
Identifying the targets and functions of N-linked protein glycosylation in Campylobacter jejuni
Joel A.
Cain
ab,
Ashleigh L.
Dale
 ab,
Zeynep
Sumer-Bayraktar
ab,
Nestor
Solis†
a and
Stuart J.
Cordwell
*abcd
ab,
Zeynep
Sumer-Bayraktar
ab,
Nestor
Solis†
a and
Stuart J.
Cordwell
*abcd
aSchool of Life and Environmental Sciences, The University of Sydney, 2006, Australia
bCharles Perkins Centre, The University of Sydney, Level 4 East, The Hub Building (D17), 2006, Australia. E-mail: stuart.cordwell@sydney.edu.au; Tel: +612-9351-6050
cDiscipline of Pathology, School of Medical Sciences, The University of Sydney, 2006, Australia
dSydney Mass Spectrometry, The University of Sydney, 2006, Australia
First published on 22nd April 2020
Abstract
Campylobacter jejuni is a major cause of bacterial gastroenteritis in humans that is primarily associated with the consumption of inadequately prepared poultry products, since the organism is generally thought to be asymptomatic in avian species. Unlike many other microorganisms, C. jejuni is capable of performing extensive post-translational modification (PTM) of proteins by N- and O-linked glycosylation, both of which are required for optimal chicken colonization and human virulence. The biosynthesis and attachment of N-glycans to C. jejuni proteins is encoded by the pgl (protein glycosylation) locus, with the PglB oligosaccharyltransferase (OST) enabling en bloc transfer of a heptasaccharide N-glycan from a lipid carrier in the inner membrane to proteins exposed within the periplasm. Seventy-eight C. jejuni glycoproteins (represented by 134 sites of experimentally verified N-glycosylation) have now been identified, and include inner and outer membrane proteins, periplasmic proteins and lipoproteins, which are generally of poorly defined or unknown function. Despite our extensive knowledge of the targets of this apparently widespread process, we still do not fully understand the role N-glycosylation plays biologically, although several phenotypes, including wild-type stress resistance, biofilm formation, motility and chemotaxis have been related to a functional pgl system. Recent work has described enzymatic processes (nitrate reductase NapAB) and antibiotic efflux (CmeABC) as major targets requiring N-glycan attachment for optimal function, and experimental evidence also points to roles in cell binding via glycan–glycan interactions, protein complex formation and protein stability by conferring protection against host and bacterial proteolytic activity. Here we examine the biochemistry of the N-linked glycosylation system, define its currently known protein targets and discuss evidence for the structural and functional roles of this PTM in individual proteins and globally in C. jejuni pathogenesis.
 Joel A. Cain | Joel Cain received a Bachelor of Science with Honours in 2012 and is currently completing a PhD degree at The University of Sydney, Australia. He was awarded an Australian Postgraduate Award for these studies. He has also worked as a Research Assistant in Sydney Mass Spectrometry. His current work focuses on the contributions of post-translational modifications, such as targeted protein degradation and N-glycosylation, towards the pathogenic mechanisms of the enteric pathogen Campylobacter jejuni. |
 Ashleigh L. Dale | Ashleigh Dale received a Bachelor of Science with Honours at the University of Sydney at the end of 2018 and commenced her PhD candidature in biochemistry the following year. Her current work involves implementing cross-linking mass spectrometry approaches to define the interactome of C. jejuni and characterize protein–protein interactions in the hope of elucidating mechanisms of pathogenesis in this organism. |
 Zeynep Sumer-Bayraktar | Zeynep Sumer-Bayraktar received a PhD in biochemistry from Macquarie University, Sydney Australia in 2016. Her research focuses on protein post-translational modifications, specifically the glycan-components of N- and O-linked glycosylation using liquid chromatography and mass spectrometry-based approaches. In her current work, Zeynep investigates the role of protein glycosylation in Campylobacter jejuni human gut colonization and the glyco/proteomic responses of the human host against C. jejuni infection. |
 Nestor Solis | Nestor Solis obtained his PhD in microbial proteomics in 2014 from The University of Sydney, Australia, where he explored the identification of cell-surface proteins from Staphylococcus species using cell-shaving proteomics. He joined the laboratory of Professor Christopher Overall in 2014 to further expand on proteomic methods to study N- and C-terminomes in macrophages. He was awarded two fellowships in 2016: a Michael Smith Foundation for Health Research postdoctoral fellowship from British Columbia, Canada, and a CJ Martin Early Career Fellowship from the National Health and Medical Research Council of Australia. |
 Stuart J. Cordwell | Stuart Cordwell is Professor of Analytical Biochemistry in the School of Life and Environmental Sciences and the Discipline of Pathology, School of Medical Sciences at the University of Sydney. He is also the Director of Sydney Mass Spectrometry. His research focusses on post-translational modifications of proteins and their roles in the virulence mechanisms of pathogenic bacteria. His group is also increasingly using multi-omics strategies to decipher these mechanisms to better understand the proteome-phenotype nexus. |
Introduction
Campylobacter jejuni is a Gram negative enteric pathogen with helical cell morphology. C. jejuni is also microaerophilic and typically requires oxygen levels to be no greater than 10% for growth.1 Gastrointestinal infection caused by this organism was first characterized in the late 1970s2 and C. jejuni is now considered the most common causative agent of gastroenteritis in the developed world, with an estimated 400 million people infected worldwide annually.3 Infection in humans is acquired though consumption of contaminated water or food, particularly under-cooked or inappropriately handled poultry products (with estimates suggesting between 75–90% of supermarket chicken is contaminated with the organism4), since C. jejuni is generally considered an asymptomatic commensal in avian species.5 While the differences in human and avian response to C. jejuni infection are largely unknown, there is evolving evidence that differences in host mucin O-glycan composition, particularly sulfated O-glycans, may play a role in colonization.6 Furthermore, chicken mucins from different regions of the gastrointestinal tract can inhibit human epithelial cell virulence,7 providing further evidence for glycan recognition in the establishment of host-specific niches.8Human disease is generally self-limiting and symptoms present as fever and abdominal cramping that progress from mild to, in some cases, severe diarrhoea.2,3 Relapse is possible in the absence of medical intervention, and is likely due to gut persistence for up to 3 weeks.9C. jejuni infection is also an established antecedent for an increasing number of debilitating conditions including Guillain–Barré Syndrome (GBS), Miller–Fisher Syndrome (MFS), immunoproliferative small intestine disease, reactive arthritis and Sweet's syndrome.10,11 The basis for these post-acute immune-mediated disorders is thought to be largely based on cross-reactivity between antibodies directed against C. jejuni surface lipooligosaccharide (LOS) and human cell surface gangliosides, and this relationship has been reviewed extensively.12–14
Several C. jejuni genomes have been sequenced from laboratory-adapted and clinical strains and several features remain consistent; the organism encodes ∼1620–1650 genes, a large proportion of which encode membrane-associated proteins that are poorly functionally annotated.15–17 Human infection is not completely understood but involves bacterial adherence to gut epithelial cells, followed by invasion and subsequent toxin production. Several factors are critical in C. jejuni host colonization, including flagellar-based motility, cell shape, chemosensing and chemotaxis mediated by transducer-like proteins (Tlps), as well as a number of adhesins including the fibronectin-binding proteins Campylobacter adherence factor CadF and fibronectin-like protein FlpA, the surface-exposed lipoprotein JlpA and the PEB antigens [reviewed in18–20]. The ability to survive in the hostile environment encountered during gut infection, consisting of for example low pH, presence of bile salts and competitive factors from established microflora, is paramount to establishing disease and C. jejuni is adapted to utilize nutrients, such as amino and organic acids as primary carbon sources, that are in rich supply in the gut micro-environment (e.g. serine and proline from mucins, organic acids produced as a by-product of metabolism by resident microorganisms).21,22C. jejuni lacks typical virulence-associated type III/IV secretion systems (T3SS/T4SS) employed by many other enteric bacteria to secrete toxins and proteases that directly interact with host cells, although it is now well-established that extracellular virulence factors (e.g. the Campylobacter invasion antigens [Cia] and cytolethal distending toxin [CDT]) are secreted via the flagellar export apparatus that acts as a pseudo-T3SS.20,23 Another mechanism by which C. jejuni virulence determinants can interact with host cells is via their packaging into outer membrane vesicles (OMVs;24–26). Finally, despite the somewhat small size of the genome, C. jejuni devotes considerable resources to post-translational modification (PTM) of proteins by N- and O-linked glycosylation, both of which are considered established virulence determinants.
Protein glycosylation in C. jejuni
Despite their ability to synthesize large polysaccharides, bacteria were long thought to be enzymatically incapable of modifying proteins with glycans. In the past two decades however, this opinion has largely been overturned with the identification of conserved bacterial N- and O-linked glycosylation systems in many microorganisms.27–30 Such systems are almost universally biologically important and associated with pathogenic processes including cell–cell recognition and binding,31 however their ultimate purpose and functions remain to be determined. C. jejuni was the first bacterium to be recognized as containing a ‘general glycosylation system’ that could widely modify proteins.32–34 Since then, our understanding of the biochemistry, targets and putative functions of these PTMs has increased rapidly. While our knowledge remains incomplete, recent advances in glycoproteomics-focused mass spectrometry (MS) have generated large-scale site identifications in C. jejuni35 and other organisms, which have enabled directed functional studies to elucidate the roles of these modifications in bacterial phenotypes associated with pathogenicity.O-Glycosylation in C. jejuni
C. jejuni modifies its flagella by O-glycosylation of the flagellin structural protein with derivatives of the bacterial-specific sialic acid-like monosaccharides, pseudaminic acid or the closely related legionaminic acid.36–41 At least 19–23 serine/threonine sites are modified on the FlaA flagellin depending on the strain employed,42–46 and O-glycan attachment is essential for both chicken and human infection phenotypes, including motility, autoagglutination, chicken colonization, and human epithelial cell adherence and invasion.47–49 Unlike N-glycosylation (see below), there is considerably more structural heterogeneity with respect to the attached glycan in individual strains, including both chemical and steriometric differences. Synthesis of pseudaminic and legionaminic acid occur independently of one another, beginning with nucleotide-linked precursors; pseudaminic acid as a uridine diphosphate (UDP)-linked and legionaminic acid as a guanosine diphosphate (GDP)-linked precursor. Enzymatic affinities for specific nucleotide precursors are critical for differentiation of the two pathways and this prevents competition for intermediates.38 Synthesis of pseudaminic acid from UDP-N-acetylglucosamine (UDP-GlcNAc) is performed by the actions of PseB, PseC and PseH, which act sequentially to form UDP-2,4-diNAc-6-deoxy-altropyranose.50–52 Following release of UDP by PseG, this sugar serves as the substrate of PseI to form pseudaminic acid.50,52–54 Synthesis of legionaminic acid and derivatives mirrors pseudaminic acid in many respects; from a GDP-GlcNAc precursor, formation of UDP-2,4-diNAc-6-deoxy-glucose is catalyzed by the sequential activities of LegB, LegC and LegH.38 In principal, the lack of initial LegB epimerase activity creates the structural distinction between pseudaminic and legionaminic acids – instead this activity is performed by the hydrolysing 2-epimerase LegG alongside nucleotide release to form 2,4-diNAc-6-deoxy-mannose, which is then used by the legionaminic acid synthase LegI to form legionaminic acid.38,47 Both pseudaminic and legionaminic acid are subsequently conjugated onto cytodine monophosphate (CMP) nucleotides by the CMP-sugar synthetases PseF and LegF, respectively, prior to attachment onto FlaA by an undefined glycosyltransfersase.38,50 Unlike other Gram negative bacteria,55–57 in C. jejuni there appears to be no other substrates of O-glycosylation (at least those modified with the flagellar glycan; see below) beyond FlaA. There is conjecture that given the role of the flagellar apparatus as a T3SS-like export apparatus that proteins secreted via this pathway may also be O-glycosylated in a similar manner to the FlaA flagellin. No studies however, have been able to globally identify extracellular proteins from this organism, mostly due to the very specific requirements (presence of confounding serum or host cells) needed to induce secretion in C. jejuni.It has been suggested that the major outer membrane protein (MOMP), which accounts for ∼40–50% of the total membrane protein in C. jejuni,17 can also be O-glycosylated58 with a glycan unrelated to the flagellin modification described above. MOMP may be modified at Thr-268 with the tetrasaccharide Gal-β1,3-GalNAc-β1,4-GalNAc-β1,4-GalNAc-α1, although intact glycan-peptide MS validation is yet to be generated. MOMP modification was further indicated by Whitworth and colleagues in C. jejuni strain 81–176 by galactose oxidase (GalO)-mediated selective biotinylation and subsequent enrichment of GalNAc containing cell surface glycoconjugates.59 Site-directed mutagenesis of Thr-268 indicated that this residue is important for autoagglutination, biofilm formation and colonization of both human Caco-2 cells and chickens,58 a phenotype also consistent with observations on the roles of FlaA glycosylation. It remains to be seen whether additional O-glycoproteins are present in C. jejuni and whether this PTM occurs as a widespread presence on proteins secreted by the organism during infection.
The pgl-encoded N-glycosylation system in C. jejuni
C. jejuni was the first bacterium demonstrated to possess the ability to N-glycosylate proteins. Proteins are modified by the N-linked addition of a heptasaccharide glycan (GalNAc-α1,4-GalNAc-α1,4-[Glcβ1,3]-GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,3-diNAcBac-β1; where diNAcBac is N′,N′-diacetylbacillosamine [2,4-diacetamido-2,4,6 trideoxyglucopyranose])33 at the consensus sequon Asp (D)/Glu (E)-X1-Asn (N)-X2-Ser (S)/Thr (T) (where X1,2 ≠ Pro), with Asn (N) being the attachment site.60,61 The synthesis of the N-glycan and attachment to proteins are encoded by the 16 kb pgl (protein glycosylation) gene cluster;32,34,62 the pgl cluster is highly conserved among members of the order Campylobacteriales,63,64 although N-glycan composition and structure, as well as the genomic location of the pgl locus (including being split into multiple loci), can differ between species.63 Gene transfer of the complete pgl cluster into otherwise N-glycosylation incompetent bacterial species (e.g. most commonly E. coli) is sufficient to confer the ability to N-glycosylate co-expressed acceptor proteins.62 Therefore, the pgl system has become a model for the production of glycoconjugate vaccines in recombinant expression procedures [reviewed in65–67].Biosynthesis and transfer of the N-glycan to proteins (Fig. 1) involves the actions of 10 Pgl proteins (an eleventh member of the locus, pglG, does not appear to contribute to the process and remains functionally undefined68) and begins with the cytoplasmic synthesis of nucleotide-activated (uridine diphosphate; UDP) UDP-diNAcBac from UDP-GlcNAc, which is catalyzed by the activities of (in order) the PglF dehydratase (conferring the rate-limiting step in the Pgl pathway), PglE aminotransferase and PglD acetyltransferase.69–74 Synthesis of diNAcBac has been reviewed extensively elsewhere.75,76 The potential for cross-talk between the N- and O-linked pathways is evidenced by shared nucleotide-activated precursors and by the activity of PglD, which can form intermediates from within the legionaminic acid biosynthetic pathway, albeit at substantially reduced catalysis compared with LegH.38 DiNAcBac is attached to the cytoplasmic side of an inner membrane spanning lipid carrier (undecaprenyl-pyrophosphate [Und-P]) by the PglC glycosyl-1-phosphate transferase, and Und-P then serves as the carrier for the nascent N-glycan.77,78 Continued synthesis of the glycan on Und-P-diNAcBac involves the sequential addition of 5 N-acetylgalactosamine (GalNAc) residues by three pgl-encoded glycosyltransferases (the first by PglA, the second by PglJ and the final three by PglH).79 Glycan length is controlled by increased competitive inhibition of the PglH active site relative to the number of GalNAc residues, and is considered limited by the final GalNAc(x5) product.80 The PglH tertiary structure also contains a novel ‘ruler helix’ that binds the pyrophosphate of Und-P and limits PglH catalysis to 3 GalNAc.81 Glycan synthesis is completed by the PglI glucosyltransferase, which adds a single glucose (Glc) branch to the third GalNAc in the N-glycan.68 This last Glc residue is not a strict requirement for N-glycosylation as, unlike all previous steps, addition of the complete N-glycan (without the Glc branch) to proteins still occurs in the absence of pglI,77,82 albeit at lower catalytic efficiency. Deletion of other pgl genes results in either complete loss of the N-glycan or the presence of significantly truncated N-glycans (e.g. pglD82), as well as compromised protein transfer efficiency.77 Once the heptasaccharide has been completed, the PglK flippase translocates the Und-P-linked glycan from the cytoplasm into the periplasmic space utilizing a mechanism dependent on the hydrolysis of two molecules of ATP.83,84 The mature glycan is then transferred en bloc from Und-P onto target proteins by the PglB oligosaccharyltransferase (OST),68,85 which recognizes both the Und-P-N-glycan complex and peptide acceptor as substrates.86
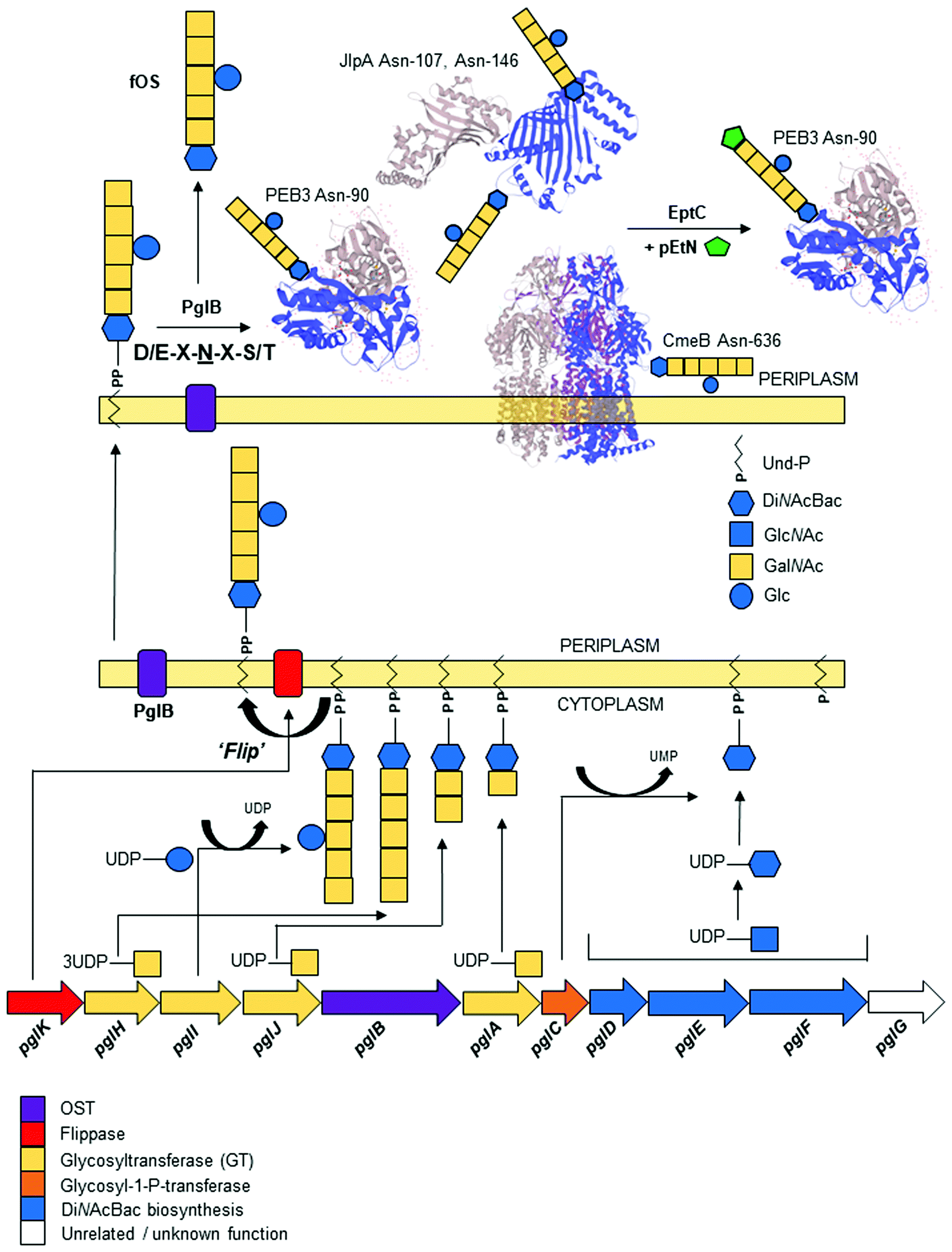 | ||
Fig. 1 Schematic of the biosynthesis and attachment of the heptasaccharide N-glycan to C. jejuni proteins. (lower) The pgl gene cluster colour coded by functional class; (middle) glycan biosynthesis begins with the ordered actions of PglFED that convert UDP-GlcNAc to diNAcBac. The PglC glycosyl-1-phosphate transferase adds diNAcBac-P to the membrane-bound lipid carrier undecaprenylphosphate (Und-P). Glycan assembly continues in the cytoplasm with the sequential addition of 5 GalNAc (1 by PglA, a second by PglJ and the final 3 by PglH). PglI adds a glucose (Glc) to the third GalNAc. Following assembly, the PglK flippase flips the N-glycan into the periplasm; (upper) the PglB OST transfers the N-glycan to proteins predominantly at the consensus sequon D/E-X1-![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) -X2-S/T (where X1,2 cannot be proline); structures of known glycoproteins PEB3, JlpA and CmeB are shown with the glycan positioned at known glycosites; the pEtN transferase EptC can further modify some glycoproteins with pEtN at the terminal GalNAc of the heptasaccharide. PglB can also liberate a free glycan (fOS). -X2-S/T (where X1,2 cannot be proline); structures of known glycoproteins PEB3, JlpA and CmeB are shown with the glycan positioned at known glycosites; the pEtN transferase EptC can further modify some glycoproteins with pEtN at the terminal GalNAc of the heptasaccharide. PglB can also liberate a free glycan (fOS). | ||
The PglB OST is also capable of releasing the N-glycan from Und-P into the periplasm as a ‘free oligosaccharide’ (fOS),82,87,88 although the exact proportion of N-glycan as Asn-bound:fOS remains a point of contention. Nothaft et al. reported a ratio favouring high fOS at ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, while Scott et al. reported a distribution of 4.5:1 in favour of protein-bound N-glycan.82,89 While there are a number of technical considerations that may help explain this discrepancy,89,90 there may also be dynamic control of fOS production based on environmental conditions and the kinetics of PglB. The fOS itself has been shown to provide protection against osmotic stress, further supporting the notion that the cellular fate of the N-glycan may be determined to some degree by environmental sensing.82 Unlike protein N-glycosylation, the free N-glycan is highly dependent on the synthesis of the complete heptasaccharide, as pglI deficient strains produce ∼55% less fOS.90 The N-glycan itself can also be further modified with a phosphoethanolamine (pEtN) group, which is added to the terminal GalNAc of the heptasaccharide at a small number of glycosites by the sole C. jejuni pEtN transferase, EptC.91 An inability to detect pEtN-modified fOS suggests that variation of the glycan by EptC occurs post-attachment to protein targets.91
:10, while Scott et al. reported a distribution of 4.5:1 in favour of protein-bound N-glycan.82,89 While there are a number of technical considerations that may help explain this discrepancy,89,90 there may also be dynamic control of fOS production based on environmental conditions and the kinetics of PglB. The fOS itself has been shown to provide protection against osmotic stress, further supporting the notion that the cellular fate of the N-glycan may be determined to some degree by environmental sensing.82 Unlike protein N-glycosylation, the free N-glycan is highly dependent on the synthesis of the complete heptasaccharide, as pglI deficient strains produce ∼55% less fOS.90 The N-glycan itself can also be further modified with a phosphoethanolamine (pEtN) group, which is added to the terminal GalNAc of the heptasaccharide at a small number of glycosites by the sole C. jejuni pEtN transferase, EptC.91 An inability to detect pEtN-modified fOS suggests that variation of the glycan by EptC occurs post-attachment to protein targets.91
Attachment of the N-glycan to proteins in C. jejuni
Modification of protein substrates by PglB at the C. jejuni N-glycosylation consensus motif is driven by a tryptophan–tryptophan–aspartic acid (WWD) motif, which is common to eukaryotic OST STT3 homologs.85,92 The OST WWD (WWDYG in C. jejuni PglB) motif interacts by hydrogen bonding with residues at the +2 position (Ser/Thr) in the N-glycosylation sequon, while isoleucine 572 (Ile-572) of PglB also contacts the Thr methyl group in target sequons and Ile-572 point mutants have reduced activity.92 The additional specificity of the C. jejuni sequon (requiring D/E at the −2 position compared to N-X-S/T in eukaryotic N-glycosylation61) is conferred at least partly by PglB Arg-331, which interacts with these acidic residues.85 In yeast OST STT3, this residue is an Asp, which explains the shorter eukaryotic-like N-X-S/T glycosylation motif based on charge repulsion. The distorted conformation of the peptide in the PglB OST active site is not capable of accommodating Pro at either the −1 or +1 positions,85 and this conformational constraint may also contribute to the preference of PglB for targeting unstructured regions of its protein substrates.93 Mechanistically, it is thought that the carboxamide group of the modified Asn is twisted through hydrogen bond interactions with PglB residues Asp-56 and Glu-319, which prime it for nucleophilic attack of the lipid-linked heptasaccharide.94 A large, partially disordered periplasmic external loop region (known as ‘EL5’) was also identified that contains a C-terminal portion (including Glu-319) involved in sequon binding, and disengagement of EL5 allows release of the glycosylated substrate from PglB.85 A conserved Tyr residue (Tyr-293) in the N-terminal region of EL5 was found to be essential for PglB catalysis but did not influence sequon binding, yet rather was associated with the interaction with the lipid-linked N-glycan donor.95 Additional studies have suggested a further conserved motif (475DGGK478) in PglB may contribute to Und-P binding, and that this region could be essential for function, as PglB orthologs that do not contain this motif are unable to glycosylate proteins or produce fOS.96Despite our knowledge of the structure and function of PglB and other OSTs, a number of elements still remain poorly understood. Firstly, observations of N-glycosylation at non-canonical sequons89,97 are not consistent with the above model, particularly considering that it has previously been demonstrated that such substitutions are catalytically unfavourable.92 It has been suggested that these atypical or non-canonical occupied sequons may reflect observations that peptide binding is not necessarily the rate-limiting step in the PglB reaction.95 Evidence for this can be seen in similar turnover rates between sequons containing Thr or Ser at the +2 position despite an apparent 4-fold reduced affinity of PglB for Ser,92 and this is further supported by similar propensities for the two amino acids at this position in vivo.89 Additionally, while PglB can modify glutamine (Gln) at very low rates in in vitro peptide-based assays,94 no glycosite at Gln has been demonstrated in any C. jejuni glycopeptide identified thus far, with the very small number of non-canonical sequons limited to differences at the −2 and +2 positions.89 Additionally, the exquisite sensitivity of MS-based approaches for glycopeptide identification may mean that even experimentally verified non-canonical sequons could occur at extremely low occupancy and have potentially little biological value. Finally, the current model does not address how PglB is able to perform fOS release given the catalytic importance of also binding a peptide substrate. In yeast, purified STT3 can generate fOS by hydrolyzing the lipid (dolichol rather than Und-P) linked oligosaccharide irrespective of peptide binding,98 however an observation that the WWD motif is required for PglB-mediated fOS release82 suggests a peptide substrate is necessary in Campylobacter.
Addition of the N-glycan does not appear to be coupled to any particular membrane translocation pathway as CmeA was modified when shuttled into the periplasm via either the secretory (Sec) or twin-arginine translocation (Tat) bacterial translocation systems.93 Kowarik et al. however, did demonstrate differences in N-glycosylation site occupancy when proteins were transported via these different systems in E. coli,93 and consistent with their findings that showed lower N-glycosylation efficiency with Tat-translocated proteins, only 1 identified C. jejuni N-glycoprotein is predicted (by SignalP99) to be translocated using this system.100
Identification of proteins modified by N-glycosylation in C. jejuni
In early studies, lectin affinity approaches employing soybean agglutinin (SBA) specific for the GalNAc residues of the C. jejuni N-linked glycan were undertaken in combination with gel electrophoresis for separation of SBA-bound proteins. Gel-separated proteins could also be highlighted by Western blotting using glycan-specific antisera.33,34 In each case, significant problems were encountered, including the lack of separation of very hydrophobic membrane-associated proteins on gels, the difficulty in performing post-separation analysis and confirmation of glycosylation sites for blotted proteins; and finally, even for those proteins that could be gel separated, the incompatibility of the Asn-N-glycan bond (most likely due to the structure of diNAcBac) with protein-N-glycosidase F (PNGase F) digestion and chemical β-elimination that meant intact glycopeptide analysis was needed for site verification.35Site-specific glycopeptide analysis firstly relied on collision-induced dissociation (CID) MS-based fragmentation, however the highly labile nature of the glycosidic bonds resulted in very poor peptide backbone sequence coverage and therefore an inability to identify the modified sites. The advent of higher energy collisional dissociation (HCD) fragmentation enabled switching between CID (for glycan confirmation) and HCD for peptide fragmentation and sequencing,35 while concurrent advances in hydrophilic interaction liquid chromatography (HILIC) facilitated better enrichment and separation of glycosylated C. jejuni peptides compared to previous studies employing SBA affinity and gel electrophoresis. An optimized workflow employing HCD tandem MS (MS/MS) provides glycan-derived diagnostic oxonium ions from the C. jejuni N-glycan (e.g. GalNAc, 204.08 mass:charge [m/z]) and peptide sequence.89,91 In addition to improvements in MS-based glycan site identification, glycoprotein analysis can also be coupled to a multi-protease digestion strategy (e.g. employing alternatives to trypsin, including pepsin and chymotrypsin) that improve N-glycosite coverage and provide independent site verification in many cases.35
This approach has now yielded the identification of 134 sites of C. jejuni N-glycosylation from 78 membrane-associated proteins that have been experimentally confirmed (predominantly by MS), including periplasmic proteins, lipoproteins, inner membrane proteins and at least one protein that is thought to be surface-exposed (the lipoprotein JlpA101) across 5 C. jejuni strains,33,35,61,63,89 meaning that C. jejuni is likely to be the most complete glycoproteome yet described in the literature (Table 1). Some glycoproteins are modified at multiple sites; for example, the Cj0152c putative membrane protein (which shares significant sequence similarity with the Helicobacter pylori neuraminidase/sialidase) contains 6 occupied canonical sites, as well as a single atypical site (Fig. 2A). Cj0152c also contains an additional pseudo-sequon (70ENNPT74) that is not occupied, and that is predicted to be located in the cytoplasmic region of the protein. Cj0610c (encoding the peptidoglycan O-acetyltransferase PatB) is potentially the most ‘modified’ protein in C. jejuni since it contains 5 confirmed N-glycosites and 10 N-sequons in total, all of which are predicted to be located within the periplasm; structural elucidation of this protein could be particularly useful in determining the three-dimensional constraints involved in N-glycan site occupancy (see below). A further 5 proteins (Cj0114, Cj0592c, Cj0843c, Cj1013c and Cj1670c) each contain 4 verified N-glycosites (Table 1). Additionally, eight proteins have been identified with the pEtN-modified N-glycan attached.91 Although the function of the pEtN-glycan remains completely unknown, the proteins displaying this modification are amongst the most immunogenic in C. jejuni, including the major antigen PEB3 (Cj0289c), and the previously identified immunogens CjaC (Cj0734c), CjaA (Cj0982c) and JlpA (Cj0983c).17,34,101–103 Despite this, deletion of the eptC pEtN transferase responsible for pEtN modification of the N-glycan did not influence the reactivity of these proteins with human serum.91 Further work is required to better understand the occupancy levels of non- to pEtN-modified N-glycan on these glycosites and thus to assist in determining the biological role of the pEtN group in this context. It is also important to note that a second Campylobacter species, C. gracilis, exclusively modifies proteins with an N-glycan displaying a terminal pEtN group,63 however again, the role of this modification remains to be elucidated.
| Cj | 81–176 | Gene | Identification | Sequence/site# | Location | Topology |
|---|---|---|---|---|---|---|
| #Where glycosylation site was identified only in another strain this sequence is shown in (brackets), +non-canonical sequon denoted by underlining of atypical amino acid at −2 or +2 position, Asn (N) highlighted in bold and shaded in italicized bold is also modified by pEtN-modified N-glycan; location, predicted subcellular localization as determined by PSORTb (vers. 3.0.2.)106 and Lipo P 1.0,139 (x) number of predicted transmembrane regions (TMR), or presence of signal peptide (SP), unless experimentally proven all lipoproteins were considered anchored to OM or IM (dependent on Lipo P use of the ‘+2 rule’, Asp at +2 from lipo-Cys predicts IM anchoring, all other amino acids predict OM anchoring) with protein facing into the periplasm; topology, predicted location of the N-glycosylation site as determined by TmPred (https://embnet.vital-it.ch/software/TMPRED) and TOPCONS.140 Cyto, cytoplasm; E, extracellular; IM, inner membrane; LP, lipoprotein; OM, outer membrane; PP, periplasm; SE, surface exposed; Unk, unknown. ^Site identified by expression in E. coli containing the pgl cluster and over-expression in C. jejuni [H. M. Frost, PhD Thesis, University of Manchester, 2015], not seen in any wild-type C. jejuni glycoproteome studies.a Cj0017c localization depends on correct prediction of orientation for N- and C-terminus of protein.b Cj0371 co-localises to the poles of C. jejuni cells and thus co-localises with flagella.134 Thus, the protein is potentially surface-exposed (SE).c Cj0592c PSORT b predicts unknown localization; Lipo P predicts a lipoprotein signal peptide with OM anchor (Asp at +2 position to SpII cleavage site); protein is described as ‘putative periplasmic protein’.d Cj0599 PSORT b predicts unknown localization; protein contains C-terminal OmpA domain suggesting OM localization and therefore topology could be PP or SE.e Cj0776c PSORT b predicts cytoplasmic localization; 1 predicted TMR; TOPCONS predicts 1 TMR with the majority of the protein localized to the periplasm.f Cj0864 Ding et al.141 reported this sequence as DMoxNVS (where the methionine is methionine sulfoxide), however the NCTC11168 sequence indicates the −2 position is an alanine. This sequence was also low scoring as discussed in the text.g Cj0944c PSORT b predicts cytoplasmic localization; Lipo P and TOPCONS predict 1 SP and periplasmic location.h Cj0982c PSORT b predicts periplasmic localization; TOPCONS and Lipo P predict lipoprotein with IM anchoring. Experimental evidence in ref. 103.i Cjj81176_1263 was originally described in ref. 89 and 91 as CJE1384. | ||||||
| Cj0011c | 0037 | cj0011c | Putative non-specific DNA-binding protein (competence ComEA-like; natural transformation protein) | 49EANFT53 | IM (1) | PP |
| Cj0017c | 0044 | dsbI | Disulfide bond formation protein DsbI | 3EINKT7 | IM (5) | Cytoa |
| Cj0081 | 0118 | cydA | Cytochrome bd oxidase subunit I | 283DNNES287 | IM (9) | PP |
| 351EN(S)NDT355 | PP | |||||
| Cj0089 | 0124 | cj0089 | Putative lipoprotein (TPR tetricopeptide repeat-like helical domain protein) | 73DFNKS77 | LP/IM (SP) | PP |
| Cj0114 | 0149 | cj0114 | Putative periplasmic protein (TPR tetricopeptide repeat-like helical domain protein; putative Tol-Pal system protein YbgF/putative cell division coordinator CpoB) | 99ENNFT103 | OM | PP |
| 153DA(V)NLS157 | PP | |||||
| 171DSNST175 | PP | |||||
| 177ENNNT181 | PP | |||||
| Cj0131 | 0166 | cj0131 | Putative peptidase M23 family protein/putative zinc metallopeptidase (putative Gly–Gly endopeptidase) | 73DDNTS75 | Unk (1) | PP |
| Cj0143c | 0179 | znuA | Putative periplasmic ABC transport solute-binding protein (zinc-binding ABC transporter ZnuA) | 26E(D)QNTS30 | PP | PP |
| Cj0152c | 0188 | cj0152c | Putative membrane protein (45.3% similarity to H. pylori sialidase A/neuraminidase) | 126EQNNT130 | Unk (1) | PP |
157DNNK![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) 161+ 161+ |
PP | |||||
| 163ETNRT167 | PP | |||||
| 182DKNIS186 | PP | |||||
| 188ENNIS192 | PP | |||||
| 193ENNTT197 | PP | |||||
| 250DFNIS254 | PP | |||||
| Cj0158c | 0194 | cj0158c | Putative haem-binding lipoprotein (cytochrome c oxidase Cbb3-like protein) | 119DKNHS123 | LP/OM (SP) | PP |
| Cj0168c | 0204 | cj0168c | Putative periplasmic protein | 26DVNQT30 | PP (SP) | PP |
| Cj0176c | 0212 | cj0176c | Putative lipoprotein | 29DLNKT33 | LP/OM (SP) | PP |
| Cj0177 | ND | ctuA/chaN | Putative iron transport protein (putative iron-regulated lipoprotein) | 83EGNLS87^ | IM (1) | PP |
| Cj0182 | 0213 | cj0182 | Putative transmembrane transport protein (ABC transporter transmembrane family; long chain fatty acid ABC transport protein; peptide antibiotic transport protein SbmA) | 58DSNST62 | IM | PP |
| 70ENNAT74 | PP | |||||
| Cj0199c | 0230 | Putative periplasmic protein | 126DINLS130 | Unk (1) | PP | |
| Cj0200c | 0231 | cj0200c | Putative periplasmic protein | 33DNNKT37 | Unk (SP) | PP |
| Cj0235c | 0260 | secG | Uncharacterized protein (preprotein translocase subunit SecG) | 87ENNNT91 | IM (2) | PP |
| 118DVNSS122 | PP | |||||
| Cj0238 | 0263 | cj0238 | Putative mechanosensitive ion channel family protein (MscS family membrane integrity protein) | 24DANIS28 | IM (5) | PP |
| 56DENSS60 | PP | |||||
| Cj0256 | 0283 | eptC | Putative sulfatase family protein (phosphoethanolamine transferase EptC; lipid A/lipooligosaccharide pEtN transferase EptC) | 213ENNHT217 | IM (5) | PP |
| Cj0268c | 0295 | cj0268c | Putative transmembrane protein (SPFH domain/band 7 family protein; FtsH protease regulator HflC) | 274EANAT278 | Unk (1) | PP |
| Cj0277 | 0304 | mreC | Homolog of E. coli rod-shape determining protein MreC | 91DQNST95 | Unk (1) | PP |
| Cj0289c | 0315 | peb3 | Major antigenic peptide PEB3 (thiosulfate/sulfate-binding protein) | 88DFNVS92 | Unk (SP) | PP |
| Cj0313 | 0335 | cj0313 | Putative integral membrane protein (putative lipooligosaccharide export ABC transporter permease LptG) | 173DLNLS177 | IM (6) | PP |
| 196DGNIT200 | PP | |||||
| Cj0365c | 0388 | cmeC | Outer membrane channel protein CmeC (multi-drug antibiotic efflux system CmeABC protein) | 30EANYS34 | OM (SP) | PP |
| 47ENNSS51 | PP | |||||
| Cj0366c | 0389 | cmeB | Efflux pump membrane transporter CmeB (Multi-drug antibiotic efflux system CmeABC protein) | 634DRNVS638 | IM (12) | PP |
| Cj0367c | 0390 | cmeA | Periplasmic fusion protein CmeA (multi-drug antibiotic efflux system CmeABC protein) | 121DFNRS125 | IM (1) | PP |
| 271DNNNS275 | PP | |||||
| Cj0371 | 0395 | cj0371 | UPF0323 lipoprotein Cj0371 (putative secreted protein involved in flagellar motility) | 75DLNGT79 | LP/OM (SP) | PP/SEb |
| Cj0376 | 0400 | cj0376 | Putative periplasmic protein | 50DKNQT54 | Cyto | PP |
| Cj0397c | 0420 | cj0397c | Uncharacterized protein | 105DFNNT109 | Unk (1) | PP |
| Cj0399 | 0422 | cvpA | Colicin V production protein homolog CvpA | 179DLNNT183 | IM (4) | PP |
| Cj0404 | 0428 | dedD | Putative transmembrane protein (SPOR sporulation domain-containing protein; putative cell division protein DedD) | 101EQNNT105 | Unk (1) | PP |
| Cj0454c | 0479 | cj0454c | Putative membrane protein | 91ENNKS95 | IM (1) | PP |
| Cj0455c | 0480 | cj0455c | Putative membrane protein | 60![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) QNQT64+ QNQT64+ |
IM (1) | PP |
| Cj0494 | 0515 | cj0494 | Putative exporting protein | 26DNNIT30 | Unk | PP/SE |
| Cj0508 | 0536 | pbpA | Penicillin-binding protein PbpA (penicillin-binding protein 1A; peptidoglycan transpeptidase PBP1A) | 312DANLS316 | IM (1) | PP |
| Cj0511 | 0539 | ctpA | Putative secreted protease (protease family S41; carboxy-terminal protease CtpA) | 67DQNIS71 | IM (1) | PP |
| Cj0515 | 0543 | cj0515 | Putative periplasmic protein | 207ELNAT211 | IM (3) | PP |
| 234DFNAS238 | PP | |||||
| Cj0530 | 0555 | cj0530 | Putative periplasmic protein (AsmA family protein DUF3971 domain) | 519DFNAS523 | OM (1) | PP/SE |
| 617DSNKT621 | PP/SE | |||||
| Cj0540 | 0565 | cj0540 | Putative exporting protein | 173ENNNS177 | Unk (0) | PP/SE |
| Cj0587 | 0615 | cj0587 | Putative integral membrane protein | 282DNNLS286 | IM (8) | PP |
| Cj0592c | 0620 | cj0592c | Putative periplasmic protein (putative lipoprotein; Cj0591 paralog) | 96DINQS100 | Unk (SP)c | PP |
| 103ENNES107 | PP | |||||
| 127ENNQS131 | PP | |||||
| 137DVNMT141 | PP | |||||
| Cj0599 | 0627 | cj0599 | Putative OmpA family membrane protein (putative chemotaxis protein MotB; Putative flagellar motor motility protein MotB; Cj0336c MotB paralog) | 97EANIT101 | Unk (1) | PP/SEd |
| 109DLNST113 | PP/SE | |||||
| 168DNNIT172 | PP/SE | |||||
| Cj0608 | 0637 | cj0608 | Putative outer membrane efflux protein (putative TolC-like outer membrane protein; putative antibiotic efflux CmeC paralog) | 35DLNLT39 | OM (2) | PP |
| Cj0610c | 0639 | cj0610c | Putative periplasmic protein (SNGH family hydrolase; putative lipase/esterase; peptidoglycan O-acetyltransferase PatB) | 82DENLS86 | Unk (1) | PP |
| 98DENTS102 | PP | |||||
| 113DANIS117 | PP | |||||
| 296ENNRS300 | PP | |||||
| 331EENAS335 | PP | |||||
| Cj0633 | 0661 | cj0633 | Putative periplasmic protein (putative polysaccharide deacetylase; putative glycoside hydrolase/deacetylase) | 73DNNKS77 | Cyto (1) | PP |
| 123DTNLT127 | PP | |||||
| 129DQNLT133 | PP | |||||
| Cj0648 | 0676 | cj0648 | Putative membrane protein (putative lipooligosaccharide transport system substrate-binding protein LptC) | 49ESNTS53 | IM (1) | PP |
| 103EGNVT107 | PP | |||||
| Cj0652 | 0680 | pbpC | Penicillin-binding protein PbpC (pencillin-binding protein PBP2; peptidoglycan transpeptidase PBP2) | 99DLNAS103 | IM (1) | PP |
| 467ENNNT471 | PP | |||||
| Cj0694 | 0717 | ppiD | Putative periplasmic protein (SurA domain-containing outer membrane protein folding protein; peptidyl-prolyl cis/trans isomerase PpiD) | 132DFNKT136 | IM (1) | PP |
| 306DQNIS310 | PP | |||||
| 426DQNSS430 | PP | |||||
| Cj0734c | 0757 | hisJ | Probable histidine-binding protein (periplasmic lipoprotein CjaC; solute transport protein HisJ) | 26EN(S)NAS30 | IM (PP) | PP |
| Cj0776c | 0797 | cj0776c | Putative periplasmic protein | 87DENQS91 | Cyto (1)e | PP |
| 103ENNQS107 | PP | |||||
| 111DTNTS115 | PP | |||||
| Cj0780 | 0801 | napA | Periplasmic nitrate reductase NapA (catalytic subunit of the NapAB complex) | 385DDNES389 | IM (PP) | PP |
| Cj0783 | 0804 | napB | Periplasmic nitrate reductase NapB (electron transfer subunit of the NapAB complex) | 48EANFT52 | IM (PP) | PP |
| Cj0843c | 0859 | slt | Putative secreted transglycosylase (soluble lytic murein peptidoglycan transglycosylase) | 97DANLT101 | IM (PP) | PP |
| 173DLNTG(S)177 | PP | |||||
| 327DANAS331 | PP | |||||
| 374DYNKT378 | PP | |||||
| Cj0846 | 0862 | cj0846 | Uncharacterized metallophosphoesterase (Ser/Thr phosphatase family protein) | 280DLNTS284 | IM (3) | PP |
| Cj0864 | 0880 | cj0864 | Putative periplasmic protein (putative thiol: disulfide interchange protein DsbA homolog) | 50MNVS54+f |
IM (PP) | PP |
| Cj0906c | 0915 | pgp2 | Putative periplasmic protein (peptidoglycan L-D-carboxypeptidase Pgp2) | 53DKNIS57 | IM (SP) | PP |
| Cj0944c | 0968 | cj0944c | Putative periplasmic protein (putative flagellar protein FliL; chemotaxis-associated protein) | 219ENNAS223 | Cyto (0)g | PP |
| 238DENST242 | ||||||
| Cj0958c | 0981 | yidC | Membrane protein insertase YidC (integral membrane protein assembly/folding protein YidC) | 40EQNIT44 | IM (5) | PP |
48![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif) QNTS52+ QNTS52+ |
PP | |||||
| 154DENGS158 | PP | |||||
| Cj0982c | 1001 | cjaA | Putative amino acid transporter periplasmic solute-binding protein CjaA | 137DSNIT141 | IM (PP/LP)h | PP |
| Cj0983 | 1002 | jlpA | Uncharacterized lipoprotein Cj0983 (surface-exposed lipoprotein JlpA) | 105E(K)ANAS109 | OM (SE) | SE |
| 144DINAS148 | SE | |||||
| Cj1007c | 1025 | cj1007c | Putative mechanosensitive ion channel family protein (MscS family osmotic stress resistance protein) | 17DVNRT21 | IM (4) | PP |
| Cj1013c | 1032 | cj1013c | Putative cytochrome c biogenesis protein CcmF/CycK/CcsA family protein CcsB | 178ENNNS182 | IM (14) | PP |
| 230DENLT234 | PP | |||||
| 530DLNST534 | PP | |||||
| 731DGNWT(I)735 | PP | |||||
| Cj1032 | 1051 | cmeE | Membrane fusion component of antibiotic efflux system CmeDEF | 199DQNGT203 | IM (1) | PP |
| Cj1053c | 1073 | cj1053c | Putative integral membrane protein (amino acid/carbohydrate/antibiotic transport permease motifs protein; lipooligosaccharide ligase-like motif protein) | 75DINVS79 | IM (2) | PP |
| 96DNNQS100 | PP | |||||
| Cj1055c | 1075 | cj1055c | Putative sulfatase family protein (putative arylsulfatase; putative phosphoglycerol transferase lipooligosaccharide synthesis protein homolog) | 616ESNDT620 | IM (5) | PP |
| Cj1126c | 1143 | pglB | Undecaprenyl-diphosphooligosaccharide-protein glycosyltransferase (PglB oligosaccharyltransferase) | 532DYNQS536 | IM (12) | PP |
| Cj1219c | 1232 | cj1219c | Putative periplasmic protein (uncharacterized protein involved in outer membrane biogenesis assembly) | 47DVNIT51 | OM (1) | PP |
| Cj1345c | 1344 | pgp1 | Putative periplasmic protein (peptidoglycan D-L-carboxypeptidase Pbp1) | 59DYNIT63 | Cyto (1) | PP |
| 159EINAS163 | PP | |||||
| 348DGNET352 | PP | |||||
| Cj1373 | 1376 | cj1373 | Putative integral membrane protein (antibiotic resistance sterol-sensing domain protein; RND superfamily export protein MmpL family) | 134DINRT138 | IM (12) | PP |
| 497DQNTS501 | PP | |||||
| Cj1444c | 1438 | kpsD | Capsule polysaccharide export system periplasmic protein KpsD | 37DQNLS41 | IM (PP) | PP |
| 50ENNLT54 | PP | |||||
| Cj1496c | 1488 | cj1496c | Putative periplasmic protein (putative magnesium transporter MgtE-like protein; putative motility chaperone MotE; putative flagellar protein FliG) | 71EVNAT75 | Cyto (PP) | PP |
| 167DNNAS171 | PP | |||||
| Cj1565c | 1550 | pflA | Paralysed flagellar motility protein A PflA | 456DNNAS460 | Cyto (PP) | PP |
| 495EGNFS499 | PP | |||||
| Cj1621 | 1608 | cj1621 | Putative periplasmic protein | 197DLNKT201 | E (1) | PP |
| Cj1661 | 1652 | cj1661 | Putative ABC transport system permease (putative antibiotic macrolide export protein MacB; putative cell division protein FtsX) | 188ENNQS192 | IM (4) | PP |
| Cj1670c | 1666 | cgpA | Putative periplasmic protein (campylobacter glycoprotein A; AMIN-domain containing protein, membrane protein assembly protein) | 26DQNIT30 | Unk (0) | PP |
| 71DVNKS75 | PP | |||||
| 104EKNSS108 | PP | |||||
| 111ESNST115 | PP | |||||
| ND | 0063 | sirA | Dissimilatory sulfite reductase SirA/MccA | 213DGNLS217 | IM (1) | PP |
| ND | 0701 | kdpC | Potassium-transporting ATPase KdpC subunit | 83DTNES87 | IM (1) | PP |
| ND | 1263 | 1263 | Uncharacterized proteini | 26EQNGS30 | Unk (SP) | PP |
| VirB10 | pVir0003 | virB10 | Type IV secretion system protein VirB10 | 30EENVS34 | OM (SP) | PP |
| 95DNNIT99 | PP | |||||
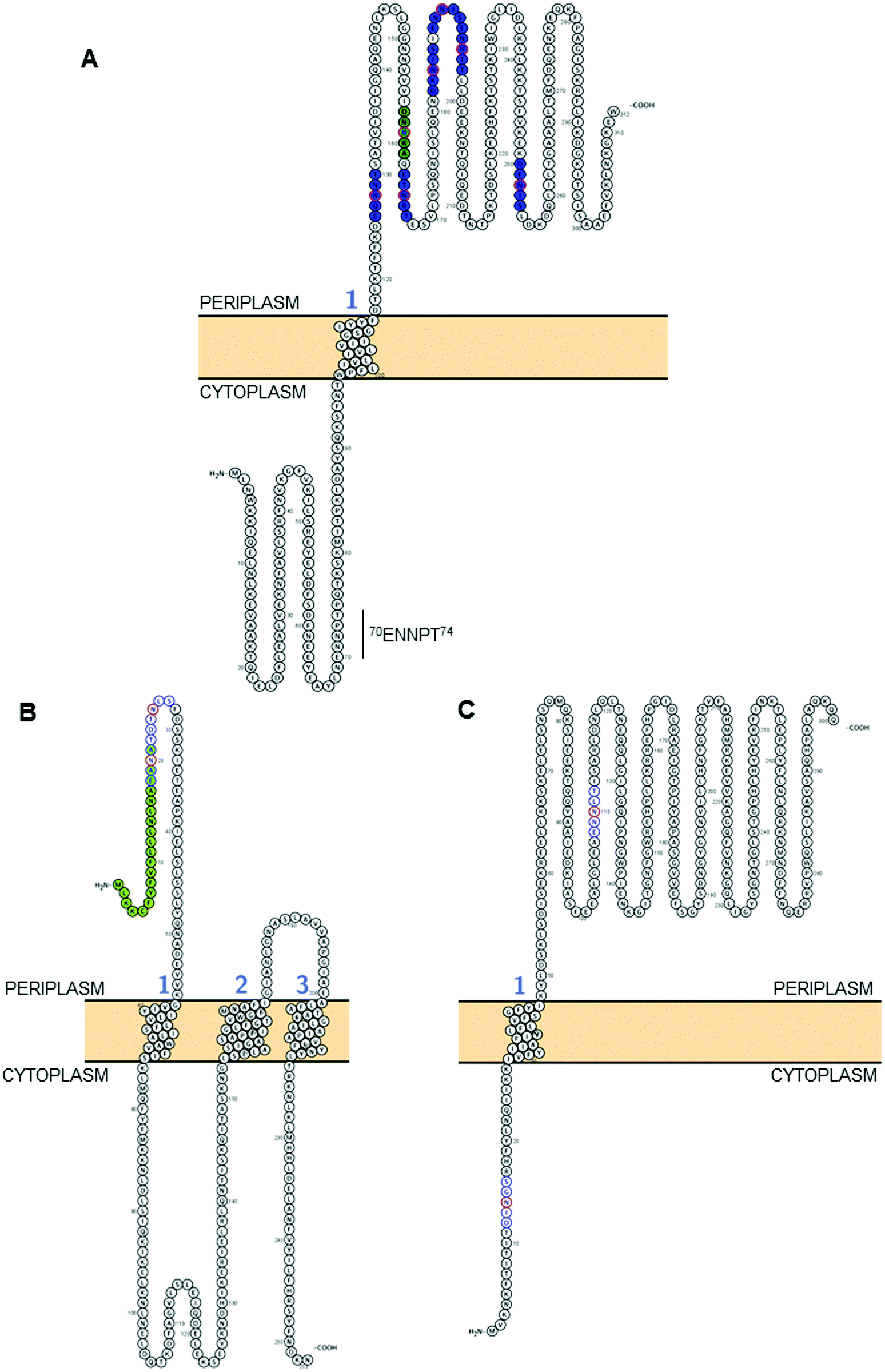 | ||
| Fig. 2 Modelling of predicted surface topologies of 3 C. jejuni N-glycoproteins. (A) Cj0152c; positions of experimentally verified N-glycosites (Asn; N) are shown in red circles with occupied sequons shown in blue fill, the position of a non-canonical, but occupied sequon is shown in green fill; (B) Cj0179 (ExbB1); positions of two sequons (not experimentally verified) are shown in blue with the Asn residues in red, the N-terminal signal peptide that overlaps the first sequon is in green; (C) Cj1087c; positions of two sequons (not experimentally verified) are shown in blue with the Asn residues in red, the sequon at position 12DINGS16 is predicted to reside in the cytoplasm and hence cannot be glycosylated. All topologies were visualized using Protter.142 | ||
As discussed above, despite PglB showing a preference for Thr at the +2 position,92 there is no obvious bias towards Thr in the identified N-glycosites; in fact only 60 of 134 identified sites contain sequons with a Thr in this position (44.8%), with 73 containing Ser (54.5%) and the final sequon displaying alanine (Ala) in a non-canonical sequon (Table 1).89 Conversely, there is clear preference for Asp at the −2 position with 84 sequons displaying this amino acid (62.7%) compared with only 47 displaying Glu (35.1%). The final 3 sequons were non-canonical (Table 1). These data align with previous studies that have tested various sequon compositions and their glycosylation efficiency by the PglB OST and found DQNAT to be the optimal sequon, as well as an ∼5-fold preference for Asp, rather than Glu, at the −2 position.104
Structural constraints of N-glycosylation
C. jejuni contains ∼500 N-glycosylation sequons within the translated genome sequence, depending on the strain examined (for example, 510 sequons are found in strain HB93-1335), and ∼370 of these are found in >260 predicted membrane-associated proteins (or proteins of unknown localization) suggesting that there are evolutionary constraints associated with maintenance of the sequon in proteins connected with this sub-cellular localization; similar sequon bias (albeit against the presence of the sequon) has been observed for the HMW system in Haemophilus influenzae.105 Realistically, the modifiable N-glycoproteome is likely to be considerably smaller, given that both topological and structural constraints likely play a crucial role in the ability of the PglB OST to modify a given sequon. While tools such as PSORTb106 provide predicted sub-cellular localization for a given protein sequence, it is absolutely critical to understand that the topology of the protein defines PglB sequon accessibility. This is particularly important for proteins associated with the cytoplasmic/inner membrane in Gram negative organisms, since regions within these proteins can be cytoplasmic, and thus any sequons contained within those regions will not be amenable to the catalytic activity of PglB. For the 134 N-glycosites shown in Table 1, we employed a variety of localization and topology tools that show 133 (99.2%) are predicted to localize to the periplasm (Fig. 2A and Table 1). Only a single site, 3EIKT7 from DsbI (Cj0017c), is predicted to localize to the cytoplasm. This site is challenging to accurately predict given its proximity to the N-terminus and the difficulty in orienting termini into the inner membrane (inside or outside). Furthermore, the site was identified only in a single study35 and was based on a low scoring, manually validated and very short glycopeptide sequence; an approach no longer valid due to many improvements in computational intact glycopeptide analysis (another site identified in a similar manner is the non-canonical 50AMVS54 from Cj0864). Despite the overwhelming association of experimentally verified N-glycosites with periplasmic localization, many sequons in membrane-associated proteins that have not been experimentally identified are predicted to localize to the cytoplasm (or be located within cleaved signal peptides) and therefore cannot be glycosylated (for examples, see Fig. 2B and C). Fundamentally, this means that the theoretical N-glycoproteome of C. jejuni may only comprise between 200–250 possible sites.
Beyond localization and topology, the next major influence on sequon occupancy is the tertiary conformation of the protein, with the three-dimensional structure of both the target protein and PglB itself dictating site accessibility.85,93,107 Unlike in eukaryotes, where N-glycosylation occurs in the endoplasmic reticulum (with further processing in the Golgi apparatus) prior to or during folding (and hence partially dictates the final conformation), the prevailing viewpoint is that the C. jejuni N-glycan is added to already, or at least partially, folded substrates,93,100 meaning that existing tertiary structural constraints are a major factor in the final attachment and kinetics of the modification. Sequons buried within the tertiary structure are therefore inaccessible to PglB and cannot be modified, irrespective of their sub-cellular location. The earliest structural consideration of C. jejuni N-glycosylation was based on the crystal structure of the major antigen PEB3 (Cj0289c),108 which showed the N-glycosite at 88DFVS92 occurs in a flexible exposed loop region readily accessible to the PglB OST. Therefore, without determining structures of glycoproteins it remains difficult to predict which sequons will be occupied and the likely level of site occupancy, and there are only very few N-glycoproteins for which three-dimensional structures are currently available. In addition to PEB3, and PglB itself,85 structures of components of the tripartite antibiotic efflux system CmeABC (Cj0365c–Cj0367c) have also been elucidated,109,110 and all 3 are N-glycoproteins (Table 1). CmeA is the periplasmic membrane fusion family protein, with 2 N-glycosites both predicted to be located within the periplasm (Table 1). CmeC is the outer membrane channel and examination of the crystal structure109 shows that both experimentally verified glycosylated sequons (30EAYS34 and 47ENSS51) are located in a periplasmic disordered exposed loop region that leads from the membrane-embedded N-terminal lipidated cysteine (following removal of the signal peptide) to the first structured part of the protein. Therefore both sequons are consistent with the known structural requirements for N-glycosylation.93,99,108
CmeB, which is the inner membrane efflux pump, contains one well characterized N-glycosite (634DRVS638). A second sequon (653DRNAS657) is located proximal to this confirmed site, but no experimental evidence exists for this site being occupied in any C. jejuni strain, and hence CmeB is the only protein with both an occupied and unoccupied glycosite for which structural information can currently be determined. These two sites are also of interest since their sequons are near identical and hence, any effects of differences at the −2 and +2 positions, as described above, are likely to be negligible (indeed the arginine [Arg] at the −1 position is shared, while the +1 position is a semi-conservative substitution from valine [Val] to alanine [Ala], which are both aliphatic amino acids) and most likely do not influence site occupancy. Interrogation of the CmeB tertiary structure shows that both sequons are located in the large periplasmic section of the protein located between the sixth and seventh transmembrane-spanning regions (TMR; residues 554–867, with CmeB predicted to contain 11 TMR, excluding the N-terminal signal peptide) and are found in short disordered exposed loop regions separated by a small alpha-helix (Fig. 3A). Tertiary structure modelling shows that 634DRVS638 is located close to the membrane and the modified Asn is highly solvent accessible, while 653DRNAS657 is located further into the periplasm. Although solvent accessible, Asn-655 is partially occluded by Arg-654 (Fig. 3A). The CmeB structure was next modelled in protein complex with the PglB OST, using the model sequon DQNAT104 to provide the PglB binding conformation. CmeB/PglB docking clearly demonstrated a preference for the Asn-636 site, consistent with the identification of this site in several MS-based studies (Fig. 3B and C), while the Asn-655 site does not appear to readily interact with the PglB model, and hence therefore is likely to either not be glycosylated or glycosylated at only very low site stoichiometry.
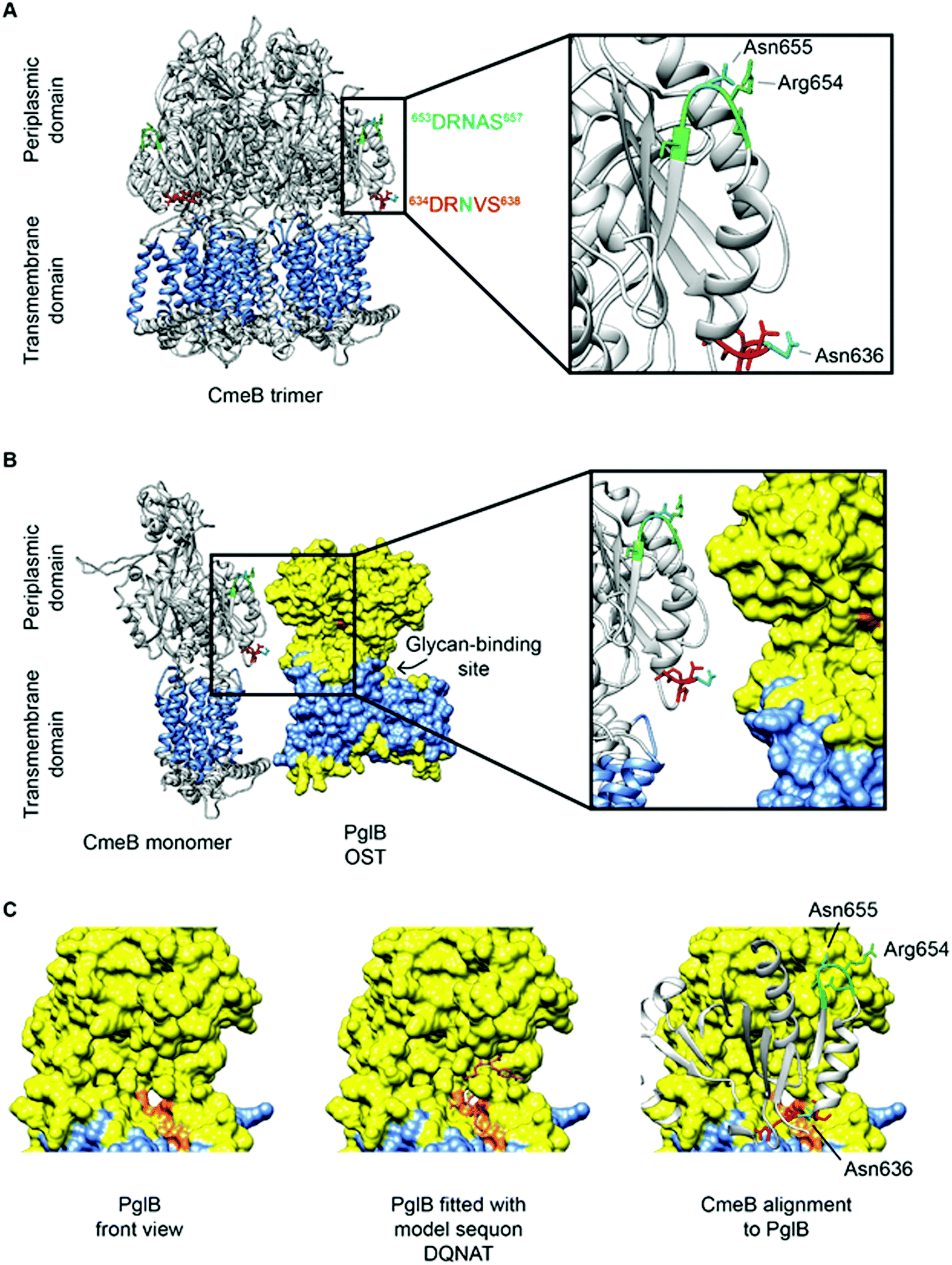 | ||
| Fig. 3 CmeB modeling with PglB highlighting N-glycosylation sequons. (A) CmeB trimer (Protein Data Bank [PDB]: 5LQ3) has a transmembrane domain (highlighted in blue with 11 TMR) and periplasmic domain. The experimentally validated sequon (634DRVS638) is labelled red and the non-identified sequon (653DRNAS657) is labelled green, with both Asn labelled cyan. Both Asn are located on the periplasmic side on exposed loops and are solvent-accessible with Asn-636 more accessible than Asn-655; (B) The PglB OST (PDB: 3RCE) shown in yellow has a transmembrane spanning domain (highlighted in blue with 12 TMR) and a larger periplasmic region where the catalytic domain is located. The sequon recognition site is highlighted in orange and facing towards CmeB with the glycan-binding site located behind. Sequon 634DRVS638 is in closer proximity and has better accessibility to the PglB catalytic site; (C) (left) PglB viewed from the front (90° counter-clockwise rotation to upper panels) reveals the sequon-binding surface in orange, (Middle) PglB fitted with the model peptide mimic DQNAT, (right) alignment of CmeB to PglB (90° counter-clockwise rotation to panel B) reveals that sequon 634DRVS638 is more spatially likely to fit into the active site of PglB suggesting this sequon is more readily glycosylated than 653DRNAS657. Analysis was performed in UCSF Chimera 1.14 (build 42094). | ||
Further evidence for structural constraints determining optimal glycosylation have been shown for the doubly glycosylated surface-exposed glycoprotein JlpA.101 Scott et al. showed that JlpA must be glycosylated at one site (144DIAS148) before a second site (105EAAS109) can be glycosylated, inferring that structural modifications to JlpA conferred by Asn-146 glycosylation open the protein conformation and allow PglB to add the N-glycan to the second site. These structural constraints have since been confirmed using structural predictions and crystallography.111 Finally, nuclear magnetic resonance (NMR) analysis of a recombinant C. jejuni CmeA domain indicates that the N-glycan itself adopts a rigid rod conformation112 that appears to fold back over the exposed protein (thus suggesting a role in protection from proteolysis), although it remains to be seen how well conserved this is in vivo. Although no examples have been shown in the literature, the converse may also be true in that the N-glycan itself may hinder accessibility of a second site in a given protein to the PglB OST. Despite this possibility, proteins such as Cj0152c (Fig. 2A) have multiple sites in close sequence space; occupied sequons are found at 7 sites, with 3 (Asn-184, Asn-190 and Asn-195) located within 20 amino acids. To determine if N-glycan steric hindrance of PglB occurs, better understanding of individual site occupancy, in the context of tertiary structures, is needed.
A final structural/topological consideration is the role of N-glycosylation in OMVs that have been associated with C. jejuni virulence.24–26 OMVs package cytoplasmic, periplasmic, outer membrane-associated and N-glycoproteins in a ‘bleb’-like structure.24 PglB is located in the cytoplasmic/inner membrane, which is not typically associated with OMVs. It is possible however, that inner membrane fragments may also be packaged into OMVs, and all C. jejuni OMV proteomics studies have demonstrated the identification of integral cytoplasmic membrane proteins (e.g. CmeB24). Packaging of PglB into OMVs may enable glycosylation of sites not typically found in the membrane; however despite this, we and others have observed no such cytoplasmic N-glycosites, even at low levels, which may imply that PglB does not occur in OMVs, or that OMVs are not induced (or collected) under the culture conditions employed in the N-glycosite discovery studies conducted thus far. C. jejuni OMV composition is however, altered in pgl-negative compared with wild-type C. jejuni,113 suggesting N-glycosylation does impact protein packaging, although no differences were observed in the ability of OMVs from either pgl positive or negative bacteria to induce an immune response.113
Putative functions of C. jejuni protein N-glycosylation
Deletion of genes from the pgl cluster (except pglI) results in C. jejuni that are poorly able to colonize chickens and display reduced adherence to, and invasion of, human epithelial cells.114,115 Additional recent modelling of C. jejuni virulence in a human small intestine-like gut-immune co-culture model also revealed that pgl-negative C. jejuni (in this case, pglE deletion) were vastly deficient (∼100 times less) in adherence and invasion.113 Therefore, N-glycosylation is considered a fundamental virulence determinant in this organism. Despite this, until recently116,117 there have been very few studies that have broadly characterized pgl-associated phenotypes, and the function(s) of the N-glycosylation system in general, and even more so the role of the N-glycan on individual proteins, remains almost completely unknown. Genome-wide and functional screens have shown an association between glycosylation and biofilm formation, amongst other traits, and pgl-negative C. jejuni are attenuated in some, but not all, models of biofilm growth.116,118C. jejuni pgl deletion strains incapable of N-glycosylating proteins display a number of additional altered traits including a reduced capacity for natural transformation,119 altered antibiotic resistance,117 greater susceptibility to host-derived proteases,120 impaired motility116 and modified binding to host cell surface lectins.121 Proteomics-based approaches have been employed in an attempt to globally characterize phenotypes associated with N-glycosylation.116,117pgl negative C. jejuni displayed evidence of induction of the stress response and were attenuated for survival at temperature extremes,82,116 particularly survival at lower temperatures, which has further implications for the pgl system acting as an interventional target to limit the presence of the organism on supermarket chicken for human consumption. Loss of glycosylation influenced metabolism and nutrient transport, as well as chemoattraction towards some of these substrates.116 Finally, pgl-negative C. jejuni displayed very strongly reduced abundance of proteins involved in respiration using alternative electron acceptors under low oxygen conditions; a phenotype paramount to C. jejuni colonization of all hosts, and potentially associated with a loss of proton motive force.116 This phenotype could be linked to reduced nitrate reductase (NapA/NapB) activity, which was shown in both PglB OST (ΔpglB) and glycan biosynthesis (ΔpglFED) negative C. jejuni.116 Since both NapA and NapB are experimentally proven glycoproteins (Table 1), this loss of activity may be associated with a requirement for glycosylation in formation of the NapAB complex, in generating a structural conformation that maximizes Nap catalysis, or in providing stability against protein degradation.The membrane-associated targets of the pgl N-glycosylation system are largely functionally uncharacterized ‘putative’ proteins. The remaining proteins share some degree of sequence identity with well characterized proteins from other organisms, while only a very small number have been experimentally validated. Examination of the relationships between glycoprotein identifications (Table 1) highlight several clusters of potentially functionally related classes of protein, including those involved in antibiotic resistance (all 3 members of the CmeABC antibiotic efflux system are glycosylated, as is CmeE of the CmeDEF efflux system), and antibiotic resistance has been strongly associated with the pgl system.117 Additionally, proteins with putative functions, or sequence similarity to proteins, involved in peptidoglycan biosynthesis, modification and C. jejuni helical cell morphology (Pgp1, Pgp2, MreC, PatB [Cj0610c], Cj0843c and the penicillin-binding proteins PbpA and PbpC), LOS and capsular polysaccharide (CPS) transport and assembly (Cj0313/LptG, Cj0648/LptC, Cj1053c, Cj1055c and KpsD), and membrane protein translocation and assembly (SecG, Cj0238, PpiD, YidC, Cj1219c, CgpA) are also enriched in the 78 identified N-glycoproteins, however these phenotypes have not yet been tested in pgl negative C. jejuni or N-glycosite mutants.
While several of the above studies have examined phenotypes from the perspective of pgl negative and positive C. jejuni, comparatively fewer studies have attempted to exploit site-directed mutagenesis to understand the role of the N-glycan in individual proteins. This is mainly due to the difficulty in generating site mutants in C. jejuni, which is considered poorly tractable and somewhat recalcitrant to molecular biology approaches considered standard in species such as E. coli. Despite this, a limited number of studies have been performed.122–124N-Glycosite point mutants in cmeA (CmeA contains 2 glycosites; Table 1) have increased susceptibility to several antimicrobials including bile salts and ciprofloxacin, and are attenuated for chicken colonization.125 The PglB OST is also capable of transferring the N-glycan to itself,61 however recombinant PglB expressed in otherwise non-glycosylating E. coli remains capable of catalyzing the transfer of N-glycans to proteins,62 suggesting PglB does not strictly require modification with the heptasaccharide to maintain function. Plasmid encoded VirB10 (as well as CmeA, discussed above) was reported to require N-glycosylation to perform its function (in natural transformation) at wild-type levels.123 VirB10 is not universally distributed among strains of C. jejuni, however observations of impaired natural transformation in the absence of N-glycosylation have also been observed in studies of the Cj0011c N-glycoprotein.126 Several confirmed N-glycoproteins (including DsbI, JlpA, PEB3, EptC, Cj0268c, Cj0371, Cj0454c, Cj0511c/CtpA, Cj0587 and Pgp1/Pgp2) have been associated with host colonization;127–134 however, these focused studies of individual glycoproteins have only rarely attempted to provide evidence of a contribution from the N-glycan, rather than testing gene-specific deletion mutants. In vitro expression and functional analysis of C. jejuni N-glycoproteins in non-pgl-containing E. coli suggest that N-glycosylation is not required for the function of a number of glycoproteins,128,130,135,136 however, without site mutants or comparative expression in pgl-positive expression systems, it is not possible to compare the functional efficiency of these proteins when glycosylated.
While evidence that C. jejuni protein N-glycosylation occurs on folded substrates indicates that the modification is not a driver of protein folding, there is a mounting body of evidence to suggest that the N-glycan may be important for protein stability. Mansell et al. demonstrated that the glycoproteins PEB3, CjaA and PatB/Cj0610c displayed differences in protein stability in an N-glycosylation competent, pgl system-containing E. coli.137 These proteins also showed altered folding when glycosylated, further supporting the JlpA evidence that indicates glycan attachment can alter conformational state.111 Similarly, Min et al. showed an increase in thermostability for recombinant expressed PEB3 engineered to have an additional N-glycosylation site in comparison to an unmodified variant.138 Finally, Alemka et al. showed that a pgl-negative strain displayed reduced viability when cultured under physiological levels of human- and chicken-derived proteases,120 which also supports the notion that N-glycosylation is involved in conferring protein stability.
Conclusions
N- and O-linked glycosylation in C. jejuni are fundamental requirements for virulence. Interventions targeting the biosynthesis of unique bacterial sugars may be useful in the future to reduce severity of human infection, and in particular to limit serious immune-mediated complications. Additionally, knowledge of the biochemistry, structural biology and the many peptide targets of these pathways provides a unique opportunity to better understand the functional roles of these PTM in conferring organism-wide phenotypes, and in specific protein functions. The overall lack of data regarding the function of N-glycosylation on C. jejuni proteins means that the association with virulence remains poorly understood, and could reflect a general requirement for glycosylation in a global, C. jejuni-specific process (such as protection against proteolytic degradation) and/or that the effect is protein-specific. For the latter, comprehensive knowledge of glycosylation sites is still required despite our advances in understanding the glycoproteome in this organism. Therefore, several approaches are needed; (i) a full phenotypic characterization of different pgl mutants that are attenuated for virulence, coupled with multi-omics approaches to determine affected pathways; (ii) a comprehensive analysis of occupied and unoccupied N-glycosylation sequons, and their occupancy, that can be quantified across many changes in environmental or host-specific conditions to create a knowledge bank of sites suitable for mutational analysis; and (iii) testable hypotheses regarding the role of the N-glycan that can be examined by interventional approaches. Ultimately, it remains likely that the pgl N-glycosylation system plays a multi-factorial role in C. jejuni biology that is imperative in environmental, avian and human niches occupied by the organism.Conflicts of interest
There are no conflicts to disclose.Acknowledgements
This work was supported in part by the National Health and Medical Research Council (NHMRC) of Australia (Project Grant APP1106878 to S. J. C.). J. A. C. and A. L. D. are supported by Australian Government Research Training Program (RTP) Stipends. A. L. D. is additionally supported by the William G. Murrell Postgraduate Scholarship in Microbiology and a University of Sydney Merit Award Supplementary Scholarship. N. S. is supported by an NHMRC Early Career Postdoctoral Fellowship.References
- P. L. Griffiths and R. W. Park, J. Appl. Bacteriol., 1990, 69, 281–301 CrossRef CAS PubMed.
- M. B. Skirrow, Br. Med. J., 1977, 2, 9–11 CrossRef CAS.
- J. P. Butzler, Clin. Microbiol. Infect., 2004, 10, 868–876 CrossRef PubMed.
- P. D. Allan, C. Palmer, F. Chan, R. Lyons, O. Nicholson, M. Rose, S. Hales and M. G. Baker, BMC Public Health, 2018, 18, 414 CrossRef PubMed.
- W. A. Awad, C. Hess and M. Hess, Avian Pathol., 2018, 47, 352–363 CrossRef PubMed.
- W. B. Sruwe, R. Gough, M. E. Gallagher, D. T. Kenny, S. D. Carrington, N. G. Karlsson and P. M. Rudd, Mol. Cell. Proteomics, 2015, 14, 1464–1477 CrossRef PubMed.
- A. Alemka, S. Whelan, R. Gough, M. Clyne, M. E. Gallagher, S. D. Carrington and B. Bourke, J. Med. Microbiol., 2010, 59, 898–903 CrossRef CAS PubMed.
- R. Janssen, K. A. Krogfelt, S. A. Cawthraw, W. van Pelt, J. A. Wagenaar and R. J. Owen, Clin. Microbiol. Rev., 2008, 21, 505–518 CrossRef PubMed.
- M. J. Blaser, D. N. Taylor and R. A. Feldman, Epidemiol. Rev., 1983, 5, 157–176 CrossRef CAS PubMed.
- M. Koga, M. Kishi, T. Fukusako, N. Ikuta, M. Kato and T. Kanda, J. Neurol., 2019, 266, 1655–1662 CrossRef.
- E. F. Wijdicks and C. J. Klein, Mayo Clin. Proc., 2017, 92, 467–479 CrossRef.
- V. Phongsisay, Immunobiology, 2016, 221, 535–543 CrossRef CAS PubMed.
- J. A. Goodfellow and H. J. Willison, Nat. Rev. Neurol., 2016, 12, 723–731 CrossRef.
- F. Yoshida, H. Yoshinaka, H. Tanaka, S. Hanashima, Y. Yamaguchi, M. Ishihara, M. Saburomaru, Y. Kato, R. Saito, H. Ando, M. Kiso, A. Imamura and H. Ishida, Chem. – Eur. J., 2019, 25, 796–805 CrossRef CAS PubMed.
- J. Parkhill, B. W. Wren, K. Mungall, J. M. Ketley, C. Churcher, D. Basham, T. Chillingworth, R. M. Davies, T. Feltwell, S. Holroyd, K. Jagels, A. V. Karlyshev, S. Moule, M. J. Pallen, C. W. Penn, M. A. Quail, M. A. Rajandream, K. M. Rutherford, A. H. M. van Vliet, S. Whitehead and B. G. Barrell, Nature, 2000, 403, 665–668 CrossRef CAS PubMed.
- C. P. Skarp, O. Akinrinade, A. J. Nilsson, P. Ellström, S. Myllykangas and H. Rautelin, Sci. Rep., 2015, 5, 17300 CrossRef CAS.
- S. J. Cordwell, A. C. Len, R. G. Touma, N. E. Scott, L. Falconer, D. Jones, A. Connolly, B. Crossett and S. P. Djordjevic, Proteomics, 2008, 8, 122–139 CrossRef CAS PubMed.
- V. Korolik, Curr. Opin. Microbiol., 2019, 47, 32–37 CrossRef CAS PubMed.
- D. J. Bolton, Food Microbiol., 2015, 48, 99–108 CrossRef PubMed.
- P. M. Burnham and D. R. Hendrixson, Nat. Rev. Microbiol., 2018, 16, 551–565 CrossRef CAS PubMed.
- M. Stahl, J. Butcher and A. Stintzi, Front. Cell. Infect. Microbiol., 2012, 2, 5 Search PubMed.
- D. Hofreuter, Front. Cell. Infect. Microbiol., 2014, 4, 137 Search PubMed.
- M. E. Konkel, J. D. Klena, V. Rivera-Amill, M. R. Monteville, D. Biswas, B. Raphael and J. Mickelson, J. Bacteriol., 2004, 186, 3296–3303 CrossRef CAS PubMed.
- A. Elmi, E. Watson, P. Sandu, O. Gundogdu, D. C. Mills, N. F. Inglis, E. Manson, L. Imrie, M. Bajaj-Elliott, B. W. Wren, D. G. Smith and N. Dorrell, Infect. Immun., 2012, 80, 4089–4098 CrossRef CAS.
- A. Elmi, A. Dorey, E. Watson, H. Jagatia, N. F. Inglis, O. Gundogdu, M. Bajaj-Elliott, B. W. Wren, D. G. E. Smith and N. Dorrell, Cell. Microbiol., 2018, 20, 3 CrossRef.
- N. Taheri, M. Fällman, S. N. Wai and A. Fahlgren, J. Proteomics, 2019, 195, 33–40 CrossRef CAS.
- C. M. Szymanski and B. W. Wren, Nat. Rev. Microbiol., 2005, 3, 225–237 CrossRef CAS PubMed.
- H. Nothaft and C. M. Szymanski, Nat. Rev. Microbiol., 2010, 8, 765–778 CrossRef CAS PubMed.
- M. Koomey, Curr. Opin. Struct. Biol., 2019, 56, 198–203 CrossRef CAS PubMed.
- A. H. Bhat, S. Maity, K. Giri and K. Ambatipudi, Crit. Rev. Microbiol., 2019, 45, 82–102 CrossRef CAS PubMed.
- J. Poole, C. J. Day, M. von Itzstein, J. C. Paton and M. P. Jennings, Nat. Rev. Microbiol., 2018, 16, 440–452 CrossRef CAS PubMed.
- C. M. Szymanski, R. Yao, C. P. Ewing, T. J. Trust and P. Guerry, Mol. Microbiol., 1999, 32, 1022–1030 CrossRef CAS PubMed.
- N. M. Young, J. R. Brisson, J. Kelly, D. C. Watson, L. Tessier, P. H. Lanthier, H. C. Jarrell, N. Cadotte, F. St. Michel, E. Aberg and C. M. Szymanski, J. Biol. Chem., 2002, 277, 42530–42539 CrossRef CAS PubMed.
- D. Linton, E. Allan, A. V. Karlyshev, A. D. Cronshaw and B. W. Wren, Mol. Microbiol., 2002, 43, 497–508 CrossRef CAS PubMed.
- N. E. Scott, B. L. Parker, A. M. Connolly, J. Paulech, A. V. Edwards, B. Crossett, L. Falconer, D. Kolarich, S. P. Djordjevic, P. Højrup, N. H. Packer, M. R. Larsen and S. J. Cordwell, Mol. Cell. Proteomics, 2011, 10, M000031 CrossRef PubMed.
- P. Thibault, S. M. Logan, J. F. Kelly, J. R. Brisson, C. P. Ewing, T. J. Trust and P. Guerry, J. Biol. Chem., 2001, 276, 34862–34870 CrossRef CAS PubMed.
- D. J. McNally, A. J. Aubry, J. P. Hui, N. H. Khieu, D. Whitfield, C. P. Ewing, P. Guerry, J. R. Brisson, S. M. Logan and E. C. Soo, J. Biol. Chem., 2007, 282, 14463–14475 CrossRef CAS PubMed.
- I. C. Schoenhofen, E. Vinogradov, D. M. Whitfield, J. R. Brisson and S. M. Logan, Glycobiology, 2009, 19, 715–725 CrossRef CAS PubMed.
- A. I. M. Salah Ud-Din and A. Roujeinikova, Cell. Mol. Life Sci., 2018, 75, 1163–1178 CrossRef CAS PubMed.
- H. S. Chidwick and M. A. Fascione, Org. Biomol. Chem., 2020, 18, 799–809 RSC.
- N. Zebian, A. Merkx-Jacques, P. P. Pittock, S. Houle, C. M. Dozois, G. A. Lajoie and C. Creuzenet, Glycobiology, 2016, 26, 386–397 CrossRef CAS PubMed.
- M. Schirm, I. C. Schoenhofen, S. M. Logan, K. C. Waldron and P. Thibault, Anal. Chem., 2005, 77, 7774–7782 CrossRef CAS PubMed.
- G. N. Ulasi, A. J. Creese, S. X. Hui, C. W. Penn and H. J. Cooper, Proteomics, 2015, 15, 2733–2745 CrossRef CAS.
- C. G. Zampronio, G. Blackwell, C. W. Penn and H. J. Cooper, J. Proteome Res., 2011, 10, 1238–1245 CrossRef CAS PubMed.
- C. P. Ewing, E. Andreishcheva and P. Guerry, J. Bacteriol., 2009, 191, 7086–7093 CrossRef CAS PubMed.
- S. M. Logan, J. P. Hui, E. Vinogradov, A. J. Aubry, J. E. Melanson, J. F. Kelly, H. Nothaft and E. C. Soo, FEBS J., 2009, 276, 1014–1023 CrossRef CAS PubMed.
- S. L. Howard, A. Jagannathan, E. C. Soo, J. P. Hui, A. J. Aubry, I. Ahmed, A. Karlyshev, J. F. Kelly, M. A. Jones, M. P. Stevens, S. M. Logan and B. W. Wren, Infect. Immun., 2009, 77, 2544–2556 CrossRef CAS PubMed.
- P. Guerry, C. P. Ewing, M. Schirm, M. Lorenzo, J. Kelly, D. Pattarini, G. Majam, P. Thibault and S. Logan, Mol. Microbiol., 2006, 60, 299–311 CrossRef CAS PubMed.
- C. D. Carrillo, E. Taboada, J. H. Nash, P. Lanthier, J. Kelly, P. C. Lau, R. Verhulp, O. Mykytczuk, J. Sy, W. A. Findlay, K. Amoako, S. Gomis, P. Willson, J. W. Austin, A. Potter, L. Babiuk, B. Allan and C. M. Szymanski, J. Biol. Chem., 2004, 279, 20327–20338 CrossRef CAS PubMed.
- I. C. Schoenhofen, D. J. McNally, E. Vinogradov, D. Whitfield, N. M. Young, S. Dick, W. W. Wakarchuk, J. R. Brisson and S. M. Logan, J. Biol. Chem., 2006, 281, 723–732 CrossRef CAS PubMed.
- W. S. Song, M. S. Nam, B. Namgung and S. I. Yoon, Biochem. Biophys. Res. Commun., 2015, 458, 843–848 CrossRef CAS PubMed.
- D. J. McNally, J. P. Hui, A. J. Aubry, K. K. Mui, P. Guerry, J. R. Brisson, S. M. Logan and E. C. Soo, J. Biol. Chem., 2006, 281, 18489–18498 CrossRef CAS.
- E. S. Rangarajan, A. Proteau, Q. Cui, S. M. Logan, Z. Potetinova, D. Whitfield, E. O. Purisima, M. Cygler, A. Matte, T. Sulea and I. C. Schoenhofen, J. Biol. Chem., 2009, 284, 20989–21000 CrossRef CAS PubMed.
- W. K. Chou, S. Dick, W. W. Wakarchuk and M. E. Tanner, J. Biol. Chem., 2005, 280, 35922–35928 CrossRef CAS PubMed.
- D. Vorkapic, F. Mitterer, K. Pressler, D. R. Leitner, J. H. Anonsen, L. Liesinger, L. M. Mauerhofer, T. Kuehnast, M. Toeglhofer, A. Schulze, F. G. Zingl, M. F. Feldman, J. Reidl, R. Birner-Gruenberger, M. Koomey and S. Schild, Front. Microbiol., 2019, 10, 2780 CrossRef PubMed.
- Y. Fathy Mohamed, N. E. Scott, A. Molinaro, C. Creuzenet, X. Ortega, G. Lertmemongkolchai, M. M. Tunney, H. Green, A. M. Jones, D. DeShazer, B. J. Currie, L. J. Foster, R. Ingram, C. De Castro and M. A. Valvano, J. Biol. Chem., 2019, 294, 13248–13268 CrossRef.
- J. A. Iwashkiw, A. Seper, B. S. Weber, N. E. Scott, E. Vinogradov, C. Stratilo, B. Reiz, S. J. Cordwell, R. Whittal, S. Schild and M. F. Feldman, PLoS Pathog., 2012, 8, e1002758 CrossRef CAS.
- J. Mahdavi, N. Pirinccioglu, N. J. Oldfield, E. Carlsohn, J. Stoof, A. Aslam, T. Self, S. A. Cawthraw, L. Petrovska, N. Colborne, C. Sihlbom, T. Borén, K. G. Wooldridge and D. A. Ala'Aldeen, Open Biol., 2014, 4, 130202 CrossRef PubMed.
- G. E. Whitworth and B. Imperiali, Glycobiology, 2015, 25, 756–766 CrossRef CAS PubMed.
- M. Nita-Lazar, M. Wacker, B. Schegg, S. Amber and M. Aebi, Glycobiology, 2005, 15, 361–367 CrossRef CAS.
- M. Kowarik, N. M. Young, S. Numao, B. L. Schulz, I. Hug, N. Callewaert, D. C. Mills, D. C. Watson, M. Hernandez, J. F. Kelly, M. Wacker and M. Aebi, EMBO J., 2006, 25, 1957–1966 CrossRef CAS.
- M. Wacker, D. Linton, P. G. Hitchen, M. Nita-Lazar, S. M. Haslam, S. J. North, M. Panico, H. R. Morris, A. Dell, B. W. Wren and M. Aebi, Science, 2002, 298, 1790–1793 CrossRef CAS.
- H. Nothaft, N. E. Scott, E. Vinogradov, X. Liu, R. Hu, B. Beadle, C. Fodor, W. G. Miller, J. Li, S. J. Cordwell and C. M. Szymanski, Mol. Cell. Proteomics, 2012, 11, 1203–1219 CrossRef.
- A. J. Jervis, A. G. Wood, J. A. Cain, J. A. Butler, H. Frost, E. Lord, R. Langdon, S. J. Cordwell, B. W. Wren and D. Linton, Glycobiology, 2018, 28, 233–244 CrossRef CAS.
- J. D. Valderrama-Rincon, A. C. Fisher, J. H. Merritt, Y. Y. Fan, C. A. Reading, K. Chhiba, C. Heiss, P. Azadi, M. Aebi and M. P. DeLisa, Nat. Chem. Biol., 2012, 8, 434–436 CrossRef CAS.
- V. S. Terra, D. C. Mills, L. E. Yates, S. Abouelhadid, J. Cuccui and B. W. Wren, J. Med. Microbiol., 2012, 61, 919–926 CrossRef CAS PubMed.
- H. Nothaft and C. M. Szymanski, Curr. Opin. Chem. Biol., 2019, 53, 16–24 CrossRef CAS.
- J. Kelly, H. Jarrell, L. Millar, L. Tessier, L. M. Fiori, P. C. Lau, B. Allan and C. M. Szymanski, J. Bacteriol., 2006, 188, 2427–2434 CrossRef CAS.
- N. B. Oliver, M. M. Chen, J. R. Behr and B. Imperiali, Biochemistry, 2006, 45, 13659–13669 CrossRef PubMed.
- M. Demendi and C. Creuzenet, Biochem. Cell Biol., 2009, 87, 469–483 CrossRef CAS PubMed.
- M. J. Morrison and B. Imperiali, J. Biol. Chem., 2013, 288, 32248–32260 CrossRef CAS PubMed.
- N. B. Olivier and B. Imperiali, J. Biol. Chem., 2008, 283, 27937–27946 CrossRef CAS PubMed.
- A. S. Riegert, J. B. Thoden, I. C. Schoenhofen, D. C. Watson, N. M. Young, P. A. Tipton and H. M. Holden, Biochemistry, 2017, 56, 6030–6040 CrossRef CAS PubMed.
- A. S. Riegert, N. M. Young, D. C. Watson, J. B. Thoden and H. M. Holden, Protein Sci., 2015, 24, 1609–1616 CrossRef CAS PubMed.
- M. J. Morrison and B. Imperiali, Biochemistry, 2014, 53, 624–638 CrossRef CAS PubMed.
- N. B. Olivier, M. M. Chen, J. R. Behr and B. Imperiali, Biochemistry, 2006, 45, 13659–13669 CrossRef CAS PubMed.
- D. Linton, N. Dorrell, P. G. Hitchen, S. Amber, A. V. Karlyshev, H. R. Morris, A. Dell, M. A. Valvano, M. Aebi and B. W. Wren, Mol. Microbiol., 2005, 55, 1695–1703 CrossRef CAS.
- K. J. Glover, E. Weerapana, M. M. Chen and B. Imperiali, Biochemistry, 2006, 45, 5343–5350 CrossRef CAS PubMed.
- K. J. Glover, E. Weerapana and B. Imperiali, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14255–14259 CrossRef CAS PubMed.
- J. M. Troutman and B. Imperiali, Biochemistry, 2009, 48, 2807–2816 CrossRef CAS PubMed.
- A. S. Ramírez, J. Boilevin, A. R. Mehdipour, G. Hummer, T. Darbre, J. L. Reymond and K. P. Locher, Nat. Commun., 2018, 9, 445 CrossRef PubMed.
- H. Nothaft, X. Liu, D. J. McNally, J. Li and C. M. Szymanski, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15019–15024 CrossRef CAS PubMed.
- C. Perez, S. Gerber, J. Boilevin, M. Bucher, T. Darbre, M. Aebi, J. L. Reymond and K. P. Locher, Nature, 2015, 524, 433–438 CrossRef CAS PubMed.
- C. Perez, A. R. Mehdipour, G. Hummer and K. P. Locher, Structure, 2019, 27, 669–678 CrossRef CAS PubMed.
- C. Lizak, S. Gerber, S. Numao, M. Aebi and K. P. Locher, Nature, 2011, 474, 350–355 CrossRef CAS PubMed.
- M. Napiórkowska, J. Boilevin, T. Darbre, J. L. Reymond and K. P. Locher, Sci. Rep., 2018, 8, 16297 CrossRef PubMed.
- X. Liu, D. J. McNally, H. Nothaft, C. M. Szymanski, J. R. Brisson and J. Li, Anal. Chem., 2006, 78, 6081–6087 CrossRef CAS PubMed.
- H. Nothaft, X. Liu, J. Li and C. M. Szymanski, Virulence, 2010, 1, 546–550 CrossRef PubMed.
- N. E. Scott, N. B. Marzook, J. A. Cain, N. Solis, M. Thaysen-Andersen, S. P. Djordjevic, N. H. Packer, M. R. Larsen and S. J. Cordwell, J. Proteome Res., 2014, 13, 5136–5150 CrossRef CAS.
- R. Dwivedi, H. Nothaft, B. Reiz, R. M. Whittal and C. M. Szymanski, Biopolymers, 2013, 99, 772–783 CrossRef CAS PubMed.
- N. E. Scott, H. Nothaft, A. V. Edwards, M. Labbate, S. P. Djordjevic, M. R. Larsen, C. M. Szymanski and S. J. Cordwell, J. Biol. Chem., 2012, 287, 29384–29396 CrossRef CAS PubMed.
- S. Gerber, C. Lizak, G. Michaud, M. Bucher, T. Darbre, M. Aebi, J. L. Reymond and K. P. Locher, J. Biol. Chem., 2013, 288, 8849–8861 CrossRef CAS PubMed.
- M. Kowarik, S. Numao, M. F. Feldman, B. L. Schulz, N. Callewaert, E. Kiermaier, I. Catrein and M. Aebi, Science, 2006, 314, 1148–1150 CrossRef CAS.
- C. Lizak, S. Gerber, G. Michaud, M. Schubert, Y. Y. Fan, M. Bucher, T. Darbre, M. Aebi, J. L. Reymond and K. P. Locher, Nat. Commun., 2013, 4, 2627 CrossRef PubMed.
- C. Lizak, S. Gerber, D. Zinne, G. Michaud, M. Schubert, F. Chen, M. Bucher, T. Darbre, R. Zenobi, J. L. Reymond and M. Aebi, J. Biol. Chem., 2014, 289, 735–746 CrossRef CAS.
- Y. Barre, H. Nothaft, C. Thomas, X. Liu, J. Li, K. K. S. Ng and C. M. Szymanski, Glycobiology, 2017, 27, 978–989 CrossRef CAS.
- F. Schwarz, C. Lizak, Y. Y. Fan, S. Fleurkens, M. Kowarik and M. Aebi, Glycobiology, 2011, 21, 45–54 CrossRef CAS.
- Y. Harada, R. Buser, E. M. Ngwa, H. Hirayama, M. Aebi and T. Suzuki, J. Biol. Chem., 2013, 288, 32673–32684 CrossRef CAS.
- J. J. A. Armenteros, K. D. Tsirigos, C. K. Sønderby, T. N. Petersen, O. Winther, S. Brunak, G. von Heijne and H. Nielsen, Nat. Biotechnol., 2019, 37, 420–423 CrossRef.
- J. M. Silverman and B. Imperiali, J. Biol. Chem., 2016, 291, 22001–22010 CrossRef CAS.
- N. E. Scott, D. R. Bogema, A. M. Connolly, L. Falconer, S. P. Djordjevic and S. J. Cordwell, J. Proteome Res., 2009, 8, 4654–4664 CrossRef CAS PubMed.
- A. Wyszyńska, K. Tomczyk and E. K. Jagusztyn-Krynicka, Acta Biochim. Pol., 2007, 54, 143–150 CrossRef.
- A. Wyszyńska, J. Zycka, R. Godlewska and E. K. Jagusztyn-Krynicka, Curr. Microbiol., 2008, 57, 181–188 CrossRef.
- M. M. Chen, K. J. Glover and B. Imperiali, Biochemistry, 2007, 46, 5579–5585 CrossRef CAS.
- J. A. Gawthorne, N. Y. Tan, U. M. Bailey, M. R. Davis, L. W. Wong, R. Naidu, K. L. Fox, M. P. Jennings and B. L. Schulz, Biochem. Biophys. Res. Commun., 2014, 445, 633–638 CrossRef CAS.
- N. Y. Yu, J. R. Wagner, M. R. Laird, G. Melli, S. Rey, R. Lo, P. Dao, S. C. Sahinalp, M. Ester, L. J. Foster and F. S. L. Brinkman, Bioinformatics, 2010, 26, 1608–1615 CrossRef CAS PubMed.
- M. Wacker, M. F. Feldman, N. Callewaert, M. Kowarik, B. R. Clarke, N. L. Pohl, M. Hernandez, E. D. Vines, M. A. Valvano, C. Whitfield and M. Aebi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7088–7093 CrossRef CAS PubMed.
- E. S. Rangarajan, S. Bhatia, D. C. Watson, C. Munger, M. Cygler, A. Matte and N. M. Young, Protein Sci., 2007, 16, 990–995 CrossRef CAS PubMed.
- C. C. Su, A. Radhakrishnan, N. Kumar, F. Long, J. R. Bolla, H. T. Lei, J. A. Delmar, S. V. Do, T. H. Chou, K. R. Rajashankar, Q. Zhang and E. W. Yu, Protein Sci., 2014, 23, 954–961 CrossRef CAS.
- C. C. Su, L. Yin, N. Kumar, L. Dai, A. Radhakrishnan, J. R. Bolla, H. T. Lei, T. H. Chou, J. A. Delmar, K. R. Rajashankar, Q. Zhang, Y. K. Shin and E. W. Yu, Nat. Commun., 2017, 8, 171 CrossRef PubMed.
- F. Kawai, S. Paek, K. J. Choi, M. Prouty, M. I. Kanipes, P. Guerry and H. J. Yeo, J. Struct. Biol., 2012, 177, 583–588 CrossRef CAS PubMed.
- V. Slynko, M. Schubert, S. Numao, M. Kowarik, M. Aebi and F. H. Allain, J. Am. Chem. Soc., 2009, 131, 1274–1281 CrossRef CAS PubMed.
- C. Y. Zamora, E. M. Ward, J. C. Kester, W. L. K. Chen, J. G. Velazquez, L. G. Griffith and B. Imperiali, Glycobiology, 2020 DOI:10.1093/glycob/cwz105.
- C. M. Szymanski, D. H. Burr and P. Guerry, Infect. Immun., 2002, 70, 2242–2244 CrossRef CAS PubMed.
- A. V. Karlyshev, P. Everest, D. Linton, S. Cawthraw, D. G. Newell and B. W. Wren, Microbiology, 2004, 150, 1957–1964 CrossRef CAS PubMed.
- J. A. Cain, A. L. Dale, P. Niewold, W. P. Klare, L. Man, M. Y. White, N. E. Scott and S. J. Cordwell, Mol. Cell. Proteomics, 2019, 18, 715–734 CrossRef CAS PubMed.
- S. Abouelhadid, S. J. North, P. Hitchen, P. Vohra, C. Chintoan-Uta, M. Stevens, A. Dell, J. Cuccui and B. W. Wren, mBio, 2019, 10, e00297 CrossRef CAS PubMed.
- B. Pascoe, G. Méric, S. Murray, K. Yahara, L. Mageiros, R. Bowen, N. H. Jones, R. E. Jeeves, H. M. Lappin-Scott, H. Asakura and S. K. Sheppard, Environ. Microbiol., 2015, 17, 4779–4789 CrossRef CAS PubMed.
- C. S. Vegge, L. Brøndsted, M. Ligowska-Marzęta and H. Ingmer, PLoS One, 2012, 7, e45467 CrossRef CAS PubMed.
- A. Alemka, H. Nothaft, J. Zheng and C. M. Szymanski, Infect. Immun., 2013, 81, 1674–1682 CrossRef CAS PubMed.
- N. M. van Sorge, N. M. Bleumink, S. J. van Vliet, E. Saeland, W. L. van der Pol, Y. van Kooyk and J. P. van Putten, Cell. Microbiol., 2009, 11, 1768–1781 CrossRef CAS PubMed.
- T. Kakuda and V. J. DiRita, Infect. Immun., 2006, 74, 4715–4723 CrossRef CAS PubMed.
- J. C. Larsen, C. Szymanski and P. Guerry, J. Bacteriol., 2004, 186, 6508–6514 CrossRef CAS PubMed.
- L. M. Davis, T. Kakuda and V. J. DiRita, J. Bacteriol., 2009, 191, 1631–1640 CrossRef CAS PubMed.
- R. K. Dubb, H. Nothaft, B. Beadle, M. R. Richards and C. M. Szymanski, Glycobiology, 2020, 30, 105–119 CrossRef PubMed.
- B. Jeon and Q. Zhang, J. Bacteriol., 2007, 189, 7399–7407 CrossRef CAS PubMed.
- S. Jin, A. Joe, J. Lynett, E. K. Hani, P. Sherman and V. L. Chan, Mol. Microbiol., 2001, 39, 1225–1236 CrossRef CAS PubMed.
- E. Frirdich, J. Biboy, C. Adams, J. Lee, J. Ellermeier, L. D. Gielda, V. J. DiRita, S. E. Girardin, W. Vollmer and E. C. Gaynor, PLoS Pathog., 2012, 8, e1002602 CrossRef CAS PubMed.
- E. Frirdich, J. Vermeulen, J. Biboy, F. Soares, M. E. Taveirne, J. G. Johnson, V. J. DiRita, S. E. Girardin, W. Vollmer and E. C. Gaynor, J. Biol. Chem., 2014, 289, 8007–8018 CrossRef CAS PubMed.
- A. V. Karlyshev, G. Thacker, M. A. Jones, M. O. Clements and B. W. Wren, FEBS Open Bio, 2014, 4, 468–472 CrossRef CAS PubMed.
- V. Novik, D. Hofreuter and J. E. Galán, Infect. Immun., 2008, 78, 3540–3553 CrossRef.
- A. M. Lasica, A. Wyszyńska, K. Szymanek, P. Majewski and E. K. Jagusztyn-Krynicka, J. Appl. Genet., 2010, 51, 383–393 CrossRef CAS.
- A. M. Tareen, C. G. Lüder, A. E. Zautner, U. Groß, M. M. Heimesaat, S. Bereswill and R. Lugert, PLoS One, 2013, 9, e107076 Search PubMed.
- X. Du, N. Wang, F. Ren, H. Tang, X. Jiao and J. Huang, Front. Microbiol., 2016, 7, 1094 Search PubMed.
- T. W. Cullen and M. S. Trent, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5160–5165 CrossRef CAS PubMed.
- X. Zeng, B. Gillespie and J. Lin, Front. Microbiol., 2015, 6, 1292 Search PubMed.
- T. J. Mansell, C. Guarino and M. P. DeLisa, Biotechnol. J., 2013, 8, 1445–1451 CrossRef CAS.
- T. Min, M. Vedadi, D. C. Watson, G. A. Wasney, C. Munger, M. Cygler, A. Matte and N. M. Young, Biochemistry, 2009, 48, 3057–3067 CrossRef CAS.
- O. Rahman, S. P. Cummings, D. J. Harrington and I. C. Sutcliffe, World J. Microbiol. Biotechnol., 2008, 24, 2377–2382 CrossRef CAS.
- K. D. Tsirigos, C. Peters, N. Shu, L. Käll and A. Elofsson, Nucleic Acids Res., 2015, 43, W401–W407 CrossRef CAS.
- W. Ding, H. Nothaft, C. M. Szymanski and J. Kelly, Mol. Cell. Proteomics, 2009, 8, 2170–2185 CrossRef CAS.
- U. Omasits, C. H. Ahrens, S. Müller and B. Wollscheid, Bioinformatics, 2014, 30, 884–886 CrossRef CAS PubMed.
Footnote |
| † Current address: Centre for Blood Research, Department of Oral Biological and Medical Sciences, University of British Columbia, Vancouver, Canada. |
| This journal is © The Royal Society of Chemistry 2020 |