
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
γ-Alumina-supported Pt17 cluster: controlled loading, geometrical structure, and size-specific catalytic activity for carbon monoxide and propylene oxidation†
Yuichi
Negishi
 *ab,
Nobuyuki
Shimizu
a,
Kanako
Funai
a,
Ryo
Kaneko
a,
Kosuke
Wakamatsu
a,
Atsuya
Harasawa
a,
Sakiat
Hossain
a,
Manfred E.
Schuster
c,
Dogan
Ozkaya
c,
Wataru
Kurashige
d,
Tokuhisa
Kawawaki
ab,
Seiji
Yamazoe
*e and
Shuhei
Nagaoka
*d
*ab,
Nobuyuki
Shimizu
a,
Kanako
Funai
a,
Ryo
Kaneko
a,
Kosuke
Wakamatsu
a,
Atsuya
Harasawa
a,
Sakiat
Hossain
a,
Manfred E.
Schuster
c,
Dogan
Ozkaya
c,
Wataru
Kurashige
d,
Tokuhisa
Kawawaki
ab,
Seiji
Yamazoe
*e and
Shuhei
Nagaoka
*d
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.kagu.tus.ac.jp
bPhotocatalysis International Research Center, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
cJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading RG4 9NH, UK
dJohnson Matthey Japan, G.K., 5123-3, Kitsuregawa, Sakura, Tochigi 329-1492, Japan. E-mail: Shuhei.Nagaoka@mattheyasia.com
eDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji-shi, Tokyo 192-0397, Japan. E-mail: yamazoe@tmu.ac.jp
First published on 3rd December 2019
Abstract
Although Pt is extensively used as a catalyst to purify automotive exhaust gas, it is desirable to reduce Pt consumption through size reduction because Pt is a rare element and an expensive noble metal. In this study, we successfully loaded a Pt17 cluster on γ-alumina (γ-Al2O3) (Pt17/γ-Al2O3) using [Pt17(CO)12(PPh3)8]Cln (n = 1, 2) as a precursor. In addition, we demonstrated that Pt is not present in the form of an oxide in Pt17/γ-Al2O3 but instead has a framework structure as a metal cluster. Moreover, we revealed that Pt17/γ-Al2O3 exhibits higher catalytic activity for carbon monoxide and propylene oxidation than γ-Al2O3-supported larger Pt nanoparticles (PtNP/γ-Al2O3) prepared using the conventional impregnation method. Recently, our group discovered a simple method for synthesizing the precursor [Pt17(CO)12(PPh3)8]Cln. Furthermore, Pt17 is a Pt cluster within the size range associated with high catalytic activity. By combining our established synthesis and loading methods, other groups can conduct further research on Pt17/γ-Al2O3 to explore its catalytic activities in greater depth.
Introduction
With rapid advances in science and technology, automobiles have become indispensable in our daily lives. Because Pt can catalytically eliminate harmful substances contained in exhaust gas, this metal along with Rh and Pd are extensively used to treat exhaust gas.1 However, Pt is a rare element and an expensive precious metal. Therefore, it is essential to reduce the amount of consumed Pt.Many attempts have been made to develop catalysts without Pt. However, previous studies have implied that the activity and durability of Pt are superior to those of non-precious metals. To reduce the amount of Pt consumed while taking advantage of its characteristic features, it is essential to improve its activity and performance per unit weight of the catalyst. The size reduction of Pt nanoparticles/clusters (hereinafter: Ptn clusters) increases the proportion of surface atoms2,3 and enables the creation of new geometrical/electronic structures;4–10 thus, this approach can efficiently reduce Pt consumption.11–13
Meanwhile, the geometrical/electronic structures and chemical properties of Ptn clusters in the fine size range vary considerably depending on the number of constituent atoms.14 Therefore, it is important to load Ptn clusters with a controlled number of constituent atoms on a substrate to create a highly active supported Pt catalyst using fine Ptn clusters while elucidating the catalytic activity and performance of the clusters. Using a vacuum device with a mass selector,2,6,10,15–19 it is possible to load controlled Ptn clusters onto a substrate. In fact, magnesium-oxide-supported Ptn clusters (Ptn/MgO; n = 8–20) and titanium-dioxide-supported Ptn/TiO2 (n = 4, 7–10, 15) have been prepared with precisely controlled numbers of Pt atoms using these types of experiments. These studies also revealed that fine supported Ptn clusters exhibit high catalytic activity for the oxidation of carbon monoxide (CO).2,19 However, for the practical use of supported Pt catalysts, issues remain regarding device manufacturing costs and loading efficiency for the preparation of supported Ptn clusters using such vacuum equipment.
Recently, it has become possible to precisely synthesize various noble-metal and noble-metal-alloy clusters with atomic accuracy.20–49 Ptn clusters can be synthesized with atomic accuracy using CO as a ligand or two types of ligand, CO and phosphine.48 In addition, Ptn clusters can be precisely synthesized using special dendrimers as templates.50,51 When these Ptn clusters are adsorbed on a substrate followed by the removal of the ligands, Ptn clusters with a controlled number of constituent atoms can be loaded on the substrate without the issues of device construction cost and loading efficiency (Fig. 1). However, currently, there are few examples of controlled loading of Ptn clusters on a substrate using this approach. For the synthesis of the former ligand-protected Ptn clusters, it is essential to carry out the reaction under a CO atmosphere. For the synthesis of the latter dendrimer-protected Ptn clusters, a special dendrimer synthesis technique is needed. Therefore, few research groups are capable of conducting these precise syntheses, and fundamental and applied research on fine supported Ptn clusters is currently limited.
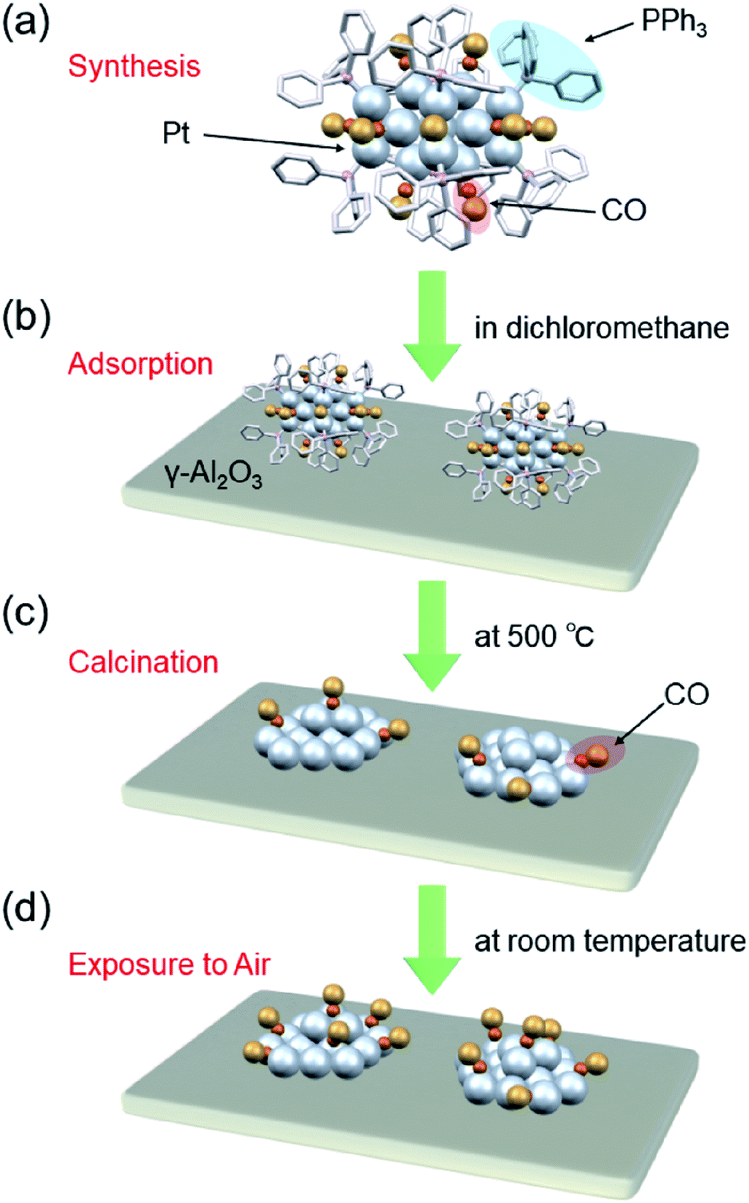 | ||
| Fig. 1 Schematic of preparation of Pt17/γ-Al2O3: (a) precise synthesis of [Pt17(CO)12(PPh3)8]Cln, (b) adsorption of [Pt17(CO)12(PPh3)8]Cln on γ-Al2O3 (Pt17(CO)12(PPh3)8/γ-Al2O3), (c) calcination of ligands while maintaining the cluster size (Pt17/γ-Al2O3), and (d) exposure of Pt17/γ-Al2O3 to air. | ||
We recently discovered a very simple method for synthesizing Pt17 clusters protected with CO and triphenylphosphine (PPh3) ([Pt17(CO)12(PPh3)8]Cln; n = 1, 2; Fig. 2(a)).52 In our synthesis method, first, the Ptn(CO)m(PPh3)l cluster, which is mainly composed of [Pt17(CO)12(PPh3)8]Cln, is prepared by mixing the reagents and heating the solvent in the atmosphere. Then, the main product, [Pt17(CO)12(PPh3)8]Cln, is separated from the obtained mixture with high purity using the difference in solubility. This method does not require special synthesis equipment or dendrimer synthesis techniques. If we could establish the loading method of the Pt17 cluster using [Pt17(CO)12(PPh3)8]Cln as a precursor, many research groups would be able to obtain fine supported Pt17 catalysts.
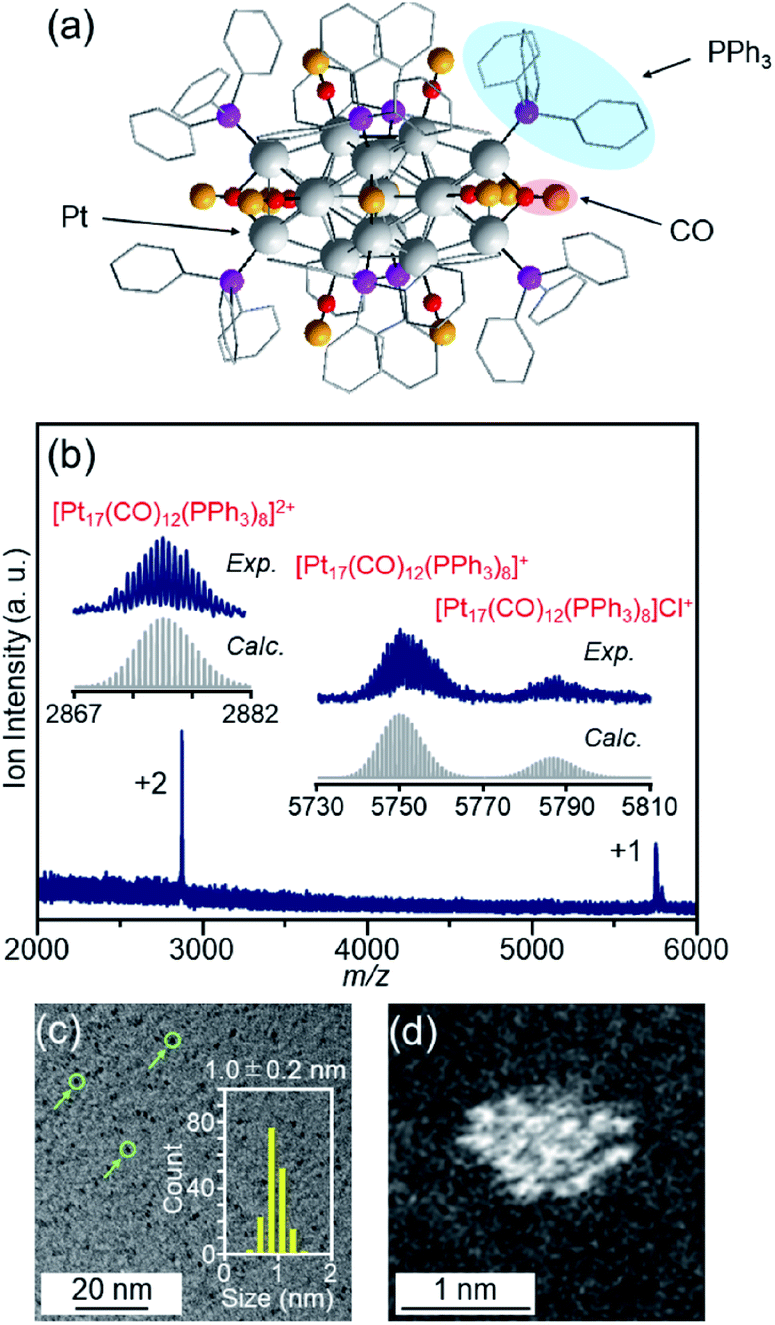 | ||
| Fig. 2 (a) Geometrical structure of [Pt17(CO)12(PPh3)8]+ determined by single-crystal X-ray crystallography. The geometrical structure (a) was reproduced from ref. 51. (b) Positive-ion ESI mass spectrum of [Pt17(CO)12(PPh3)8]Cln synthesized in this work. The spectrum indicates that both [Pt17(CO)12(PPh3)8]Cln (n = 1, 2) are contained in this sample, similar to the previous work.52 (c) TEM and (d) HAADF-STEM images of [Pt17(CO)12(PPh3)8]Cln. In (c), the green circles indicate the particles and the histogram estimated from the TEM images is also provided. Copyright 2017 American Chemical Society. | ||
In this study, the following three goals were addressed with the final objective of using supported Ptn clusters as catalysts to treat automotive exhaust gas: (i) establishment of a precise loading method of Pt17 on γ-alumina (γ-Al2O3); (ii) structural analysis of the obtained Pt17/γ-Al2O3; and (iii) evaluation of the catalytic activity of Pt17/γ-Al2O3 against the oxidation reaction of CO and propylene (C3H6). As a result, we successfully determined the conditions for loading Pt17 on γ-Al2O3 while preserving the size of Pt17 (Fig. 1). We observed that the supported Pt17 is not present in the form of an oxide53 but has a framework structure as a metal cluster in the obtained Pt17/γ-Al2O3. Furthermore, Pt17/γ-Al2O3 exhibited higher catalytic activity against the oxidation of CO and C3H6 than γ-Al2O3-supported larger Pt nanoparticles (PtNP/γ-Al2O3) prepared using the conventional impregnation method.
Results and discussion
Loading of the Pt17 cluster on γ-Al2O3
[Pt17(CO)12(PPh3)8]Cln (Fig. 2(a)) was synthesized using our previously reported method (see the Experimental section).52 In this method, Ptx(CO)y(PPh3)z clusters containing both CO and PPh3 were synthesized by mixing the reagents and heating the solvent in the atmosphere. Because CO, which is one of the ligands, can be generated by the oxidation of ethylene glycol,54 this method does not require equipment for preparing a CO atmosphere. Specifically, an ethylene glycol solution containing a Pt salt (H2PtCl6) and sodium hydroxide (NaOH) was heated at 120 °C in the atmosphere, and then PPh3 was added at room temperature to obtain Ptx(CO)y(PPh3)z clusters. [Pt17(CO)12(PPh3)8]Cln was separated from the obtained mixture using the difference in solubility in the solvent (see the Experimental section; Scheme S1†). The electrospray ionization (ESI) (Fig. 2(b)) and matrix-assisted laser desorption/ionization (MALDI) (Fig. S1†) mass spectra of the product indicate that the product contained high-purity [Pt17(CO)12(PPh3)8]Cln. The transmission electron microscopy (TEM) (Fig. 2(c)) and high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) results (Fig. 2(d)) of the product were also consistent with the results of mass spectrometry (Fig. 2(b) and S1†).The resulting [Pt17(CO)12(PPh3)8]Cln was first adsorbed onto γ-Al2O3 (Fig. 1(b)). In an aprotic solvent, the metal oxide has a permanent dipole moment on the surface.55 As reported by Tsukuda et al., when this surface comes into contact with a metal cluster containing a functional group with a high dielectric constant (e.g., a phenyl group) in the ligand, an induced dipole moment is generated in the ligand layer, and the metal clusters are adsorbed on the surface of the metal oxide via a dipole–induced dipole interaction.56 In the [Pt17(CO)12(PPh3)8]Cln used in this study, the ligand layer contained a large amount of phenyl groups, and [Pt17(CO)12(PPh3)8]Cln was thus adsorbed onto γ-Al2O3via a dipole–induced dipole interaction. Dichloromethane was used as the aprotic solvent. The concentration of [Pt17(CO)12(PPh3)8]Cln in solution was carefully controlled by inductively coupled plasma mass spectrometry (ICP-MS) such that the weight of Pt17 was 0.15 wt% relative to γ-Al2O3. The solution changed from brown to colorless and transparent after 2 h of stirring, indicating that practically all of the [Pt17(CO)12(PPh3)8]Cln was adsorbed onto γ-Al2O3 (Pt17(CO)12(PPh3)8/γ-Al2O3).
The particle diameter of Pt17(CO)12(PPh3)8/γ-Al2O3 obtained using this approach was estimated by HAADF-STEM measurement. In the HAADF-STEM image in Fig. 3(a), particles (0.94 ± 0.16 nm) with sizes similar to that of [Pt17(CO)12(PPh3)8]Cln (1.0 ± 0.2 nm; Fig. 2(d) and S2†) were observed with a narrow distribution (Fig. S3†). The HAADF-STEM image of Pt17(CO)12(PPh3)8/γ-Al2O3 (Fig. 3(a)) shows that it contained many particles with shapes similar to that of the Pt17 core of the precursor [Pt17(CO)12(PPh3)8]Cln (Fig. 3(b)). In the diffuse reflection (DR) spectrum of Pt17(CO)12(PPh3)8/γ-Al2O3, a peak structure similar to that of [Pt17(CO)12(PPh3)8]Cln was observed (Fig. 4(a) and (b)). These results indicate that aggregation of Pt17(CO)12(PPh3)8 hardly occurred during the adsorption process and that Pt17(CO)12(PPh3)8 after adsorption retained the geometrical/electronic structure of the precursor [Pt17(CO)12(PPh3)8]Cln.
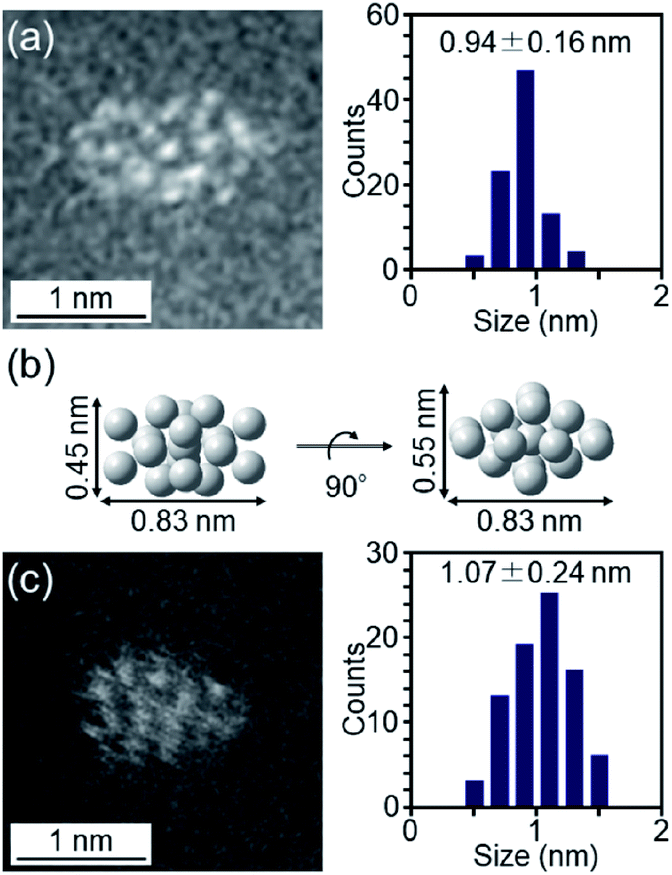 | ||
| Fig. 3 (a) HAADF-STEM image of Pt17(CO)12(PPh3)8/γ-Al2O3. (b) Pt17-core structure (two-angle views) of [Pt17(CO)12(PPh3)8]Cln (Fig. 2(a)) on the same scale as (a) and (c). (c) HAADF-STEM image of Pt17/γ-Al2O3. In (a) and (c), the histogram and average diameters estimated from the various HAADF-STEM images (Fig. S3 and S6†) are also included. | ||
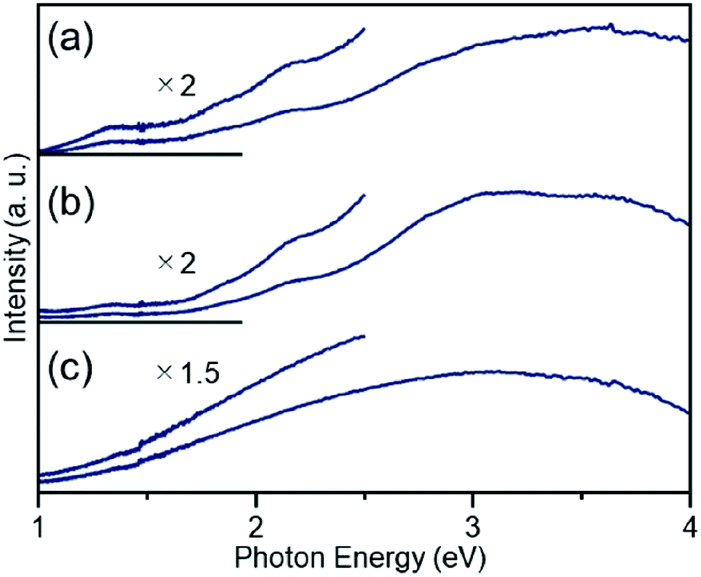 | ||
| Fig. 4 DR spectra of (a) [Pt17(CO)12(PPh3)8]Cln, (b) Pt17(CO)12(PPh3)8/γ-Al2O3, and (c) Pt17/γ-Al2O3. For (b) and (c), the spectra were obtained by the subtraction of γ-Al2O3. The DR spectral profile of [Pt17(CO)12(PPh3)8]Cln (a) differs slightly from that reported in our previous work.52 This difference likely results from the different ratios of the two kinds of charged clusters, [Pt17(CO)12(PPh3)8]Cln (n = 1 or 2), in the two studies. | ||
Then, the PPh3 ligands were removed from Pt17(CO)12(PPh3)8/γ-Al2O3 by calcination57–61 (Fig. 1(c)). Based on thermogravimetric mass spectrometry (TG-MS) analysis, a temperature of approximately 400 °C is required for PPh3 removal (Fig. S4†).62 Thus, PPh3 was removed from the Pt17 cluster by calcination at 500 °C. In the DR spectra of the sample after calcination (Fig. 4(c)), the peaks of [Pt17(CO)12(PPh3)8]Cln (Fig. 4(a)) and Pt17(CO)12(PPh3)8/γ-Al2O3 (Fig. 4(b)) were not observed. In the X-ray photoelectron spectrum after calcination (Fig. S5†), the P 2p peak was not observed. In the HAADF-STEM image of the sample after calcination (Fig. 3(c)), particles (1.07 ± 0.24 nm) with sizes similar to that of Pt17(CO)12(PPh3)8/γ-Al2O3 (0.94 ± 0.16 nm; Fig. 3(a)) were observed with a narrow distribution (Fig. S6†). These results indicate that the PPh3 ligands were removed from the cluster by calcination and that Pt17 did not aggregate during this process. Pt forms a relatively strong bond with O compared with other noble metals (318.4 ± 6.7 kJ mol−1 for Pt–O vs. 223 ± 21.1 kJ mol−1 for Au–O).63 Furthermore, as γ-Al2O3 has a complicated structure in which Al atoms are arranged octahedrally or tetrahedrally, cationic sites are present because of the surface defects in γ-Al2O3.64 Pt clusters could be strongly immobilized on γ-Al2O3 by the interaction between Pt atoms and these cationic sites.53 For these reasons, it is considered that Pt17 did not aggregate on γ-Al2O3 during calcination. To confirm the weight of Pt loaded on γ-Al2O3, Pt17/γ-Al2O3 was mixed with aqua regia, and the amount of dissolved Pt was measured using ICP optical emission spectroscopy (ICP-OES). The results confirmed that 0.15 wt% Pt was actually loaded on γ-Al2O3. The results of temperature-programmed reaction measurements indicate that the surface of the supported Pt17 was covered by CO at normal temperature (Fig. 1(d), S7 and S8†). This result is most likely due to the existence of uneliminated CO (Fig. 1(c)) as well as the adsorption of CO from the atmosphere.
Structural analysis of Pt17/γ-Al2O3
To understand deeply the charge state and geometrical structure of the obtained Pt17/γ-Al2O3, the charge state and geometry of the Pt17 cluster were investigated using X-ray absorption fine structure (XAFS) analysis.The Pt L3-edge X-ray absorption near-edge structure (XANES) spectra of [Pt17(CO)12(PPh3)8]Cln, Pt17(CO)12(PPh3)8/γ-Al2O3, and Pt17/γ-Al2O3 are shown in Fig. 5(a) together with those of Pt foil and PtO2 for comparison. The white-line intensities of [Pt17(CO)12(PPh3)8]Cln, Pt17(CO)12(PPh3)8/γ-Al2O3, and Pt17/γ-Al2O3 are similar to that of Pt foil and very different from that of PtO2. This result indicates that Pt is not present as an oxide in Pt17.53 Among the three samples, the white-line intensity increases in the order of [Pt17(CO)12(PPh3)8]Cln → Pt17(CO)12(PPh3)8/γ-Al2O3 → Pt17/γ-Al2O3. This result indicates that the number of holes in the d orbital of Pt17 increases, namely the electron density of Pt17 decreases, in this order.
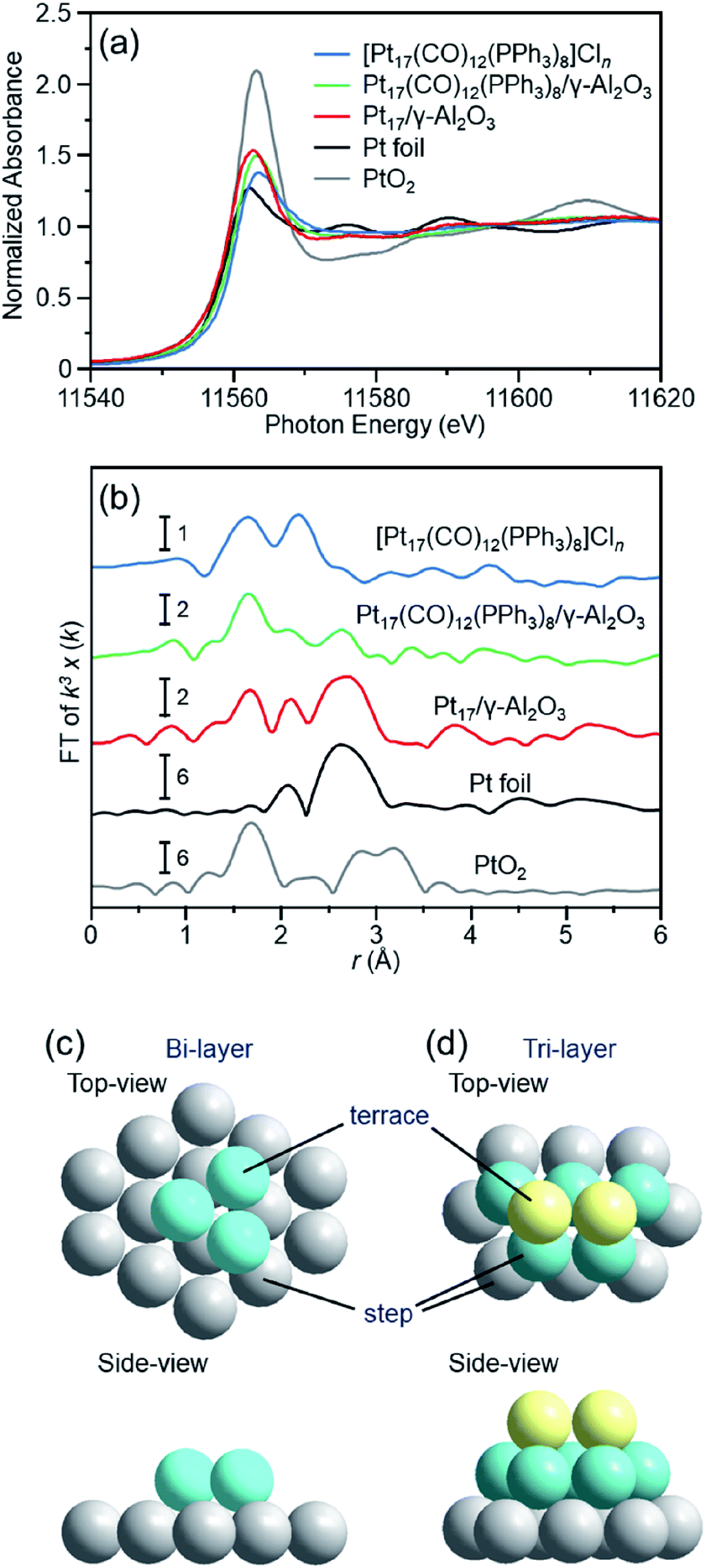 | ||
| Fig. 5 (a) Pt L3-edge XANES and (b) Pt L3-edge FT-EXAFS spectra of [Pt17(CO)12(PPh3)8]Cln, Pt17(CO)12(PPh3)8/γ-Al2O3, and Pt17/γ-Al2O3 together with those of Pt foil and PtO2 for comparison. (c) and (d) two proposed structures for Pt17 on γ-Al2O3, which were estimated based on the HAADF-STEM images (Fig. 3(c) and S6†) of Pt17/γ-Al2O3 (Fig. S11†). In (b), the peak at ∼2.3 Å in the spectrum of Pt foil is attributed to the satellite peak of the Pt–Pt bond. In (c) and (d), both top and side views are shown. | ||
Fig. 5(b) shows the Pt L3-edge Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) spectra of [Pt17(CO)12(PPh3)8]Cln, Pt17(CO)12(PPh3)8/γ-Al2O3, and Pt17/γ-Al2O3 (Tables S1–S3 and Fig. S9†). In the FT-EXAFS spectrum of [Pt17(CO)12(PPh3)8]Cln, the peaks attributed to the Pt–C and Pt–P bonds appear at ∼1.7 and ∼2.3 Å, respectively. For Pt17(CO)12(PPh3)8/γ-Al2O3, the intensity of the peak at ∼1.7 Å increased and that at ∼2.3 Å decreased, and the peak attributed to the Pt–Pt bond appeared at ∼2.8 Å. As described above, there is no significant difference in the optical absorption between [Pt17(CO)12(PPh3)8]Cln and Pt17(CO)12(PPh3)8/γ-Al2O3 (Fig. 4(a) and (b)). Therefore, it is assumed that the Pt17 cluster maintains the metal-core structure as a whole even during adsorption (Fig. 3(a) and (b)). However, the FT-EXAFS spectrum indicates that the adsorption causes a slight change in the structure of the ligand layer that covers Pt17. For the appearance of the Pt–Pt bond in the spectrum, a plausible explanation is that the variation in the Pt–Pt bond length (Fig. S10†) decreases or the fluctuation of the Pt–Pt bond decreases65 with adsorption on the substrate. The decrease in the electron density of the d orbital of Pt17 (Fig. 5(a)) caused by adsorption can also likely be attributed to the structural change of the ligand layer. In the spectrum of Pt17/γ-Al2O3 after calcination, a peak at ∼2.8 Å clearly appears, and its satellite peak (in the FT-EXAFS spectrum of the Pt foil in Fig. 5(b)) is also observed at ∼2.3 Å.66 This result indicates that the variation in the Pt–Pt bond length and/or the fluctuation of the Pt–Pt bond further decrease with the PPh3 removal and/or the structural change of the Pt17 cluster from the icosahedral-based structure (Fig. 2(a)) to the structure shown in Fig. 5(c) and (d) (see below). In this spectrum, a peak was also observed at ∼1.7 Å. As described above, the surface of supported Pt17 is covered by CO at normal temperature. The peak at ∼1.7 Å is attributed to the generated Pt–C or Pt–O bond at the Pt17/γ-Al2O3 interface.
Thus, it was observed that Pt does not form an oxide53 and that Pt17 has a framework structure like a metal cluster in Pt17/γ-Al2O3. Based on the HAADF-STEM image, the supported Pt17 is assumed to have a bi-layer2 or tri-layer structure, as shown in Fig. 5(c), (d) and S11.† Previous studies have suggested that CO and O2 are activated on the terrace Pt and step Pt, respectively, during the oxidation reaction of CO.2,3,18Fig. 5(c) and (d) show that most of the terrace Pt is located near the step Pt in Pt17/γ-Al2O3. Thus, the reaction of CO and O2, i.e., the oxidation of CO, is expected to proceed effectively over Pt17/γ-Al2O3.
Catalytic activity of Pt17/γ-Al2O3 against the oxidation reaction of CO and C3H6
We examined the catalytic activity of the obtained Pt17/γ-Al2O3. Industrially used supported Pt catalysts are frequently prepared by the impregnation method. Therefore, in this study, PtNP/γ-Al2O3, in which 0.15 wt% Pt was loaded by the impregnation method, was used as a comparative Pt catalyst. The amount of Pt was confirmed by mixing PtNP/γ-Al2O3 with aqua regia and measuring the concentration of dissolved Pt using ICP-OES. The HAADF-STEM image shown in Fig. 6 indicates that PtNP/γ-Al2O3 has an average particle size of 3.10 ± 3.14 nm.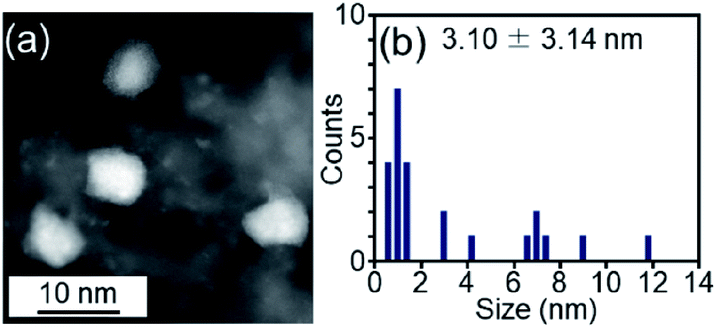 | ||
| Fig. 6 (a) Representative HAADF-STEM image and (b) histogram estimated from various HAADF-STEM images of PtNP/γ-Al2O3 prepared for comparison. | ||
The obtained Pt17/γ-Al2O3 and PtNP/γ-Al2O3 were examined for their catalytic activity against the oxidation of CO and C3H6, which are the main components in automobile gas.1 In an actual automobile, the catalysts are coated on a honeycomb substrate made of cordierite ceramic. Thus, in this study, Pt17/γ-Al2O3 and PtNP/γ-Al2O3 were coated on a honeycomb substrate to evaluate their catalytic performance in a state similar to the actual vehicle mounting conditions (Scheme S2†).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L h−1 while increasing the temperature of the honeycomb substrate to 400 °C at a rate of 20 °C min−1 (Table S4†). The conversion ratio of CO over the catalyst was estimated by evaluating the components of the mixed gas before and after circulation using an exhaust gas analyzer (Scheme 1).
000 L h−1 while increasing the temperature of the honeycomb substrate to 400 °C at a rate of 20 °C min−1 (Table S4†). The conversion ratio of CO over the catalyst was estimated by evaluating the components of the mixed gas before and after circulation using an exhaust gas analyzer (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of the estimation of CO and C3H6 conversions over Pt17/γ-Al2O3 or PtNP/γ-Al2O3 coated on a cordierite honeycomb substrate. | ||
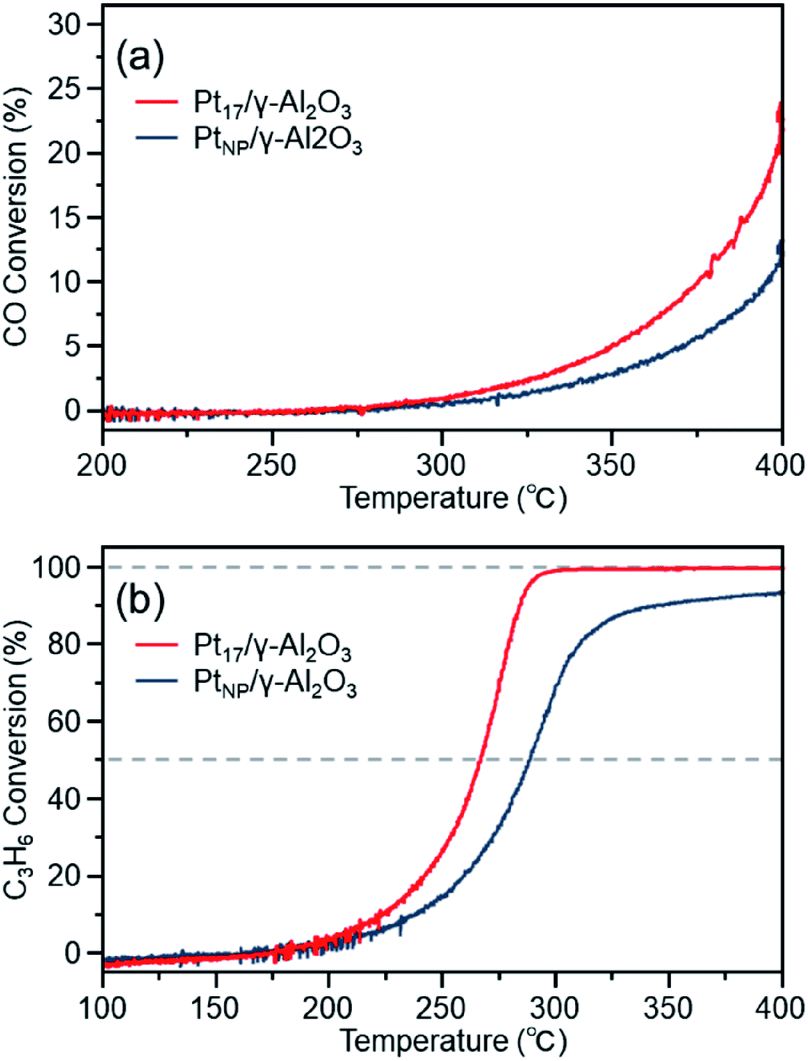
Fig. 7(a) shows the CO conversion for each catalyst (Pt17/γ-Al2O3 or PtNP/γ-Al2O3) estimated using this approach. When PtNP/γ-Al2O3 was used as the catalyst, the catalytic activity started to appear at approximately 270 °C, and the conversion reached 50% at approximately 350 °C (light-off temperature) and nearly 100% at approximately 370 °C. However, when Pt17/γ-Al2O3 was used, the catalytic activity started to manifest at approximately 240 °C, and the conversion reached 50% at approximately 330 °C and almost 100% at approximately 350 °C. These results indicate that Pt17/γ-Al2O3 exhibits higher catalytic activity at each temperature than PtNP/γ-Al2O3 and thus that Pt17/γ-Al2O3 can treat CO at lower temperatures than PtNP/γ-Al2O3. Currently, the issue of enhanced activity of exhaust-gas-treating catalysts at low temperatures must be overcome with the spread of vehicles that frequently stop and restart their engines (e.g., hybrid vehicles).1 These results strongly suggest that Pt17/γ-Al2O3 could be used as an exhaust gas-treating catalyst to overcome this issue.
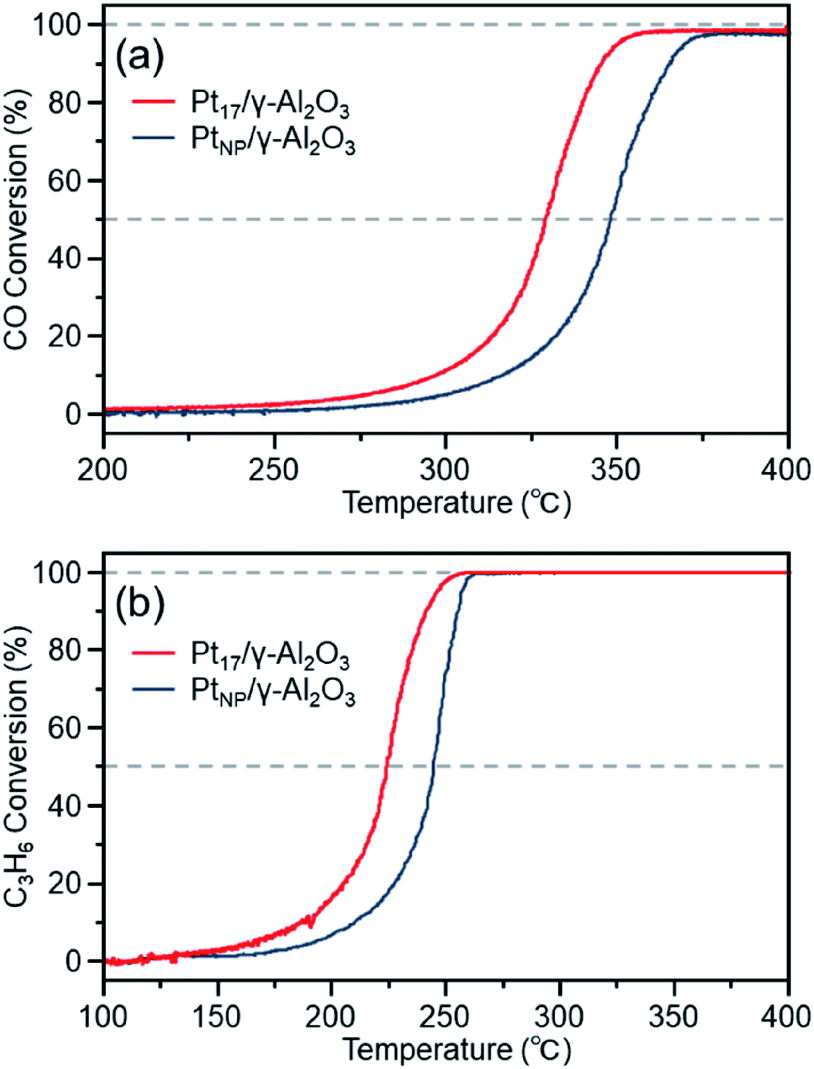 | ||
| Fig. 7 (a) CO and (b) C3H6 conversions over Pt17/γ-Al2O3 or PtNP/γ-Al2O3. | ||
The higher activity of Pt17/γ-Al2O3 than PtNP/γ-Al2O3 is considered to be associated with their respective geometrical structures.67 Although the geometrical structures of Pt17/γ-Al2O3 and PtNP/γ-Al2O3 before the reaction experiments are shown in Fig. 5 and 6, these geometrical structures should change as the catalytic reaction progresses and have not yet been elucidated.68 However, there should be more combinations composed of the terrace and step Pt in Pt17/γ-Al2O3 than in PtNP/γ-Al2O3 (Fig. 5(c) and (d)). These geometrical effects appear to make the reaction between CO and O2 more likely to occur in Pt17/γ-Al2O3, resulting in higher CO conversion of Pt17/γ-Al2O3 at any temperature. In addition, Pt17 in Pt17/γ-Al2O3 should be more susceptible to the fluctuation of the geometrical/electronic structure than PtNP in PtNP/γ-Al2O3. The ease of fluctuation of their geometrical/electronic structures may also contribute to the high activity of Pt17/γ-Al2O3.9 Furthermore, CO adsorbed on fine Ptn supported clusters generally has a longer C–O bond than that adsorbed on the larger Ptn supported nanoparticles, which promotes the oxidation reaction.69 In addition to the geometric factors, it is assumed that such a difference in CO activation caused by the difference in electronic states between the two supported clusters also contributes to the high activity of Pt17/γ-Al2O3.
000 L h−1 while increasing the temperature of the honeycomb substrate to 400 °C at a rate of 20 °C min−1 (Table S4†). The conversion ratio of C3H6 over the catalyst was estimated by evaluating the components of the mixed gas before and after circulation using an exhaust gas analyzer (Scheme 1).
Fig. 7(b) shows the C3H6 conversion for each catalyst (Pt17/γ-Al2O3 or PtNP/γ-Al2O3) estimated using this approach. When PtNP/γ-Al2O3 was used as a catalyst, the catalytic activity started to manifest at approximately 160 °C, and the conversion reached 50% at approximately 245 °C and nearly 100% at approximately 260 °C. However, when Pt17/γ-Al2O3 was used, the catalytic activity started to manifest at approximately 130 °C, and the conversion reached 50% at approximately 225 °C and nearly 100% at approximately 250 °C. These results indicate that Pt17/γ-Al2O3 exhibits higher catalytic activity at each temperature than PtNP/γ-Al2O3 for oxidizing C3H6. Currently, the mechanism of C3H6 oxidation is not as well understood as that of CO oxidation.70 Therefore, it is difficult to discuss the origin of the difference between the two activities. However, there should be a large difference between Pt17/γ-Al2O3 and PtNP/γ-Al2O3 in the number of surface Pt atoms that can participate in the reaction. It appears that this factor is responsible for the difference in activity of the two types of catalysts.
Fig. 8(a) shows the CO conversion of each catalyst (Pt17/γ-Al2O3 or PtNP/γ-Al2O3) after the aging treatment. The CO conversion rate decreased significantly for both catalysts compared with that before the aging treatment (Fig. 7(a)). A similar phenomenon was observed for the C3H6 conversion. These results indicate that the previously described procedure results in deterioration of the performance of both catalysts. However, comparing the conversion over Pt17/γ-Al2O3 and PtNP/γ-Al2O3, the use of Pt17/γ-Al2O3 resulted in higher conversion than the use of PtNP/γ-Al2O3 for both reactions. This result indicates that Pt17/γ-Al2O3 exhibits higher activity than PtNP/γ-Al2O3 even after the aging treatment.
 | ||
| Fig. 8 (a) CO and (b) C3H6 conversions over Pt17/γ-Al2O3 or PtNP/γ-Al2O3 after the aging treatment. | ||
The decrease in activity after aging is generally induced by the aggregation of the supported Pt catalyst.12,71 In fact, the aggregation of the Pt catalyst was observed after the aging treatment for both Pt17/γ-Al2O3 and PtNP/γ-Al2O3 (Fig. S12†). However, the average particle size of Pt17/γ-Al2O3 and PtNP/γ-Al2O3 after the aging treatment was 25.3 ± 19.4 and 77.5 ± 29.9 nm, respectively. Thus, the average particle size of the former was smaller than that of the latter even after the aging treatment. It is considered that because the original Pt17/γ-Al2O3 had a smaller particle size than the original PtNP/γ-Al2O3, Pt17/γ-Al2O3 had a smaller average particle size than PtNP/γ-Al2O3 after aggregation, resulting in its higher activity even after the aging treatment.
Conclusions
In this study, we successfully developed a method for producing Pt17/γ-Al2O3 using [Pt17(CO)12(PPh3)8]Cln as a precursor. Characterization of the obtained Pt17/γ-Al2O3 revealed that Pt17 is not present in the form of an oxide but has a framework structure as a metal cluster. Furthermore, it was determined that Pt17/γ-Al2O3 exhibits better catalytic activity for CO and C3H6 oxidation as well as better durability than PtNP/γ-Al2O3 prepared using the conventional impregnation method. The precursor [Pt17(CO)12(PPh3)8]Cln can be isolated with atomic precision only by mixing the reagents, heating the solvent in the atmosphere, and operating a simple separation process. The supported Pt17 is a Ptn cluster within the size range associated with high catalytic activity.18 It is expected that by using the loading method established in this study, many research groups can conduct further investigations on Pt17/γ-Al2O3 to obtain a deeper understanding of this catalyst and find new ways of using Pt17/γ-Al2O3 and Pt17 supported on other oxides. However, for practical use of the catalyst, it is necessary to investigate the catalytic activity and durability under the actual operating conditions of the exhaust gas mixing ratio.18,72–74 In addition, the loading weight also needs to be increased to that used under the actual operating conditions (Fig. S13†). We are currently attempting the measurements under such conditions with collaborations between academia and industry.Experimental
Chemicals
All the chemicals used in this study were commercially obtained and used without further purification. Hydrogen hexachloroplatinate hexahydrate (H2PtCl6·6H2O) was purchased from Tanaka Kikinzoku. Sodium hydroxide (NaOH), triphenylphosphine (PPh3), platinum (Pt) standard solution (1000 ppm), and bismuth (Bi) standard solution (100 ppm) were obtained from FUJIFILM Wako Pure Chemical Co. Acetone, acetonitrile, dichloromethane, ethylene glycol, hydrochloric acid, methanol, nitric acid, and toluene were sourced from Kanto Chemical Co., Inc. Pt nitrate (Pt(NO3)4) was obtained from Johnson Matthey. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was purchased from Tokyo Chemical Industry. Cordierite honeycomb (25.4 mm φ × 50 mm L, 400 cells per in2) and γ-Al2O3 (Puralox SCFa-160, Sasol) were obtained from NGK Insulators, Ltd. Pure Milli-Q water (18.2 MΩ cm) was generated using a Merck Millipore Direct 3 UV system.Synthesis of [Pt17(CO)12(PPh3)8]Cln
[Pt17(CO)12(PPh3)8]Cln was synthesized using the method reported in our previous paper52 with a slight modification (Scheme S1†). First, H2PtCl6·6H2O (0.10 mmol) and NaOH (2.2 mmol) were dissolved in ethylene glycol (25 mL). NaOH was used to control the pH of the solution and thereby suppress the particle size obtained by the polyol reduction.54,75 Then, the mixture was heated at 120 °C for 10 min to reduce the Pt ions and produce CO catalyzed by Pt ions. The color of the solution changed from yellow to dark brown. After cooling to room temperature (25 °C), acetone (10 mL) containing PPh3 (0.52 g, 2.0 mmol) was added to this solution at once. After several minutes, toluene (∼20 mL) and water (∼20 mL) were added to the reaction solution. The Pt clusters including Pt17(CO)12(PPh3)8 were transferred into the organic phase. Then, the organic phase was separated from the water phase and dried with a rotary evaporator. The dried product was washed with water and then methanol to eliminate ethylene glycol and excess PPh3. At this stage, the product was still a mixture of clusters of several sizes. The product was dried, and the by-products were then washed with a mixture of acetonitrile/toluene (1:1).
Preparation of catalysts
Characterization
ESI mass spectrometry was performed using a reflectron time-of-flight mass spectrometer (Bruker, micrOTOF II). In these measurements, a cluster solution with a concentration of ∼10 μg mL−1 in dichloromethane was electrosprayed at a flow rate of 180 μL h−1.MALDI mass spectra were collected using a spiral time-of-flight mass spectrometer (JEOL, JMS-S3000) with a semiconductor laser. DCTB76 was used as the MALDI matrix (cluster:matrix = 1:1000).
TEM images were recorded with a JEM-2100 electron microscope (JEOL) operating at 200 kV, typically using a magnification of 600000.
HAADF-STEM images were recorded using a JEOL ARM200CFE fitted with an aberration corrector. The catalyst powders of Pt17/γ-Al2O3 or PtNP/γ-Al2O3 were ground between two glass slides and dusted onto a holey carbon-coated Cu TEM grid.
DR spectra were acquired at ambient temperature using a V-670 spectrometer (JASCO). The wavelength-dependent optical data (I(w)) were converted to the energy-dependent data (I(E)) using the following equation that conserved the integrated spectral areas: I(E) = I(w)/|∂E/∂w| ∝ I(w) × w2.
ICP-MS was performed using an Agilent 7500c spectrometer (Agilent Technologies, Tokyo, Japan). Bi was used as the internal standard. The ICP-MS measurements were performed for the supernatant obtained after mixing [Pt17(CO)12(PPh3)8]Cln with γ-Al2O3 to estimate the unadsorbed Pt content. The adsorption efficiency and the Pt amount on γ-Al2O3 were estimated using this value.
ICP-OES was performed using an Agilent Technologies 700 series spectrometer to determine the Pt content in Pt17/γ-Al2O3 or PtNP/γ-Al2O3 after completely dissolving the sample using aqua regia.
TG-MS was performed with an STA 2500 Regulus (NETZSCH) and a JMS-Q 1500GC (JEOL) at a heating rate of 5 °C min−1 under an Ar atmosphere over the temperature range 25–900 °C using ∼3 mg sample of Pt17(CO)12(PPh3)8/γ-Al2O3.
XPS data were collected using an electron spectrometer (JEOL, JPS-9010MC) equipped with a chamber at a base pressure of ∼2 × 10−8 Torr. X-rays from the Mg-Kα line (1253.6 eV) were used for excitation.
Pt L3-edge XAFS measurements were performed at beamline BL01B1 of the SPring-8 facility of the Japan Synchrotron Radiation Research Institute (proposal numbers 2018B1422 and 2019A0944). The incident X-ray beam was monochromatized using a Si(111) double-crystal monochromator. As references, the XAFS spectra of Pt foil and solid PtO2 were recorded in transmission mode using ionization chambers. The Pt L3-edge XAFS spectra of the samples were measured in fluorescence mode using a 19-element Ge solid-state detector at room temperature. The X-ray energies for the Pt L3-edges were calibrated using Au foil. The XANES and EXAFS spectra were analyzed using the xTunes program77 as follows. The χ spectra were extracted by subtracting the atomic absorption background using cubic spline interpolation and normalized to the edge height. The normalized data were used as the XANES spectra. The k3-weighted χ spectra in the k range 3.0–14.0 Å−1 for the Pt L3-edge were Fourier transformed into r space for structural analysis. The curve fitting analysis was performed in the range of 1.2–3.0 Å for the Pt L3-edge. In the curve fitting analysis, the phase shifts and backscattering amplitude function of Pt–C, Pt–P, and Pt–Pt were calculated using the FEFF8.5L program.
Measurements of catalytic activity
Catalytic activity tests on the honeycomb catalysts of Pt17/γ-Al2O3 or PtNP/γ-Al2O3 were performed using a flow reactor. The honeycomb catalysts were fixed in a tubular reactor, and their catalytic activity was evaluated by supplying a gas mixture containing a reducing gas (CO or C3H6), an oxidation gas (O2), and a carrier gas (N2) at a space velocity (SV) of 50000 L h−1 (Table S4†). The conversions of CO or C3H6 were monitored using an exhaust gas analyzer (MEXA-ONE-D1, HORIBA) from 100 °C to 400 °C at a heating rate of 20 °C min−1. Before each catalytic activity test, the honeycomb catalyst was pre-treated at 400 °C for 0.5 h under a flow of the gas mixture used for the catalytic activity test.
Aging treatment for durability tests
The aged catalyst samples were prepared by hydrothermal redox aging under a flow of perturbed reduction gas (3% of H2, 3% of CO and 10% of H2O) and oxidation gas (3% of O2 and 10% of H2O) with N2 balance (Table S5†). The perturbation cycle was for 3 min, the aging temperature was 1000 °C, and the duration was 4 h.Author contributions
Y. Negishi and S. Nagaoka designed the experiments and conducted the measurement with the help of N. Shimizu, K. Funai, R. Kaneko, K. Wakamatsu, A. Harasawa, S. Hossain, W. Kurashige and T. Kawawaki. S. Yamazoe conducted XAFS experiments at SPring-8. M. E. Schuster and D. Ozkaya conducted HAADF-STEM experiments in England. Y. Negishi and S. Nagaoka co-wrote the paper and all authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Juri Maekawa and Ms Sayaka Hashimoto for technical assistance. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers JP16H04099 and 16K21402 and Grants-in-Aid for Scientific Research on Innovative Areas “Coordination Asymmetry” (grant number 17H05385) and “Innovations for Light-Energy Conversion” (grant number 18H05178). Funding from Asahi Glass Foundation is also gratefully acknowledged.References
- R. M. Heck, R. J. Farrauto and S. T. Gulati, Catalytic Air Pollution Control: Commercial Technology, John WILEY & Sons, Canada, 3rd edn, 2009 Search PubMed.
- Y. Watanabe, X. Wu, H. Hirata and N. Isomura, Catal. Sci. Technol., 2011, 1, 1490–1495 RSC.
- C. Yin, F. R. Negreiros, G. Barcaro, A. Beniya, L. Sementa, E. C. Tyo, S. Bartling, K.-H. Meiwes-Broer, S. Seifert, H. Hirata, N. Isomura, S. Nigam, C. Majumder, Y. Watanabe, A. Fortunelli and S. Vajda, J. Mater. Chem. A, 2017, 5, 4923–4931 RSC.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed.
- J. H. Kang, L. D. Menard, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2006, 128, 12068–12069 CrossRef CAS PubMed.
- A. Bettac, L. Köller, V. Rank and K. H. Meiwes-Broer, Surf. Sci., 1998, 402–404, 475–479 CrossRef CAS.
- B. R. Cuenya, A. I. Frenkel, S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono and Q. Wang, Phys. Rev. B, 2010, 82, 155450 CrossRef.
- U. Heiz, A. Sanchez, S. Abbet and W.-D. Schneider, Eur. Phys. J. D, 1999, 9, 35–39 CrossRef CAS.
- S. I. Sanchez, L. D. Menard, A. Bram, J. H. Kang, M. W. Small, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2009, 131, 7040–7054 CrossRef CAS PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS PubMed.
- S. Bonanni, K. Aït-Mansour, W. Harbich and H. Brune, J. Am. Chem. Soc., 2014, 136, 8702–8707 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- T. Imaoka, Y. Akanuma, N. Haruta, S. Tsuchiya, K. Ishihara, T. Okayasu, W.-J. Chun, M. Takahashi and K. Yamamoto, Nat. Commun., 2017, 8, 688 CrossRef PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- S. Nagaoka, T. Matsumoto, K. Ikemoto, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2007, 129, 1528–1529 CrossRef CAS PubMed.
- U. Heiz, A. Sanchez, S. Abbet and W.-D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- F. S. Roberts, M. D. Kane, E. T. Baxter and S. L. Anderson, Phys. Chem. Chem. Phys., 2014, 16, 26443–26457 RSC.
- T. Tsukuda and H. Häkkinen, Protected Metal Clusters: From Fundamentals to Applications, Elsevier B.V., Amsterdam, The Netherlands, 1st edn, 2015 Search PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS PubMed.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS PubMed.
- Y. Niihori, S. Hashimoto, Y. Koyama, S. Hossain, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2019, 123, 13324–13329 CrossRef CAS.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS PubMed.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- Y. Pei, P. Wang, Z. Ma and L. Xiong, Acc. Chem. Res., 2019, 52, 23–33 CrossRef CAS PubMed.
- B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104–3113 CrossRef CAS PubMed.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- Q. Tang, G. Hu, V. Fung and D.-e. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 4134–4142 CrossRef CAS PubMed.
- Y. Song, S. Wang, J. Zhang, X. Kang, S. Chen, P. Li, H. Sheng and M. Zhu, J. Am. Chem. Soc., 2014, 136, 2963–2965 CrossRef CAS PubMed.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- Q.-F. Zhang, X. Chen and L.-S. Wang, Acc. Chem. Res., 2018, 51, 2159–2168 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Coord. Chem. Rev., 2018, 355, 27–38 CrossRef CAS.
- E. G. Mednikov, M. C. Jewell and L. F. Dahl, J. Am. Chem. Soc., 2007, 129, 11619–11630 CrossRef CAS PubMed.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- L. V. Nair, S. Hossain, S. Wakayama, S. Takagi, M. Yoshioka, J. Maekawa, A. Harasawa, B. Kumar, Y. Niihori, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2017, 121, 11002–11009 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS.
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef CAS PubMed.
- N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278–14280 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 13457–13461 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- V. Machek, V. Ružička, M. Šourková, J. Kunz and L. Janáček, React. Kinet. Catal. Lett., 1982, 21, 13–16 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, United States of America, 2007 Search PubMed.
- S. Blonski and S. H. Garofalini, Surf. Sci., 1993, 295, 263–274 CrossRef CAS.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS PubMed.
- B.-K. Teo and P. A. Lee, J. Am. Chem. Soc., 1979, 101, 2815–2832 CrossRef CAS.
- J. Singh, E. M. C. Alayon, M. Tromp, O. V. Safonova, P. Glatzel, M. Nachtegaal, R. Frahm and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2008, 47, 9260–9264 CrossRef CAS PubMed.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176–184 CrossRef CAS PubMed.
- O. S. Alexeev, S. Y. Chin, M. H. Engelhard, L. Ortiz-Soto and M. D. Amiridis, J. Phys. Chem. B, 2005, 109, 23430–23443 CrossRef CAS PubMed.
- M. J. Hazlett, M. Moses-Debusk, J. E. Parks II, L. F. Allard and W. S. Epling, Appl. Catal., B, 2017, 202, 404–417 CrossRef CAS.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Am. Chem. Soc., 2010, 132, 7968–7975 CrossRef CAS PubMed.
- V. Matsouka, M. Konsolakis, R. M. Lambert and I. V. Yentekakis, Appl. Catal., B, 2008, 84, 715–722 CrossRef CAS.
- J. Březina, P. Boutikos, A. B. Arvajová, R. Pečinka and P. Kočí, Top. Catal., 2019, 62, 252–258 CrossRef.
- S. B. Kang, C. Kalamaras, V. Balakotaiah and W. Epling, Ind. Eng. Chem. Res., 2017, 56, 13628–13633 CrossRef CAS.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467–2473 CrossRef CAS.
- A. Dass, A. Stevenson, G. R. Dubay, J. B. Tracy and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 5940–5946 CrossRef CAS PubMed.
- H. Asakura, S. Yamazoe, T. Misumi, A. Fujita, T. Tsukuda and T. Tanaka, Radiat. Phys. Chem. DOI:10.1016/j.radphyschem.2019.04.020.
Footnote |
| † Electronic supplementary information (ESI) available: Data of XAFS analysis, protocols, MALDI mass spectra, HAADF-STEM images, the XPS spectrum, the TPR curve, EXAFS curves of [Pt17(CO)12(PPh3)8]Cln, Pt17(CO)12(PPh3)8/γ-Al2O3, or Pt17/γ-Al2O3. See DOI: 10.1039/c9na00579j |
| This journal is © The Royal Society of Chemistry 2020 |