 10 h). Such low temperature decomposition (as discussed below) is associated with amine-exchange at the dithiocarbamate backbone.22,39
(iv) Decomposition mechanism
Despite the widespread use of dithiocarbamate complexes as SSPs, relatively little is known regarding the decomposition mechanism(s).22,23,36–39 Here we first address the molecular transformations of the precursors. It is clear from our XAS measurements that dissolution of 1 leads to a breakdown of the dimeric solid-state structure to afford a tetrahedral monomer and in oleylamine this binds one equivalent of amine to afford 5-coordinate [Zn(S2CNMe2)2(RNH2)] (1.RNH2). As detailed above, Lee and co-workers studied the decomposition of 1 in octylamine by NMR spectroscopy22 and suggest that the key decomposition species is [Zn(S2CNMe2)2(RNH2)2] (R = octyl), in which free and bound octylamine are in rapid exchange. They isolated this as a waxy solid and studied its thermal decomposition in d6-dmso and d8-toluene by variable temperature NMR spectroscopy. We question assignment of this waxy solid as [Zn(S2CNMe2)2(RNH2)2] since we have no evidence of a bis(adduct) in our XAS work and favour formation of 5-coordinate 1.RNH2 which at higher temperatures loses the amine; as might be expected on entropic grounds.In Lee's experiments on the decomposition of putative [Zn(S2CNMe2)2(RNH2)2] zwitterionic complexes X and Y, resulting from nucleophilic addition of the primary amine to the backbone carbon of the dithiocarbamate, are suggested to be observed by NMR spectroscopy (Scheme 2). From our work on the decomposition of [Ni(S2CNBui2)2] in oleylamine39 we suggest that resonances assigned to X and Y are actually associated with the thioureas, Me2NC(S)NH(Oct) and (Oct)NHC(S)NH(Oct) respectively. This supposition is consistent with the work of Reedijk and co-workers who isolated thioureas, Me2NC(S)NH(Hex) and (Hex)NHC(S)NH(Hex), from the decomposition of 1 in hexylamine.23 Nevertheless, the key premise of Lee and co-workers, namely that the amine attacks the backbone carbon and this leads to exchange NMe2 for NHR, is in accord with the findings of Reedijk22 and our experiments on nickel.39 Thus the key decomposition intermediates are [Zn(S2CNMe2)(S2CNHR)] and [Zn(S2CNHR2)2] resulting from exchange of one or both NMe2 groups. While the former have never been isolated, O'Brien and co-workers have prepared and crystallographically characterised [Zn(S2CNHHex2)2] which decomposes to nanoparticulate zinc blende at 300 °C in hexadecylamine.19
 | ||
| Scheme 2 Lee's proposed intramolecular amine attack at the dithiocarbamate carbons. | ||
Based on our current observations and those of others we propose a mechanism for the molecular transformations leading to the degradation of 1 (and other zinc dithiocarbamate complexes with secondary amine backbones) in solutions of primary amines (Scheme 3). Initial binding of the primary amine to zinc affords a 5-coordinate amine complex; which at higher temperatures (ca. 70 °C) is reversible. At ca. 90 °C, nucleophilic attack of primary amine at a backbone carbon leads to amine-exchange to afford [Zn(S2CNMe2)(S2CNHR)] and (depending upon the rate of decomposition of this) possibly the bis(primary amine) derivatives [Zn(S2CNHR2)2]. We have previously suggested that for related nickel complexes amine-exchange and decomposition are in competition but for zinc it appears that decomposition is rapid once a single amine-exchange has occurred.39 Thus formation of [Zn(S2CNMe2)(S2CNHR)] alone is enough to trigger ZnS formation; deprotonation of the dithiocarbamate leading to a zwitterionic dithiocarbimate which can extrude isothiocyanate.
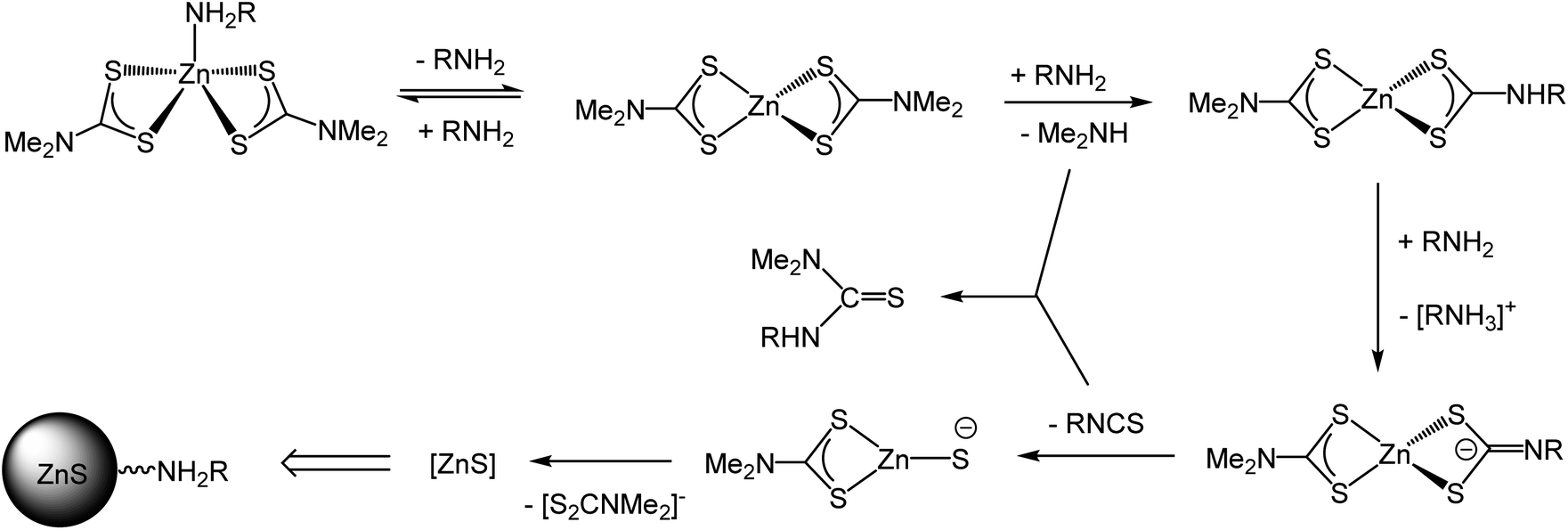 | ||
| Scheme 3 Proposed mechanism for the formation of ZnS from the thermal decomposition of 1 in primary amines. | ||
(v) Nanoparticle morphology
A wide range of different nanoparticulate forms of ZnS have been prepared2 and it is not obvious why decomposition of [Zn(S2CNR2)2] in primary amines gives rise to high aspect nanowires. The amine (and indeed other solvents) plays multiple roles in the decomposition of SSPs including; interacting with molecular species, controlling the rate of growth and final particle size distribution, preventing particle aggregation and interacting with surface sites to passivate surface defects.40 The XAS studies show that the amine binds to [Zn(S2CNR2)2] precursors, and most likely all other molecular species, as Zn(II) is a good Lewis acid. Most likely the surface of the formed nanowires will also have close to 100% amine coverage as the small size of primary amines allows much higher surface coverage than other solvent-capping agents such as tri-n-octylphosphine oxide (TOPO).41 Three separate explanations have been put forward for the formation of high aspect ZnS nanowires from [Zn(S2CNR2)2]/RNH2 mixtures. Wang11 has proposed that nanowire formation is due to the selective absorption of the amine onto the (100) planes of the developing material, thus inhibiting growth in this dimension; supporting evidence coming from the formation of much wider wires upon addition of oleic acid to the decomposition mixture. Jia and Banin have attributed nanowire formation with the dissolution of monomers from the end facets of the initially formed nanoparticles, being followed by their secondary growth onto the side facets of the particles.12 Certainly monomers on the end of the wires will have lower coordination numbers than those in the bulk and hence might be expected to be more labile. Shen and co-workers offer a third view, namely that nanowires result from the loose aggregation of initially formed ellipse-like nanostructures,14 finding that formation of ultrathin nanowires is highly dependent upon the reaction medium; addition of PPh3 to the amine giving longer nanowires, while in neat PPh3 spherical nanoparticles result. We favour this explanation and note that in non-coordinating solvents, or in amine solutions at lower temperatures, the same precursors give spherical nanoparticles.15–17Summary and conclusions
The synthesis of ultrathin ZnS nanowires continues to attract considerable interest11–14,42,43 and the decomposition of zinc dithiocarbamate complexes in primary amines at temperatures of ca. 250 °C is proving to be a simple and valuable strategy to these materials.11–14 Here we add a new dithiocarbamate complex, [Zn(S2CNiBu2)2] (2), to the available SSPs and show that its decomposition in oleylamine at 230 °C cleanly affords high aspect ratio wurtzite nanowires with an average particle length of 34.3 nm and width of 3.3 nm. Addition of the thiuram disulfide, (S2CNiBu2)2 (3) results in a slight lengthening (av. 45.0 nm) of the wires, while the width remained unaffected. In order to gain further insight into the role of the amine in this process, in situ XAS studies were carried out on the simplest homologue, [Zn(S2CNMe2)2] (1), showing that five-coordinate [Zn(S2CNMe2)2(RNH2)] (1.RNH2) predominated in the amine solution, being stable up to 70 °C but decomposing above 90 °C. Combining our own observations with those of others22,23 has allowed us to piece together a molecular decomposition mechanism, the key feature of which is rate-determining amine-exchange at the backbone carbon of the dithiocarbamate to yield complexes [Zn(S2CNR2)(S2CNHR)] and [Zn(S2CNHR2)2] with one and two primary dithiocarbamate ligands respectively. Most likely once a single exchange has occurred decomposition, promoted via deprotonation of the ligand and extrusion of isothiocyanate, will be rapid. Thus onset of ZnS formation (ca. 90 °C) is far below that of the nanowire formation and this is likely a result of the aggregation of the initially formed spherical nanoparticles.In previous work we have shown that the amine-exchange process is key to the facile decomposition of nickel SSPs, [Ni(S2CNR2)2]39 and others have also previously shown that the primary amine dithiocarbamate complexes, [M(S2CNHR)n], have significantly lower decomposition temperatures than their secondary amine counterparts.19,39 Thus, the rate of decomposition of different [Zn(S2CNR2)2] SSPs in primary amines most likely depends upon their relative rates of secondary for primary amine exchange, which relates to both the electrophility of the backbone carbon and (likely most importantly) to the steric size of the secondary amine substituents as in the transition state the backbone carbon becomes sp3-hydridised.39 Thus the primary amine plays multiple roles in the decomposition process; (i) solubilises the SSP, (ii) acts as a heat sink, (iii) coordinates to the Lewis acid Zn(II) in all molecular species, (iv) acts as a nucleophile resulting in amine exchange, (v) acts as a base to deprotonate the primary amine dithiocarbamate, (vi) drives the reaction by combining with the extruded isothiocyanate and (vii) acts as a capping agent for the generated nanomaterials.
From this work and our previous work on nickel39 it is clear that it is not the secondary amine dithiocarbamate complexes [M(S2CNR2)n] that are the actual precursors to nanoparticulate metal sulfides but rather these are precursors to species with primary amine dithiocarbamate ligands. Indeed there is mounting evidence that this is the case44 and given this then further study on the synthesis, stability and decomposition of primary amine dithiocarbamate complexes [M(S2CNHR)n] is warranted, and this is a direction of our current research. In this context an interesting recent development from Prasad and co-workers should be noted.45 They generated zwitterionic [OleylNH3][OleylNHCS2] in situ upon addition of carbon disulfide to oleylamine solutions, it reacting rapidly with the metal salt to generate in situ primary amine dithiocarbamate complexes, [M(S2CNHOleyl)n], which decompose readily and cleanly to nanoparticulate metal sulfides.
Experimental
All 1H and 13C{1H} NMR spectra were obtained on either a Bruker Avance III 400 or Avance 600 spectrometer, the latter being equipped with a cryoprobe. All spectra were recorded using CDCl3 which was dried and degassed over molecular sieves prior to use; 1H and 13C{1H} chemical shifts are reported relative to SiMe4. The mass spectra were obtained using either Micromass 70-SE spectrometer using Electron Ionisation (EI) or a Thermo Finnigan MAT900xp spectrometer using Fast Atom Bombardment (FAB). Elemental analysis was carried using Elemental Analyser (CE-440) (Exeter Analytical Inc).PXRD patterns were measured on a Brucker AXS D4 diffractometer using Cu Kα1 radiation. The diffraction patterns obtained were compared to database standards. For TEM characterisation a 4 μL droplet of nanoparticle suspension (chloroform) was placed on a holey carbon-coated copper TEM grid and allowed to evaporate in air under ambient laboratory conditions for several minutes. TEM images were obtained using a JEOL-1010 microscope at 100 kV equipped with a Gatan digital camera. HRTEM measurements were collected using a Jeol 2100 TEM with a LaB6 source operating at an acceleration voltage of 200 kV. Micrographs were taken on a Gatan Orius charge-coupled device (CCD).
Synthesis of precursors
[Zn(S2CNMe2)2] (1)46 and (S2CNiBu2)2 (3)39 were prepared by literature procedures. While [Zn(S2CNiBu2)2] (2) is in the literature46,47 full synthetic details and characterisation data are not clearly presented. iBu2NH (2.62 mL, 15 mmol) was added to NaOH (0.60 g, 15 mmol) dissolved in water (100 mL). To this mixture CS2 (0.90 mL, 15 mmol) was added dropwise over 10 minutes and the mixture stirred overnight. ZnSO4·7H2O (1.44 g, 5 mmol) was added yielding an off-white powder, which was filtered, washed with water and dried. 2.09 g, 88%. Anal. calc. for C18H36N2S4Zn: C, 45.60; H, 7.65; N, 5.91. Found: C, 46.61; H, 7.97; N, 5.59. 1H NMR (CDCl3): δ 0.97 (d, J = 6.6 Hz, 24H, CH3), 2.39 (m, J = 6.8 Hz, 4H, CH), 3.70 (d, J = 7.5 Hz, 8H, CH2). 13C{1H} NMR (CDCl3): 20.3 (CH3), 27.1 (CH), 62.2 (CH2), 204.5 (CS2) ppm. MS: m/z 472 [M+], 268 [M+ − S2CNiBu2], 383 [ZnS(SCNiBu2)2 − iBu].Decomposition studies
[Zn(S2CNiBu2)2] (5 mM) was added to oleylamine (20 mL) in a three-neck round bottom flask attached to a condenser and evacuated and refilled with nitrogen repeatedly for 15 minutes. The solution was heated to 230 °C and held there for 1 h. The mixture was allowed to cool to room temperature slowly, whereupon methanol (80 mL) was added with stirring. The mixture was centrifuged and then the solution decanted leaving behind the resultant nanoparticles. This procedure was repeated three times and finally the material was allowed to dry in air.XAS studies
XAS spectra were acquired at the iron K-edge (7112 eV) on the Dutch-Belgian EXAFS beamline, BM26A.48 Monochromatic radiation was supplied by a double Si(111) crystal, ion chambers were used to measure incident and transmitted beam intensities (I0 and It), and X-ray fluorescence was measured using a 9 element germanium solid state detector. Solid 1 was diluted with polyvinylpyrrolidone, pelletized and placed in the beam. Solutions of 1 were held within in situ liquid cells. The in situ liquid cell used was developed at UCL for synchrotron based experiments on liquid samples. Cartridge heaters embedded into the conductive cell body allow temperatures to reach up to 200 °C, subject to pressure buildup of the system. The Kapton sealed reaction chamber holds 400 μL of solution with a fixed path length of 2 mm. The in situ liquid microtron cell was developed by Sample Environment at the ESRF for liquid experiments up to 260 °C. With quartz cell windows and a 4 mm path length, the cell was used for measurements in transmission. XAS data was normalized and background subtracted using Horae Athena software.49 Linear combination analyses were also performed using Horae Athena. Detailed EXAFS analyses were performed on Excurve Version 9.273.50Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding and beamline staff at DUBBLE, ESRF for their technical support and the Netherlands Organisation for Scientific Research (NWO) for making beam time available on the DUBBLE beamlines at the ESRF. WB's contribution is based upon work supported by Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the U.S. Department of Energy.References
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- (a) X. Fang, L. Wu and L. Hu, Adv. Mater., 2011, 23, 585–598 CrossRef CAS PubMed; (b) L.-W. Yin, Y. Bando, J.-H. Zhan, M.-S. Li and D. Golberg, Adv. Mater., 2005, 17, 1972–1977 CrossRef CAS; (c) S. Hamad, S. M. Woodley and C. R. A. Catlow, Mol. Simul., 2009, 35, 1015–1032 CrossRef CAS; (d) A. Tiwari and S. J. Dhoble, Cryst. Growth Des., 2017, 17, 381–407 CrossRef CAS; (e) A. Tiwari and S. J. Dhoble, RSC Adv., 2016, 6, 64400–64420 RSC; (f) Z. Jiang, H. Sun, Z. Qin, X. Jiao and D. Chen, Chem. Commun., 2012, 48, 3620–3622 RSC; (g) W.-H. Zhang and W.-D. Zhang, Mater. Lett., 2013, 98, 5–7 CrossRef CAS; (h) C. Feigl, S. P. Russo and A. S. Barnard, J. Mater. Chem., 2010, 20, 4971–4980 RSC.
- (a) M.-R. Gao, J. Jiangand and S.-H. Yu, Small, 2012, 8, 13–27 CrossRef CAS PubMed; (b) D. Fan, M. Afzaal, M. A. Mallik, C. Q. Nguyen, P. O'Brien and P. J. Thomas, Coord. Chem. Rev., 2007, 251, 1878–1888 CrossRef CAS; (c) M. Afzaal, M. A. Malik and P. O'Brien, J. Mater. Chem., 2010, 20, 4031–4040 RSC; (d) L. G. Bloor, C. J. Carmalt and D. J. Pugh, Coord. Chem. Rev., 2011, 255, 1293–1318 CrossRef CAS; (e) J. van Embden, A. S. R. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS; (f) K. Ramasamy, M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2013, 25, 3551–3569 CrossRef CAS; (g) N. L. Pickett and P. O'Brien, Chem. Rec., 2001, 1, 467–479 CrossRef CAS PubMed; (h) P. O'Brien, M. A. Malik, M. Chunggaze, T. Trindade, J. R. Walsh and A. C. Jones, J. Cryst. Growth, 1997, 170, 23–29 CrossRef; (i) M. A. Azad, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef PubMed; (j) N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS; (k) M. Lazell, P. O'Brien, D. J. Otway and J.-H. Park, Dalton Trans., 2000, 4479–4486 RSC.
- G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71–561 CrossRef CAS.
- (a) R. Nomura, K. Miyawaki, T. Toyosaki and H. Matsuda, Chem. Vap. Deposition, 1996, 2, 174–179 CrossRef CAS; (b) M. Kemmler, M. Lazell, P. O'Brien, D. J. Otway, J.-H. Park and J. R. Walsh, J. Mater. Sci.: Mater. Electron., 2002, 13, 531–535 CrossRef CAS; (c) M. J. Almond, H. Redman and D. A. Rice, J. Mater. Chem., 2000, 10, 2842–2846 RSC; (d) A. Birri, B. Harvey, G. Hogarth, E. Subasi and F. Ugur, J. Organomet. Chem., 2007, 692, 2448–2455 CrossRef CAS; (e) F. Srouji, M. Afzaal, J. Waters and P. O'Brien, Chem. Vap. Deposition, 2005, 11, 91–94 CrossRef CAS; (f) P. O'Brien, D. J. Otway and J.-H. Park, Mater. Res. Soc. Symp. Proc., 2000, 606, 133–138 CrossRef; (g) O. C. Monteiro, H. I. S. Nogueira and T. Trindade, Chem. Mater., 2001, 13, 2103–2111 CrossRef CAS; (h) P. O'Brien, D. J. Otway and J. R. Walsh, Thin Solid Films, 1998, 315, 57–61 CrossRef.
- (a) D. M. Frifo, O. F. Z. Khan and P. O'Brien, J. Cryst. Growth, 1989, 96, 989–992 CrossRef; (b) J. Wang, J. R. Hyde, J. W. Wilson, K. Mallik, P. J. Sazio, P. O'Brien, M. A. Malik, M. Afzaal, C. Q. Nguyen, M. W. George, S. M. Howdle and D. C. Smith, Adv. Mater., 2009, 21, 4115–4119 CrossRef; (c) B. Ludolph, M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Commun., 1998, 1849–1850 RSC.
- (a) W. Han and M. Y. Gao, Cryst. Growth Des., 2008, 8, 1023–1030 CrossRef CAS; (b) Y. Zhang, Y. Du and Q. Wang, CrystEngComm, 2010, 12, 3658–3663 RSC; (c) M. Akhtar, J. Akhter, M. A. Malik, P. O'Brien, F. Tuna, J. Raftery and M. Helliwell, J. Mater. Chem., 2011, 21, 9737–9745 RSC; (d) M. Akhtar, A. L. Abdelhady, M. A. Malik and P. O'Brien, J. Cryst. Growth, 2012, 346, 106–112 CrossRef CAS.
- (a) A. Adeogun, M. Afzaal and P. O'Brien, Chem. Vap. Deposition, 2006, 12, 597–599 CrossRef CAS; (b) Y. Lou, A. C. S. Samia, J. Cowen, K. Banger, X. Chen, H. Lee and C. Burda, Phys. Chem. Chem. Phys., 2003, 5, 1091–1095 RSC; (c) K. Ramasamy, V. L. Kuznetsov, K. Gopal, M. A. Malik, J. Raftery, P. P. Edwards and P. O'Brien, Chem. Mater., 2013, 25, 266–276 CrossRef CAS; (d) M. Afzaal, C. L. Rosenberg, M. A. Malik, A. J. P. White and P. O'Brien, New J. Chem., 2011, 35, 2773–2780 RSC; (e) E. P. C. Higgins, S. G. McAdams, D. G. Hopkinson, C. Byrne, A. S. Walton, D. J. Lewis and R. A. W. Dryfe, Chem. Mater., 2019, 31, 5384–5391 CrossRef CAS; (f) N. Zeng, D. G. Hopkinson, B. F. Spencer, S. G. McAdams, A. A. Tedstone, S. J Haigh and D. J. Lewis, Chem. Commun., 2019, 55, 99–102 RSC.
- (a) P. O'Brien and J. Waters, Chem. Vap. Deposition, 2006, 12, 620–626 CrossRef; (b) H. Cui, R. D. Pike, R. Kershaw, K. Dwight and A. Wold, J. Solid State Chem., 1992, 101, 115–118 CrossRef CAS; (c) G. H. Singhal, R. I. Botto, L. D. Brown and K. S. Colle, J. Solid State Chem., 1994, 109, 166–171 CrossRef CAS; (d) X. Chen, Z. Wang, X. Wang, J. Wan, J. Liu and Y. Qian, Chem. Lett., 2004, 33, 1294–1295 CrossRef CAS; (e) R. Chauhan, M. Trivedi, J. Singh, K. C. Molloy, G. Kociok-Köhn, U. P. Mulik, D. P. Amalnerkar and A. Kumar, Inorg. Chim. Acta, 2014, 415, 69–74 CrossRef CAS.
- (a) M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2001, 13, 913–920 CrossRef CAS; (b) T. Trindade, P. O'Brien and X.-M. Zhang, Chem. Mater., 1997, 9, 523–530 CrossRef CAS; (c) D. C. Onwudiwe and P. A. Ajibade, Int. J. Mol. Sci., 2011, 12, 5538–5551 CrossRef CAS PubMed; (d) D. C. Onwudiwe and P. A. Ajibade, Mater. Lett., 2011, 65, 3258–3261 CrossRef CAS; (e) P. A. Ajibade, D. C. Onwudiwe and M. J. Moloto, Polyhedron, 2011, 30, 246–252 CrossRef CAS; (f) E. Y. M. Lee, N. H. Tran and R. N. Lamb, Appl. Surf. Sci., 2005, 241, 493–496 CrossRef CAS; (g) M. Motevalli, P. O'Brien, J. R. Walsh and I. M. Watson, Polyhedron, 1996, 15, 2801–2808 CrossRef CAS; (h) P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart and S. R. P. Silva, J. Cryst. Growth, 1996, 167, 133–142 CrossRef; (i) R. Nomura, T. Murai, T. Topyosaki and H. Matsuda, Thin Solid Films, 1995, 271, 4–7 CrossRef CAS; (j) P. O'Brien, J. R. Walsh, I. M. Watson, M. Motevalli and L. Henrikson, J. Chem. Soc., Dalton Trans., 1996, 2491–2496 RSC; (k) D. Zeng, M. J. Hampden-Smith, T. M. Alam and A. L. Rheingold, Polyhedron, 1994, 13, 2715–2730 CrossRef CAS; (l) R. D. Pike, H. Cui, R. Kershaw, K. Dwight, A. Wold, T. N. Blanton, A. A. Wernberg and H. J. Gysling, Thin Solid Films, 1992, 224, 221–226 CrossRef CAS.
- Y. Zhang, H. Xu and Q. Wang, Chem. Commun., 2010, 46, 8941–8943 RSC.
- G. Jia and U. Banin, J. Am. Chem. Soc., 2014, 136, 11121–11127 CrossRef CAS PubMed.
- C. J. Barrelet, Y. Wu, D. C. Bell and C. M. Lieber, J. Am. Chem. Soc., 2003, 125, 11498–11499 CrossRef CAS PubMed.
- G. Zhu, S. Zhang, Z. Xu, J. Ma and X. Shen, J. Am. Chem. Soc., 2011, 133, 15605–15612 CrossRef CAS PubMed.
- L. D. Nyamen, A. A. Nejo, V. S. R. Pullabhotla, P. T. Ndifon, M. A. Malik, J. Akhtar, P. O'Brien and N. Revaprasadu, Polyhedron, 2014, 67, 129–135 CrossRef CAS.
- B. A. Prakasam, M. Lahtinen, A. Peuronen, M. Muruganandham, E. Kolehmainen, E. Haapaniemi and M. Sillanpää, Mater. Lett., 2015, 144, 19–21 CrossRef CAS.
- (a) M. Hrubaru, D. C. Onwudiwe and E. Hosten, J. Sulfur Chem., 2016, 37, 37–47 CrossRef CAS; (b) P. A. Ajibade, D. C. Onwudiwe and M. J. Moloto, Polyhedron, 2011, 30, 248–252 CrossRef; (c) D. C. Onwudiwe and P. A. Ajibade, Mater. Lett., 2011, 65, 3258–3261 CrossRef CAS.
- G. Zhu, J. Yang, C. Bao, X. Zhang, Z. Ji, S. Wu and X. Shen, J. Colloid Interface Sci., 2016, 468, 138–144 Search PubMed.
- A. A. Memon, M. Afzaal, M. A. Malik, C. Q. Nguyen, P. O'Brien and J. Raftery, Dalton Trans., 2006, 4499–4505 RSC.
- (a) K. Ramasamy, M. A. Malik and P. O'Brien, Chem. Commun., 2012, 48, 5703–5714 RSC; (b) K. Ramasamy, M. A. Malik and P. O'Brien, Chem. Sci., 2011, 2, 1170–1172 RSC; (c) O. Stroyuk, A. Raevskaya, O. Selyshchev, V. Dzhagen, N. Gaponik, D. R. T. Zahn and A. Eychmüller, Sci. Rep., 2018, 8, 1–10 CrossRef CAS PubMed; (d) A. Khare, A. W. Wills, L. M. Ammerman, D. J. Norris and E. S. Aydil, Chem. Commun., 2011, 47, 11721–11723 RSC; (e) M. D. Regulacio, C. Ye, S. H. Lim, M. Bosman, E. Ye, S. Chen, Q.-H. Xu and M.-Y. Han, Chem.–Eur. J., 2012, 18, 3127–3131 CrossRef CAS PubMed; (f) B. D. Chernomordik, A. E. Beland, D. D. Deng, L. F. Francis and S. Eray, Chem. Mater., 2014, 26, 3191–3201 CrossRef CAS; (g) A. Roffey, N. Hollingsworth and G. Hogarth, Nanoscale Adv., 2019, 1, 3056–3066 RSC; (h) B. D. Chernomordik, A. E. Béland, N. D. Trejo, A. A. Gunawan, D. D. Deng, K. A. Mkhoyan and E. S. Aydil, J. Mater. Chem. A, 2014, 2, 10389–10395 RSC; (i) H. Zheng, X. Li, K. Zong, Y. Sun, J. Liu, H. Wang and H. Yan, Mater. Lett., 2013, 110, 1–3 CrossRef CAS.
- (a) J. R. Dethlefsen and A. Døssing, Nano Lett., 2011, 11, 1964–1969 CrossRef CAS PubMed; (b) A. Polovitsyn, Z. Dang, J. L. Movilla, B. Martín-García, A. H. Khan, G. H. V. Bertrand, R. Brescia and I. Moreels, Chem. Mater., 2017, 29, 5671–5680 CrossRef CAS; (c) H.-J. Pan, C.-W. Lai, S.-W. Chou and P.-T. Chou, Mater. Express, 2012, 2, 224–232 CrossRef CAS; (d) L. Xi, D.-Y. Cho, M. Duchamp, C. B. Boothroyd, J. Y. Lek, A. Besmehn, R. Waser, Y. M. Lam and B. Kardynal, ACS Appl. Mater. Interfaces, 2014, 6, 18233–18242 CrossRef CAS PubMed; (e) J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598–1601 CrossRef CAS PubMed; (f) J. Ziegler, S. Xu, E. Kucur, F. Meister, M. Batentschuk, F. Gindele and T. Nann, Adv. Mater., 2008, 20, 4068–4073 CrossRef CAS; (g) S. Xu, J. Ziegler and T. Nann, J. Mater. Chem., 2008, 18, 2653–2656 RSC; (h) X. Yang, D. Zhao, K. S. Leck, S. T. Tan, Y. X. Tang, J. Zhao, H. V. Demir and X. W. Sun, Adv. Mater., 2012, 24, 4180–4185 CrossRef CAS PubMed; (i) D. Chen, F. Zhao, H. Qi, M. Rutherford and X. Peng, Chem. Mater., 2010, 22, 1437–1444 CrossRef CAS; (j) W. Zhang, G. Chen, J. Wang, B.-C. Ye and X. Zhong, Inorg. Chem., 2009, 48, 9723–9731 CrossRef CAS PubMed; (k) H. Wang, H. Nakamura, M. Ushara, Y. Yamaguchi, M. Miyazaki and H. Maeda, Adv. Funct. Mater., 2005, 15, 603–608 CrossRef CAS; (l) S. Shen, Y. Zhang, L. Peng, Y. Du and Q. Wang, Angew. Chem., Int. Ed., 2011, 50, 7115–7118 CrossRef CAS PubMed; (m) M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Mater., 2002, 14, 2004–2010 CrossRef CAS.
- Y. K. Jung, J. I. Kim and J. K. Lee, J. Am. Chem. Soc., 2010, 132, 178–184 CrossRef CAS PubMed.
- A. Dirksen, P. J. Nieuwenhuizen, M. Hoogenraad, J. G. Haasnoot and J. Reedijk, J. Appl. Polym. Sci., 2001, 79, 1074–1083 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- A. Roffey, N. Hollingsworth, H.-U. Islam, W. Bras, G. Sankar, N. H. de Leeuw and G. Hogarth, Nanoscale Adv., 2019, 1, 2965–2978 RSC.
- (a) C. C. Ashworth, N. A. Bailey, M. Johnson, J. A. McCleverty, N. Morrison and B. Tabbiner, J. Chem. Soc., Chem. Commun., 1976, 743–744 RSC; (b) J. A. McCleverty, N. Spencer, N. A. Bailey and S. L. Shackleton, J. Chem. Soc., Dalton Trans., 1980, 1939–1944 RSC.
- K. Hagen, C. J. Holwill and D. A. Rice, Inorg. Chem., 1989, 28, 3239–3242 CrossRef CAS.
- H. Klug, Acta Crystallogr., 1966, 21, 536–546 CrossRef.
- A. Decken, R. A. Gossage, M. Y. Chan, C. Lai and E. R. T. Tiekink, Appl. Organomet. Chem., 2004, 18, 101–102 CrossRef CAS.
- A. M. Malik, M. Motavalli and P. O'Brien, Polyhedron, 1999, 18, 1259–1262 CrossRef.
- A. V. Ivanov, A. S. Zaeva, E. V. Novikova, A. V. Gerasimenko and W. Forsling, Russ. J. Coord. Chem., 2007, 33, 233–243 CrossRef CAS.
- G. M. C. Higgins and B. Saville, J. Chem. Soc., 1963, 2812–2817 RSC.
- E. Coates, B. Rigg, B. Saville and D. Skelton, J. Chem. Soc., 1965, 5613–5620 RSC.
- A. V. Ivanov, A. S. Zaeva, A. V. Gerasimenko and T. A. Rodina, Russ. J. Coord. Chem., 2008, 34, 688–698 CrossRef CAS.
- (a) A. Manohar, V. Venkatachalam, K. Ramalingham, S. Thirumaran, G. Bocelli and A. Cantoni, J. Chem. Crystallogr., 1998, 28, 861–866 CrossRef CAS; (b) S. Thirumaran, K. Ramalingam, G. Bocelli and A. Cantoni, Polyhedron, 1999, 18, 925–930 CrossRef CAS; (c) R. F. Klevtsova, L. A. Glinskaya, S. M. Zemskova and S. V. Larionov, Polyhedron, 1999, 18, 3559–3565 CrossRef CAS.
- (a) G. Xie, Z.-P. Qiao, M.-H. Zeng, X.-M. Chen and S.-L. Gao, Cryst. Growth Des., 2004, 4, 513–516 CrossRef CAS; (b) M. Green, L. Sandiford, K. M. Anderson and Y. Ma, ChemPlusChem, 2012, 77, 192–195 CrossRef CAS; (c) G. Barone, T. Chaplin, T. G. Hibbert, A. T. Kana, M. F. Mahon, K. C. Molloy, I. D. Worsley, I. P. Parkin and S. L. Price, J. Chem. Soc., Dalton Trans., 2002, 1085–1092 RSC; (d) M. Chunggaze, J. McAlesse, P. O'Brien and D. J. Otway, J. Chem. Soc., Chem. Commun., 1998, 833–834 RSC; (e) M. Chunggaze, M. A. Malik and P. O'Brien, J. Mater. Chem., 1999, 9, 2433–2437 RSC.
- L. H. van Poppel, T. L. Groy and M. T. Caudle, Inorg. Chem., 2004, 43, 3180 CrossRef CAS PubMed.
- A. Roffey, N. Hollingsworth, H.-U. Islam, M. Mercy, C. R. A. Catlow, G. Sankar, N. H. de Leeuw and G. Hogarth, Nanoscale, 2016, 8, 11067–11075 RSC.
- N. Hollingsworth, A. Roffey, H.-U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. de Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- C. Bullen and P. Mulvaney, Langmuir, 2006, 22, 3007–3013 CrossRef CAS.
- M. Green, J. Mater. Chem., 2010, 20, 5797–5809 RSC.
- S. Kumar, A. Sinhamahapatra, N. Sutradhar, H. C. Bajaj and A. B. Panda, Chem. Commun., 2012, 48, 850–852 RSC.
- (a) Z. Deng, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 8695–8698 CrossRef CAS PubMed; (b) S. K. Pahari, A. Sinhamahapatra, N. Sutradhar, H. C. Bajaj and A. B. Panda, Chem. Commun., 2012, 48, 850–852 RSC; (c) Y. Jaing, X. M. Meng, J. Liu, Z. R. Hong, C. S. Lee and S. T. Lee, Adv. Mater., 2003, 15, 1195–1198 CrossRef; (d) X. S. Fang, Y. Bando, G. Z. Shen, C. H. Ye, U. K. Gautam, P. M. F. J. Cosat, C. Y. Zhi, C. C. Tang and D. Golberg, Adv. Mater., 2007, 19, 2593–2596 CrossRef CAS.
- (a) N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS; (b) L. van Poppel, T. L. Groy and M. T. Caudle, Inorg. Chem., 2004, 43, 3180–3188 CrossRef CAS PubMed; (c) A. W. Wills, M. S. Kang, A. Khare, W. L. Gladfelter and D. J. Norris, ACS Nano, 2010, 4, 4523–4530 CrossRef CAS PubMed; (d) J. J. Grenland, C. Lin, K. Gong, D. F. Kelley and A. M. Kelley, J. Phys. Chem. C, 2017, 121, 7056–7061 CrossRef CAS; (e) A. M. Munro, C. Chandler, M. Garling, D. Chai, V. Popovich, L. Lystrom and S. Kilina, J. Phys. Chem. C, 2016, 120, 29455–29462 CrossRef CAS.
- (a) A. Bera, B. Mandal, P. N. Goswami, A. K. Rath and B. L. V. Prasad, Langmuir, 2018, 34, 5788–5797 CrossRef CAS PubMed; (b) A. Bera, B. Busupalli and B. L. V. Prasad, ACS Sustainable Chem. Eng., 2018, 6, 12006–12016 CrossRef CAS.
- A. M. Bond and A. F. Hollenkamp, Inorg. Chem., 1990, 29, 284–289 CrossRef CAS.
- (a) D. Oktavec, E. Beinrohr, B. Siles, J. Stefanec and J. Garaj, Collect. Czech. Chem. Commun., 1980, 45, 1495–1501 CrossRef CAS; (b) M. Tarique, E-J. Chem., 2011, 8, 2020–2023 CrossRef CAS.
- S. Nikitenko, A. M. Beale, A. M. J. van der Eerden, S. D. M. Jacques, O. Leynaud, M. G. O'Brien, D. Detollenaere, R. Kaptein, B. M. Weckhuysen and W. Bras, J. Synchrotron Radiat., 2008, 15, 632–640 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- N. Binsted, EXCURV98, CCLRC Daresbury Laboratory Computer Program, 1998 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |