
Serial in-solution digestion protocol for mass spectrometry-based glycomics and proteomics analysis†
Manveen K.
Sethi
 ,
Margaret
Downs
and
Joseph
Zaia
*
,
Margaret
Downs
and
Joseph
Zaia
*
Boston University School of Medicine, Boston University, Department of Biochemistry, Boston, 02118, USA. E-mail: jzaia@bu.edu
First published on 6th April 2020
Abstract
Advancement in mass spectrometry has revolutionized the field of proteomics. However, there remains a gap in the analysis of protein post-translational modifications (PTMs), particularly for glycosylation. Glycosylation, the most common form of PTM, is involved in most biological processes; thus, analysis of glycans along with proteins is crucial to answering important biologically relevant questions. Of particular interest is the brain extracellular matrix (ECM), which has been called the “final Frontier” in neuroscience, which consists of highly glycosylated proteins. Among these, proteoglycans (PGs) contain large glycan structures called glycosaminoglycans (GAGs) that form crucial ECM components, including perineuronal nets (PNNs), shown to be altered in neuropsychiatric diseases. Thus, there is a growing need for high-throughput methods that combine GAG (glycomics) and PGs (proteomics) analysis to unravel the complete biological picture. The protocol presented here integrates glycomics and proteomics to analyze multiple classes of biomolecules. We use a filter-aided sample preparation (FASP) type serial in-solution digestion of GAG classes, including hyaluronan (HA), chondroitin sulfate (CS), and heparan sulfate (HS), followed by peptides. The GAGs and peptides are then cleaned and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). This protocol is an efficient and economical way of processing tissue or cell lysates to isolate various GAG classes and peptides from the same sample. The method is more efficient (single-pot) than available parallel (multi-pot) release methods, and removal of GAGs facilitates the identification of the proteins with higher peptide-coverage than using conventional-proteomics. Overall, we demonstrate a high-throughput & efficient protocol for mass spectrometry-based glycomic and proteomic analysis (data are available via ProteomeXchange with identifier PXD017513).
1. Introduction
Over the years, a number of methods for integrated mass spectrometry-based glycomics and proteomics have been introduced.1,2 With the advent of new experimental techniques, methods, technologies, and instrumentation, the integrated effort to analyze glycans (glycomics), and glycoproteins (glycoproteomics) along with proteins (proteomics) has gained momentum.2–7 Still, insufficient attention has been paid to glycosaminoglycans (GAGs) and proteoglycans (PGs), which are multi-anionic and highly sulfated and, thus, immensely challenging to study by liquid chromatography-tandem-mass spectrometry (LC-MS/MS). Examples of analytical limitations include low-PG-sensitivity on LC-systems, low PG-peptide-sequence coverage, and significant sulfate losses in tandem-MS.8 Thus, PG profiling remains immature relative to other ‘omics (genomics/proteomics/N-or O-glycomics) approaches,9 impeding advances in our understanding of the structural and biological roles of GAGs and PGs in various biosystems. We developed a mass spectrometry-compatible method for the extraction of GAGs from wet tissue.10 In order to combine glycomics and proteomics, we next demonstrated serial extraction and analysis of multiple classes of GAGs and peptides from tissue slides,11,12 a method that allows precise targeting of regions in tissue slides. We now describe a new streamlined, filter aided sample preparation-based method optimized for the recovery of GAG saccharides and peptides from biological samples that must be analyzed as a wet tissue.Proteoglycans (PGs) are glycoproteins containing GAG structures that are linear polysaccharides constituting of sulfated disaccharide repeating units. Because the biosynthetic reactions do not go to completion, the GAG chains are heterogeneous concerning chain length and modifications. GAGs can be either protein-bound or present in the free form.13 Thus, they may play essential roles in all areas of physiology through their biophysical properties and capacities to bind growth factors and growth factor receptors. There is a growing interest in documenting the roles of GAGs and PGs and their binding partners in neurological disease mechanisms.14 Based on differences in the type of monosaccharide units and their modification, GAGs can be divided into categories based on the repeated disaccharide unit, i.e., heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS) and hyaluronic acid (or hyaluronan) (HA). HS consists of repeating disaccharide units of uronic acid (glucuronic or iduronic acid) with N-acetylglucosamine; CS/DS consists of N-acetylgalactosamine with glucuronic or iduronic acid; HA is unsulfated and present in free form without any core protein; KS consists of repeating units of N-acetylglucosamine with galactose.13,15Fig. 1 shows structures for HA, CS/DS, HS, and KS.
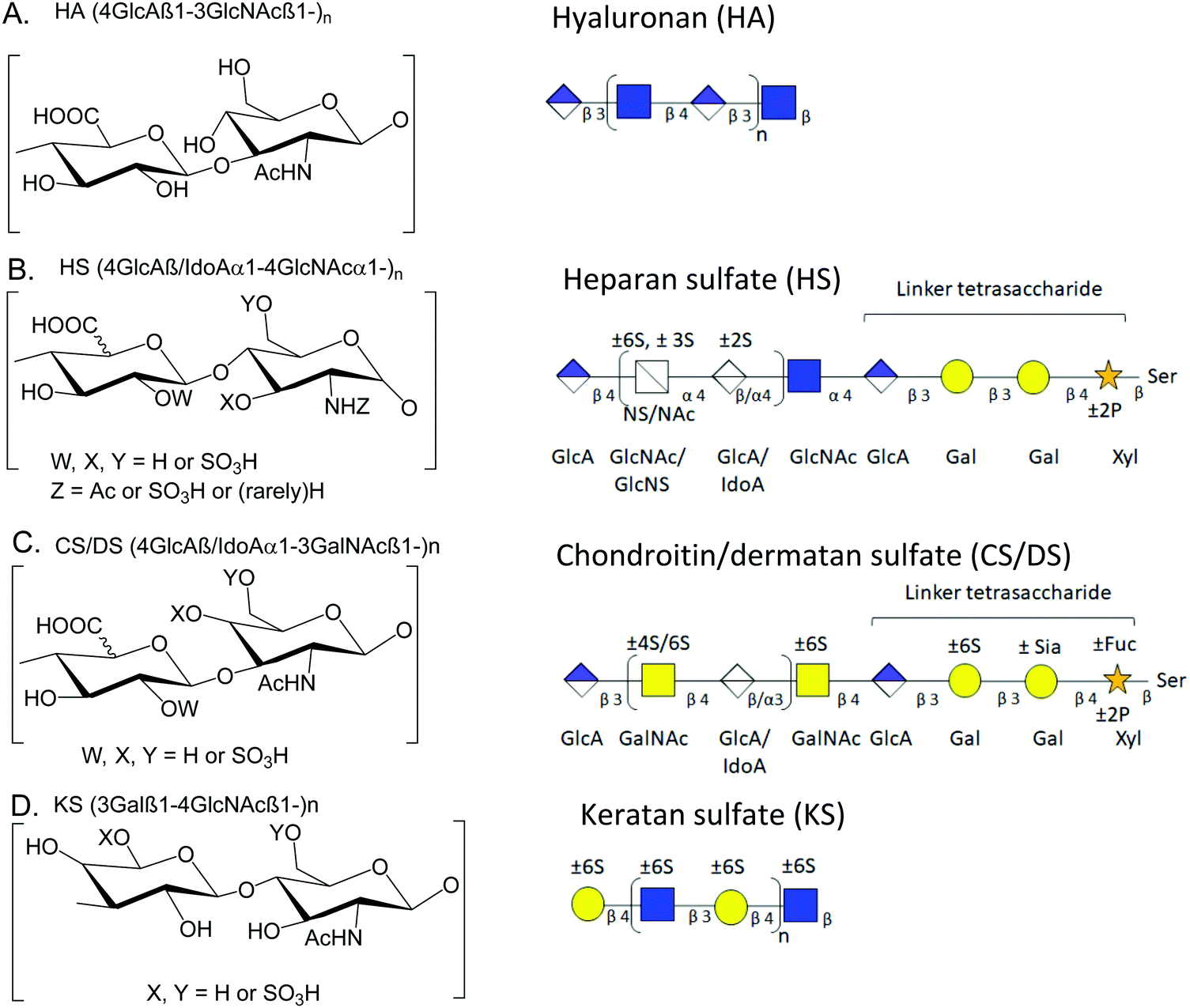 | ||
| Fig. 1 Structure of key GAGs. (A) Hyaluronic acid (HA) disaccharide units, HA chains. (B) Heparan sulfate (HS) disaccharide unit, HS chains, and linker tetrasaccharide structure. (C) Chondroitin, and dermatan sulfate (CS/DS) disaccharide unit, CS/DS chains, and linker tetrasaccharide structure. (D) Keratan sulfate (KS) disaccharide units, KS chains. Abbreviations: Fuc, fucose; GlcA, glucuronic acid; IdoA, iduronic acid; GalNAc, N-acetylgalactosamine; GlcNAc N-acetylglucosamine; Sia, sialic acid; Xyl, xylose; Ser, serine; Ac, acetyl; S, sulfate (SO3H). | ||
The extracellular matrix (ECM) consists of a network of glycoproteins, PGs, collagens, and HA. These molecules form an organized network that acts as a scaffold, mediates cell-to-cell communication, binds secreted proteins such as growth factors, and regulates the activity of protein complexes. Many matrix proteins have lectin domains that recognize glycan epitopes, forming robust molecular networks. The ECM also contains enzymes that modify glycosylation patterns on proteins,16 but the exact glycan makeup of mature matrix glycoproteins and PGs, and their spatial and temporal variations, remain ill-defined. Antibody-based and staining studies have shown spatial and temporal regulation of glycosylation in various organs17,18 that indicate the presence or absence of a particular glycan epitope but do not define explicitly the underlying structure. Established mass spectrometry-based proteomics methods suffice for qualitative assignment of glycosites but do not provide complete glycosylation coverage.19
We seek to establish a streamlined protocol to analyze PG GAG chains, N-glycans, and core proteins in the brain or any other mammalian tissue. Exhaustive digestion of tissue using GAG lyase enzymes releases disaccharides that can be quantified using LC-MS. The abundances of the disaccharides released by specific lyase enzymes can be used to estimate the domain structure of the GAG chains.20,21 Compared to our on-slide digestion protocol that provides a selection of target area on a tissue slides11,12,22,23 and MALDI-imaging method for glycans that provides higher spatial resolution,24–26 this method can be applied to free-floating or frozen tissues and provides a higher recovery of glycans and proteins than the on-slide method. Moreover, widely used MALDI-imaging dissociates fragile glycan substituent's including sulfates, and is not recommended for analysis of GAGs.27 The serial in-solution digestion platform suggested here is more rapid (less sample-processing time), and efficient (single-pot)28,29 than the currently used parallel approach, i.e., a multi-pot simultaneous enzymes application methods.10,28–30 The removal of GAGs facilitates protein identification of the remaining deglycosylated PGs with higher peptide-coverage,31 compared to current studies achieving only low PG-coverage.32,33 In addition, the removal of GAGs creates a linker-glycosite that leaves a linker tetrasaccharide plus one disaccharide to the protein/peptide, assisting with the identification of site-specific glycosylation of PGs.31,34Table 1 provides a comparison of our method, including starting material, sample processing time, derivatisation/enrichment steps, loading amounts, and detection range for proteins, GAGs, and N-glycans, with other tissue digestion protocols available for integrated (or individual) glycomics and proteomics.
| Method | Starting material | Sample processing time | Total number of samples processed at a given time | Derivatisation/fractionation/enrichment prior to LC-MS/MS | Injection volume | Detection range (proteins, GAGs, N-glycans) | Ref. |
|---|---|---|---|---|---|---|---|
| In solution digestion of GAGS and protein | 20–100 μg of standard protein or mouse brain lysate | Total time = 5 days, 1 day per biomolecule class | 24 | No | 100 ng of protein, 30% of purified resuspended GAGs | 1000–1500 proteins, all unsaturated and saturated GAG disaccharides | Presented manuscript |
| On-slide tissue digestion | 1.8 mm area on slide corresponding to 10 nL tissue volume | Total time = 5 days, 1 day per bio-molecule class | 36 | No | 10% of resuspended peptide, and N-glycans, and 30% resuspended GAGs | 1000–1200 proteins, all unsaturated GAG disaccharide, saturated GAGs are rarely observed | 12 and 46 |
| In-solution tissue digestion of protein using FASP | 100 μg of mouse brain tissue lysate | 2–3 days only for peptide/protein | Not stated | Yes | 5 μg of peptides | 1264, 2957 proteins from FASP and MED FASP, respectively, of brain tissue lysate | 47 |
| Tissue glyco capture for N-glycans | Whole mouse brain tissue | 2 days | Not stated | Yes | Aliquot representing 40 ug brain tissue | 200 N-glycans, 90 distinct N-glycan compositions | 48 |
| MALDI-IMS | FFPE tissue microarray (TMA) sections (15 × 15 mm, 4–8 μm thick), 100 μm resolution | 1–2 days per bio-molecule class | Using a 0.6 mm core allows arraying at higher density more than 500 tissue core can be arranged on a standard microscopic slide | Yes | Not stated | 100 proteins, 25–50 N-glycan | 49 |
| Filter Aided N-glycan separation (FANGS) | 25–250 μg protein | 2–3 days for derivatized N-glycans | Not stated | Yes | 400 ng of protein for proteomics, 5 μL of resuspended (100%) of N-glycan samples | 200–300 proteins, 100–150 glycoproteins, 25 N-glycans. | 50 |
Our protocol follows a filter-aided sample preparation (FASP) type serial in-solution digestion of glycosaminoglycan (GAG) classes, including hyaluronan (HA), chondroitin sulfate (CS), and heparan sulfate (HS) followed by peptides from tissue or cell lysates (Fig. 2). Filter-aided sample preparation (FASP) first described by the Mann group in 200935 that used molecular weight cut-off (MWCO) membrane as a ‘reactor’ on which complex protein mixtures were chemically modified and digested. They utilized urea to remove SDS completely, and then generated tryptic peptides from complex protein mixtures for mass spectral analysis. This method has been modified since and has also been applied for the enrichment of N-glycoproteins prior to enzymatic deglycosylation and proteomics analysis.36–38 We seek to exploit the concept of using MWCO membrane filters as a reactor to digest each GAG classes and collect it as flowthrough, and finally collect proteins and perform trypsin digestion. The abundances of the extracted GAG disaccharides and peptides are determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantifying 1 HA disaccharide, the 6 most abundant CS unsaturated disaccharides (Fig. 3; representative EICs for 6 CS disaccharide standards) and 3 saturated CS disaccharides (see Fig. S1A, ESI† for EIC s of CS saturated disaccharide structures), the 8 most abundant HS unsaturated disaccharides (Fig. 4; representative EICs for 8 HS disaccharide standards) and 3 saturated HS disaccharides (see Fig. S1B, ESI† for EICs of HS saturated disaccharide structures, and approximately 1000–1500 proteins (Fig. 5; TIC for mouse brain tissue lysate peptides). In the protocol presented here, mouse brain lysate, and various standard proteoglycans and proteins, for example, syndecan, aggrecan, neurocan, and AGP are used at different steps of the protocol to target different biomolecules (Table 2). Each enzymatic digestion was incubated overnight with enzymes applied directly on the MWCO filters, and each biomolecule takes one day of sample preparation/processing time.
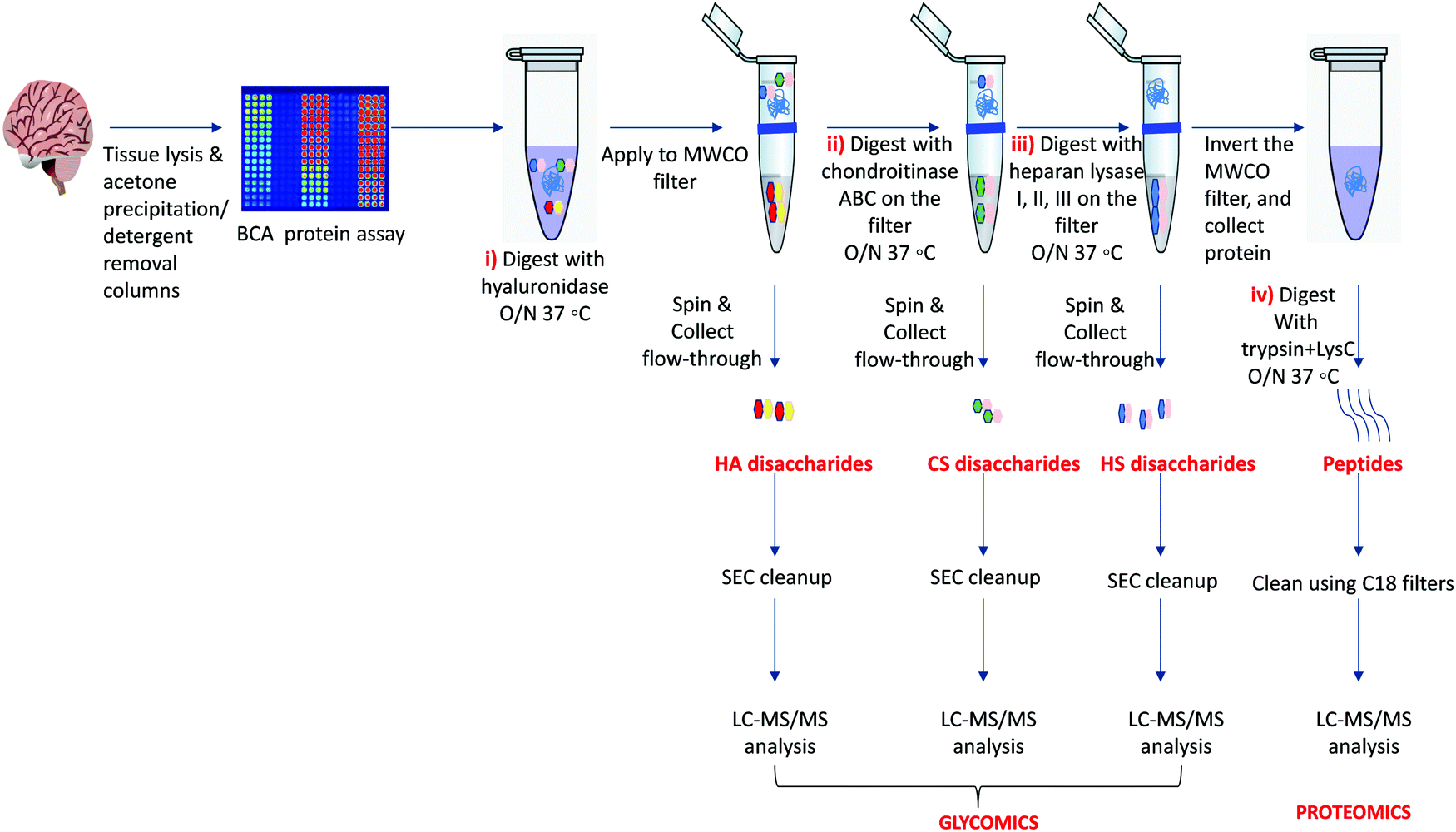 | ||
| Fig. 2 Workflow for serial in-solution digestion protocol to extract (i) hyaluronic acid (HA) disaccharides, (ii) chondroitin sulfate (CS disaccharides), (iii) heparan sulfate (HS disaccharides), and (iv) peptides that are desalted and subjected to LC-MS/MS. | ||
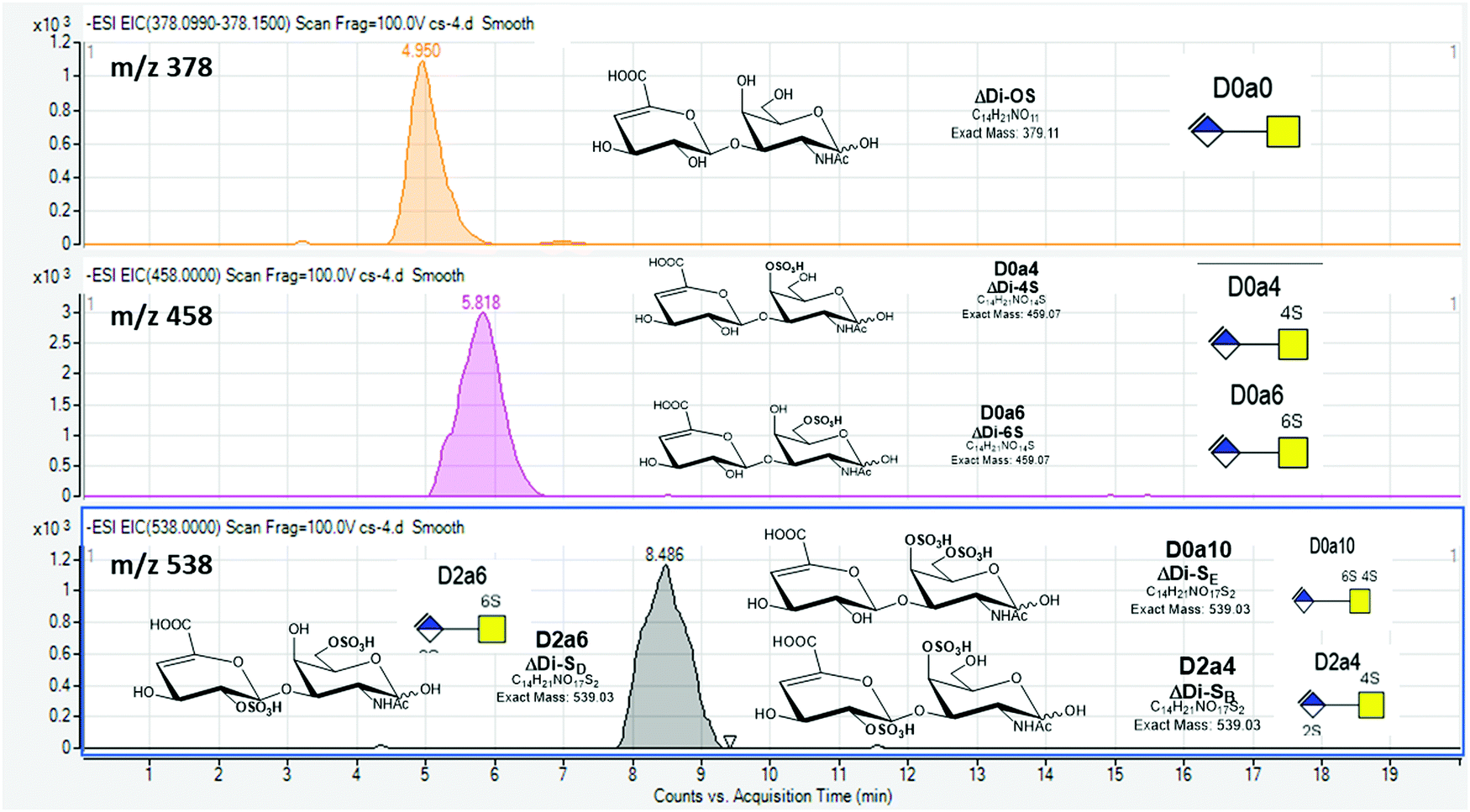 | ||
Fig. 3 Extracted ion chromatograms (EICs) for chondroitin sulfate (CS) disaccharide standards using GlycanPac AXH-1 (ThermoScientific) column mounted on an Agilent 1200 LC attached to an Agilent 6520 Q-TOF.  ; Δ-uronic acid, ; Δ-uronic acid,  ; N-acetyl galactosamine. ; N-acetyl galactosamine. | ||
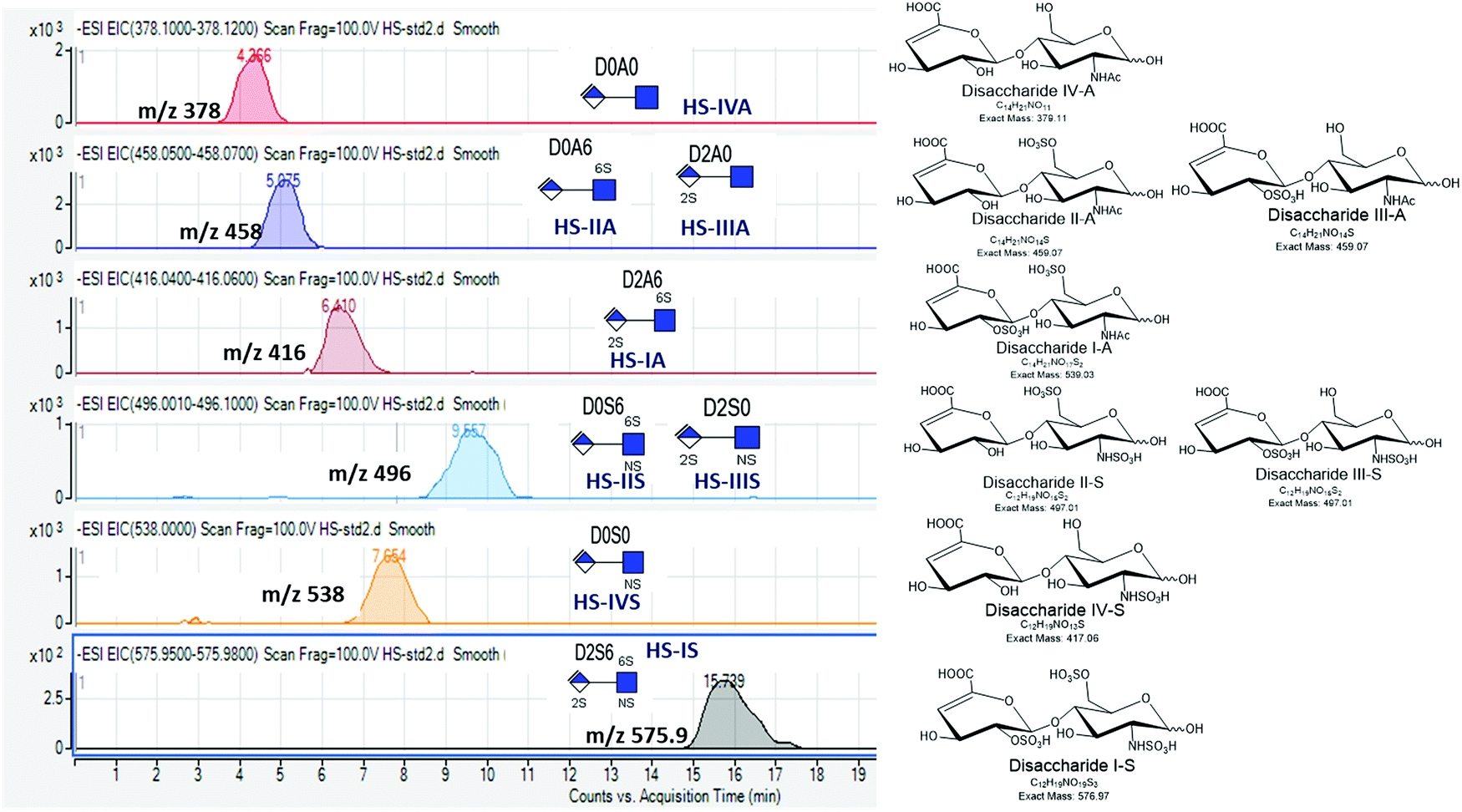 | ||
Fig. 4 Extracted ion chromatograms (EICs) for heparan sulfate (HS) disaccharide standards using GlycanPac AXH-1 (ThermoScientific) column mounted on an Agilent 1200 LC attached to an Agilent 6520 Q-TOF.  ; Δ-uronic acid, ; Δ-uronic acid,  ; N-acetyl glucosamine. ; N-acetyl glucosamine. | ||
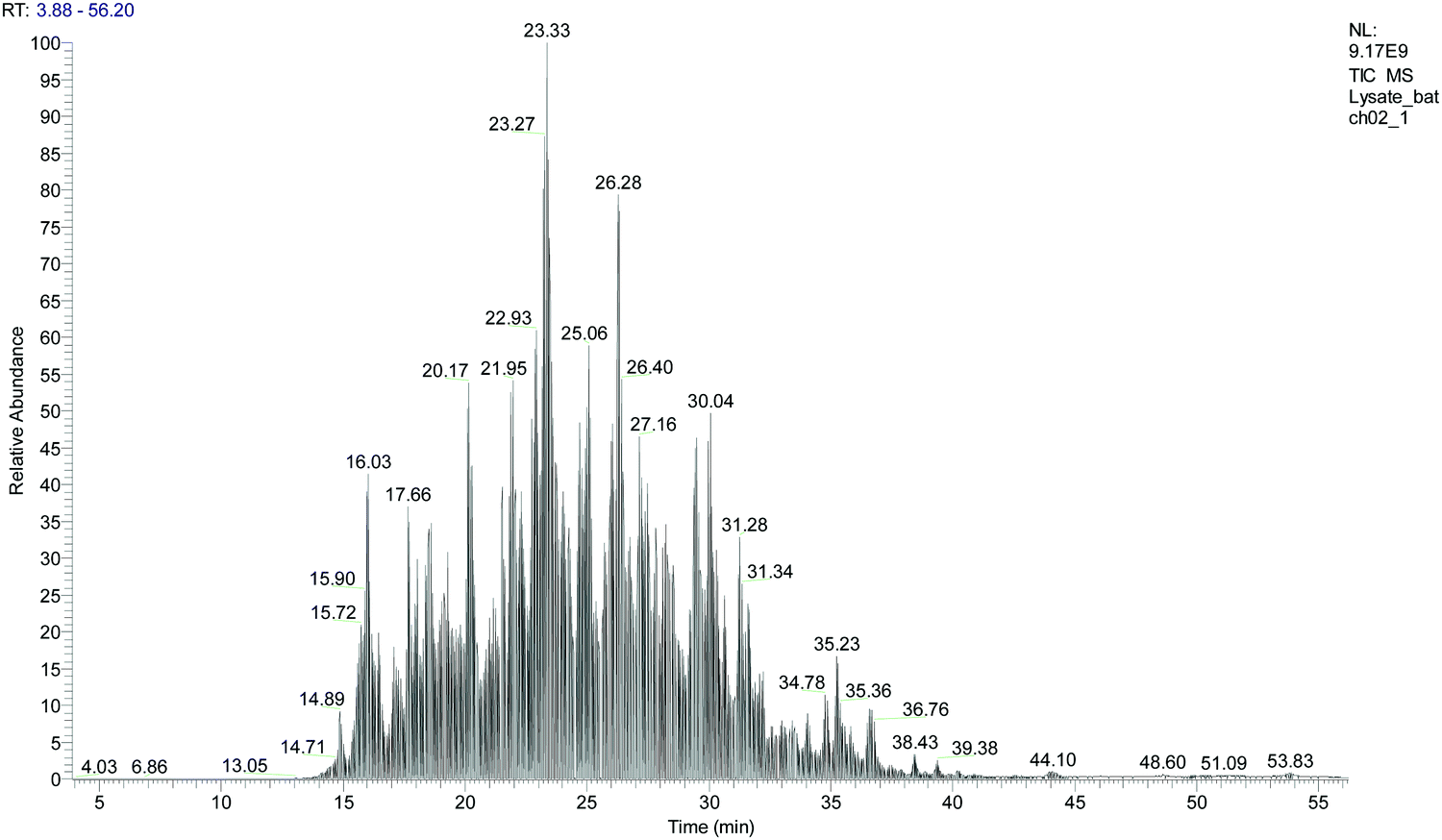 | ||
| Fig. 5 Total ion chromatogram (TIC) for mouse brain tissue lysate after serial in-solution digestion protocol. | ||
| Batch 1 | Batch 2 | Targeted biomolecules | Steps |
|---|---|---|---|
| Lysate (100 μg × 1) | Lysate (100 μg × 1) | HA, CS, HS, peptides | i, ii, iii, iv |
| Hyaluronan (100 μg × 1) | Hyaluronan (100 μg × 1) | HA | i |
| Syndecan-1 (20 μg × 1) | Syndecan-2 (20 μg × 1) | CS, HS (only for batch1), peptides | ii, iii, iv |
| Aggrecan (20 μg × 1) | Syndecan-1 (20 μg × 1) | CS, peptides | ii, iv |
| Neurocan (20 μg x 1) | — | CS, peptides | ii, iv |
| HSBK (100 μg × 1) | HSBK (100 μg × 1) | HS | iii |
| HSPIM (100 μg × 1) | HSPIM (100 μg × 1) | HS | iii |
| AGP (30 μg × 1) | AGP (30 μg × 1) | Peptides | iv |
2. Materials
2.1 Reagents
• Tris–HCl, pH 7.5 (BP1757-100, Fisher BioReagents)• Tris–HCl, pH 8.0 (BP1758-100, Fisher BioReagents)
• Triton X-100 (AC21568-2500, Acros Organics)
• Sodium dodecyl sulfate (L3771, Sigma Aldrich)
• EDTA (V4231, Promega)
• Sodium chloride
• Pierce proteinase inhibitor mini-tablets (PIA32955, Thermo-Fisher Scientific)
• Pierce micro BCA protein assay kit (PI23235, Thermo-Fisher Scientific)
• Ammonium acetate (631-61-8, Fisher Chemicals),
• Ammonium bicarbonate (A6141, Sigma Aldrich)
• Calcium chloride (C5080, Sigma Aldrich),
• Dithiothreitol (D0632, Sigma),
• Iodoacetamide (163-2109, Biorad),
• Trifluoro-acetic acid (A116-10X AMP, Fisher Scientific)
• Acetonitrile (A955-1, LC-MS grade, Fisher Scientific),
• Water (W6-1, LC-MS grade, Fisher Scientific),
• Formic acid (A117-10x-1AMP, LC-MS grade, Fisher Scientific)
• Hyaluronidase (H1136-1AMP, Sigma)
• Chondroitinase ABC (Sigma)
• Heparan lyases 1,2,3 (P0735L, P0736L, P0737L New England Biolabs)
• Trypsin (PRV5280, Promega)/trypsin–LysC (PRV5072, Promega)/Glu-C (PRV1651, Promega)
• High molecular weight hyaluronan (GLR002. R&D systems)
• Mouse brain tissue lysate (89-014-501, Abnova)
• Human neurocan recombinant protein (6508NC050, R&D systems)
• Aggrecan from bovine articular cartilage (A1960-1MG, SigmaAldrich)
• Human Syndecan-1 Protein (#7879, BioVision Incorporated)
• α1-Acid glycoprotein from human plasma (#G9885, SigmaAldrich)
• Pierce retention time calibration mixture (#88321, ThermoScientific)
• Pierce™ HeLa Protein Digest Standard (#88328, ThermoScientific)
• Heparan sulfate from porcine intestinal mucosa (HSPIM) (Celsus Laboratories, Inc.
• Heparan sulfate from bovine kidney (HSBK) (SigmaAldrich)
2.2 Equipment
• Sonic dismembrator Model 100, FisherScientific• Vortex Bouxter S1P vortex MIXER, #S8223-1
• Benchtop Centrifuge, VWR Galaxy mini
• Pierce C18 spin columns (89870, Thermo-Fisher Scientific)
• Microcon™ Centrifugal Filters for Protein and DNA Concentration: Ultracel-10 Membrane (MRCPRT010, MilliporeSigma)
• Pierce Detergent Removal Spin Columns (Thermo-Fisher Scientific, 87777)
• Incubator control for 37 °C
• 22 R microfuge centrifuge, Beckmann Coulter
• Centrifuge, Thermo IEC Micromax
• Eppendorf thermomixer (1.5 mL, 37 °C, 60 °C)
• Vacuum centrifugation (SPD1010 Speedvac system, Thermo Savant)
• LC Systems – Agilent 1200 LC (Agilent Technologies), nanoAcquity UPLC (Waters Technology)
• Mass Spectrometry systems – Agilent 6520 Q-TOF (Agilent Technologies), Q-Exactive HF mass spectrometer (Thermo-Fisher Scientific).
• Columns
• Superdex peptide (3.2/300 column, GE Healthcare Life Sciences)
• Reversed phased C-18 analytical (BEH C18, 150 μm × 100 mm) and trapping (180 μm × 20 mm), Waters technology
• GlycanPac AXH-1 (ThermoScientific) column (1.9 μm, 0.3 × 150 mm)
2.3 Software
• GlycReSoft software (http://www.bumc.bu.edu/msr/glycresoft)• Peaks Studio 8.5 or other proteomics database searching software
• Qualitative analysis software (version B.06; Agilent Technologies).
3. Experimental design
3.1 Tissue lysis
Any free-floating or fresh frozen mammalian tissues can be used to perform the protocol. Once the tissue lysis is performed, the protein estimation of the lysis is required to use an equal amount of protein for different samples for further digestion steps.3.2 Serial in-solution digestion
GAGs are released in-solution sequentially from equal amounts of tissue lysates using specific enzymes that cleave each GAG disaccharide, including hyaluronidase for HA disaccharides, chondroitinase ABC enzyme for CS disaccharides, and heparin lyase I, II, and III for HS disaccharides. After GAG removal, the peptides are released from the same sample using trypsin or trypsin + LysC. The removal of GAGs facilitates the identification of the proteins with higher peptide-coverage using conventional proteomics.16 A FASP-type protocol is utilized to apply tissue lysate to MWCO (10 kDa, 0.5 mL), followed by serial application of specific enzymes to the sample on the filter, followed by overnight incubation (37 °C) and release of each GAG class (HA, CS, and HS) as a flow-through, followed by digestion of retained protein to release peptides.3.3 Desalting for GAGs
There exist several orthogonal techniques for desalting of glycans including, size exclusion chromatography, strong anion exchange chromatography, and porous graphite carbon cleanup. We find size exclusion chromatography (SEC) as the most unbiased, efficient, and reproducible approach toward glycan desalting. The GAGs are cleaned using the SEC chromatography column (Superdex™ peptide PC 3.2/30, GE Healthcare), and they elute between 35–45 min at a UV absorbance of 232 nm.3.4 C-18 cleanup peptides
The peptides are cleaned using ThermoScientific C-18 spin columns.3.5 Data acquisition
A robust data acquisition workflow is equally important as a streamlined sample processing workflow. We analyze our GAG disaccharides on a negative ionization mode electrospray liquid chromatography (LC)-mass spectrometry (MS) system. The LC-MS conditions are optimized for higher recovery of GAGs from LC, and appropriate tuning of MS source conditions to minimize in-source sulfate losses to identify highly sulfated disaccharide. Disaccharides are separated using a 1.9 μm, 0.3 × 150 mm GlycanPac AXH-1 (ThermoScientific) column mounted on an Agilent 1200 LC (Agilent Technologies, Santa Clara, CA). A 20 min isocratic method is used and a flow rate of 7 μL min−1 (HS) and 5 μL min−1 (HA and CS). Mass spectrometry analyses for GAGs is performed using an Agilent 6520 Q-TOF (Agilent Technologies, Santa Clara, CA) using electrospray ionization. A synthetic internal standard (800 fmol quantity of ΔHexA2S–GlcNCoEt(6S) (Hd009, Iduron)) is added to all the samples before LC-MS analyses (Fig. S2, ESI†). We run a mixture of commercially available CS (Fig. 3) and HS (Fig. 4) standards disaccharides (Iduron) every day to ensure stable instrument performance. The criteria used in our laboratory for a satisfactory MS analysis of GAG disaccharides include a ≤20% coefficient of variation (CV), and ≤30% CV for CS and HS disaccharides, respectively. This is based on the observed CV values +5% that are observed in practice in our laboratory over different GAG experiments. A ≤10% variation in retention time is acceptable.For proteomics, the instrument is calibrated every week in positive ion mode using positive mode calibration mixture (ThermoScientific), producing an intensity signal of 4–5 × 108, and ≥15% of MS/MS to MS signal for m/z 524.3 under high-performance instrument conditions. A loading amount of 100 fmol of Pierce retention time calibration mixture (ThermoScientific) (Fig. S3, ESI†) and 50 ng of Pierce HeLa cell lysate (ThermoScientific) (Fig. S4, ESI†) are run day-to-day for instrument performance generating a detection signal of 1.5–2 × 109, and a 2.5–3.5 × 109 under high-performance instrument conditions. A retention time variation of ≤10% is acceptable among various LC-MS/MS runs. The peptide samples are also spiked with a 50 fmol retention time calibration mixture as an internal standard prior to LC-MS analysis. Fig. S5 (ESI†) shows peptide spiked internal standard peptide (m/z 493.7) to be consistent concerning signal intensity and retention time for various samples used in this protocol. The nano-LC-MS/MS separation of peptides is performed using a nanoAcquity UPLC (Waters Technology Corp.) and Q-Exactive HF mass spectrometer (Thermo-Fisher Scientific). Reversed phased C-18 analytical (BEH C18, 150 μm × 100 mm) and trapping (180 μm × 20 mm) columns from Waters technology are used with a 75 or 120 min method at a flow rate of 0.5 μL min−1. In a typical label-free proteomics experiment, we have a loading amount of ∼100 ng having a detection range of 3–4 × 109. We ensure that the total ion chromatograms (TICs) are within 2 fold for all the samples, and blanks or peptide retention time mixtures are run every 5–6 samples to identify any carry-over and run-to-run variability.
3.6 Data analysis
For glycomics, data are interpreted manually using the area under the curve for extracted ion chromatograms (EICs) from the raw LC-MS/MS data. The relative and absolute abundance of disaccharides are determined using standard curves. Student's t-tests are performed with two-tailed distribution using Microsoft Excel spreadsheets. For differentiating the isoforms, EIC (MS/MS), the diagnostic ions as indicated in23 for each CS and HS disaccharide isoform are extracted, and abundances are obtained from manual area calculation.For proteomics, The raw LC-MS/MS data is run against a database using available commercial software, including PEAKS (Bioinformatics Solutions, Inc., Waterloo, ON, Canada) and/or proteome discoverer. For site-specific glycosylation analysis for enriched proteoglycans (and other glycoproteins), we use our in-house publically available GlycReSoft program.31,39
4. Procedure
Samples: We used various proteoglycan and protein standards and a commercially available mouse brain lysate (Table 2) to test the protocol and individual steps. All samples (except neurocan which was acquired for batch 1 only due to unavailability of sample for batch 2, and HS for syndecan-1 was acquired for only batch 1 as the sample was lost due to autosampler failure) were performed in two batches processed a week apart. Each sample was performed as two technical replicates for LC-MS/MS analysis. We used commercially available mouse brain lysate as a complex sample to test our protocol, but this protocol applies to any tissue. Fig. 2 outlines the protocol in a stepwise manner.4.1 Tissue lysis
1. For tissue lysis, fresh or free-floating tissues (∼25 mg) are lysed with a buffer (500 μl) (for each mg of tissue use about 15–20 times buffer volume) containing 50 mM Tris–HCl (pH = 7.5), 150 mM NaCl, 2 mM EDTA, protease inhibitor cocktail tablet, 1% SDS and 0.5% Triton X-100.2. To homogenize tissue, perform ultra-sonication (sonic Dismembrator Model 100, Fisher Scientific) for 2 minutes with 20 s pulse and incubate for 30 min on ice followed by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 4 °C for 30 min.
000 rpm, 4 °C for 30 min.
3. The supernatant is transferred to separate tubes, and the protein concentration of each sample is determined using the Pierce BCA microassay (Thermo-Fisher Scientific).
Pause point: At this point, samples can be stored at −20 °C before the next step or −80 °C for extended storage.
Caution: Proteinase inhibitor cocktail tablets are only valid for one year from date of opening; it is essential to check the validity of the tablets, as an old tablet will not be effective and may cause protein degradation over time.
Caution: Make fresh lysis buffer and add proteinase tablets right before using the buffer.
4.2 In-solution digestion
4. Take an equal amount of lysis buffer, add at least 5 times acetone by volume, and leave overnight, −20 °C. Next day, centrifuge the tube at 20000 × g, 4 °C for 30 min. Discard the supernatant. Wash the protein pellet with 80% ACN/20% water, sonicate briefly, and centrifuge the tubes at 20000 × g, 4 °C for 30 min. Discard the supernatant, and air dry the pellet, keeping the tubes upside down.
Caution: To ensure complete digestion of biomolecules on applying enzymes, effective removal of detergent is required. If the lysis buffer to be used contain a high concentration of detergent or is in bulk volumes, we recommend using Pierce Detergent Removal Spin Columns (Thermo-Fisher Scientific, 87777) for complete detergent removal before going to next step.
5. Take the protein pellet, add 5 mM Ammonium acetate, vortex, spin, and add 20 milliunits of hyaluronidase enzyme, vortex, spin, and incubate at 37 °C overnight on an Eppendorf mixer at 450 rpm.
6. Next day apply the solution to MWCO filters (Microcon™ Centrifugal Filters: Ultracel-10 Membrane, MRCPRT010, MilliporeSigma), centrifuge at 14000 × g, 4 °C for 20 minutes. Wash the filter with 200 μl of water, and centrifuge at 14000 × g, 4 °C for 20 min. Repeat the step twice. Collect the flow-through; these are HA disaccharides.
Caution: Use of correct MWCO filters is essential to have an effective protein recovery. We recommend using 10 kDa filter and Microcon™ filter units with a horizontal membrane rather than Amicon filter units with vertical membrane design.
7. Put the filter from step 6 in a new Eppendorf tube and add Tris–HCl (pH = 8.0), making a final concentration of 20 mM, ammonium acetate (5 mM), chondroitinase ABC enzyme (20 millliunit), and vortex briefly, and incubate 37 °C overnight.
8. Next day, cool down samples and centrifuge the solution at 14000 × g, 4 °C for 20 min. Wash the filter with 200 μl of water, and centrifuge at 14000 × g, 4 °C for 20 min. Repeat the step twice. Collect the flow-through; these are CS disaccharides.
9. Put the filter from step 8 in a new Eppendorf tube, and add Tris–HCl (pH = 7.5) making a final concentration of 20 mM, ammonium acetate (5 mM), calcium chloride (5 mM), 20 milliunit each of heparin lyase I, II, and III, vortex briefly, and incubate 37 °C overnight.
10. Next day, cool down samples and centrifuge the solution at 14000 × g, 4 °C for 20 min. Wash the filter with 200 μl of water, and centrifuge at 14000 × g, 4 °C for 20 min. Collect the flow-through; these are HS disaccharides.
11. Put the filter on new Eppendorf, and add 200 μl 50 mM ammonium bicarbonate to buffer exchange, and centrifuge at 14000 × g for 20 min at 4 °C. Repeat the step twice and discard the flow through.
12. Invert the filter in a new tube, centrifuge at 1000 g for 3 min at 4 °C to collect the protein. Wash the filter with 50 mM ammonium bicarbonate, and pool it with the collected protein solution.
13. To the protein sample, add DTT (5 mM), and incubate 60 °C for 30 min on an Eppendorf thermomixer at 450 rpm. Cooldown the solution, and add IAA (10 mM), and incubate in the dark for 30 min at room temperature (RT).
14. Dilute the solution with 50 mM ammonium bicarbonate and add 1:30 trypsin–LysC (or trypsin), incubate at 37 °C, overnight on an Eppendorf mixer at 450 rpm. Next day stop the trypsinisation by adding 1% formic acid; these are the peptides.
15. All extracted disaccharides and peptides are dried under vacuum centrifugation (SPD1010 Speedvac system, Thermo Savant).
Pause point: Disaccharides and peptides can be stored at −20 °C before further desalting and cleanup.
4.3 Desalting/cleanup
16. Resuspend the dried disaccharides in 10 μL of water and inject onto the Superdex Peptide (3.2/300) size exclusion column, using the mobile phase (25 mM ammonium acetate, 5% acetonitrile) at a flow rate of 0.04 mL min−1. HA, CS, and HS disaccharides are collected between 35–45 min. The clean HA, CS, and HS disaccharides are dried by vacuum centrifugation.17. For peptides:
• Wash the C-18 spin columns with 200 μl of 50% ACN/50% H2O, centrifuge at 1500 × g for 1 minute.
• Equilibrate columns with 200 μl of 2% ACN/98% H2O/0.1% TFA, centrifuge at 1500 × g for 1 minute. Repeat twice.
• Reconstitute sample in 100 μl of 2% ACN/98% H2O/0.1% TFA, apply to the C-18 column, centrifuge at 1500 × g for 1 min.
Note: the flow-through should be reapplied to the spin column and centrifuged, repeating a total of two times to ensure complete peptide binding to C-18. Do not discard the flowthrough, dry, and store at −20 °C.
• Wash the filter with 200 μl of 2% ACN/98% H2O/0.1% TFA, centrifuge at 1500 × g for 1 min. Repeat thrice.
• Elute the peptides using 30 μl of 60% ACN/40% H2O/0.1% TFA, centrifuge at 1500 × g for 1 min. Repeat twice.
• Dry the clean peptides under vacuum centrifugation.
4.4 Data acquisition
18. LC-MS/MS analysis for HA, CS, and HS disaccharides GAG disaccharides are analyzed using negative mode electrospray ionization on a 6520 Q-TOF (Agilent technologies). They are separated using a 1.9 μm, 0.3 × 150 mm GlycanPac AXH-1 (ThermoScientific) column mounted on an Agilent 1200 LC (Agilent Technologies, Santa Clara, CA). A 20 min isocratic method is used (85% B) and a flow rate of 5 μL min−1 (HA, and CS) and 7 μL min−1 (HS). Solvent A is 50 mM ammonium formate pH 4.4 in 10% ACN, and solvent B is 95% ACN/5% water. Samples are constituted in 85% B, and an 800 fmol quantity of ΔHexA2S–GlcNCoEt(6S) (Iduron) is added to all the samples as an internal standard before LC-MS analyses. The targeted tandem-MS analysis is performed to differentiate between HS and CS disaccharide isoforms (Fig. 2 and 3) with a collisional induced dissociation (CID). Fixed collision energy of 20 is used. The targeted list for CS is CS was m/z 458 (z = 1) and HS is m/z 458 (charge; z = 1), and m/z 496 (z = 1). The relative and absolute abundance were determined using standard curves as previously described.5,24,2519. Nano-LC-MS/MS analysis peptides
Nano-LC-MS/MS analysis is conducted by a Q-Exactive HF mass spectrometer (Thermo-Fisher Scientific) equipped with a nano ultra-performance liquid chromatography (UPLC) (Waters Corporation) peptides are trapped on a trapping (180 μm × 20 mm) column and separated on a reversed phased C-18 analytical (BEH C18, 150 μm × 100 mm) column (Waters Corporation). We load 1 μL onto the column, and separation is achieved using a 75 min (alternatively 120 min gradient can be used) gradient of 2 to 98% acetonitrile in 0.1% FA at a flow rate of ∼500 nL min−1. Data-dependent acquisition tandem MS is acquired in the positive ionization mode for the top 20 most abundant precursor ions. The scan sequence begins with an MS1 spectrum (Orbitrap; resolution 60000; mass range 300–2000 m/z; automatic gain control (AGC) target 1 × 106; maximum injection time 100 ms). MS2 analysis consists of higher energy collisional dissociation (Orbitrap; resolution 15000; AGC 1 × 105; normalized collision energy (NCE) 30; maximum injection time 100 ms, dynamic exclusion time of 8 s).
4.5 Data analysis
20. Glycomics data analysisGlycomics data analysis is performed manually by extracting extracted ion chromatograms (EICs) for HA, CS, and HS disaccharides for each sample from the raw LC-MS/MS data using qualitative analysis software (version B.06; Agilent Technologies). The obtained abundances for each disaccharide are first normalized to a spiked internal control and then further normalized to a standard curve to obtain an absolute and relative abundance of HS or CS disaccharides (fmol) in a Microsoft Excel spreadsheet. For differentiating the isoforms, EIC (MS/MS) for diagnostic ions as indicated in ref. 23 for each CS and HS disaccharide isoform are extracted, and abundances are obtained from manual area calculation.
21. Proteomics data analysis
The raw LC-MS/MS data are converted into mzXML format using ProteoWizard msConvert.40 The data are searched using PeaksDB and PeaksPTM using Peaks Studio version 8.5 (Bioinformatics Solutions, Inc., Waterloo, ON, Canada) against the Uniprot/Swissprot database for appropriate species with a 1% false discovery rate and at least two unique peptides. A 10 ppm error tolerance for the precursor (MS1) and 0.02 Da mass error tolerance for fragment ions (MS2) are specified. A maximum of 3 missed cleavages per peptide is allowed for the database search, permitting non-tryptic cleavage at one end. Trypsin + LysC is specified as the enzyme and carbamidomethylation as a fixed modification. A peaksPTM search is performed using advanced settings of a larger set of variable modifications, including hydroxylation P, oxidation M, hydroxylation K, hydroxylation-Hex K, hydroxylation-Hex-Hex K, HexNAc ST, HexHexNAc ST, phosphorylation STY, ubiquitination K, deamidation N, methoxy K, and nitrotyrosine Y. Our in house software GlycreSoft can be further used to identify glycopeptides and linker peptides (formed after removal of GAG chains) in the proteomics data.31,39
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Step 18 | Absence of GAG disaccharides | • The enzyme was not active or present in sufficient quantity | ✓ Check for enzyme activity |
| • Detergent is still present in the samples that interfered with enzyme activity | ✓ Check for enzyme validity by the supplier | ||
| ✓ To ensure complete detergent removal, we recommend using detergent removal spin columns (ThermoScientific) | |||
| Step 18 | Absence of triply sulfated forms | • MS Source conditions are not optimal and in-source sulfate loss | ✓ Tune source for fragile tuning and sulfate loss |
| Step 19 | Low peptide/protein identification | • Peptide loss | ✓ Check flow-through from step 17, possible loss of peptides |
| • Protein loss | ✓ Make sure to use correct MWCO filters with horizontal membranes with at least 10 kDa cut off | ||
| ✓ Ensure the proteinase inhibitor used for lysis buffer was valid and active to avoid overtime degradation of the protein | |||
| Step 19 | Observe high TIC but a low number of proteins identified | • Presence of lipids contaminant from tissue | ✓ Check the number of tandem spectra acquired and the proportion of PSM matched. The expected ration should be 30% |
| • Loss in MS2 sensitivity | ✓ For MS2 sensitivity: Check the quality of ms2 spectra in peptide, retention time calibration mixture | ||
| ✓ Check for m/z 524.3 in the positive-mode calibration mixture; the intensity of MS2 peaks should be 15% higher than MS1 |
4.6 Tips for troubleshooting are provided in Table 3
5. Results & discussion
We describe herein a robust and streamlined protocol (Fig. 2) that utilizes the FASP type approach of digesting biomolecules on a MWCO filter and extracting the released biomolecules as a flowthrough. We used a commercial mouse brain lysate as a complex sample and several standard proteoglycan and proteins for testing each and/or several steps of the protocol, as shown in Table 2. We performed experiments in two different batches one week apart from each other to test the reproducibility of the protocol, except for neurocan and HS of syndecan-1, which were acquired only for batch 1. We were able to identify 1 HA, 6 CS, and 8 HS unsaturated disaccharides from the standard proteoglycans and complex sample. In the complex sample, HS D2S6 was absent or present below the detectable limit. In addition, we observed 3 CS saturated disaccharides (U0a0, U0a4/U0a6) for standard proteins, while only 2 (U0a4/U0a6) were observed for the complex sample. For HS, 3 saturated disaccharides were observed (U0A0, U0A6/U2A0) for standards, while only 1 (U0A0) was observed for the complex sample. The ion abundance counts (normalized (to internal standard m/z 552) area under the curve (AUC) ± standard deviation (SD))) are shown in Table S1 (ESI†), and coefficient of variation (% CV) for each disaccharide for all samples are shown in Table 4. The CVs for the two batches were within our specified criteria of ≤20% for CS, and ≤30% CV for HS disaccharides, illustrating a similar recovery of GAGs in the two batches.| Samples | % coefficient of variation (% CV) |
|---|---|
| D0a0 | |
| HA disaccharides | |
| Hyaluronic acid-batch 01 vs. 02 | 7 |
| Lysate-batch 01 vs. 02 | 14 |
| Samples | % coefficient of variation (% CV) | ||||
|---|---|---|---|---|---|
| D0a0 | D0a4/D0a6 | D0a10/D2a4/D2a6 | U0a0 | U0a4/U0a6/U2a0 | |
| CS disaccharides | |||||
| Aggrecan-batch 01 vs. 02 | 1 | 9 | 19 | 16 | 0.3 |
| Syndecan-batch 01 vs. 02 | 2 | 16 | 13 | 2 | 16 |
| Lysate-batch 01 vs. 02 | 11 | 20 | 6 | Not observed | 6 |
| Samples | % coefficient of variation (% CV) | |||||||
|---|---|---|---|---|---|---|---|---|
| D0A0 | DOA6/D2A0 | D2A6 | D0S0 | D0S6/D2S0 | D2S6 | U0A0 | UOA6/U2A0 | |
| HS disaccharides | ||||||||
| HSBK-batch 01 vs. 02 | 4 | 29 | 23 | 30 | 29 | 20 | 26 | 3 |
| HSPIM-batch 01 vs. 02 | 7 | 27 | 15 | 19 | 14 | 15 | 3 | 7 |
| Lysate-batch 01 vs. 02 | 22 | 16 | 4 | 14 | 16 | Not observed | 4 | Not observed |
For proteomics, syndecan and neurocan were observed with 67%, and 64% coverage, respectively, while Aggrecan was observed with 27% coverage (Table 5A and Supplemental file S1, ESI†). Aggrecan contains approximately 90 CS chains based on the total masses of core protein, CS, and the average mass of CS chains.26 With no CHABC treatment and proteolysis with trypsin + LysC, we observed 17% sequence coverage of aggrecan (Supplemental file S1, ESI†). Thus, the removal of CS chains assists in better identification of aggrecan. To increase peptide coverage for aggrecan, Glu-C digestion could be performed, followed by trypsin + Lys-C digestion as trypsin as the only protease produces extended region glycopeptides that are too large for analysis using HCD LC-tandem MS.16 A number of post-translational modifications (PTMs) for aggrecan, neurocan, and syndecan were observed including, O-glycosylation (HexNAc & HexNAcHex) on serine and threonine as identified by peaks PTM analysis (Table 5A and Fig. S6, ESI†).
| (A) Standard protein/proteoglycans | Coverage (total, unique peptides)-batch 1 | Coverage (total, unique peptides)-batch 2 | PTMs |
|---|---|---|---|
| Aggrecan | 27% (162, 158) | 28% (188, 184) | Carbamidomethylation; hydroxyproline; ubiquitation deamidation; K-methoxy; HLys; HexNAcHex; HexNAc |
| Syndecan | 67% (139, 139) | — | HexNAc, HexNAcHex hydroxyproline; deamidation; phosphorylation, K-methoxy; ubiquitation |
| Neurocan | 64% (383, 371) | — | Carbamidomethylation; hydroxyproline, phosphorylation, HexNAcHex; HexNAc deamidation N; ubiquitation, Hlys-Hex; HLys-Hex-Hex |
| AGP | 65% (53, 41) | 60% (53, 39) | Carbamidomethylation; hydroxyproline, deamidation; K-methoxy, HLys; ubiquitation, HexNAc Y-NO2, K-methoxy |
| (B) Complex mixture | Total proteins with unique peptide 1 | Total proteins with unique peptide 2 | PG and ECM related proteins observed coverage (total, unique peptide) |
|---|---|---|---|
| Mouse brain lysate | 1449-batch 1 | 747-batch 1 | Versican 0% (1, 1) |
| 1496-batch 2 | 773-batch 2 | Neurocan 1% (1, 1) | |
| Brevican 4% (3, 3) | |||
| Tenascin-R 5% (6, 6) | |||
| Disintegrin and metalloproteinase domain-containing protein 22 2% (1, 1) |
For mouse brain tissue lysate, we observed 1449 and 1496 proteins with 1 unique peptide, and 747 and 773 proteins with 2 unique peptides for batch 1 and 2, respectively. The average normalized area (normalized to total ion chromatogram (TICs)) with relative standard deviations (RSDs) between the two batches for standard proteins are shown in Table 6 (For the complete list of average normalized area and RSDs for lysate and standard proteins see Supplemental file S2, ESI†). A number of PGs and ECM related proteins were observed but with low coverage (Table 5B and Supplemental file S1, ESI†). Thus, to achieve a complete picture of ECM and proteoglycans in complex samples, an enrichment step needs to be incorporated such as using anion-exchange (SAX) chromatography followed by serial in-solution digestion as presented here and sequential proteolysis using a combination of trypsin–LysC/GluC/chymotrypsin prior to LC-MS/MS to attain more in-depth PG sequence coverage. To study proteins displaying specific PTMs enrichment is usually necessary to increase the relative analyte abundance, such as enrichment of PGs using SAX,34,41 and enrichment of glyco-peptides/-proteins using lectin affinity chromatography, hydrazide chemistry,42 and hydrophilic interaction liquid chromatography (HILIC)43,44 have been extensively used by experts in the field of glycoscience. We plan to incorporate the enrichment step in the presented protocol for more targeted workup and in-depth PG analysis in the future. When we performed the serial in-solution protocol on a human brain tissue lysed in-house rather than a commercial mouse brain, we observed 1297 proteins with 1 unique peptide, and 782 proteins with 2 unique peptides and a number of ECM related proteins and proteoglycans were observed with high coverage (Supplemental file S3, ESI†). This implies that the low coverage of PG or ECM proteins in the commercial mouse brain lysate could be sample-related.
| Protein | Normalized area batch 01 | Normalized area batch 02 | Average normalized area ± standard deviation | Relative standard deviation (%) |
|---|---|---|---|---|
| a For the two replicates in batch 01. | ||||
| Syndecan-1 | 4.10 × 109 | 4.00 × 109 | 4.05 × 109 ± 6.85× 107 | 2 |
| AGP | 2.09 × 109 | 2.37 × 109 | 2.23 × 109 ± 1.96× 108 | 9 |
| Aggrecan | 1.37 × 1010 | 1.35 × 1010 | 1.26 × 1010 ± 1.74 × 108 | 1 |
| Neurocana | 5.61 × 1010a |
5.02 × 1010a |
5.32 × 1010 ± 4.16 × 109a |
8a |
In conclusion, we present a streamlined protocol to perform integrated glycomics and proteomics on tissue samples. This protocol could be utilized for simultaneous processing of 24 samples at a given time, making it efficient and high-throughput despite a 5 day workup procedure. The end goal is to establish a robust platform for identification and characterization of GAG compositions and PG and other ECM related molecules specifically in the brain that play crucial roles in various neurological processes and understand their role in underlying molecular mechanisms in neuropsychiatric disorder.
Abbreviations
| FASP | Filter-aided sample preparation |
| GAGs | Glycosaminoglycans |
| PGs | Proteoglycans |
| ECM | Extracellular matrix |
| HS | Heparan sulfate |
| HA | Hyaluronic acid/hyaluronan |
| CS | Chondroitin sulfate |
| MWCO | Molecular weight cut off |
| TIC | Total ion chromatogram |
| EIC | Extracted ion chromatogram |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This work was supported by NIH grants U01CA221234 and R01GM133963. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE45 partner repository with the dataset identifier PXD017513 and 10.6019/PXD017513.References
- E. S. Au-Hecht, J. P. Au-McCord and D. C. Au-Muddiman, A Quantitative Glycomics and Proteomics Combined Purification Strategy, JoVE, 2016, 109, e53735 Search PubMed.
- A. D. Taylor, W. S. Hancock, M. Hincapie, N. Taniguchi and S. M. Hanash, Towards an integrated proteomic and glycomic approach to finding cancer biomarkers, Genome Med., 2009, 1(6), 57 CrossRef PubMed.
- M. Thaysen-Andersen and N. H. Packer, Advances in LC–MS/MS-based glycoproteomics: Getting closer to system-wide site-specific mapping of the N- and O-glycoproteome, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844(9), 1437–1452 CrossRef CAS PubMed.
- M. Wuhrer, M. I. Catalina, A. M. Deelder and C. H. Hokke, Glycoproteomics based on tandem mass spectrometry of glycopeptides, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 849(1), 115–128 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Xu, Q. Li, E. Goonatilleke and C. B. Lebrilla, Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses, Chem. Rev., 2018, 118(17), 7886–7930 CrossRef CAS PubMed.
- P. Rudd, N. G. Karlsson, K. H. Khoo and N. H. Packer, Glycomics and Glycoproteomics, in Essentials of Glycobiology, ed. Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., et al., Cold Spring Harbor, NY, 3rd edn, 2015, pp. 653–666 Search PubMed.
- D. J. Harvey, Proteomic analysis of glycosylation: structural determination of N- and O-linked glycans by mass spectrometry, Expert Rev. Proteomics, 2005, 2(1), 87–101 CrossRef CAS PubMed.
- G. O. Staples and J. Zaia, Analysis of Glycosaminoglycans Using Mass Spectrometry, Curr. Proteomics, 2011, 8(4), 325–336 CrossRef CAS PubMed.
- J. Zaia, Glycosaminoglycan glycomics using mass spectrometry, Mol. Cell. Proteomics, 2013, 12(4), 885–892 CrossRef CAS PubMed.
- X. Shi and J. Zaia, Organ-specific heparan sulfate structural phenotypes, J. Biol. Chem., 2009, 284(18), 11806–11814 CrossRef CAS PubMed.
- L. Turiak, C. Shao, L. Meng, K. Khatri, N. Leymarie, Q. Wang, H. Pantazopoulos, D. R. Leon and J. Zaia, Workflow for combined proteomics and glycomics profiling from histological tissues, Anal. Chem., 2014, 86(19), 9670–9678 CrossRef CAS PubMed.
- R. Raghunathan, M. K. Sethi and J. Zaia, On-slide tissue digestion for mass spectrometry based glycomic and proteomic profiling, MethodsX, 2019, 6, 2329–2347 CrossRef PubMed.
- U. Lindahl, J. Couchman, K. Kimata, J. D. Esko: Proteoglycans and Sulfated Glycosaminoglycans. in Essentials of Glycobiology, ed. Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., et al., Cold Spring Harbor, NY, 3rd edn, 2015, pp. 207–221 Search PubMed.
- P. D. Smith, V. J. Coulson-Thomas, S. Foscarin, J. C. Kwok and J. W. Fawcett, “GAG-ing with the neuron”: The role of glycosaminoglycan patterning in the central nervous system, Exp. Neurol., 2015, 274(Pt B), 100–114 CrossRef CAS PubMed.
- J. L. Funderburgh, Mini Review Keratan sulfate: structure, biosynthesis, and function, Glycobiology, 2000, 10(10), 951–958 CrossRef CAS PubMed.
- R. O. Hynes and A. Naba, Overview of the matrisome–an inventory of extracellular matrix constituents and functions, Cold Spring Harbor Perspect. Biol., 2012, 4(1), a004903 Search PubMed.
- H. Pantazopoulos, A. Boyer-Boiteau, E. H. Holbrook, W. Jang, C. G. Hahn, S. E. Arnold and S. Berretta, Proteoglycan abnormalities in olfactory epithelium tissue from subjects diagnosed with schizophrenia, Schizophr. Res., 2013, 150(2–3), 366–372 CrossRef PubMed.
- J. van Horssen, P. Wesseling, L. P. van den Heuvel, R. M. de Waal and M. M. Verbeek, Heparan sulphate proteoglycans in Alzheimer's disease and amyloid-related disorders, Lancet Neurol., 2003, 2(8), 482–492 CrossRef CAS PubMed.
- R. Raghunathan, M. K. Sethi, J. A. Klein and J. Zaia, Proteomics, glycomics, and glycoproteomics of matrisome molecules, Mol. Cell. Proteomics, 2019, 18(11), 2138–2148 CrossRef PubMed.
- H. Desaire, T. L. Sirich and J. A. Leary, Evidence of block and randomly sequenced chondroitin polysaccharides: sequential enzymatic digestion and quantification using ion trap tandem mass spectrometry, Anal. Chem., 2001, 73(15), 3513–3520 CrossRef CAS PubMed.
- J. L. Spencer, J. A. Bernanke, J. A. Buczek-Thomas and M. A. Nugent, A computational approach for deciphering the organization of glycosaminoglycans, PLoS One, 2010, 5(2), e9389 CrossRef PubMed.
- R. Raghunathan, N. K. Polinski, J. A. Klein, J. D. Hogan, C. Shao, K. Khatri, D. Leon, M. E. McComb, F. P. Manfredsson and C. E. Sortwell, et al., Glycomic and Proteomic Changes in Aging Brain Nigrostriatal Pathway, Mol. Cell. Proteomics, 2018, 17(9), 1778–1787 CrossRef CAS PubMed.
- C. Shao, X. Shi, J. J. Phillips and J. Zaia, Mass spectral profiling of glycosaminoglycans from histological tissue surfaces, Anal. Chem., 2013, 85(22), 10984–10991 CrossRef CAS PubMed.
- R. R. Drake, T. W. Powers, E. E. Jones, E. Bruner, A. S. Mehta and P. M. Angel, MALDI Mass Spectrometry Imaging of N-Linked Glycans in Cancer Tissues, Adv. Cancer Res., 2017, 134, 85–116 CrossRef CAS PubMed.
- R. R. Drake, T. W. Powers, K. Norris-Caneda, A. S. Mehta and P. M. Angel, In Situ Imaging of N-Glycans by MALDI Imaging Mass Spectrometry of Fresh or Formalin-Fixed Paraffin-Embedded Tissue, Curr. Protoc. Protein Sci., 2018, 94(1), e68 CrossRef PubMed.
- R. R. Drake, C. A. West, A. S. Mehta and P. M. Angel, MALDI Mass Spectrometry Imaging of N-Linked Glycans in Tissues, Adv. Exp. Med. Biol., 2018, 1104, 59–76 CrossRef CAS PubMed.
- P. Juhasz and K. Biemann, Utility of non-covalent complexes in the matrix-assisted laser desorption ionization mass spectrometry of heparin-derived oligosaccharides, Carbohydr. Res., 1995, 270(2), 131–147 CrossRef CAS PubMed.
- C. Shao, X. Shi, M. White, Y. Huang, K. Hartshorn and J. Zaia, Comparative glycomics of leukocyte glycosaminoglycans, FEBS J., 2013, 280(10), 2447–2461 CrossRef CAS PubMed.
- J. E. Turnbull, R. L. Miller, Y. Ahmed, T. M. Puvirajesinghe and S. E. Guimond, Glycomics profiling of heparan sulfate structure and activity, Methods Enzymol., 2010, 480, 65–85 CAS.
- J. Chen, T. Kawamura, M. K. Sethi, J. Zaia, V. Repunte-Canonigo and P. P. Sanna, Heparan sulfate: Resilience factor and therapeutic target for cocaine abuse, Sci. Rep., 2017, 7(1), 13931 CrossRef PubMed.
- J. A. Klein, L. Meng and J. Zaia, Deep sequencing of complex proteoglycans: a novel strategy for high coverage and site-specific identification of glycosaminoglycan-linked peptides, Mol. Cell. Proteomics, 2018, 17(8), 1578–1590 CrossRef CAS PubMed.
- D. C. Hondius, P. van Nierop, K. W. Li, J. J. Hoozemans, R. C. van der Schors, E. S. van Haastert, S. M. van der Vies, A. J. Rozemuller and A. B. Smit, Profiling the human hippocampal proteome at all pathologic stages of Alzheimer's disease, J. Alzheimer's Dis., 2016, 12(6), 654–668 Search PubMed.
- L. E. Donovan, L. Higginbotham, E. B. Dammer, M. Gearing, H. D. Rees, Q. Xia, D. M. Duong, N. T. Seyfried, J. J. Lah and A. I. Levey, Analysis of a membrane-enriched proteome from postmortem human brain tissue in Alzheimer's disease, Proteomics Clin Appl, 2012, 6(3-4), 201–211 CrossRef CAS PubMed.
- F. Noborn, A. Gomez Toledo, A. Green, W. Nasir, C. Sihlbom, J. Nilsson and G. Larson, Site-specific identification of heparan and chondroitin sulfate glycosaminoglycans in hybrid proteoglycans, Sci. Rep., 2016, 6(1), 34537 CrossRef CAS PubMed.
- J. R. Wiśniewski, A. Zougman, N. Nagaraj and M. Mann, Universal sample preparation method for proteome analysis, Nat. Methods, 2009, 6(5), 359 CrossRef PubMed.
- J. R. Wiśniewski, D. F. Zielinska and M. Mann, Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method, Anal. Biochem., 2011, 410(2), 307–309 CrossRef PubMed.
- J. Potriquet, M. Laohaviroj, J. M. Bethony and J. Mulvenna, A modified FASP protocol for high-throughput preparation of protein samples for mass spectrometry, PLoS One, 2017, 12(7), e0175967 CrossRef PubMed.
- J. Erde, R. R. Loo and J. A. Loo, Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments, J. Proteome Res., 2014, 13(4), 1885–1895 CrossRef CAS PubMed.
- J. Klein, L. Carvalho and J. Zaia, Application of network smoothing to glycan LC-MS profiling, Bioinformatics, 2018, 34(20), 3511–3518 CrossRef CAS PubMed.
- D. Kessner, M. Chambers, R. Burke, D. Agus and P. Mallick, ProteoWizard: open source software for rapid proteomics tools development, Bioinformatics, 2008, 24(21), 2534–2536 CrossRef CAS PubMed.
- F. Noborn, A. Gomez Toledo, C. Sihlbom, J. Lengqvist, E. Fries, L. Kjellen, J. Nilsson and G. Larson, Identification of chondroitin sulfate linkage region glycopeptides reveals prohormones as a novel class of proteoglycans, Mol. Cell. Proteomics, 2015, 14(1), 41–49 CrossRef CAS PubMed.
- H. Hu, K. Khatri, J. Klein, N. Leymarie and J. Zaia, A review of methods for interpretation of glycopeptide tandem mass spectral data, Glycoconjugate J., 2016, 33(3), 285–296 CrossRef CAS PubMed.
- C. Zhang, Z. Ye, P. Xue, Q. Shu, Y. Zhou, Y. Ji, Y. Fu, J. Wang and F. Yang, Evaluation of Different N-Glycopeptide Enrichment Methods for N-Glycosylation Sites Mapping in Mouse Brain, J. Proteome Res., 2016, 15(9), 2960–2968 CrossRef CAS PubMed.
- S. Mysling, G. Palmisano, P. Højrup and M. Thaysen-Andersen, Utilizing Ion-Pairing Hydrophilic Interaction Chromatography Solid Phase Extraction for Efficient Glycopeptide Enrichment in Glycoproteomics, Anal. Chem., 2010, 82(13), 5598–5609 CrossRef CAS PubMed.
- Y. Perez-Riverol, A. Csordas, J. Bai, M. Bernal-Llinares, S. Hewapathirana, D. J. Kundu, A. Inuganti, J. Griss, G. Mayer and M. Eisenacher, et al., The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res., 2019, 47(D1), D442–D450 CrossRef CAS PubMed.
- R. Raghunathan, M. K. Sethi, J. A. Klein and J. Zaia, Proteomics, Glycomics, and Glycoproteomics of Matrisome Molecules, Mol. Cell. Proteomics, 2019, 18(11), 2138–2148 CrossRef PubMed.
- J. R. Wisniewski, Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols, Anal. Chem., 2016, 88(10), 5438–5443 CrossRef CAS PubMed.
- I. J. Ji, S. Hua, D. H. Shin, N. Seo, J. Y. Hwang, I. S. Jang, M. G. Kang, J. S. Choi and H. J. An, Spatially-resolved exploration of the mouse brain glycome by tissue glyco-capture (TGC) and nano-LC/MS, Anal. Chem., 2015, 87(5), 2869–2877 CrossRef CAS PubMed.
- R. Casadonte and R. M. Caprioli, Proteomic analysis of formalin-fixed paraffin-embedded tissue by MALDI imaging mass spectrometry, Nat. Protoc., 2011, 6(11), 1695–1709 CrossRef CAS PubMed.
- E. S. Hecht, J. P. McCord and D. C. Muddiman, A Quantitative Glycomics and Proteomics Combined Purification Strategy, J. Visualized Exp., 2016,(109), e53735 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mo00019a |
| This journal is © The Royal Society of Chemistry 2020 |