
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal DNA detection realized by peptide based carbon nanotube biosensors†
Wenjun
Li‡
a,
Yubo
Gao‡
b,
Jiaona
Zhang
b,
Xiaofang
Wang
b,
Feng
Yin
 *ac,
Zigang
Li
*ac and
Min
Zhang
*b
*ac,
Zigang
Li
*ac and
Min
Zhang
*b
aState Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, P. R. China. E-mail: yinfeng@pkusz.edu.cn; lizg@pkusz.edu.cn
bSchool of Electronic and Computer Engineering, Peking University Shenzhen Graduate School, Shenzhen, 518055, China. E-mail: zhangm@ece.pku.edu.cn
cPingshan Translational Medicine Center, Shenzhen Bay Laboratory, Shenzhen, 518055, P. R. China
First published on 21st November 2019
Abstract
Although DNA recognition has been achieved using numerous biosensors with various sensing probes, the utilization of bio-interaction between DNA and biomolecules has seldom been reported in universal DNA detection. Peptides as natural molecules have the unique ability to bind to universal DNA and excellent selectivity for DNA after being functionalized with specific groups. In this work, we report a peptide based carbon nanotube (CNT) thin-film-transistor (TFT) biosensor, which can achieve sensitive sequence-independent DNA detection. In the presence of DNA, a significant increase of ΔIon could be observed within 5 minutes, which was considered to be due to the electrostatic adsorption between the DNA and peptide of opposite zeta potential. With the gradual increase of the concentration of DNA, the ΔIon signals agree with the Hill–Langmuir model (R2 = 0.98), indicating a negatively cooperative interaction between the peptide and DNA (the Hill coefficient n < 1). Compared with the former reported universal DNA bio-detector and NanoDrop (a spectrometer from Thermo Scientific™), this unique peptide based CNT-DNA sensor demonstrated a broader sensing range from nearly 1.6 × 10−4 to 5 μmol L−1 and a much lower detection limit of approximately 0.88 μg L−1. For the quantification of cDNA from T47D cancer cells, this unique peptide based CNT sensor could achieve efficient cDNA detection. To the best of our knowledge, this is the first report on the utilization of a peptide as a sensing element in the design of CNT based DNA biosensors, which enables highly efficient universal DNA detection.
1 Introduction
The detection and quantification of nucleic acids are always a desire for clinical diagnosis,1 forensic investigation2 and biological research.3 For instance, in the screening of early-stage cancer, a simple, fast and efficient DNA biosensor is urgently required.4 To achieve this, various DNA biosensors have been developed with high accuracy and efficiency in the past decade based on colorimetric5,6 fluorescent7,8 chemiluminescent9,10 and electronic detectors.11,12Among these detectors, the electronic detector could achieve simple and real-time readout of DNA signals, as the transducing elements (such as nanoparticles,13–15 organic conductive materials16–18 and carbon-based materials19–21) could amplify the micro response to a readable signal in seconds. Benefiting from the miniaturization and portability, the electronic based DNA detectors showed promising potential for further commercial application.22,23
Among sensor devices, the graphene based transistor is one of the well-developed biosensing platforms with similar physicochemical properties to CNT-TFT.24–26 For instance, Mohanty et al. utilized a modified graphene based transistor to achieve DNA-hybridization detection.27 However, as graphene has no bandgap, it needs extra chemical and geometric manipulation to control its bandgap, which increases the difficulty of transistor manufacture. Comparatively, transistor manufacture with purified carbon nanotubes is relatively simple. Besides, many other FET based DNA sensors could also achieve efficient DNA sensing and detection.28–33
The carbon nanotube thin film transistor (CNT-TFT) is another well-developed biosensing platform and can capture tiny signals for bio-analysis.34,35 The target molecules are recognized and captured on the active CNT channel, resulting in the amplification of a tiny response for further detection.36,37 For the detection and quantification of DNA molecules, various CNT based biosensors have been developed. For instance, Star et al. immobilized synthetic oligonucleotides to specifically recognize the H63D sequence of the HFE gene.38 Martínez et al. utilized the polymer NO6 for the immobilization of sensing element and achieved efficient DNA detection.20 These studies could detect specific types of DNA based on sequencing.
However, a CNT based sequence-independent DNA detector is also desired in some applications, like the quantification of the total amount of DNA (regardless of sequences) in genomic samples, which has seldom been reported. Until recently, Dai et al. utilized the instantaneously electrostatic attraction (IEA) between Ru(NH3)63+ and DNA molecules to design an immobilization free DNA detector.37 The introduced Ru(NH3)63+ directly captured the free DNA, resulting in an instantaneous change of electrical signals with further amplification. This unique universal DNA sensing detector displayed its capability for detecting genomic DNA. In fact, there are various DNA interacting biomolecules that sense DNA and moderate its biofunction in cells. This bio-interaction has the potential to be utilized for the sensing and detection of DNA molecules. However, most DNA binding biomolecules are difficult to prepare and may be inactive during the further modification with CNTs.39,40
Recently, we have found a peptide (with the sequence Fmoc-RRMEHRMEW) which has a special bio-interaction with tiny nucleic acids and causes a significant decrease in zeta potential.41,42 Taking this exciting decrease of zeta potential into consideration, we envisioned that this change of surface potential (which resulted from the interaction between the peptide and nucleic acids) could be greatly amplified and further detected as a sequence-independent DNA biosensor. Based on this, herein, we report a CNT based DNA biosensor with biological molecules as the receptor to achieve the efficient detection and quantification of nucleic acids. Compared with the former reported universal DNA detection strategy and NanoDrop (a spectrometer from Thermo Scientific™), this peptide based DNA sensor displays a broader sensing range from nearly 1.6 × 10−4 to 5 μmol L−1 and a lower detection limit of 0.88 μg L−1.37 To the best of our knowledge, this is the first reported CNT based DNA biosensor with a peptide as the sensing element.
In order to incorporate the peptide into the CNT detector, an appropriate connection between the DNA responsive peptide and carbon nanotubes should be guaranteed. As shown in Fig. 1, N-(1-pyrene)maleimide was utilized to modify the CNTs with active maleimide groups by hydrophobic interaction between 1-pyrene butyric acid and the side wall of the CNTs. Then, through the bio-orthogonal reaction between –SH and maleimide, the connection between CNTs and the decapeptide was realized with the coupling of maleimides on the side wall surfaces. Subsequently, a significant change of ΔIon could be observed after incubation with a DNA solution for 5 min, indicating that these peptide based CNTs could be utilized as a DNA biosensor. And the ΔIon signal followed the Hill–Langmuir equation with the additional concentration of the DNA solution (R2 = 0.98).
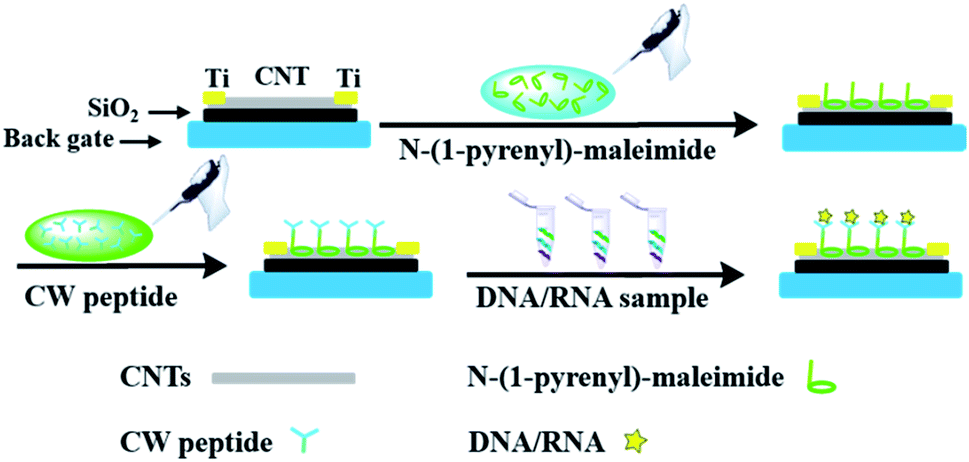 | ||
| Fig. 1 Schematic representation of the novel peptide based CNT biosensor. | ||
Furthermore, this unique peptide based DNA sensor could achieve efficient cDNA quantification from T47D cells. These results demonstrated that this unique peptide based CNT bio-detector could be utilized for the detection and quantification of DNA.
2 Experimental
2.1 Reagents and materials
The 99% semiconducting purity nanotube powder was purchased from Sigma-Aldrich Company. The 1,2-dichloroethane ≥99.9% (GC) solvent was purchased from Aladdin Company. N-(1-Pyrene)maleimide and other reactive reagents were purchased from Beijing Bailingwei Company. DNA and RNA were purchased from Shanghai GenePharma Company (the sequence of DNA: GAAATGTGGCAACTCGTC; the sequence of RNA: AAGGAAAGCUAGAAGAAAATT).2.2 Preparation of the DNA sensing peptide
The preparation of CW peptides (Fmoc-RRMEHRMEW) was conducted based on the standard Fmoc-based solid phase peptide synthesis (SPPS). The protocol of the SPPS is summarized in the literature and described below.43 Notably, for the further bio-orthogonal conjugation of –SH and maleimide, cysteine (Cys) was introduced into the original peptide sequence to form the CW peptide (Fmoc-RRMEHRMEW). MBHA resin (loading capacity: 0.37 mmol g−1) was swelled with NMP for 30 min. Then 50% (vol/vol) morpholine in DMF was used to deprotect the –Fmoc group from MBHA resin for 30 min ×2. After washing with DCM and DMF 3 times, an amino acid coupling mixture (the Fmoc-protected amino acids (5.0 equiv.), HCTU (4.9 equiv.), and DIPEA (10.0 equiv.)), which is dissolved in DMF, was added for coupling for 2.5 h, followed by washing with DCM and DMF 3 times. After this, 50% (vol/vol) morpholine in DMF was added to deprotect the Fmoc group for the later amino acid coupling.After coupling all of the amino acids to MBHA resin, further purification was needed for peptides. The resin was treated with a mixture of TFA/H2O/TIS (95/2.5/2.5) for 2 h and dried by nitrogen blowing. After precipitation with hexane/Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume) at 4 °C, the mixture was further dissolved in 42% (vol/vol) acetonitrile/water and purified by HPLC with UV detection at 220 nm or 280 nm and then identified by LC-MS.
:1 in volume) at 4 °C, the mixture was further dissolved in 42% (vol/vol) acetonitrile/water and purified by HPLC with UV detection at 220 nm or 280 nm and then identified by LC-MS.
2.3 Preparation of the CNT-TFT for the DNA biosensor
2.4 Modification and functionalization of the CNT active channel
The device was incubated with a 6 mM N-(1-pyrenyl)-maleimide solution and gently rotated for 4 hours in a shaker at room temperature. After that, DMF and ddH2O were utilized to wash the device several times to remove the residual N-(1-pyrenyl)-maleimide and DMF. The chips were then incubated in ddH2O containing selective decapeptides and rotated for another 16 hours in the shaker. The entire shaking process was maintained at 25 °C and a speed of 60 rpm, ensuring sufficient reaction and effective retention of CNTs. In order to compare the differences before and after CNT functionalization, peptide connection, and DNA capture, the electrical characteristics of the original sensor were measured after every step. As shown in Fig. S1 and S2,† the amount of CNT film was unchanged after the modification and washing steps. After the CNT functionalization was completed, the devices were washed three times with ddH2O and blown dry using N2, which could remove the unfixed decapeptide and eliminate the effect of water molecules on the current.46 Finally, the electrical signals were measured as the initial characteristics of the sensor using an Agilent B1500A Semiconductor Device Analyzer.2.5 Electrical measurement of nucleic acids
Nucleic acids (DNA or RNA) were dissolved in ddH2O or DEPC water and diluted to different gradient concentrations. A small drop of nucleic acid was added to the devices and incubated for 4 minutes, and then washed off and dried with deionized water and nitrogen three times. Finally, the response electrical signals were detected using an Agilent B1500A Semiconductor Device Analyzer. For the establishment of standard curves, all the electrical signals were recorded three times. The error bars represent the standard error of mean square root values from two independent experiments.2.6 cDNA quantification from T47D cells
T47D cells were cultured in 1640 medium with 10% serum and 1% PS. After washing by PBS for three times, TRIzol reagent (Invitrogen) was added and incubated with the T47D cells for 5 min at room temperature. After mixing with 100 μL chloroform and leaving undisturbed for 5 min, the sample was centrifuged at 4 °C at 12000 rpm for 15 min. Then the supernatant liquid was carefully collected in a clear tube without RNase and mixed with isopropanol of the same volume. After incubating on ice for 10 min, the sample was centrifuged at 4 °C at 12000 rpm for 15 min. Then, the supernatant was discarded and the sediment was washed with 75% ethyl alcohol two times. After that, the supernatant was discarded and the sediment was kept on ice for 10 min to remove the remaining ethanol. Finally, 20 μL DEPC water was added to dissolve the sediment (RNA). After that, 2 μL extracted RNA was taken from the T47D cells, and 1 μL Oligo-dT (Invitrogen) was added. After incubating the mixture at 65 °C for 5 min, the mixture was placed on ice immediately.47,48 The solution was mixed with RNase inhibitor, dNTP, and reverse transcriptase and its buffer, and PCR was used for reverse transcription amplification. Finally, the PCR product cDNA from T47D cells was collected and stored at −20 °C before dilution and further detection by the peptide based CNT biosensor.
3 Results and discussion
3.1 Design of the peptide based DNA biosensor
As shown in Fig. 2a, Ti electrodes were designed at both ends with a CNT channel positioned in the middle. Besides, the scanning electron microscope (SEM) image of the middle active channel region clearly shows that the CNTs formed a uniform random network with a density of about 10 tubes per μm (Fig. 2b). As shown in Fig. S1a and S2a,† amounts of carbon nanotubes could be observed on the CNT with relatively uniform morphology, which enables further functionalization.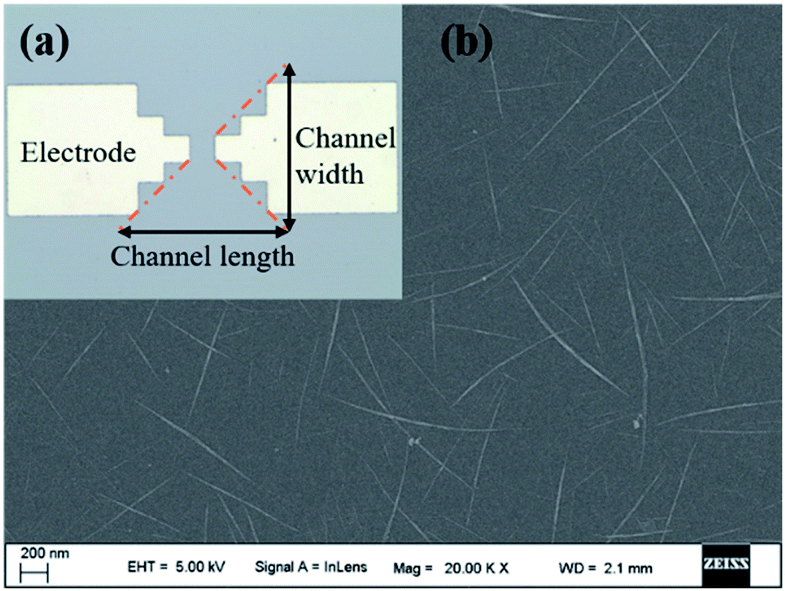 | ||
| Fig. 2 (a) The optical microscope image of CNT TFT with Ti electrodes and CNTs; (b) the SEM image of the uniform CNT network. | ||
Then N-(1-pyrene)maleimide incubation was performed on the CNT transistor for the further connection of the DNA sensitive peptide. And this functionalization process could be monitored by the change of the transfer characteristic curves (Ids–Vgs). As shown in Fig. 3a, the initial signal (black line) exhibited representative p-type characteristics at a drain voltage Vds of −0.1 V with a width and length both of 50 μm. After the incubation with N-(1-pyrene)maleimide, the value of Ids (red line) showed a significant decrease at Vgs = −10 V compared with the initial curve (black line), indicating the formation of π-stacking interaction of N-(1-pyrene)maleimide and the side walls of CNTs.49 Subsequently, the incubation of the decapeptide (Fmoc-RRMEHRMEW) further resulted in a clear reduction of the Ids signal, indicating the formation of a connection between the peptide and CNTs. These results demonstrated that the decapeptide could be successfully modified on the CNTs through the linkage of –SH and maleimide. And these changes in the electrical characteristics verified the effectiveness of the design, which displayed promising potential for further DNA detection.
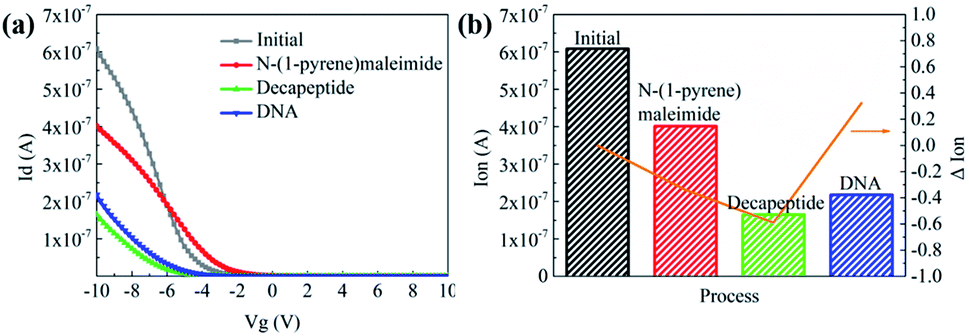 | ||
| Fig. 3 (a) Ids–Vgs characteristics at Vds = −0.1 V with W, L = 50 μm and (b) significant changes of Ion at subsequent stages of functionalization and detection. The characteristics of the initial stage (black) and incubation stages with N-(1-pyrene)maleimide (red), decapeptide (green), and DNA (blue) were measured using an Agilent B1500A Semiconductor Device Analyzer. Ion is defined as the drain current at Vgs = −10 V and ΔIon′ = (Ion′ − Ion0)/Ion0 represents the relative responses of Ion compared with prior stages. | ||
3.2 Response to the DNA molecule
To further investigate the electronic response of decapeptide modified CNTs, a 10 μM DNA solution was incubated for 5 min (Fig. S3†). Excitingly, an unambiguous increase of Ids could be observed in the Ids–Vgs curve (blue line), as shown in Fig. 3a. Compared with various electrical parameters extracted from multiple experiments, Ion was identified with the most significant changes, considering its high sensitivity and stability simultaneously. Thus, ΔIon was selected to evaluate the response of each stage in every former step. As shown in Fig. 3b, after connection with N-(1-pyrene)maleimide and decapeptide, Ion of the CNT TFT markedly decreased by approximately 40% and 60%, respectively, while, the DNA incubation resulted in an increase of Ion by about 30%. These changes of the electrical characteristics demonstrated that the functionalization of CNTs could be successfully achieved by the incubation of N-(1-pyrene)maleimide and the DNA response peptide. And the decapeptide modified CNTs could further serve as a DNA biosensor.Two control biosensors with/without peptide functionalization were also fabricated to further verify that the increase of ΔIon was attributed to the peptide based responsive elements. As shown in Fig. 4, no obvious increase of the Ion signal could be observed in the CNT TFT sample without the peptide, indicating that the free DNA could not be detected if no sensing element of peptide is connected with CNTs, while, in the presence of the peptide, a significant improvement of the Ion signal could be observed after the incubation of the DNA solution. These results demonstrate that the sensitive response of Ion is attributed to the existence of the DNA responsive peptide, and the sensor platform with the peptide shows an obvious response when detecting DNA.
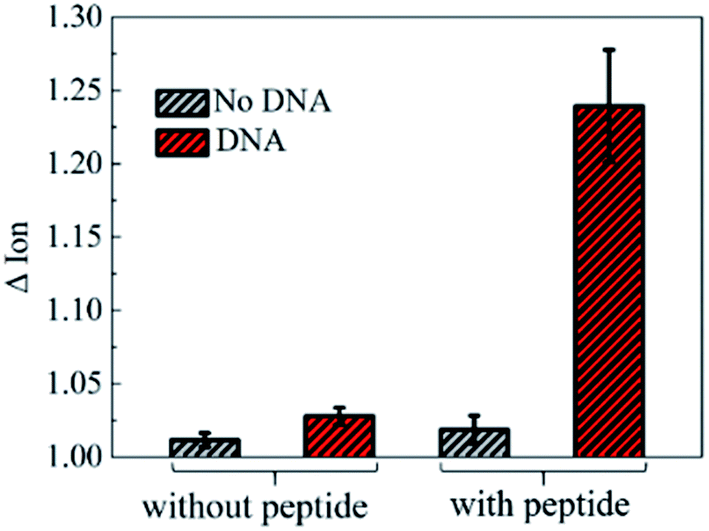 | ||
| Fig. 4 Comparison of responses to DNA for biosensors with and without the peptide. The increase in current only occurred in sensors with the peptide and the response signal of this biosensor in the blank control buffer was around 0.018. The error bars represent the standard error of the mean square root values from two independent experiments. | ||
Besides, different lengths of single stranded DNA were detected by this peptide-CNT biosensor. And their ΔIon signals displayed a similar and consistent electronic response, indicating that the sensor really can detect different lengths of DNA with stable ΔIon signal responses (Fig. S4†). Furthermore, DNA with different GC contents and double stranded DNA were also investigated using this peptide based CNT biosensor. As shown in Fig. S5 and S6,† both DNA with a high GC content and double stranded DNA could be detected resulting in a similar ΔIon response. These results demonstrated that this peptide based CNT biosensor could achieve universal DNA detection with a stable ΔIon response.
3.3 Analysis of the detection range, limits and detection mechanism
Furthermore, the detection of DNA solutions with different concentrations from 5 × 10−5 to 5 μmol L−1 was carried out to explore the detection range of this novel peptide based biosensor. As shown in Fig. 5, the responses of the Ion signal were gradually improved with the increase of the DNA concentration. The fitted curve shows a Hill–Langmuir relationship with the degree of the fitting correlation coefficient (R2) reaching 0.98. These results further demonstrate that this decapeptide modified CNT TFT can potentially be utilized for the quantification of DNA.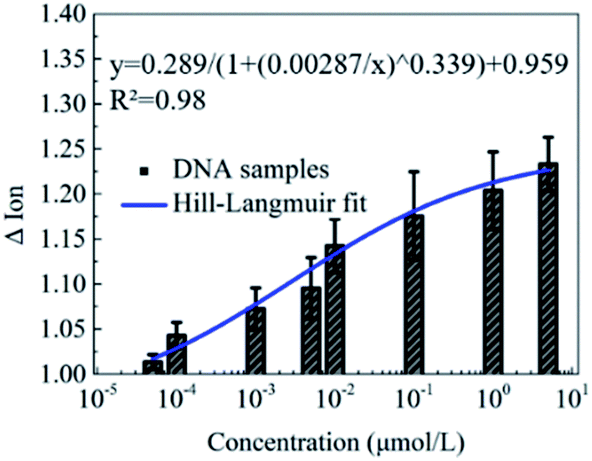 | ||
| Fig. 5 The ΔIon signal responses to DNA concentrations from 5 × 10−5 to 5 μmol L−1. The relationship between ΔIon and the DNA concentration could be fitted with the Hill–Langmuir model with a fitting correlation coefficient R2 of approximately 0.98. The error bars represent the standard error of the mean square root values from two independent experiments. | ||
According to the electrostatic adsorption mechanism, the curve was fitted with the Hill–Langmuir model, which was used to describe the degree of cooperativity of the ligand binding to the receptor.50
| ΔIon = A/(1 + (KA/c)n) + Z |
Besides, benefiting from the sensitive response to nucleic acids, this unique peptide based detector could also be utilized for the quantification of RNA. As shown in Fig. S7,† this peptide based biosensor displayed a significant Ion response in the presence of RNA solutions with different concentrations from 1–0.0001 μmol L−1. With the increase of RNA concentration, the Ion response was also fitted with the Hill–Langmuir relationship with an R2 about 0.99. These results demonstrated that this unique peptide based CNT biosensor could be used for universal nucleic acid detection.
Furthermore, a zeta potential assay was conducted to investigate the potential mechanism of this unique ΔIon signal increase in the peptide based CNT biosensor. As shown in Fig. 6a, in the absence of DNA, the zeta potential of the peptide-DNA complex displayed a significant decrease. Notably, the formed complex had the same negative ZP with DNA, indicating that once the DNA molecule was incorporated into the decapeptide, the complex may display a reduced absorption towards DNA. These results were consistent with the Hill–Langmuir model, especially with the negatively cooperative interaction situation (n < 1).
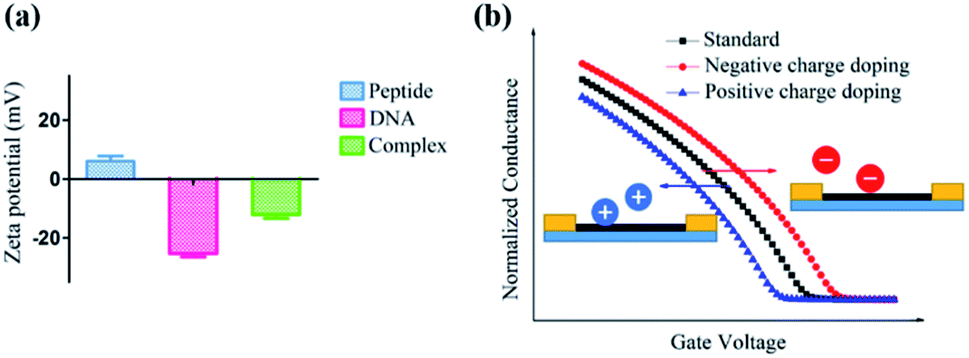 | ||
| Fig. 6 (a) Zeta potential values of the CW peptide, DNA, and their complex. The electrostatic adsorption of the peptide and DNA occurred because of their opposite ZP and the formed complex had the same ZP as DNA, which hindered the continuous adsorption with DNA. (b) Illustration of the current change sensitive to charge doping. The negative charge doping induced a right shift of the characteristic curve, resulting in an enhanced current and vice versa. | ||
As for the peptide based CNT biosensor, the DNA molecule with a negative surface potential could serve as the centralized carrier.51 Thus, attributed to the selectivity of the peptide, the DNA molecule could be captured, further leading to a significant change in the zeta potential, which could be amplified and detected with the ΔIon signals. The following simulation of the charge effect on the p-type transistor supported this hypothesis. As shown in Fig. 6b, the increase of the negative charge resulted in the reduction of the current, while the enhancement of the positive charge led to the increase of the current, which is consistent with the experimental observations.
3.4 cDNA quantification from T47D cells
As this unique peptide based DNA biosensor could achieve sequence-independent DNA detection, we further applied this novel peptide based CNT detector to cDNA quantification of T47D cells. As shown in Fig. 7, the detected concentration of cDNA from T47D cells was 7.32 ng μL−1 using peptide based CNTs, which was consistent with the results using a nanodrop of 10.36 ng μL−1. These results demonstrated that this novel peptide based DNA biosensor could also be utilized for the quantification of cDNA from the T47D cancer cell line, regardless of the sequence. Furthermore, as our peptide based CNT biosensor displayed a broader sensing range and low detection limit of 8.8 × 10−4 ng μL−1, it could be potentially utilized for tracing the amount of DNA in the future.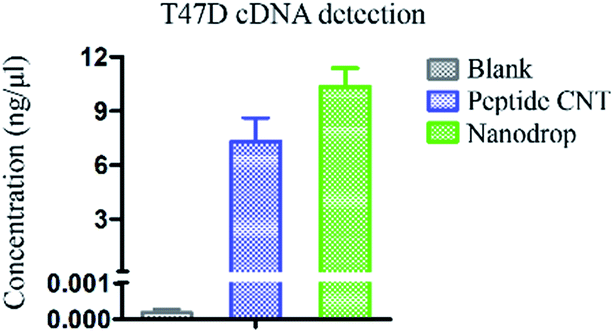 | ||
| Fig. 7 Detection of cDNA from T47D cells using the peptide based CNT biosensor, with ddH2O as the blank and NanoDrop as the control. The concentration of cDNA detected by peptide CNTs was 7.32 ng μL−1. The concentration of cDNA detected by NanoDrop was 10.36 ng μL (The concentration of cDNA was quantified by the Hill–Langmuir equation: y = 0.289/(1 + 0.01586/x)0.339 + 0.959, where the unit of concentration was converted to ng μL−1). The error bars represent the standard error of the mean square root values from two independent experiments. | ||
4 Conclusions
Although numerous DNA detection strategies have been developed with various sensing elements for DNA capture, the utilization of a biomolecule for universal sequence-independent DNA quantification has seldom been reported. In this work, we have realized a universal DNA detection platform by incorporating the DNA sensitive CW peptide into the CNT TFT platform to amplify and monitor sensitive electronic signals. In the presence of DNA, a significant increase of ΔIon could be observed for the peptide based CNT bio-detector. With the gradual increase of the DNA concentration, the ΔIon signal followed the Hill–Langmuir relationship with an R2 about 0.989 (n < 1). Compared with the former reported sequence-independent DNA detection strategy and NanoDrop, the sensing range of this peptide based biosensor was more than 12-times broader (from nearly 1.6 × 10−4 to 5 μmol L−1) and the detection limit was 4300-times lower to only 0.88 μg L−1.37 To the best of our knowledge, this is the first reported CNT TFT based DNA biosensor with a peptide as the sensing element to realize universal detection. We believe that, with the wider sensing range and better detection limit, the proposed sensor could greatly reduce the amount of the nucleic acid sample of interest and be applied for detecting DNA isolated from PB (peripheral blood) or tissues in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Shenzhen Science and Technology Innovation Committee, JCYJ20180507181702150 and JCYJ20170817172023838, and National Natural Science Foundation of China grants 21778009 and 81701818.References
- L. A. Diaz Jr and A. Bardelli, J. Clin. Oncol., 2014, 32(6), 579–586 CrossRef PubMed.
- R. Lehmann-Werman, D. Neiman, H. Zemmour, J. Moss, J. Magenheim, A. Vaknin-Dembinsky, S. Rubertsson, B. Nellgård, K. Blennow and H. Zetterberg, et al. , Proc. Natl. Acad. Sci. U. S. A., 2016, 113(13), E1826–E1834 CrossRef CAS PubMed.
- C. Abbosh, N. J. Birkbak and C. Swanton, Nat. Rev. Clin. Oncol., 2018, 15, 577–586 CrossRef CAS PubMed.
- C. Abbosh, N. J. Birkbak, G. A. Wilson, M. Jamal-Hanjani, T. Constantin, R. Salari, J. Le Quesne, D. A. Moore, S. Veeriah and R. Rosenthal, et al. , Nature, 2017, 545, 446–451 CrossRef CAS PubMed.
- H. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(39), 14036–14039 CrossRef CAS PubMed.
- W. Xu, X. Xue, T. Li, H. Zeng and X. Liu, Angew. Chem., Int. Ed., 2009, 48(37), 6849–6852 CrossRef CAS PubMed.
- X. Shu, Y. Liu and J. Zhu, Angew. Chem., Int. Ed., 2012, 51(44), 11006–11009 CrossRef CAS PubMed.
- L. Zhang, W. Zheng, R. Tang, N. Wang, W. Zhang and X. Jiang, Biomaterials, 2016, 104, 269–278 CrossRef CAS PubMed.
- R. Freeman, X. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133(30), 11597–11604 CrossRef CAS PubMed.
- S. Zhang, H. Zhong and C. Ding, Anal. Chem., 2008, 80(19), 7206–7212 CrossRef CAS PubMed.
- J. Mei, Y.-T. Li, H. Zhang, M.-M. Xiao, Y. Ning, Z.-Y. Zhang and G.-J. Zhang, Biosens. Bioelectron., 2018, 110, 71–77 CrossRef CAS PubMed.
- S. Sorgenfrei, C.-Y. Chiu, R. L. Gonzalez Jr, Y.-J. Yu, P. Kim, C. Nuckolls and K. L. Shepard, Nat. Nanotechnol., 2011, 6, 126 CrossRef CAS PubMed.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47(40), 7602–7625 CrossRef CAS PubMed.
- B. M. Reinhard, M. Siu, H. Agarwal, A. P. Alivisatos and J. Liphardt, Nano Lett., 2005, 5(11), 2246–2252 CrossRef CAS PubMed.
- L. Wu, E. Xiong, X. Zhang, X. Zhang and J. Chen, Nano Today, 2014, 9(2), 197–211 CrossRef CAS.
- P. Lin, X. Luo, I.-M. Hsing and F. Yan, Adv. Mater., 2011, 23(35), 4035–4040 CrossRef CAS PubMed.
- T. T. K. Nguyen, T. N. Nguyen, G. Anquetin, S. Reisberg, V. Noël, G. Mattana, J. Touzeau, F. Barbault, M. C. Pham and B. Piro, Biosens. Bioelectron., 2018, 113, 32–38 CrossRef CAS PubMed.
- Q. Zhang and V. Subramanian, Biosens. Bioelectron., 2007, 22(12), 3182–3187 CrossRef CAS PubMed.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48(26), 4785–4787 CrossRef CAS PubMed.
- M. T. Martínez, Y.-C. Tseng, N. Ormategui, I. Loinaz, R. Eritja and J. Bokor, Nano Lett., 2009, 9(2), 530–536 CrossRef PubMed.
- Q. Zheng, H. Wu, Z. Shen, W. Gao, Y. Yu, Y. Ma, W. Guang, Q. Guo, R. Yan, J. Wang and K. Ding, Analyst, 2015, 140(19), 6660–6670 RSC.
- H. Jiang and E.-C. Lee, Biosens. Bioelectron., 2018, 118, 16–22 CrossRef CAS PubMed.
- V. Schroeder, S. Savagatrup, M. He, S. Lin and T. M. Swager, Chem. Rev., 2019, 119(1), 599–663 CrossRef CAS PubMed.
- Z. Yin, Q. He and X. Huang, et al. , Nanoscale, 2012, 4(1), 293–297 RSC.
- T. Chen, P. T. K. Loan and C. Hsu, et al. , Biosens. Bioelectron., 2013, 41, 103–109 CrossRef CAS PubMed.
- R. Stine, J. T. Robinson and P. E. Sheehan, et al. , Adv. Mater., 2010, 22(46), 5297–5300 CrossRef CAS PubMed.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8(12), 4469–4476 CrossRef CAS PubMed.
- S. Xu, S. Jiang and C. Zhang, et al. , Appl. Surf. Sci., 2018, 427, 1114–1119 CrossRef CAS.
- S. Xu, S. Jiang and J. Wang, et al. , Sens. Actuators, B, 2016, 222, 1175–1183 CrossRef CAS.
- S. Xu, C. Zhang and S. Jiang, et al. , Sens. Actuators, B, 2019, 284, 125–133 CrossRef CAS.
- S. Xu, B. Man and S. Jiang, et al. , ACS Appl. Mater. Interfaces, 2015, 7(20), 10977–10987 CrossRef CAS PubMed.
- S. Xu, J. Zhan and B. Man, et al. , Nat. Commun., 2017, 8(1), 14902 CrossRef CAS PubMed.
- S. Xu, B. Man and S. Jiang, et al. , Nanotechnology, 2014, 25(16), 165702 CrossRef PubMed.
- B. L. Allen, P. D. Kichambare and A. Star, Adv. Mater., 2007, 19(11), 1439–1451 CrossRef CAS.
- G. O. Silva, Z. P. Michael, L. Bian, G. V. Shurin, M. Mulato, M. R. Shurin and A. Star, ACS Sens., 2017, 2(8), 1128–1132 CrossRef CAS PubMed.
- F. Li, J. Peng, J. Wang, H. Tang, L. Tan, Q. Xie and S. Yao, Biosens. Bioelectron., 2014, 54, 158–164 CrossRef CAS PubMed.
- S. Dai, W. Lu, Y. Wang and B. Yao, Biosens. Bioelectron., 2019, 127, 101–107 CrossRef CAS PubMed.
- A. Star, E. Tu, J. Niemann, J.-C. P. Gabriel, C. S. Joiner and C. Valcke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(4), 921–926 CrossRef CAS PubMed.
- W. H. Hudson and E. A. Ortlund, Nat. Rev. Mol. Cell Biol., 2014, 15(11), 749–760 CrossRef CAS PubMed.
- A. J. P. Smith and S. E. Humphries, J. Mol. Biol., 2009, 385(3), 714–717 CrossRef CAS PubMed.
- W. Li, D. Wang, X. Shi, J. Li, Y. Ma, Y. Wang, T. Li, J. Zhang, R. Zhao, Z. Yu, F. Yin and Z. Li, Mater. Horiz., 2018, 5(4), 745–752 RSC.
- Y. Ma, W. Li, Z. Zhou, X. Qin, D. Wang, Y. Gao, Z. Yu, F. Yin and Z. Li, Bioconjugate Chem., 2019, 30(3), 536–540 CrossRef CAS PubMed.
- Y.-W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761 CrossRef CAS PubMed.
- Z. Liu, J. Zhao, W. Xu, L. Qian, S. Nie and Z. Cui, ACS Appl. Mater. Interfaces, 2014, 6(13), 9997–10004 CrossRef CAS PubMed.
- J.-O. Lee, C. Park, J.-J. Kim, J. Kim, J. W. Park and K.-H. Yoo, J. Phys. D: Appl. Phys., 2000, 33(16), 1953–1956 CrossRef CAS.
- W. Kim, A. Javey, O. Vermesh, Q. Wang, Y. Li and H. Dai, Nano Lett., 2003, 3(2), 193–198 CrossRef CAS.
- F. Yin, C. Yang, Q. Wang, S. Zeng, R. Hu, G. Lin, J. Tian, S. Hu, R. Lan, H. Yoon, F. Lu, K. Wang and K. Yong, Theranostics, 2015, 5, 818 CrossRef CAS PubMed.
- A. Eguchi, B. R. Meade, Y. C. Chang, C. T. Fredrickson, K. Willert, N. Puri and S. F. Dowdy, Nat. Biotechnol., 2009, 27, 567 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50(6), 1371–1374 CrossRef CAS PubMed.
- J. Boyle, Biochem. Mol. Biol. Educ., 2005, 33(1), 74–75 CrossRef CAS.
- Y. Wang, W. Zhang, J. Zhang, W. Sun, R. Zhang and H. Gu, ACS Appl. Mater. Interfaces, 2013, 5(20), 10337–10345 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00625g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |