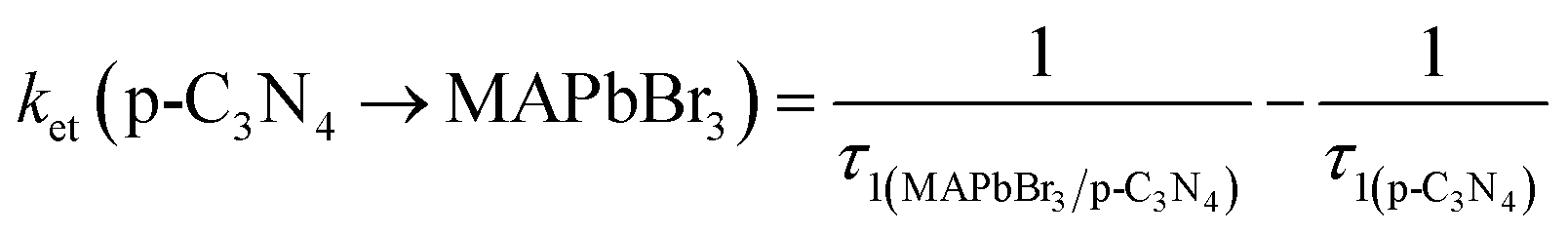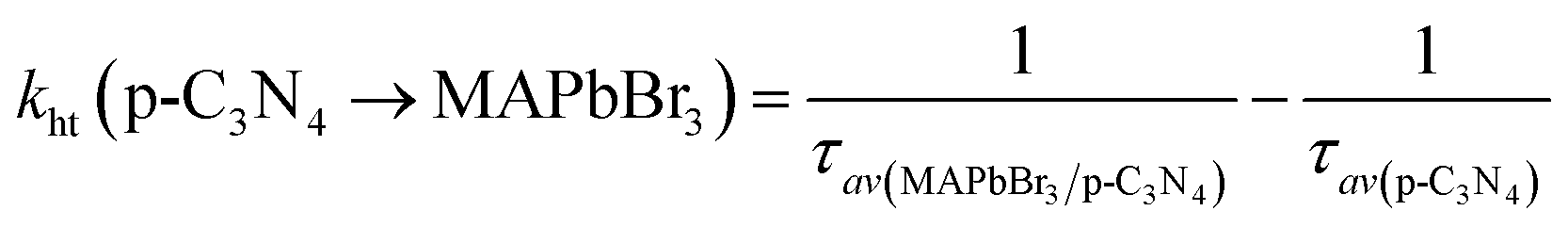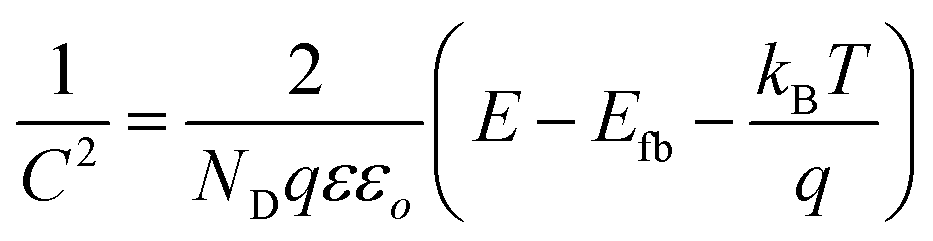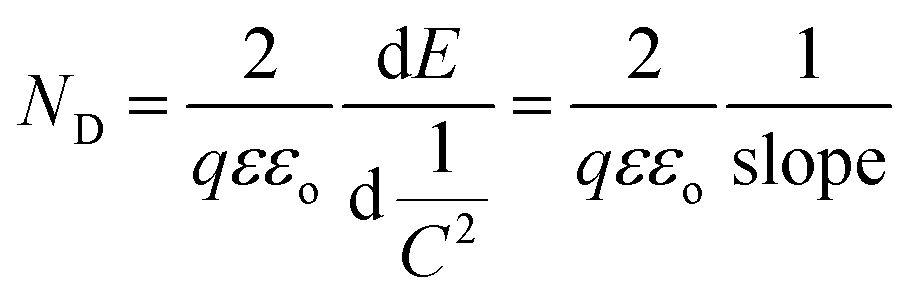DOI:
10.1039/C9NA00634F
(Paper)
Nanoscale Adv., 2020,
2, 274-285
Isopropanol-assisted synthesis of highly stable MAPbBr3/p-g-C3N4 intergrowth composite photocatalysts and their interfacial charge carrier dynamics†
Received
8th October 2019
, Accepted 20th November 2019
First published on 21st November 2019
Abstract
Two phase photocatalysts can be intergrown with each other, resulting in superior photocatalytic properties. Herein, methylamine lead bromide (MAPbBr3) wrapped/entrapped protonated graphitic carbon nitride (p-g-C3N4) intergrowth microcrystals were fabricated by mixing a pervoskite precursor with p-g-C3N4 colloidal sol. A highly stable isopropanol (IPA) solvent based photocatalytic system for dye degradation was demonstrated. The composite with an optimal p-g-C3N4 mass percentage of 3.3 wt% (denoted as MAPbBr3/p-g-C3N4-1.0 mg) exhibited the highest photocatalytic degradation of malachite green (99.8%) within 10 min under visible light, which was 5.3-fold and 16-fold greater than that exhibited by its constituents separately. The strong chemical interaction and fundamental photophysical processes in MAPbBr3/p-g-C3N4 were systematically evaluated by spectroscopic and electrochemical techniques, confirming the effective separation of photogenerated electron–hole pairs and faster interfacial charge transfer behavior. Furthermore, active superoxide radicals (O2˙−) played a vital role in the catalytic reaction, because of the significant photoinduced electron transfer rate (ket) in the inverted type-I core/shell MAPbBr3/p-g-C3N4 band configuration structure. In addition, MAPbBr3/p-g-C3N4 has good cycling stability for 10 cycles and versatility for other cationic (RhB) and anionic (MO) dye pollutants, indicating the great potential for solar energy conversion into chemical energy.
1. Introduction
In 2016, Park and coworkers first introduced the dynamic equilibrium concept utilizing organic–inorganic hybrid halide perovskite (CH3NH3PbX3) catalysts.1 Subsequently, both all inorganic and hybrid perovskites with the general formula APbX3 (A = methylammonium (MA), formamidinium (FA) or cesium (Cs); X = Br, I) were explored in photocatalytic water splitting,2,3 CO2 reduction in ethyl acetate solvent4,5 and selective organic synthesis fields.6,7 Additionally, halide perovskites for the photocatalytic and photoelectrochemical degradation of organic dyes were reported.8–12 For example, CsPbX3 can decompose methyl orange (MO) within 100 min under visible light irradiation.10 Cardenas-Morcoso et al. confirmed that the photocatalytic degradation of a 2-mercaptobenzothiazole (MBT) compound resulted from hole injection from CsPbBr3 nanodots to MBT.11 Moreover, a novel alcohol-based photocatalytic system using lead-free Cs2AgBiBr6 perovskite was proposed for degradation of dyes including rhodamine B (RhB), methyl red (MR), etc.12 However, to the best of our knowledge, literature reports on halide perovskite MAPbBr3 photocatalysts for degradation of dyes in isopropanol (IPA) solvent are still rare.
Because of the ionic migration nature, the inherent instability issues of organic halide perovskites make them generally incompatible with typical photocatalytic reactions in aqueous media. Therefore, special organic alcohol-catalyzed substitutional growth processes13,14 and polymer modification15 were used to prepare highly air (up to ≈1.5 months), thermally and chemically stable halide perovskites with unique surface chemical states. In particular, large MAPbBr3 microcrystals were fabricated through an isopropanol-assisted solvothermal method in our previous report.14 Furthermore, surface modification and defect passivation strategies using carbon-based materials had been investigated to increase the chemical stability, for example, via integrating graphene oxide (GO) with MAPbI3 (ref. 3) and CsPbX3 (ref. 4), and polymeric carbon nitride (C3N4) with CsPbBr3 quantum dots5 and MAPbBr3 nanocrystals.16 The stability enhancement was attributed to the interaction and intimate contact at the interface between the nanocrystals and the encapsulation layer, facilitating strong interfacial electronic hybridization and coupling.15 Although grafting rGO onto MAPbI3 microparticles was reported,3 it is clear that hybrid perovskite microcrystals with high crystallinity will reduce defects, but at the same time the long diffusion distance and reduced active surface area are not beneficial to the photocatalytic activity relative to nanocrystal composites. Recently, two phases of cocatalysts and host photocatalysts were intergrown with each other, which resulted in superior photocatalytic properties.17 Befitting from this two phase intergrowth feature, good contact and strong coupling between cocatalysts and host photocatalysts give rise to efficient charge transfer and surface redox reaction. On the other hand, in perovskite solar cells, new concepts such as incorporating g-C3N4 into perovskite precursor solution were developed to enhance the binding strength in perovskite films and finally improve crystallization and reduce the intrinsic defect density of the perovskite layer.18,19 In addition, as another rising star, graphite-like carbon nitride (g-C3N4) is an important class of metal-free organic photocatalysts that has attracted particular research interest since its first demonstration of photocatalytic H2 production in 2009.20 Further research had been motivated by their tunable band gaps, earth-abundance, low cost of synthesis and ease of surface functionalization. In particular, g-C3N4 is an ideal choice for coupling with perovskite-type metal oxide compounds such as AgNbO3, KTa0.75Nb0.25O3 and bismuth oxyhalide-based semiconductors, and black phosphorus21–24 to form hybrid photocatalysts that greatly hinder the annihilation of charge carriers and extend the life of electrons. However, there are few reports on the preparation of highly stable MAPbBr3/C3N4 hybrid intergrowth microcrystal photocatalysts as well as investigation of interfacial charge carrier dynamics.
Herein, we designed highly crystalline MAPbBr3 microcrystal wrapped/entrapped protonated graphitic carbon nitride (p-g-C3N4) intergrowth structures by a one-step injection of C3N4 sol into the pervoskite precursor without any surfactants. The unique chemical covalent C–N–Br interaction at the interface could eliminate the defects and enhance the stability of MAPbBr3/p-g-C3N4, which facilitated the charge separation in the photocatalytic process. In addition, we systematically studied the effects of the composition and carrier dynamics behavior of the composites on photocatalytic activity for molecular oxygen activation.
2. Experimental section
2.1. Materials and preparation procedures
All chemicals and reagents were analytically pure and used without further purification. The dark-yellow g-C3N4 powder was synthesized according to a modified approach reported previously.20 g-C3N4 was treated with HBr at room temperature for 10 hours, collected, and centrifuged to obtain a light yellow powder (protonated p-g-C3N4). A series of characterization experiments with bulk b-g-C3N4 and protonated p-g-C3N4 nanosheets showed that the p-g-C3N4 nanosheets have a thickness of 4–5 nm (Fig. S1–S3†). Next, 30 mg (0.08 mmol) lead acetate (Pb(CH3COO)2·3H2O; 99.99% metal basis; Aladdin Industrial Corporation), 5 mL HBr solution (Z ≥ 47.0%; AR; Aladdin Industrial Corporation), and 450 μL CH3NH2 alcohol solution (30–33%; AR; Aladdin Industrial Corporation) were added into a beaker with stirring. After stirring constantly for 15 min to get a homogeneous mixture, different amounts of p-g-C3N4 (0, 0.25/0.08 wt%, 0.5/1.6 wt%, 1/3.3 wt%, 1.5 mg/5 wt%) dissolved in isopropanol (IPA) were added. The mixture was transferred into a 50 mL stainless-steel Teflon-lined autoclave and the container was closed and maintained at 120 °C for 4 h. Then the autoclave was allowed to cool down to room temperature under ambient conditions. The resulting solid product was collected, rinsed with distilled isopropanol several times and then dried in a vacuum oven at 55 °C for 12 h.
2.2. Characterization of samples
The morphologies and microstructures were investigated using a scanning electron microscope (FESEM) (Hitachi S-4800). The Fourier transform infrared (FTIR) spectra were acquired using a Nexus 870 FTIR instrument (USA). The photoluminescence (PL) and time resolved PL decay spectra were measured using a home-built PL scanned imaging microscope coupled with a time-correlated single photon counting (TCSPC) module at 500 nm laser excitation at room temperature. Electrochemical experiments were performed using an electrochemical workstation (CHI-660E) with a three-electrode system, employing a Pt coil as the counter electrode (CE) and a Ag/AgCl electrode as the reference electrode (RE). Dichloromethane (DCM) and tetrabutyl ammonium hexafluorophosphate (Bu4NPF6) were used as the solvent and conducting electrolyte, respectively. Hall coefficients (RH) were measured in a Quantum Design Physical Property Measurement System (PPMS). The charge carrier concentration (nH) was calculated by using nH = 1/RHe, where e is the elementary charge. The Hall carrier mobility (μH) was calculated according to the relation μH = σRH.
2.3. Photocatalytic experiments
The photocatalytic activity of the samples was evaluated by the degradation of malachite green (MG) under visible light. Typically, 5.0 mg catalyst was well dispersed in 40 mL MG isopropanol solution at an initial concentration of 10 mg mL−1 (C0). Subsequently, the dispersion was stirred for 60 min in the dark to reach the adsorption equilibrium. Then, the container was irradiated under visible light from a 300 W Xe lamp with a vertical distance of 20 cm. Finally, the concentration of MG (C) was analyzed using an UV-vis spectrophotometer at given time intervals. ESR-trapping measurements were carried out in 2 mL acetonitrile mixed with 20 μL of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 4 mg samples. After being irradiated, the mixture was characterized using a Bruker EMX plus model spectrometer operating at room temperature.
3. Results and discussion
3.1. Structure and morphology of MAPbBr3/p-g-C3N4
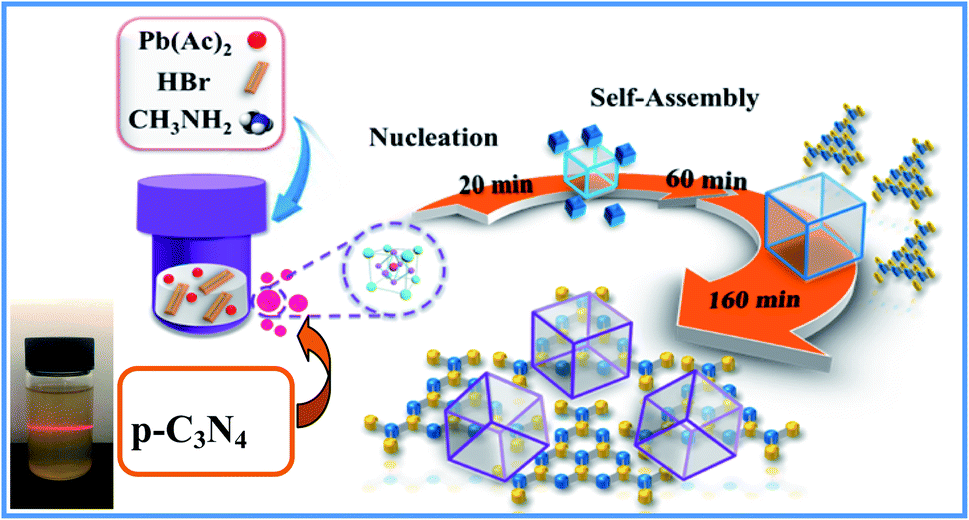
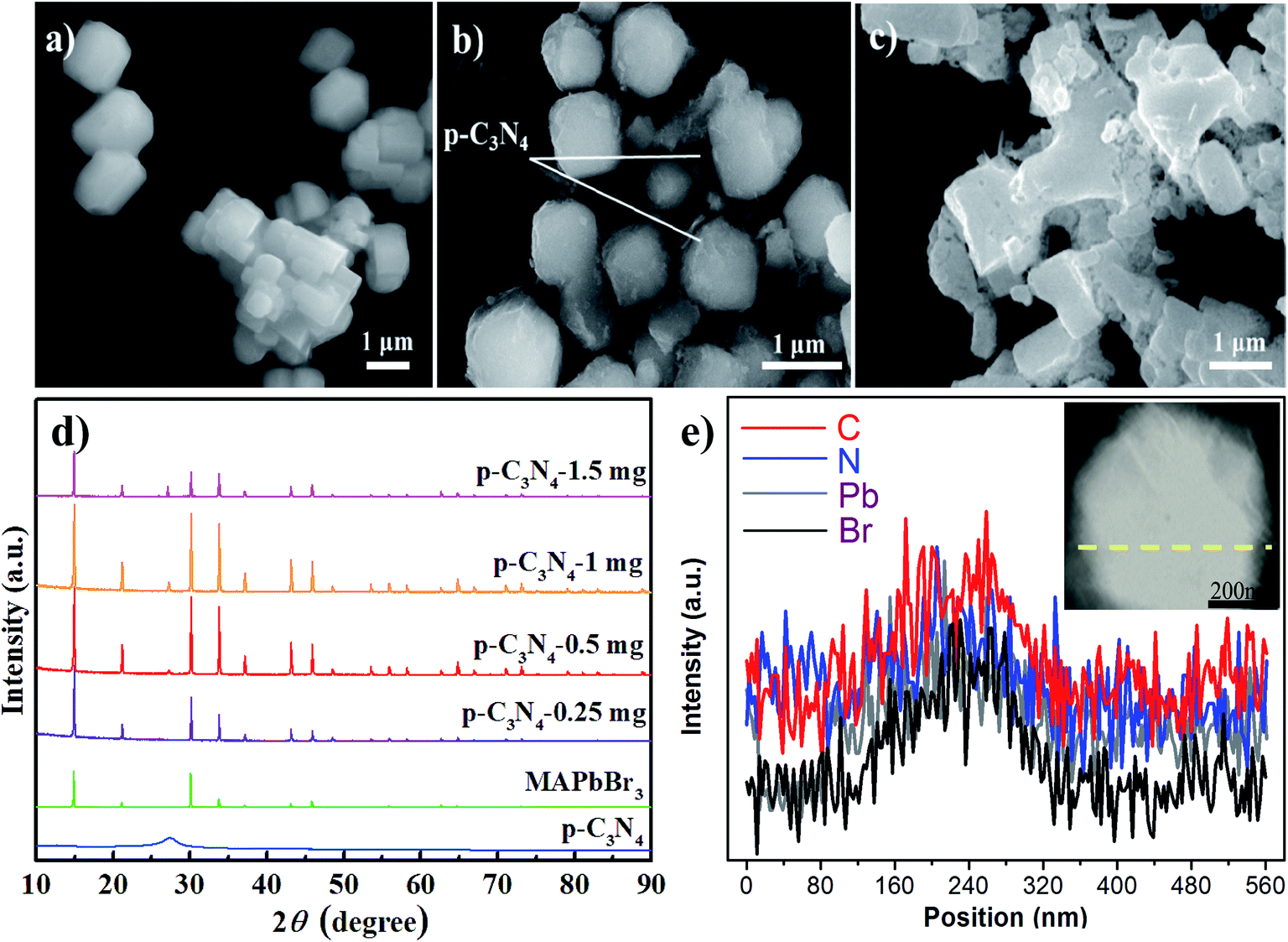
As we all know, the growth of perovskite single crystals, nanocrystals and polycrystalline films is a complicated process which strongly depends on precursor composition, solvent choice, deposition temperature, etc. Recently, the substitutional growth mechanism catalyzed by polar protic alcohols has been used for the preparation of highly chemically stable MAPbI3 powders up to ≈1.5 months. The reaction between alcohols and the precursors is initiated by the polar nature of the alcohols that can readily donate hydrogen to the reaction medium.13 On the other hand, the stable p-g-C3N4 colloidal suspension was prepared by HBr treatment methods, without using other acids such as HCl or HNO3 in order to avoid introducing impurities. Last but not least, it is reported that suitable incorporation of g-C3N4 could reduce the intrinsic defect density by passivating the charge recombination centers, improving the crystal quality of the perovskite film.19 Therefore, isopropanol and HBr are very suitable precursors for our present fabricated system. Fig. 1 shows a schematic illustration of the formation and morphological evolution process of the MAPbBr3/p-g-C3N4 samples using the stable p-g-C3N4 colloidal suspension solution. A typical Tyndall effect of p-g-C3N4 colloidal suspension is clearly evident, as indicated by the beam passing through the transparent sol solution. According to our previous research work,14 the MAPbBr3 microparticles (with an average size of about 5 μm) become rougher due to the wrapped/entrapped p-g-C3N4 nanosheets in Fig. 2a and b. With increasing the amount of 1.5 mg p-g-C3N4, the sample appeared as irregular shaped particles in Fig. 2c, indicating that the high amount of C3N4 nanosheet sol could affect the crystal nucleation and growth via modulation of the local dielectric environment and precursor concentrations during growth kinetics processes.15,19 It is important to understand the nature of the interaction between MAPbBr3 and p-g-C3N4, which influences the photo-electro-chemical properties of the composite. The composition and chemical structure of the MAPbBr3–p-g-C3N4 samples with different contents of p-g-C3N4 (denoted as MAPbBr3/p-g-C3N4-x, where x = 0, 0.25, 0.5, 1.0, and 1.5 mg) were studied by XRD and FTIR spectroscopy. The typical peaks of Fig. 2d at 2θ of 14.9°, 21.1°, 30.2°, 33.8°, 37.0°, 43.1°, and 45.6° correspond to (100), (110), (200), (210), (211), (220) and (300) planes, respectively, indicating that the product contains pure cubic phase CH3NH3PbBr3 (MAPbBr3) with a = 5.9394 Å (space group = Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Besides, the strong one at 27.4° represents the stacking of the π-conjugated p-g-C3N4 (p-C3N4) layers. No other impurity phases are discovered, reflecting the presence of a high crystal quality two-phase composite. Particularly, the intensity of XRD peaks for the MAPbBr3/p-g-C3N4-0.25 mg is stronger than that of pure MAPbBr3, suggesting that the crystallinity of the composites was improved. This is attributed to the relatively controlled nucleation and growth behavior due to p-g-C3N4 providing heterogeneous nucleation sites in the crystallization process through interaction between the hydrogen atoms of the MA and basic sites on p-g-C3N4.19Fig. 2e shows the EDS line profiles recorded from an individual MAPbBr3/p-g-C3N4 intergrowth microcrystal, indicating that the content of C and N is slight higher than that of Pb and Br due to wrapped/entrapped p-g-C3N4 nanosheets. Such microstructures could not be prepared when bulk g-C3N4 powder was used. With increasing the content of p-g-C3N4, the diffraction peak at 26.4° gradually increases, while the diffraction peak intensity of MAPbBr3 increases to the highest value for MAPbBr3–p-g-C3N4-1 mg, and then decreases for MAPbBr3–p-g-C3N4-1.5 mg. The reduced crystallinity of MAPbBr3–p-g-C3N4-1.5 mg may be attributed to the occurrence of micro-strain in the MAPbBr3 crystals, induced by crystal imperfections/structural defects including dislocations, vacancies, stacking faults, etc. It is worth noting that the peaks at 26.4° of p-g-C3N4 still exist for MAPbBr3–p-g-C3N4 after ultrasonic washing treatment, showing that the MAPbBr3/p-g-C3N4 intergrowth hybrids are very robust due to chemical interaction, which is beneficial to electron transportation.
m). Besides, the strong one at 27.4° represents the stacking of the π-conjugated p-g-C3N4 (p-C3N4) layers. No other impurity phases are discovered, reflecting the presence of a high crystal quality two-phase composite. Particularly, the intensity of XRD peaks for the MAPbBr3/p-g-C3N4-0.25 mg is stronger than that of pure MAPbBr3, suggesting that the crystallinity of the composites was improved. This is attributed to the relatively controlled nucleation and growth behavior due to p-g-C3N4 providing heterogeneous nucleation sites in the crystallization process through interaction between the hydrogen atoms of the MA and basic sites on p-g-C3N4.19Fig. 2e shows the EDS line profiles recorded from an individual MAPbBr3/p-g-C3N4 intergrowth microcrystal, indicating that the content of C and N is slight higher than that of Pb and Br due to wrapped/entrapped p-g-C3N4 nanosheets. Such microstructures could not be prepared when bulk g-C3N4 powder was used. With increasing the content of p-g-C3N4, the diffraction peak at 26.4° gradually increases, while the diffraction peak intensity of MAPbBr3 increases to the highest value for MAPbBr3–p-g-C3N4-1 mg, and then decreases for MAPbBr3–p-g-C3N4-1.5 mg. The reduced crystallinity of MAPbBr3–p-g-C3N4-1.5 mg may be attributed to the occurrence of micro-strain in the MAPbBr3 crystals, induced by crystal imperfections/structural defects including dislocations, vacancies, stacking faults, etc. It is worth noting that the peaks at 26.4° of p-g-C3N4 still exist for MAPbBr3–p-g-C3N4 after ultrasonic washing treatment, showing that the MAPbBr3/p-g-C3N4 intergrowth hybrids are very robust due to chemical interaction, which is beneficial to electron transportation.
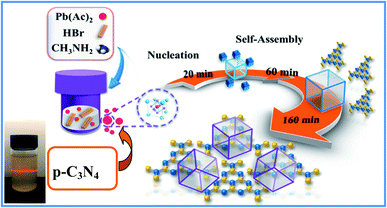 |
| | Fig. 1 Schematic illustration of the formation and morphological evolution process of MAPbBr3/p-g-C3N4 composites using a p-g-C3N4 sol solution through a solvothermal method. | |
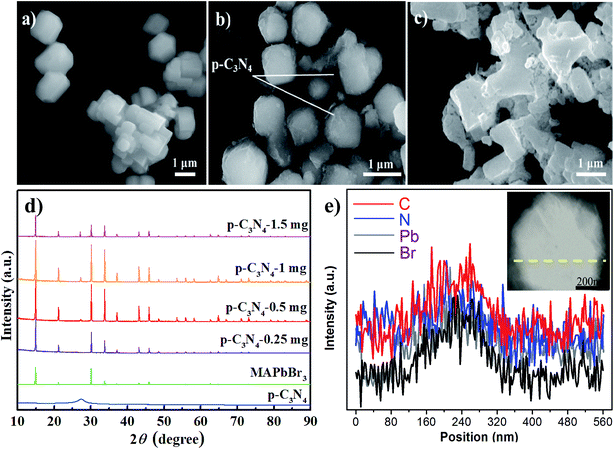 |
| | Fig. 2 (a–c) SEM images of MAPbBr3/p-C3N4-0, -1.0, and -1.5 mg samples, respectively. (d and e) The corresponding XRD patterns and EDS line scan image of an individual MAPbBr3 microcrystal encapsulated nanosheet. | |
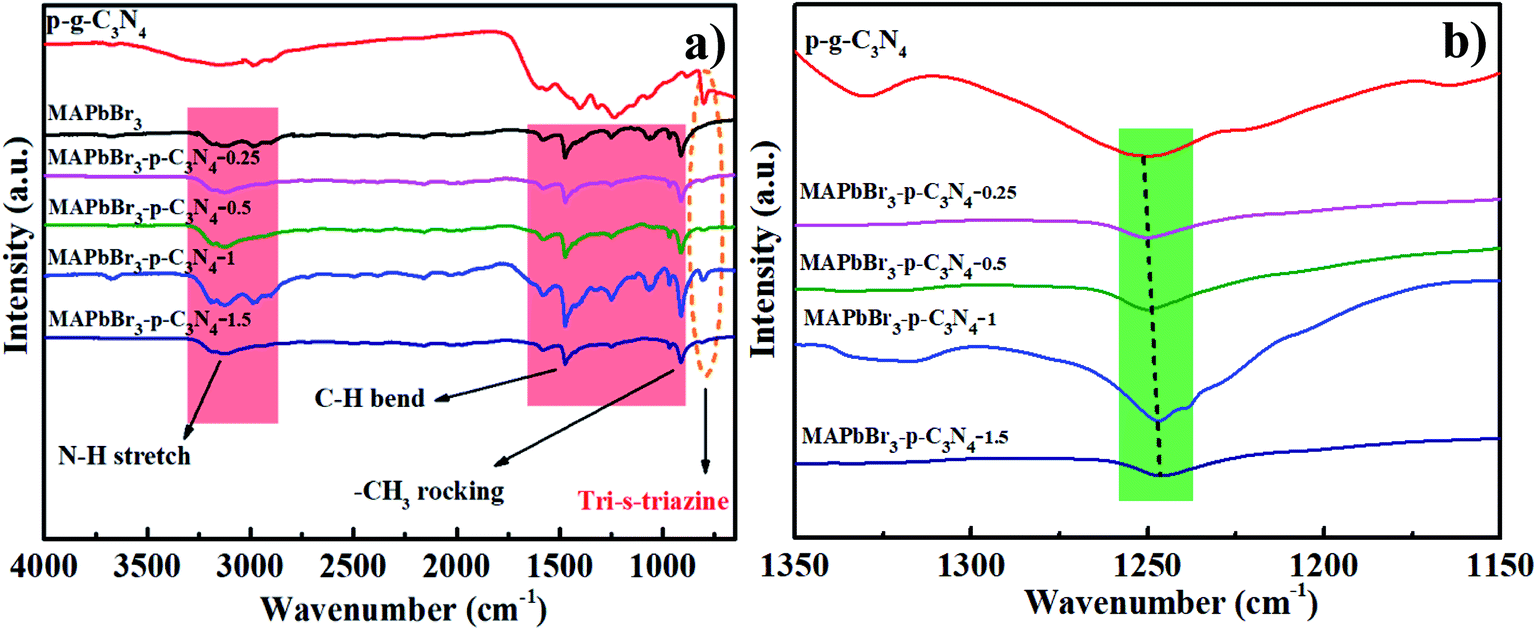
The interaction will be further discussed in the FTIR spectra. For MAPbBr3 microcrystals, the N–H stretch vibrations around 3100–3200 cm−1, and C–H bending and CH3 rocking modes at 1471 cm−1 and 916 cm−1 are in good agreement with previous results.14 Evidently, all of the bands assigned to p-g-C3N4 and MAPbBr3 are visible in MAPbBr3/p-g-C3N4 and the absorption intensity at 810 cm−1 is substantially increased and shifted toward a higher wavenumber with the introduction of greater amounts of p-g-C3N4 in Fig. 3a. It is reported that “nitrogen pots” of g-C3N4 with abundant amino sites (NHx, x = 1, 2) on the edges of heptazine units can take up the PbBr2 precursors via surface N–Br bonding interactions, forming an effective nucleation point for crystal growth.5 In contrast, it is worthy of note that when 1.5 mg p-g-C3N4 was introduced, the peak intensities of MAPbBr3/p-g-C3N4 decrease, which is possible due to greater particle agglomeration around the edges of the nanosheets. Furthermore, the typical stretching modes of aromatic C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N heterocycles in MAPbBr3/p-g-C3N4 at 1241 cm−1 are systematically shifted to lower values with the increasing content of p-g-C3N4 (Fig. 3b), which is ascribed to the high electron density of aromatic heterocycles due to C–N–Br bond chemical interaction. An analogous absorption change trend has also been observed, showing high interfacial coupling quality.5 In addition, cation–π interaction, an example of noncovalent bonding between an electron-rich π system and an adjacent cation, is proposed for pervoskite films.18,19 In brief, chemical interaction at the interface could eliminate the defects and enhance the stability of MAPbBr3/p-g-C3N4, which is also confirmed by the following photocatalytic tests.
N heterocycles in MAPbBr3/p-g-C3N4 at 1241 cm−1 are systematically shifted to lower values with the increasing content of p-g-C3N4 (Fig. 3b), which is ascribed to the high electron density of aromatic heterocycles due to C–N–Br bond chemical interaction. An analogous absorption change trend has also been observed, showing high interfacial coupling quality.5 In addition, cation–π interaction, an example of noncovalent bonding between an electron-rich π system and an adjacent cation, is proposed for pervoskite films.18,19 In brief, chemical interaction at the interface could eliminate the defects and enhance the stability of MAPbBr3/p-g-C3N4, which is also confirmed by the following photocatalytic tests.
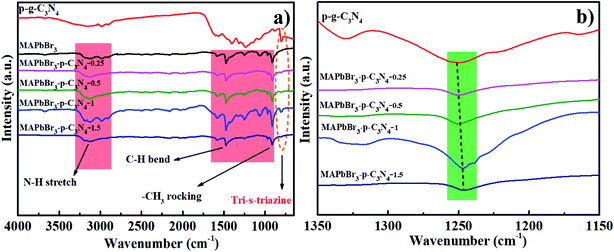 |
| | Fig. 3 (a and b) Full and partial regional FTIR spectra of products prepared with different contents of p-g-C3N4 confirming that the C–N–Br chemical bonding interaction is present in MAPbBr3/p-g-C3N4. | |
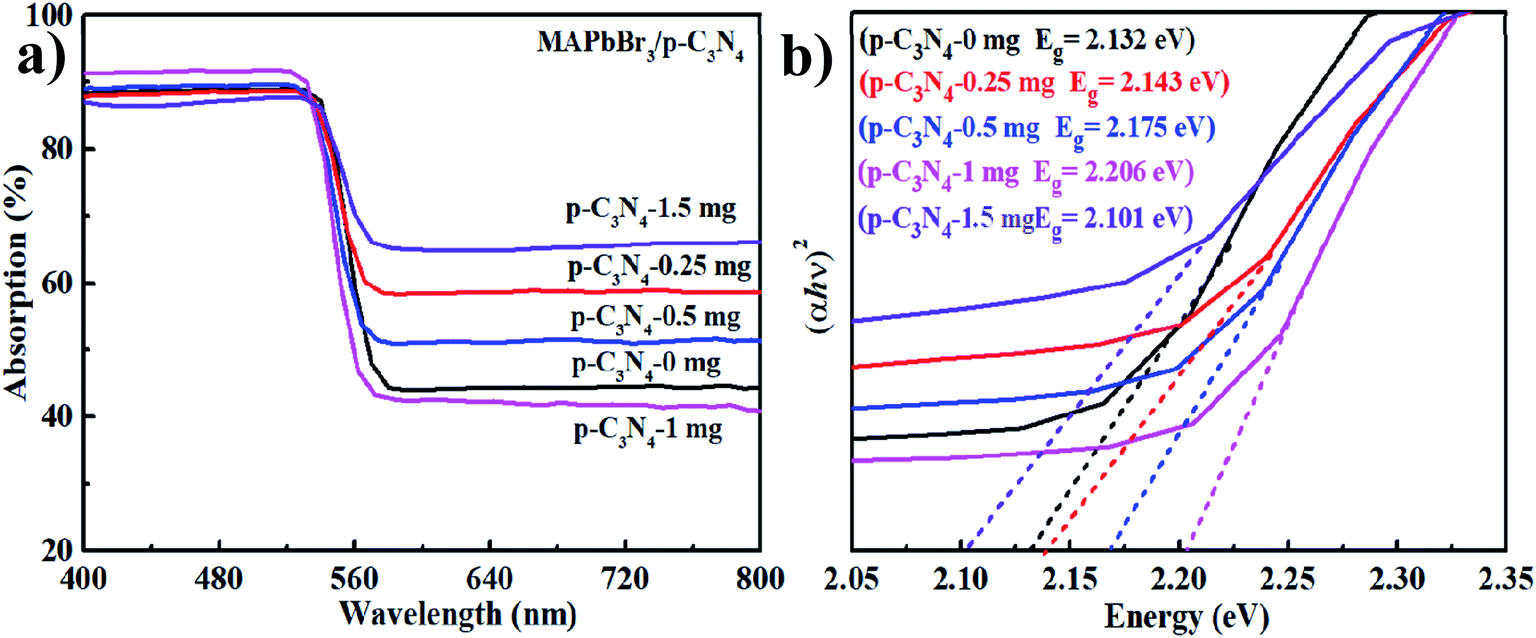
The optical properties of the samples were investigated by UV-vis diffuse reflectance spectroscopy (UV-vis DRS). As is shown in Fig. 4a, MAPbBr3–p-g-C3N4-x has an absorption edge at about 595 nm, which is the characteristic peak of MAPbBr3.2 Additionally, the optical band gaps of MAPbBr3–p-g-C3N4-x estimated from the Tauc plots using the equation αhν ∝ (h − Eg)2 are 2.132, 2.143, 2.175, 2.206, and 2.101 eV, respectively (Fig. 4b). The band gap of our sample is smaller than the previously reported optical band gap of MAPbBr3 nanoparticles (Eg = 2.39 eV),2 because the latter are nanoscale particles that can present the quantum effect. Simultaneously, the band gap values of our samples are close to the values reported for MAPbBr3 single crystals (Eg = 2.18, 2.21 eV) grown through an inverse temperature crystallization and antisolvent vapor-assisted approach.2,25–27 It is well known that the Br (4s23d104p5) 4p orbital and Pb (6s25d104f146p2) 6p orbital contribute the most to the valence band maximum (VBM) and conduction band minimum (CBM) of MAPbBr3, respectively.3 So the band gap variation with MAPbBr3/p-g-C3N4 composition is probably mainly due to the shrinkage of PbBr6 octahedra due to the chemical interaction, which is consistent with FTIR results.
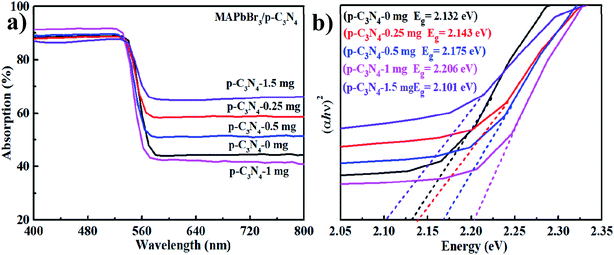 |
| | Fig. 4 (a and b) UV-vis diffuse reflectance spectra (DRS) and Tauc plots. | |
3.2. Photocatalytic degradation performance
After the structural analysis, photocatalytic degradation of malachite green (MG) in IPA solvent was employed as a model reaction to probe the structure–activity relations. MG, a cationic triphenylmethane dye (Fig. S4†), has been extensively used in the textile, leather, and pharmaceutical industries due to its low cost and high efficiency for disease prevention.8,28 Different control experiments were conducted to compare the ability of MAPbBr3, p-g-C3N4, and MAPbBr3/p-g-C3N4 composites under visible-light illumination. Fig. 5a shows the concentration–time profiles of the MG degradation over various catalysts. After irradiation for 10 min, 30%, 35%, 52%, 46%, 98% and 60% MG were reduced by MAPbBr3, p-g-C3N4, and MAPbBr3/p-g-C3N4-0.25, -0.5, -1.0 and -1.5, respectively. Remarkably, MAPbBr3/C3N4-1.0 showed significantly faster degradation rates. To the best of our knowledge, there has been no study for the photocatalytic degradation of MG within 10 min under visible light irradiation. A comparison of previously reported values for photocatalytic degradation of MG is shown in Table S3.† The time-dependent 3D absorption spectra of a malachite green solution in the presence of MAPbBr3/p-g-C3N4 are shown in Fig. 5b. During the 10 min irradiation, the characteristic absorption at about 618 nm in the MG spectra displayed a gradually declining trend, which indicated the process of N-demethylation, i.e., the non-selective attack of reactive oxygen species on the C–N bond as reported.29 The absorbance peaks at 425 nm reduce in intensity, which evidently indicates that the whole conjugated chromophore structure of MG has been destroyed.29 The surface properties of a photocatalyst is usually closely correlated with the photocatalytic activity. It is well known that a photocatalyst with a large surface area can provide more active sites and good adsorption for reactants, both of which can speed up the heterogeneous photocatalytic process. In the current case, N2-adsorption analysis indicated that the addition of a small amount of p-g-C3N4 slightly increased the surface area of samples. The BET values of MAPbBr3/p-g-C3N4, MAPbBr3, and p-g-C3N4 samples are determined to be 10.2, 7.2, and 12.2 m2 g−1, respectively. Clearly, the consistency between the BET surface area and catalytic performance is not established due to similar specific surface areas, indicating that the surface properties of the MAPbBr3/p-g-C3N4 are not the key factor.
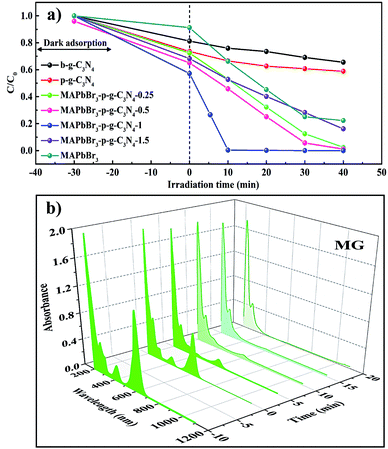 |
| | Fig. 5 (a) Photocatalytic activities of concentration–time profiles of the degradation of MG over various catalysts including b-g-C3N4, p-g-C3N4, MAPbBr3, and MAPbBr3/p-g-C3N4 samples under visible light. (b) 3D UV-vis spectra of malachite green (MG) at different irradiation times in the presence of the MAPbBr3–p-g-C3N4-1.0 mg sample. | |
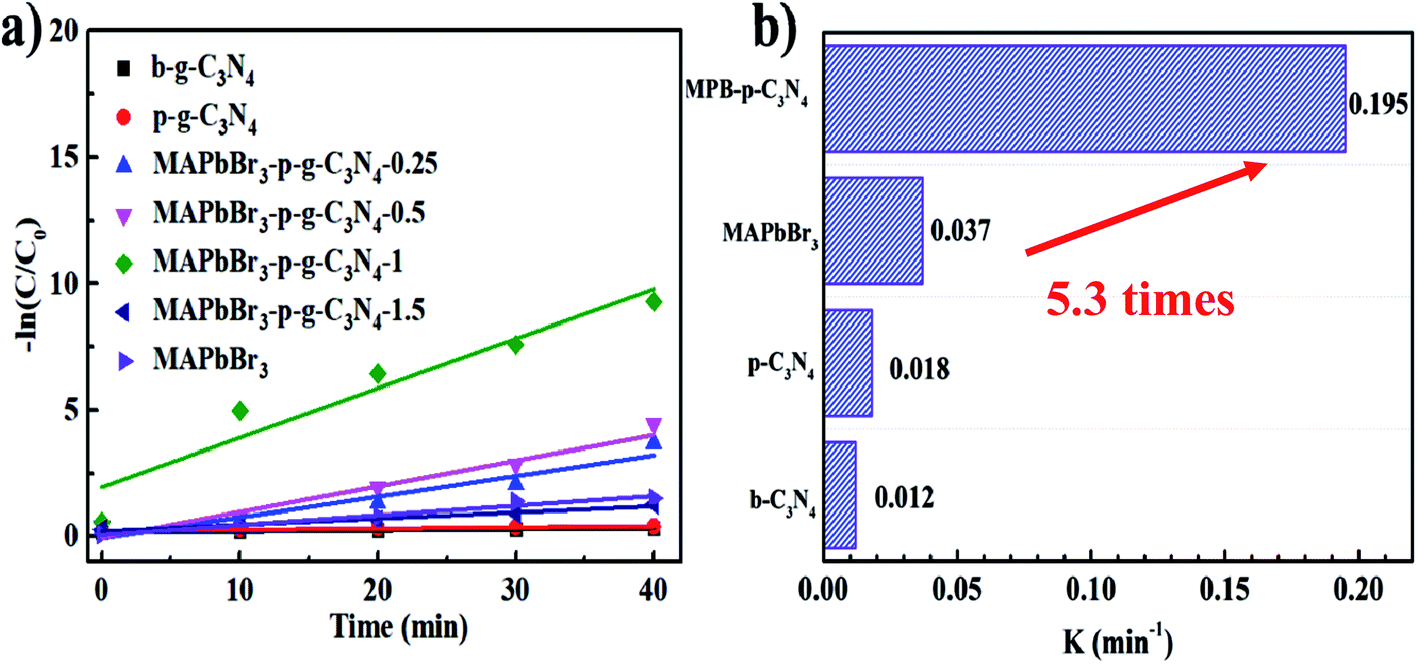
According to the kinetic curves in Fig. 6a, the rate constant k of pseudo-first-order kinetics follows the order MAPbBr3/p-g-C3N4 (0.195 min−1) > MAPbBr3 (0.037 min−1) > p-C3N4 (0.018 min−1) > b-C3N4 (0.012 min−1). Clearly, the MAPbBr3/p-g-C3N4-1.0 composite has the highest rate constant, which is almost 5.3 and 16 times higher than that of pure MAPbBr3 and C3N4, respectively (Fig. 6b). These results allow us to conclude that controlling the addition ratios of p-g-C3N4 is important for the achievement of an optimal synergistic interaction between MAPbBr3 and p-g-C3N4. Greater amounts of p-g-C3N4 nanosheets (1.5 mg) are not good for the photocatalytic performance. This can be attributed to two reasons: (1) the irregular microcrystals with low crystallinity resulted in trap states and thus reduced the efficiency of charge separation. (2) The increased p-g-C3N4 surface coverage on perovskite materials also blocked the absorption of light of MAPbBr3 and decreased the exposed surface area of MAPbBr3 to the reagent solution.4,5
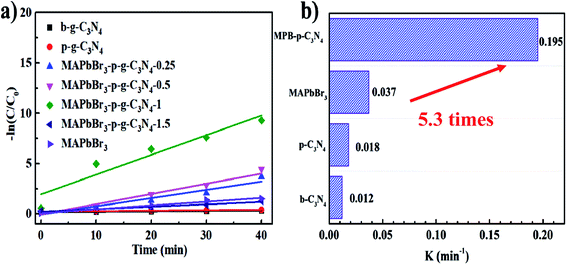 |
| | Fig. 6 (a) Photocatalytic kinetic behavior for the corresponding dependence of ln(C/C0) on irradiation time. (b) Comparison of slope values of different samples. | |
3.3. Discussion of underlying photocatalytic mechanisms
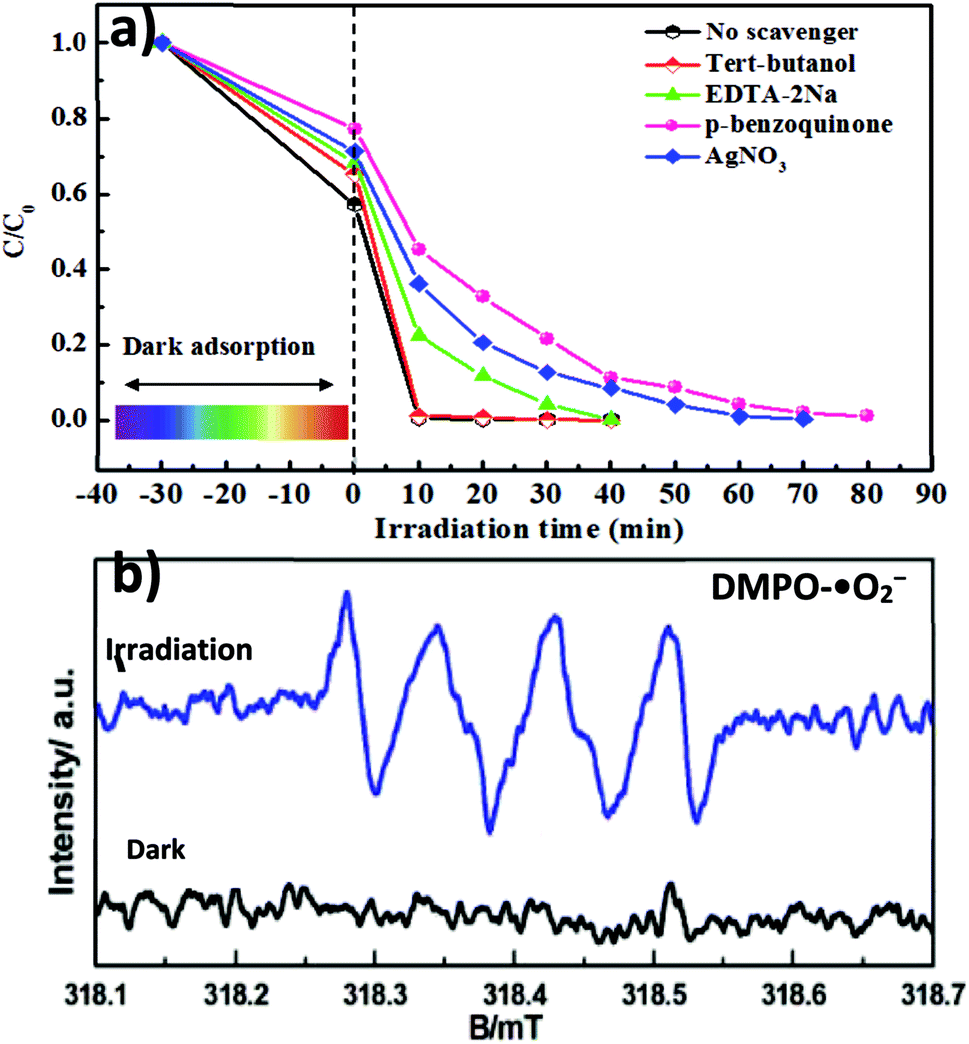
An in-depth understanding of plausible reaction mechanisms for the photodegradation of MG over semiconductors is desirable. Under visible light irradiation, the photogenerated superoxide radical (˙O2−), holes (h+) and hydroxyl radical (˙OH) play an important role in the degradation of organic dyes.8–12 For unambiguous identification of reactive oxygen species (ROS) involved in the dye degradation process, the reactive-species-trapping experiments were performed to clarify the contribution of different active species to the reaction. In general, p-benzoquinone (p-BQ), ethylenediaminetetraacetic acid disodium salt (EDTA-2Na), tert-butanol (t-BuOH) and AgNO3 are known as effective scavengers for ˙O2−, h+, ˙OH and photogenerated electrons (e−), respectively. As illustrated in Fig. 7a, the addition of t-BuOH had little effect on the photodegradation of MG. It is worth noting that isopropanol (IPA) and t-BuOH have commonly been employed to impair the hydroxyl radical (˙OH) in photocatalytic reaction systems; therefore, based on our present IPA solvent system, it is ruled out that ˙OH plays a role. However, the presence of p-BQ, AgNO3 and EDTA-2Na brought about obvious deactivation of catalysts and the photodegradation performance was decreased. Interestingly, among the scavengers employed, BQ can significantly suppress the degradation rate from 98% to 46% after 15 min, indicating that ˙O2− is a major reactive species in the system. In addition, spin-trapping electron paramagnetic resonance (EPR) experiments were also performed to give direct proof for the generation of ˙O2− over the MAPbBr3/g-C3N4-1.0 composites. In Fig. 7b, no EPR signal was observed in the dark, indicating that there is no detectable ˙O2− species formed. Upon visible-light irradiation for 5 min, a four-line EPR signal with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 was clearly observed, which is characteristic of the (5,5-dimethyl-1-pyrroline N-oxide) DMPO–˙O2− adduct, demonstrating the formation of the superoxide radical ˙O2− in the photocatalytic reaction.7 The observations strongly suggest that oxygen plays a critical role as an electron acceptor. Namely, O2 accepts the electrons from the semiconductor conduction band to form ˙O2− due to a strong thermodynamic driving force to reduce O2 (E°, (O2/˙O2−) = −0.33 V vs. Ag/AgCl at pH = 7) to superoxide ˙O2−. Very interestingly, ˙O2− was also observed in other perovskite photocatalytic systems including FAPbBr3/TiO2 and Cs2AgBiBr6.7,12 Very interestingly, ˙OH active species for CsPbX-based catalysts in an aqueous solution containing water molecules are often observed, which is attributed to the oxidation of water molecules with its negative side (δ−, O) by the photo-formed positive charges (holes) on the semiconductor surface.10,30 However, ˙O2− is the main active species in the photocatalytic dye degradation in organic alcohol solvent using organic–inorganic hybrid halide perovskites,7,12 demonstrating that the incorporation of organic solvent inhibits the generation of other ROS, especially ˙OH. Hence it can be concluded that tuning the solvent system may control the active species for organic synthesis. The phenomena may be attributed to two facts that photocatalytic reactions were performed in organic solvent media without H2O and the conjugated carbon nitride with amino nitrogen components and vacancy defects could provide more sites to adsorb O2 by Lewis acid–base interaction. In conclusion, according to the analysis above, the reactions listed below may be involved:
:1:1:1 was clearly observed, which is characteristic of the (5,5-dimethyl-1-pyrroline N-oxide) DMPO–˙O2− adduct, demonstrating the formation of the superoxide radical ˙O2− in the photocatalytic reaction.7 The observations strongly suggest that oxygen plays a critical role as an electron acceptor. Namely, O2 accepts the electrons from the semiconductor conduction band to form ˙O2− due to a strong thermodynamic driving force to reduce O2 (E°, (O2/˙O2−) = −0.33 V vs. Ag/AgCl at pH = 7) to superoxide ˙O2−. Very interestingly, ˙O2− was also observed in other perovskite photocatalytic systems including FAPbBr3/TiO2 and Cs2AgBiBr6.7,12 Very interestingly, ˙OH active species for CsPbX-based catalysts in an aqueous solution containing water molecules are often observed, which is attributed to the oxidation of water molecules with its negative side (δ−, O) by the photo-formed positive charges (holes) on the semiconductor surface.10,30 However, ˙O2− is the main active species in the photocatalytic dye degradation in organic alcohol solvent using organic–inorganic hybrid halide perovskites,7,12 demonstrating that the incorporation of organic solvent inhibits the generation of other ROS, especially ˙OH. Hence it can be concluded that tuning the solvent system may control the active species for organic synthesis. The phenomena may be attributed to two facts that photocatalytic reactions were performed in organic solvent media without H2O and the conjugated carbon nitride with amino nitrogen components and vacancy defects could provide more sites to adsorb O2 by Lewis acid–base interaction. In conclusion, according to the analysis above, the reactions listed below may be involved:| | | MAPbBr3/p-g-C3N4 + hν → e− + h+ | (1) |
| | | ˙O2− + MG → degraded products | (3) |
| | | h+ + MG → degraded products | (4) |
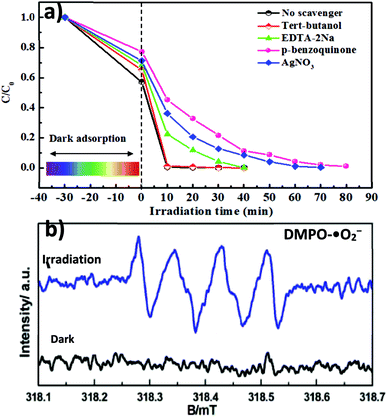 |
| | Fig. 7 (a) Effect of radical scavenger/quencher including p-benzoquinone (p-BQ), ethylenediaminetetraacetic acid disodium salt (EDTA-2Na), tert-butanol (t-BuOH) and AgNO3 on the degradation process of MG. (b) ESR spectra of the MAPbBr3/p-g-C3N4-1.0 mg sample without and with visible-light irradiation for 5 min for the detection of ˙O2− radicals. | |
3.4. Interfacial charge transfer
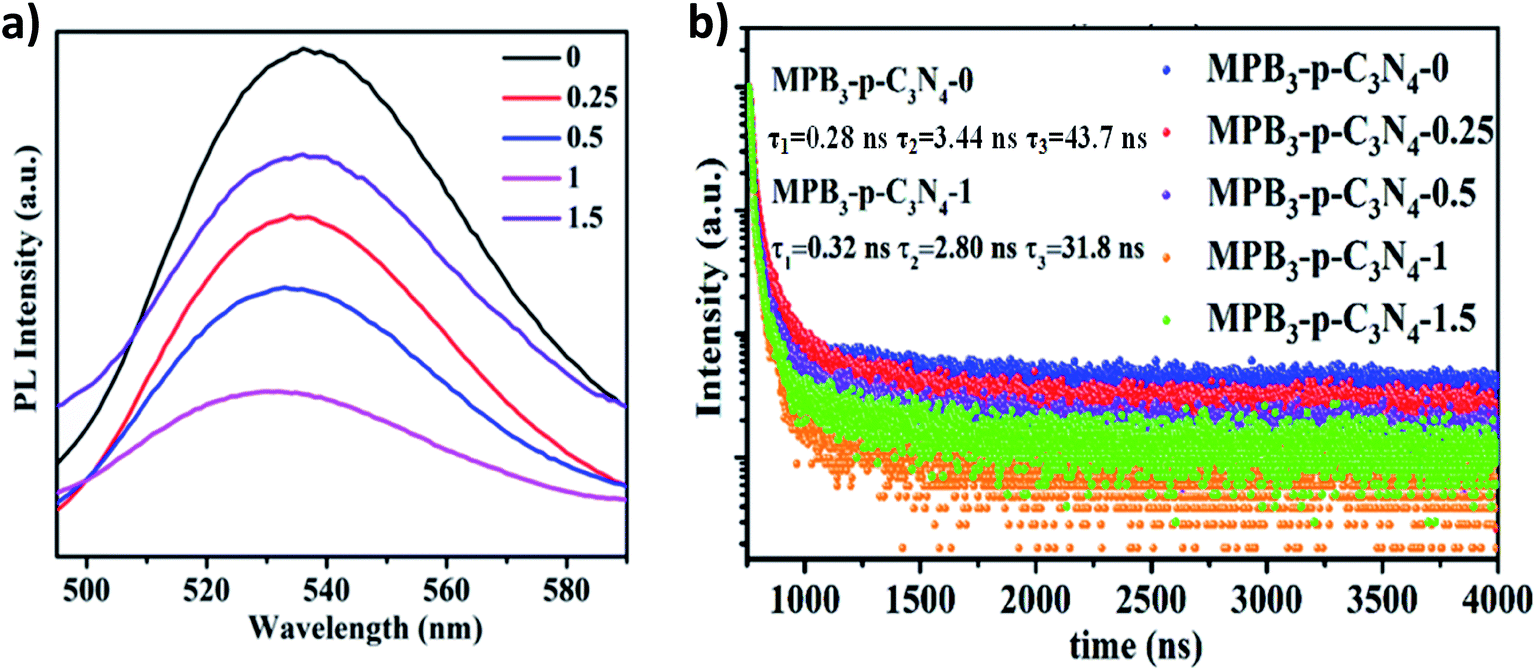
Subsequently, to further investigate the interfacial charge transfer in MAPbBr3/p-g-C3N4 intergrowth microcrystals, the PL spectra and time-resolved transient PL spectroscopy (TRPL) spectra of these samples with different loading amounts of p-g-C3N4 were recorded and are shown in Fig. 8. The PL spectrum of the MAPbBr3 crystal in Fig. 8a displayed a symmetric broad emission band with a peak at 530 nm, which is attributed to charge carrier recombination from the CB to the VB via a radiative pathway.2 With an increase in the amount of p-g-C3N4, a slight shift (521 nm) was observed, which may arise from the formation of MAPbBr3 with different structural defect densities. On the other hand, a gradual decrease in the PL intensity was found, suggesting that an additional energy-transfer pathway exists in addition to the intrinsic radiative channel for excited-state electron transfer. High crystalline quality MAPbBr3–p-g-C3N4-1 mg presented the weakest PL emission and the PL intensity is strongly quenched, indicating that the excited electrons at the interface of the MAPbBr3/p-g-C3N4 composite will rapidly be separated and recombination will be suppressed.
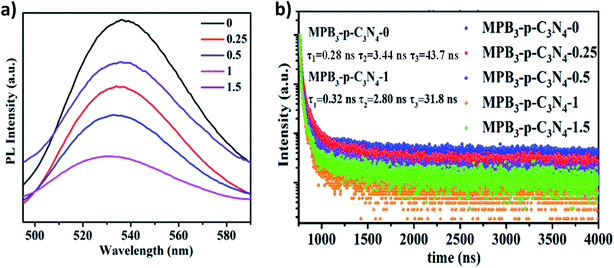 |
| | Fig. 8 (a) Steady-state PL spectra and (b) time-resolved photoluminescence decay spectra (TRPL) of MAPbBr3/p-g-C3N4 (loading amount: 0–1.5 mg) samples. | |
The decay traces of MAPbBr3 (MPB3) and MAPbBr3/p-g-C3N4-x were fitted using triexponential decay kinetics and are shown in Fig. 8b. The derivation of the three components and intensity-average lifetime (τav) was carried out using the following equation, and the results are listed in Table S1:†
| | | τav = (A1τ12 + A2τ22 + A3τ32)/(A1τ1 + A2τ2 + A3τ3) | (5) |
where
A and
τ denote the amplitudes and emission lifetimes of each component. The
τav values were determined to be 43.3 ns for MAPbBr
3, 28.2 ns for MAPbBr
3/p-g-C
3N
4-0.25, 23.1 ns for MAPbBr
3/p-g-C
3N
4-0.5, 20.8 ns for MAPbBr
3/p-g-C
3N
4-1.0 and 48.6 ns for MAPbBr
3/p-g-C
3N
4-1.5, exhibiting a PL decay time on the nanosecond scale. Significantly, the
τav value decreased to a minimum value for the MAPbBr
3/p-g-C
3N
4-1.0 sample, showing that the charge-separated state lifetime of MAPbBr
3/p-g-C
3N
4-1.0 is ∼2.08-fold longer than that of MAPbBr
3, in agreement with previous observations for MAPbI
3/rGO and CsPbBr
3/rGO composites with shorter lifetime compared to pure pervoskite.
3,4 More importantly, the contribution of the fast decay component (
τ1) underwent a significant increase from 9.70% for MAPbBr
3 to 91.1% for MAPbBr
3/p-g-C
3N
4-1.0, illustrating that more pronounced interfacial electron transfer occurred due to the existence of non-radiative quenching routes.
4 Therefore, based on the PL and TRPL results, it can be concluded that the N–Br chemical bond of the MAPbBr
3 wrapped/entrapped p-g-C
4N
3 microcrystals could allow a more efficient electron transfer, and reduce the proportion of accumulated unreactive electrons, which is presumably key to the increased photoactivity. The electron and hole transfer rate constants (
ket and
kht) can be estimated using the following expression:
16| | 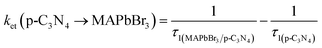 | (6) |
| | 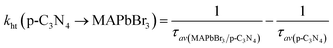 | (7) |
The calculated ket and kht values for samples of MAPbBr3/p-g-C3N4-1.0 were approximately 2.86 × 109 s−1 and 4.90 × 106 s−1, respectively. This ket value is 4.65 fold higher than the reported ket value (6.15 × 108 s−1) by Pu et al.,16 indicating that highly crystalline MAPbBr3 wrapped/entrapped p-g-C3N4 intergrowth microcrystals have great potential as new photocatalysts to convert solar energy into chemical energy.
3.5. Electrochemical analysis of MAPbBr3/p-g-C3N4
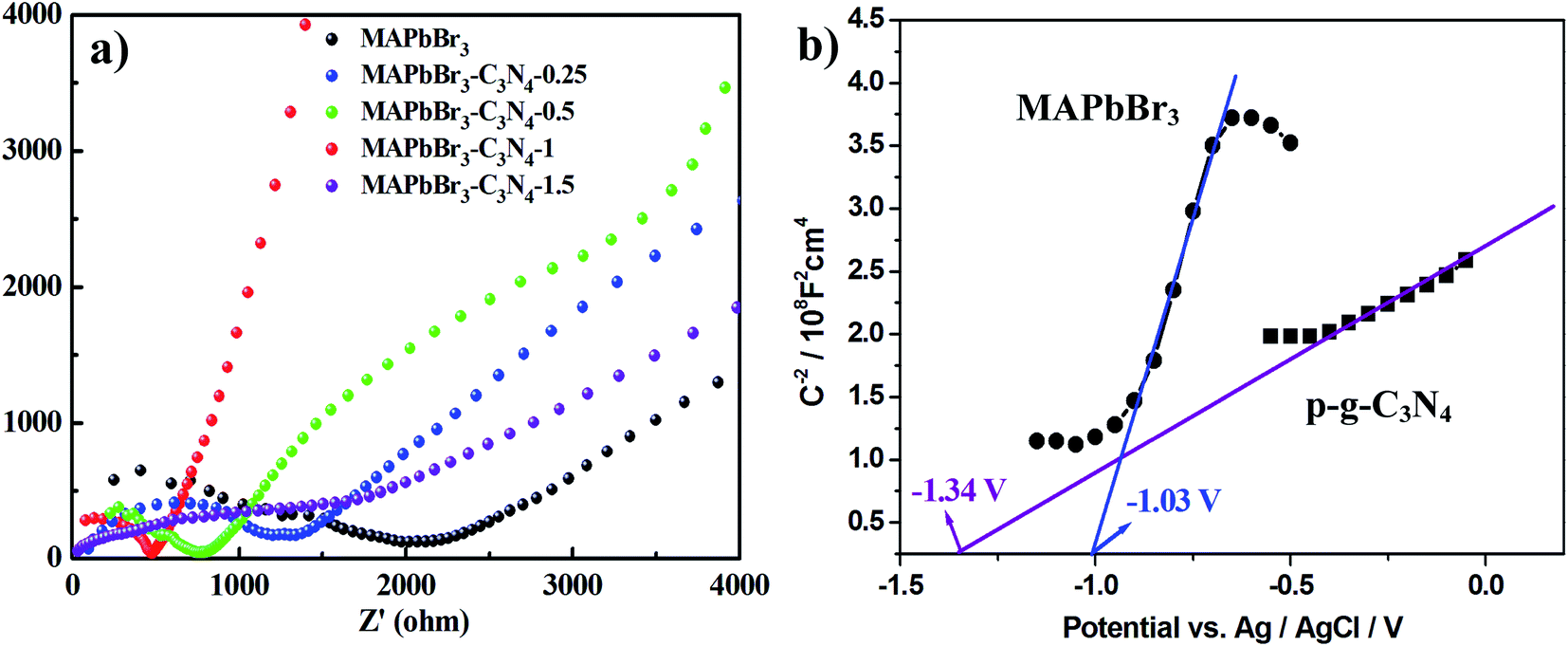
Recently, there have been some literature studies on the rich solid state chemistry aspects of perovskites through electrochemical experiments9,31–34 where defects and grain boundaries as well as chemical changes occurring at the various interfaces could be monitored. To gain deeper insight into the electron transport behaviors, electrochemical impedance spectroscopy (EIS) was conducted in dichloromethane solution to determine the charge-transfer resistance of the electrodes. The spectra depicted the interfacial charge transfer efficiency of the samples with the radius of the arc correlating with the reaction rate at the surface of the electrode.3 Hence, as shown in Fig. 9a, a smaller diameter of the Nyquist semicircle at high frequencies is realized with MAPbBr3/p-g-C3N4-1.0, implying that the charge transfer resistance is smaller than that of MAPbBr3 and the other MAPbBr3/p-g-C3N4 composites. Evidently, the pronounced decrease of the charge-transfer resistance by incorporating a suitable amount of p-g-C3N4 validates better charge transport ability and a more facilitated charge separation process of the MAPbBr3/p-g-C3N4-1.0 sample.
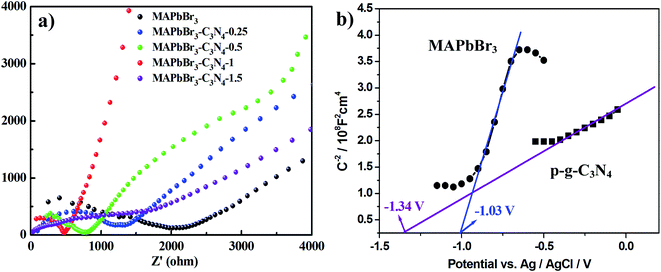 |
| | Fig. 9 (a) Electrochemical impedance spectroscopy (EIS) spectra of the MAPbBr3/p-g-C3N4 composites and (b) Mott–Schottky plot on MAPbBr3 and p-g-C3N4, showing n-type semiconducting features and flat band potentials of −1.03 V and −1.34 V vs. Ag/AgCl, respectively. | |
On the other hand, the band edge positions of the perovskite materials were obtained from electrochemical experiments. Electrochemical Mott–Schottky (MS) analysis (Fig. 9b) was also performed to estimate the flat band potential (VFB) of different optically active perovskites, which can further illustrate their band structure. The Mott–Schottky equation can be written as follows (eqn (8))
| | 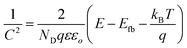 | (8) |
where
C denotes the space charge capacitance,
ND is the density of charge carriers,
q is the electronic charge (1.602 × 10
−19 C),
ε and
εo refer to the dielectric constant of the semiconductor and permittivity in a vacuum (8.85 × 10
−12 F cm
−2), respectively,
E and
Efb denote the applied potential and flat-band potential, respectively,
kB is the Boltzmann constant, and
T is the temperature in Kelvin. The value of
Efb is determined from the
x-intercept of the linear section of the Mott–Schottky curve. For instance, Gimenez's research group demonstrated that a Mott–Schottky plot on a CsPbBr
3 quantum dots/c-TiO
2/FTO film showed n-type behavior and a flat band potential of −0.109 V
vs. NHE.
9 Furthermore, Li
et al. found that the position of the valence band maximum (VBM) could be determined from the MS plots. The
VFB of MAPbBr
3 is 1.07–1.23 V
vs. NHE, which is 0.4 V more positive than
VFB of MAPbI
3.
33 Similarly, Samu
et al. reported that the VB edge of MAPbI
xBr
1−x becomes significantly more positive upon increasing Br incorporation into the lattice (0.77 → 1.15 V
vs. Ag/AgCl).
34 It is generally accepted that the CB and VB potentials in n or p-type semiconductors are approximately equal to the flat band potential and the potentials
vs. Ag/AgCl were converted to normal hydrogen electrode (NHE) potentials using the Nernst equation
35,36| | | VNHE = VAg/AgCl + VoAg/AgCl vs. NHE | (9) |
where
VoAg/AgCl vs. NHE is 0.199 V at 25 °C.
Fig. 6d shows n-type semiconducting features and the flat band potentials of MAPbBr
3 and p-g-C
3N
4 are −1.03 V and −1.34 V
vs. Ag/AgCl derived from the intersection. Gelderman
et al. have described the nature of the semiconductor–electrolyte interface, together with the Mott–Schottky equation for determining the flat-band potential which is approximately equal to that of the Fermi level.
37 The Fermi level is defined as the energy level at which the probability of occupation by an electron is 1/2, and it lies at the mid-point of the band gap in an intrinsic semiconductor. But doping could change the Fermi level due to the redistribution of electrons within the solid. The Fermi level of an n-type semiconductor lies just below the conduction band, while for a p-type semiconductor it lies just above the valence band.
37,38 Consequently, the Fermi level (flat-band potential) of a p-type catalyst is close to the VB while that of an n-type sample is located near the CB, which was also demonstrated for porous few-layer carbon nitride by Xiao
et al.36 and a NiO/g-C
3N
4 heterojunction by Tang
et al.39 Therefore, the potentials of the VB of MAPbBr
3 are calculated to be ∼1.1 eV by subtracting the optical band gap (2.13 eV) from the CBM, which is in reasonably good agreement with those reported by Li
et al.33 and Samu
et al.34 Additionally, the flat-band potential of p-g-C
3N
4–Sol was measured to be −1.34 V
versus Ag/AgCl, which is similar to the value of ultrathin g-C
3N
4 (−1.32 V
vs. Ag/AgCl)
36 and slightly smaller than the value (−1.43 V) reported by Wang
et al. So, the potentials of the VB of p-g-C
3N
4 are calculated to be ∼1.4 eV by subtracting the optical band gap (2.7 eV)
16,20 from the CB, which is slight bigger than that of MAPbBr
3 (1.1 eV).
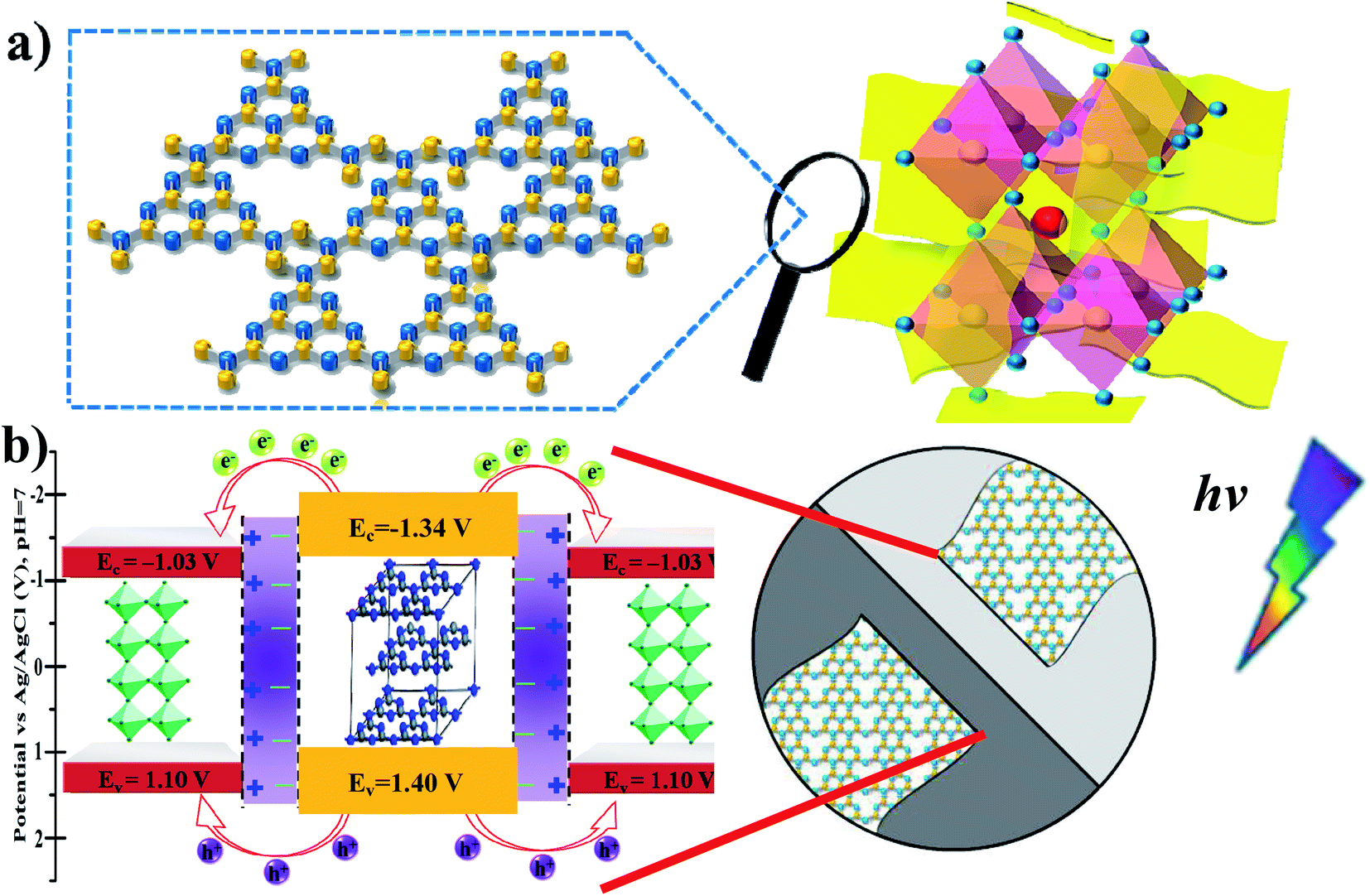
Generally, based on the bandgap and electronic energy level of the semiconductors, there are five different cases for semiconductor heterojunctions: straddling alignment (type-I), staggered alignment (type-II), Z-scheme system, p–n heterojunctions, and homojunctions.40 In type-I band alignment composed of two kinds of semiconductors, both VB and CB edges of one semiconductor are localized within the energy gap of another semiconductor. Recently, Zhao et al. reported that the Ag–CsPbBr3/CN composite has two electron transfer pathways. In fact, the first pathway is analogous to the type-I model, where the CB and VB edge potentials of pure CN are about −1.33 and 1.40 eV, and the CB and VB edge potentials of CsPbBr3 are about −1.13 and 1.1 eV, respectively.30 In our present work, the MAPbBr3/p-g-C3N4 intergrowth composites have a structure similar to this so-called inverted type-I core/shell structure, where a material with a narrower band gap is grown epitaxially around the core material with a higher band gap.40 Both electrons and holes would be rationally driven to the shell by the built-in energy potential, and the charge carriers (electrons and holes) are distributed largely in the shell region, which promotes separation of the photoexcited charge carriers and efficiently facilitates charge transfer to the shell layer surface to enhance the redox reaction. In contrast, with contact of p-type and n-type semiconductors with each other, the bands of the semiconductors will bend and the Fermi levels will equilibrate because of the formation of a space charge region after the diffusion of electrons and holes. Thus, the built-in electrical potential in the space charge region from the n-type side to the p-type side can direct the electrons and holes to quickly travel in the opposite directions, and allow more effective separation and longer lifetime of electron–hole pairs.40 These advantages endow the p–n type heterostructures with an enhanced photocatalytic performance. For example, when p-type NiO and n-type g-C3N4 are connected closely, the electrons in n-type g-C3N4 transfer to p-type NiO, leading to a positive charge; meanwhile, the holes in NiO transfer to g-C3N4, generating a negative charge. Consequently, the space charge region called the internal electric field in p–n heterojunctions is established when the electrons and holes accumulate until reaching an equilibrium, which could acts as a potential barrier to decrease the electron–hole pair recombination.39 Based on the Mott–Schottky (MS) analysis, the corresponding inverted type-I band configuration structure alignments of MAPbBr3 and p-g-C3N4 are schematically depicted in Fig. 10. In other words, in MAPbBr3 microcrystal wrapped/entrapped p-g-C3N4 microstructures, MAPbBr3 with a smaller band gap is the shell and p-g-C3N4 with a bigger band gap is the core, which is evidently different from the work of Pu et al.16 Both electrons and holes would rationally move from the p-g-C3N4 core to the MAPbBr3 shell driven by the built-in energy potential, and then diffuse to the MAPbBr3 shell layer surface to enhance the redox reaction, which promotes separation of the photoexcited charge carriers (Fig. 10b).
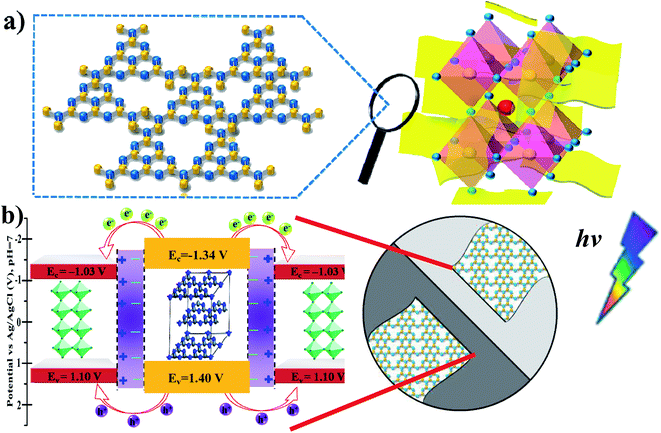 |
| | Fig. 10 (a) Schematic illustrations of the MAPbBr3/p-g-C3N4 intergrowth microcrystal composite wrapped/entrapped nanosheets on the molecular level. (b) The proposed corresponding inverted type-I band configuration structure alignments of MAPbBr3 and p-g-C3N4, which are similar to core/shell heterostructure band structures. | |
Meanwhile, based on the slopes of the Mott–Schottky plots, the density of the charge carrier, ND, can also be calculated from eqn (10).
| | 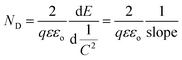 | (10) |
The relative
ε dielectric constant of a MAPbBr
3 single crystal is about 25.5.
27 Therefore, the
ND of MAPbBr
3 (5.43 × 10
16 cm
−3) was found to be comparable to previously reported values of MAPbI
3 (on the order of ∼10
16 cm
−3)
33 and was also in line with our Hall effect measurement results and smaller than the donor density of n-type CsPbBr
3 films (6.77 × 10
19 cm
−3).
9 The deviation is most probably due to the action of surface states in the polycrystalline electrode capturing and immobilizing the carriers.
Moreover, Hall effect measurements were also carried out at room temperature, showing the n-type semiconductor behavior for all MAPbBr3/p-g-C4N3 samples. Both the samples have a carrier concentration of 1016 cm−3, as shown in Table S2,† which is in agreement with the Mott–Schottky results. This mobility values are of the same order of magnitude compared to the previous result of a MAPbBr3 single crystal (up to 60 cm2 V−1 s−1).27 Our results are in accord with the fact that g-C3N4 doping increases the conductivity and carrier mobility in the perovskite film.19 With regard to MAPbBr3, another important parameter is the carrier diffusion length, LD (the distance over which the limiting photocarrier diffuses before it recombines), so we can calculate the carrier diffusion length by combining carrier lifetime with carrier mobility given by the following equation:27,28
where Boltzmann's constant
kB = 1.38 × 10
−23 J K
−1 and
T is the sample's temperature (298 K). By using the average carrier lifetime (
τav) obtained from the present work and carrier mobility
μ = 37.21–165.02 cm
2 V
−1 s
−1, the carrier diffusion lengths of
LD(MAPbBr
3) and
LD (MAPbBr
3/p-g-C
3N
4-1.0) are found to be 6.44 and 9.39 μm, respectively, which is similar to a previously reported value of 4.3 μm for MAPbBr
3,
27 and an estimated 3–17 μm in MAPbBr
3 using a shorter lifetime and lower mobility.
28 Such long carrier diffusion lengths will be favourable for photocarrier diffusion and separation for an efficient halide perovskite microcrystal photocatalyst.
3.6. Stability of the as-prepared samples
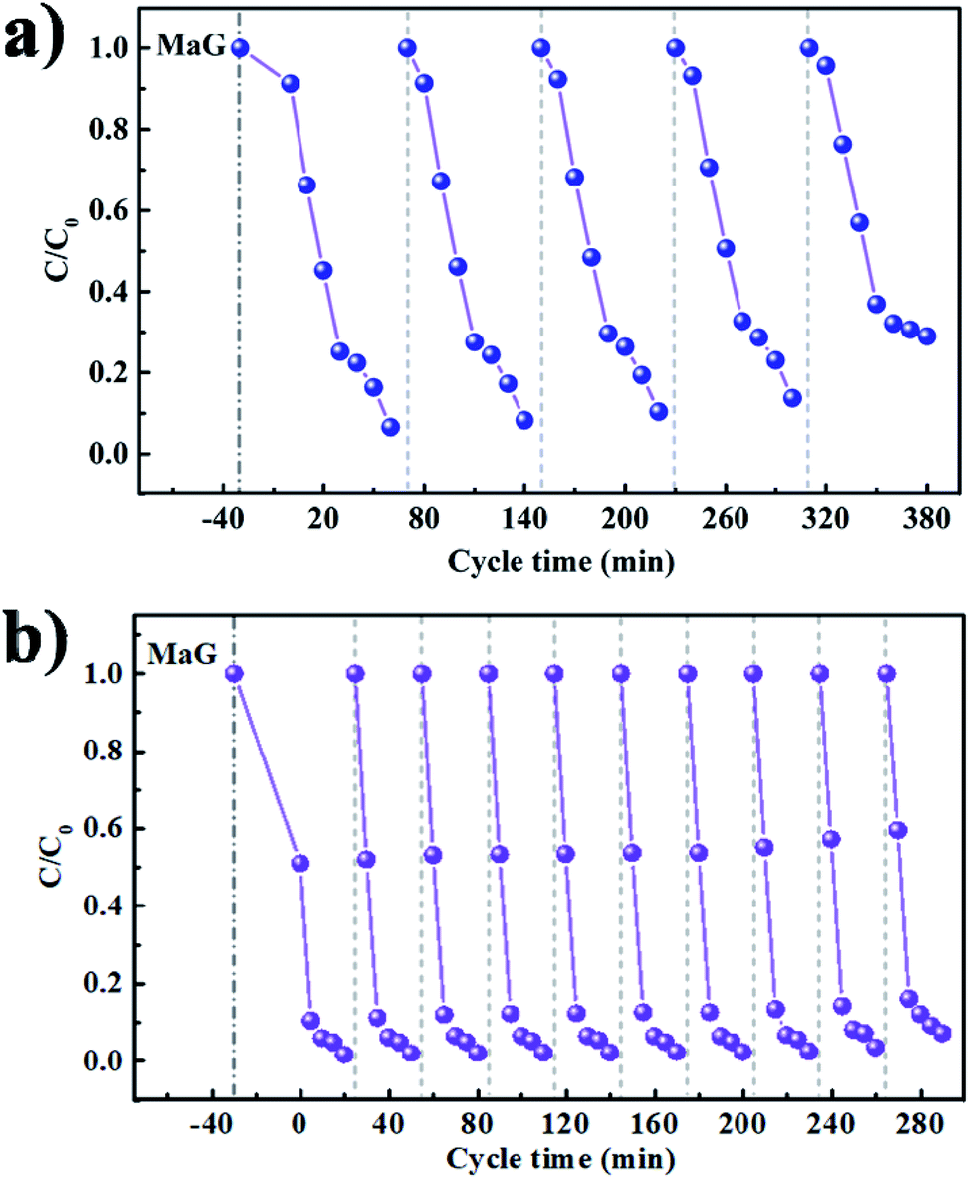
As long-term stability is also an important metric for photocatalytic systems, the recycling experiment runs for MG degradation under visible light irradiation were further tested in Fig. 11a and b. Compared with pure MAPbBr3 (Fig. 11a), no evident reduction in activity is observed in the MAPbBr3/p-g-C3N4-1.0 composite sample during at least ten reaction cycles (Fig. 11b), indicating the excellent chemical stability of the photocatalyst under testing conditions. The degradation rates were slightly decreased because of the inevitable loss of the photocatalyst mass during the recycling process. Since it is widely accepted that microcrystals are more stable than nanocrystals due to low surface Gibbs energy, it is reasonable that our samples have desirable high stability. In order to further evaluate the stability of the MAPbBr3/p-g-C3N4 samples, FTIR, XRD and SEM measurements after the recycling experiment were employed. The FTIR spectra of IPA and MAPbBr3/p-g-C3N4-1.0 were identical before and after the stability test (Fig. S5†), showing no obvious changes in chemical functional groups for the solvent and catalyst. Simultaneously, no appreciable change in the crystal structure and morphology of MAPbBr3/p-g-C3N4-1.0 can be observed, suggesting that the chemical stability of the MAPbBr3/p-g-C3N4 sample is high, and is comparable to the stability reported for double-perovskite Cs2AgBiBr6.10 Clearly, the as-synthesized MAPbBr3/p-g-C3N4 composite could thus be an effective and stable catalyst for photocatalytic reaction. On the other hand, for electrocatalyst experiments, the I–t polarization curve after many CV cycles in alkaline solution is usually used to evaluate the long-term stability. The related electrocatalyst work on the MAPbBr3/p-g-C3N4 composite is under investigation. Besides the degradation of MG, our sample exhibited a good photocatalytic performance for other cationic (RhB) and anionic (MO) dye pollutants (Fig. S6†), and the solution turned completely colorless within 20 min. To sum up, it is believed that the N–Br chemical bonding interaction between MAPbBr3 and p-g-C3N4 not only enhances charge separation and transportation efficiency, but also provides an alternative way for surface passivation of uncoordinated halides to improve their chemical stability, exhibiting huge potential for thermocatalysis, electrocatalysis and photocatalysis.
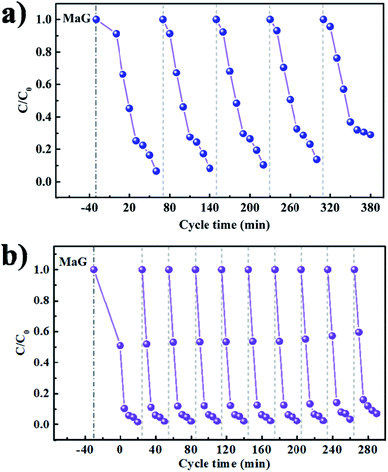 |
| | Fig. 11 (a and b) Long-term cycling stability tests of the MAPbBr3 and MAPbBr3/p-g-C3N4-1.0 under visible-light irradiation for photoreduction of malachite green. | |
4. Conclusion
In summary, we demonstrated that the construction of a MAPbBr3/p-g-C3N4 intergrowth composite photocatalyst was realized by the abundant NHx sites along the edges of p-g-C3N4 bonded with MAPbBr3. The spectroscopic and electrochemical results showed that the unique chemical covalent C–N–Br interaction at the interface could eliminate the defects and enhance the stability of MAPbBr3/p-g-C3N4, which facilitated the charge separation in the photocatalytic process. The photocatalytic activity (kMG) and interfacial charge transfer efficiency (ket) in the inverted type-I core/shell MAPbBr3/p-g-C3N4 band configuration structure were strongly dependent on the composition of p-g-C3N4 nanosheets. The MAPbBr3/g-C3N4 composites exhibited outstanding photocatalytic degradation of malachite green, with 15 and 3 times higher activity compared to their pure constituents, respectively. The photoexcited electron induced radical anions (˙O2−) were the major species for MG degradation. Furthermore, the MAPbBr3/g-C3N4 composites were found to be rather stable for at least ten cycles, revealing their promise for utilization in long-term photocatalysis. The excellent activity and good stability could enable this hybrid system to be a highly efficient visible light-driven photocatalyst for practical harvesting of energy from sunlight.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51502062, 51702067, 51572060 and 51671074) and Program for Innovation Research of Science in Harbin Institute of Technology (PIRS of HIT No B201507).
References
- S. Park, W. J. Chang, C. W. Lee, S. Park, H. Y. Ahn and K. T. Nam, Photocatalytic hydrogen generation from hydriodic acid using methylammonium lead iodide in dynamic equilibrium with aqueous solution, Nature Energy, 2016, 2, 16185–16193 CrossRef.
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, Promoting Photocatalytic H2 Evolution on Organic–Inorganic Hybrid Perovskite Nanocrystals by Simultaneous Dual-Charge Transportation Modulation, ACS Energy Lett., 2019, 4, 40–47 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M. H. Wang and B. Huang, Composite of CH3NH3PbI3 with Reduced Graphene Oxide as a Highly Efficient and Stable Visible-Light Photocatalyst for Hydrogen Evolution in Aqueous HI Solution, Adv. Mater., 2018, 30, 1–6 Search PubMed.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, A CsPbBr3 Perovskite Quantum Dot/Graphene Oxide Composite for Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Amino-assisted anchoring of CsPbBr3 perovskite quantum dots on porous g-C3N4 for enhanced photocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2018, 130, 13758–13762 CrossRef.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, Lead–Halide Perovskites for Photocatalytic alpha-Alkylation of Aldehydes, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- H. Huang, H. Yuan, K. P. F. Janssen, G. Solís-Fernández, Y. Wang, C. Y. X. Tan, D. Jonckheere, E. Debroye, J. Long, J. Hendrix, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Efficient and Selective Photocatalytic Oxidation of Benzylic Alcohols with Hybrid Organic–Inorganic Perovskite Materials, ACS Energy Lett., 2018, 3, 755–759 CrossRef CAS.
- Y. Wang, L. Luo, L. Chen, P. F. Ng, K. I. Lee and B. Fei, Enhancement of Stability and Photocatalytic Performance on Hybrid Perovskite with Aniline, ChemNanoMat, 2018, 4, 1054–1058 CrossRef CAS.
- S. Schünemann and H. Tüysüz, An Inverse Opal Structured Halide Perovskite Photocatalyst, Eur. J. Inorg. Chem., 2018, 2018, 2350–2355 CrossRef.
- G. Gao, Q. Xi, H. Zhou, Y. Zhao, C. Wu, L. Wang, P. Guo and J. Xu, Novel inorganic perovskite quantum dots for photocatalysis, Nanoscale, 2017, 9, 12032–12038 RSC.
- D. Cardenas-Morcoso, A. F. Gualdron-Reyes, A. B. Ferreira Vitoreti, M. Garcia-Tecedor, S. J. Yoon, M. S. Fuente, I. Mora-Sero and S. Gimenez, Photocatalytic and Photoelectrochemical Degradation of Organic Compounds with All-Inorganic Metal Halide Perovskite Quantum Dots, J. Phys. Chem. Lett., 2019, 10, 630–636 CrossRef CAS PubMed.
- Z. Zhang, Y. Liang, H. Huang, X. Liu, Q. Li, L. Chen and D. Xu, Stable and Highly Efficient Photocatalysis with Lead-Free Double-Perovskite of Cs2AgBiBr6, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- M. Acik, T. M. Alam, F. Guo, Y. Ren, B. Lee, R. A. Rosenberg, J. F. Mitchell, I. K. Park, G. Lee and S. B. Darling, Substitutional Growth of Methylammonium Lead Iodide Perovskites in Alcohols, Adv. Energy Mater., 2018, 8, 1701726–1701740 CrossRef.
- Q. Wang, T. Yang, H. Wang, J. Zhang, X. Guo, Z. Yang, S. Lu and W. Qin, Morphological and chemical tuning of lead halide perovskite mesocrystals as long-life anode materials in lithium-ion batteries, CrystEngComm, 2019, 21, 1048–1059 RSC.
- A. Pan, M. J. Jurow, F. Qiu, J. Yang, B. Ren, J. J. Urban, L. He and Y. Liu, Nanorod Suprastructures from a Ternary Graphene Oxide-Polymer-CsPbX3 Perovskite Nanocrystal Composite That Display High Environmental Stability, Nano Lett., 2017, 17, 6759–6765 CrossRef CAS PubMed.
- Y. C. Pu, H. C. Fan, T. W. Liu and J. W. Chen, Methylamine lead bromide perovskite/protonated graphitic carbon nitride nanocomposites: interfacial charge carrier dynamics and photocatalysis, J. Mater. Chem. A, 2017, 5, 25438–25449 RSC.
- Z. Qin, Y. Chen, X. Wang, X. Guo and L. Guo, Intergrowth of Cocatalysts with Host Photocatalysts for Improved Solar-to-Hydrogen Conversion, ACS Appl. Mater. Interfaces, 2016, 8, 1264–1272 CrossRef CAS PubMed.
- D. Wei, F. Ma, R. Wang, S. Dou, P. Cui, H. Huang, J. Ji, E. Jia, X. Jia, S. Sajid, A. M. Elseman, L. Chu, Y. Li, B. Jiang, J. Qiao, Y. Yuan and M. Li, Ion-Migration Inhibition by the Cation–π Interaction in Perovskite Materials for Efficient and Stable Perovskite Solar Cells, Adv. Mater., 2018, 30, 1707583 CrossRef PubMed.
- L. L. Jiang, Z. K. Wang, M. Li, C. C. Zhang, Q. Q. Ye, K. H. Hu, D. Z. Lu, P. F. Fang and L. S. Liao, Passivated perovskite crystallization via g-C3N4 for high-performance solar cells, Adv. Funct. Mater., 2018, 28, 1705875 CrossRef.
- J. Zhang, M. Zhang, L. Lin and X. Wang, Sol processing of conjugated carbon nitride powders for thin-film fabrication, Angew. Chem., Int. Ed., 2015, 54, 6297–6301 CrossRef CAS PubMed.
- P. Chen, P. Xing, Z. Chen, X. Hu, H. Lin, L. Zhao and Y. He,
In situ synthesis of AgNbO3/g-C3N4 photocatalyst via microwave heating method for efficiently photocatalytic H2 generation, J. Colloid Interface Sci., 2019, 534, 163–171 CrossRef CAS PubMed.
- Z. Chen, P. Chen, P. Xing, X. Hu, H. Lin, L. Zhao, Y. Wu and Y. He, Rapid fabrication of KTa0.75Nb0.25/g-C3N4 composite via microwave heating for efficient photocatalytic H2 evolution, Fuel, 2019, 241, 1–11 CrossRef CAS.
- Z. Feng, L. Zeng, Q. Zhang, S. Ge, X. Zhao, H. Lin and Y. He,
In situ preparation of g-C3N4/Bi4O5I2 complex and its elevated photoactivity in methyl orange degradation under visible light, J. Environ. Sci., 2020, 87, 149–162 CrossRef.
- Y. Zheng, Z. Yu, H. Ou, A. M. Asiri, Y. Chen and X. Wang, Black Phosphorus and Polymeric Carbon Nitride Heterostructure for Photoinduced Molecular Oxygen Activation, Adv. Funct. Mater., 2018, 28, 1705407 CrossRef.
- K. Zheng, K. Žídek, M. Abdellah, M. E. Messing, M. J. Al-Marri and T. Pullerits, Trap States and Their Dynamics in Organometal Halide Perovskite Nanoparticles and Bulk Crystals, J. Phys. Chem. C, 2016, 120, 3077–3084 CrossRef CAS.
- M. I. Saidaminov, A. L. Abdelhady, B. Murali, E. Alarousu, V. M. Burlakov, W. Peng, I. Dursun, L. Wang, Y. He, G. Maculan, A. Goriely, T. Wu, O. F. Mohammed and O. M. Bakr, High-quality bulk hybrid perovskite single crystals within minutes by inverse temperature crystallization, Nat. Commun., 2015, 6, 7586–7592 CrossRef PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. J. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- Y. Lin, S. Wu, X. Li, X. Wu, C. Yang, G. Zeng, Y. Peng, Q. Zhou and L. Lu, Microstructure and performance of Z-scheme photocatalyst of silver phosphate modified by MWCNTs and Cr-doped SrTiO3 for malachite green degradation, Appl. Catal., B, 2018, 227, 557–570 CrossRef CAS.
- Y. M. Ju, S. G. Yang, Y. C. Ding, C. Sun, A. Q. Zhang and L. H. Wang, Microwave-assisted rapid photocatalytic degradation of malachite green in TIO2 suspensions: mechanism and pathways, J. Phys. Chem. A, 2008, 112, 11172–11177 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, X. Liang, H. Shi, C. Wang, J. Fan, X. Hue and E. Liu, Enhanced photocatalytic activity of Ag-CsPbBr3/CN composite for broad spectrum photocatalytic degradation of cephalosporin antibiotics 7-ACA, Appl. Catal., B, 2019, 247, 57–69 CrossRef CAS.
- G. F. Samu, R. A. Scheidt, P. V. Kamat and C. Janáky, Electrochemistry and Spectroelectrochemistry of Lead Halide Perovskite Films: Materials Science Aspects and Boundary Conditions, Chem. Mater., 2018, 30, 561–569 CrossRef CAS PubMed.
- R. A. Scheidt, G. F. Samu, C. Janáky and P. V. Kamat, Modulation of Charge Recombination in CsPbBr3 Perovskite Films with Electrochemical Bias, J. Am. Chem. Soc., 2018, 140, 86–89 CrossRef CAS PubMed.
- Z. Li, C. C. Mercado, M. Yang, E. Palay and K. Zhu, Electrochemical Impedance Analysis of Perovskite–electrolyte Interfaces, Chem. Commun., 2017, 53, 2467–2470 RSC.
- G. F. Samu, A. Balog, F. D. Angelis, D. Meggiolaro, P. V. Kamat and C. J. ky, Electrochemical Hole Injection Selectively Expels Iodide from Mixed Halide Perovskite Films, J. Am. Chem. Soc., 2019, 141, 10812–10820 CrossRef CAS PubMed.
- F. A. Fatwa and K. Roel van de, Nature and Light Dependence of Bulk Recombination in Co-Pi-Catalyzed BiVO4 Photoanodes, J. Phys. Chem. C, 2012, 116, 9398–9404 Search PubMed.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, Molecule Self-Assembly Synthesis of Porous Few-Layer Carbon Nitride for Highly Efficient Photoredox Catalysis, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- K. Gelderman, L. Lee and S. W. Donne, Flat-band potential of a semiconductor: using the Mott–Schottky equation, J. Chem. Educ., 2007, 84, 685–688 CrossRef CAS.
- A. W. Bott, Electrochemistry of Semiconductors, Curr. Sep., 1998, 17, 87–91 CAS.
- J. Tang, R. Guo, W. Zhoua, C. Huang and W. Pan, Ball-flower like NiO/g-C3N4 heterojunction for efficient visible light photocatalytic CO2 reduction, Appl. Catal., B, 2018, 237, 802–810 CrossRef CAS.
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, State-of-the-Art Progress in Diverse Heterostructured Photocatalysts toward Promoting Photocatalytic Performance, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00634f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Sicao
Yu
a,
Wei
Qin
ab,
Sicao
Yu
a,
Wei
Qin