
The effectiveness of filtering glycopeptide peak list files for Y ions†
Robert J.
Chalkley
 *a,
Katalin F.
Medzihradszky
ab,
Zsuzsanna
Darula
b,
Adam
Pap
bc and
Peter R.
Baker
a
*a,
Katalin F.
Medzihradszky
ab,
Zsuzsanna
Darula
b,
Adam
Pap
bc and
Peter R.
Baker
a
aDepartment of Pharmaceutical Chemistry, School of Pharmacy, University of California San Francisco, USA. E-mail: chalkley@cgl.ucsf.edu
bLaboratory of Proteomics Research, Biological Research Centre, Temesvari krt. 62, H-6726 Szeged, Hungary
cDoctoral School in Biology, Faculty of Science and Informatics, University of Szeged, Kozep fasor 52, H-6726 Szeged, Hungary
First published on 6th February 2020
Abstract
Intact glycopeptide analysis is becoming more common with developments in mass spectrometry instrumentation and fragmentation approaches. In particular, collision-based fragmentation approaches such as higher energy collisional dissociation (HCD) and radical-driven fragmentation approaches such as electron transfer dissociation (ETD) provide complementary information, but bioinformatic strategies to utilize this combined information are currently lacking. In this work we adapted a software tool, MS-Filter, to search HCD peak list files for predicted Y ions based on matched EThcD results to propose additional glycopeptide assignments. The strategy proved to be extremely powerful for O-glycopeptide data, and also of benefit for N-linked data, where it allowed rescue of low confidence results from database searching.
Introduction
Intact glycopeptide analysis using mass spectrometry presents significant challenges due to the differing behavior of the peptide and glycan components in the hybrid species. Nevertheless, when analyzing a single protein, collision induced dissociation (CID) strategies have been very effective. In the characteristic fragmentation of glycopeptides using these approaches one of the most prominent fragments in O-linked glycopeptide spectra is typically the gas-phase-deglycosylated peptide (Y0), whereas for N-linked glycopeptides a similarly abundant ion is commonly the peptide retaining the core GlcNAc (Y1) [nomenclature: 1]. When analyzing an isolated protein, the masses of the unmodified versions of potentially glycosylated peptides can be calculated, so it is straightforward to use the smallest Y ion mass to identify the peptide, one can infer the mass of the glycan based on the mass difference between the observed precursor and the Y ion, then the CID fragmentation provides information on the glycan(s) composition. Resonance activation CID in ion trap yields B and Y fragments via single bond cleavages, identifying terminal groups, thus providing knowledge about branching in the oligosaccharide(s).2,3 Beam-type CID (HCD) can produce multiple bond cleavages, and thus provides some information on the direct connection of certain sugar units in form of internal oxonium ions.3,4Analysis of complex mixtures of glycoproteins is currently mostly performed by enzymatic release of glycan species then analysis of peptides and glycans separately. However, intact glycopeptide analysis for these types of samples is starting to become more common due to the development of improved fragmentation methods in mass spectrometers that allow formation of fragments from both peptide and glycan components in the same spectrum, namely EThcD and stepped HCD fragmentation.5–7 A few datasets identifying hundreds of unique glycopeptides have now been published, although there are still challenges with controlling the reliability of results from software doing these analyses.5–9
In this work we sought to investigate software strategies that make use of small Y ions to (a) increase the number of glycopeptide identifications; and (b) improve the reliability of reported results in analyses of complex glycopeptide datasets. Using Y ions to try to identify glycopeptides from CID-type fragmentation spectra is not novel. However, using EThcD data to derive the list of glycosylated peptides for querying HCD data is a new approach. Prior strategies have all employed PNGase F to deglycosylate a fraction of the sample, then analyzed this to derive a list of potential glycopeptides to consider in the intact glycopeptide analysis. Some researchers additionally performed glycomic analysis to derive a list of glycosylations to consider,10,11 whereas others have used a database of known glycans as a reference.12 These approaches require significantly more sample handling and acquisition and can only be used for N-glycosylation analysis.
We developed new features in the MS-Filter tool within Protein Prospector, then tested these on intact O-linked and N-linked glycopeptide datasets to see the effect of different filtering parameters on the resulting glycopeptide results.
Experimental methods
The O-glycopeptide data was derived from a study of intact urinary glycopeptides.5 Briefly, the peptides were enriched from a tryptic digest of human urinary proteins using Wheat Germ Agglutinin (WGA) lectin weak affinity chromatography (LWAC). The data analyzed here consisted of glycopeptides of the GlcNAc-eluted fraction of a healthy volunteer with AB blood-type. These glycopeptides were subjected to LC/MS/MS analysis on a Fusion Lumos Tribrid mass spectrometer (Thermo). HCD (28% NCE) and m/z 204.087 fragment ion-triggered EThcD (15% NCE) were used as activation methods. Only 3–5+ charged ions were permitted in the precursor ion selection. Dynamic exclusion for 30 s was enabled. Both precursor and fragments were measured in the Orbitrap. For further sample preparation and data acquisition details see ref. 5 The raw data are available at MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp), and the project's identifier is MSV000083070.Peak lists were generated using Proteome Discoverer (Thermo Scientific, v2.2.0.388). Database searches with the EThcD data were performed using Prospector Prospector v6.2.13 Two separate searches were performed where all parameters were identical except the glycan database considered. The proteins considered were all human proteins in SwissProt.2019.4.8 that was concatenated with randomized sequences for each entry (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 418 entries searched). Search parameters: the 80 most intense fragment ions were considered in each spectrum; enzyme: trypsin, 1 missed cleavage and semi-tryptic peptides were permitted; 10 pm and 20 ppm mass tolerance for precursor and fragment ions, respectively; carbamidomethylation of Cys residues was fixed; the acetylation of a protein's N-termini, Met oxidation and cyclization of N-terminal Gln were permitted. In the first search HexNAcHexNeuAc, HexNAcHexNeuAc2 and HexNAc2Hex2NeuAc2 were the only glycans considered, each was assigned as ‘common’, with two variable modifications permitted per peptide (i.e. up to two glycosylations per peptide were considered). In the other searches 126 and 37 O-glycans were specified as ‘rare’; i.e. only one glycan may decorate any given peptide. Several of the considered glycan compositions were the sum of multiple individual glycans and some of them included sodium adducts. The search results from the two larger glycan databases were merged into a single results file using Search Compare in Protein Prospector. The acceptance criteria were based on score and E-values only as reported in Table S1 (ESI†).
418 entries searched). Search parameters: the 80 most intense fragment ions were considered in each spectrum; enzyme: trypsin, 1 missed cleavage and semi-tryptic peptides were permitted; 10 pm and 20 ppm mass tolerance for precursor and fragment ions, respectively; carbamidomethylation of Cys residues was fixed; the acetylation of a protein's N-termini, Met oxidation and cyclization of N-terminal Gln were permitted. In the first search HexNAcHexNeuAc, HexNAcHexNeuAc2 and HexNAc2Hex2NeuAc2 were the only glycans considered, each was assigned as ‘common’, with two variable modifications permitted per peptide (i.e. up to two glycosylations per peptide were considered). In the other searches 126 and 37 O-glycans were specified as ‘rare’; i.e. only one glycan may decorate any given peptide. Several of the considered glycan compositions were the sum of multiple individual glycans and some of them included sodium adducts. The search results from the two larger glycan databases were merged into a single results file using Search Compare in Protein Prospector. The acceptance criteria were based on score and E-values only as reported in Table S1 (ESI†).
The dataset used for assessment of N-linked glycosylation was created by the Human Glycoproteomics Initiative. A human serum sample was reduced, alkylated and digested with trypsin, then glycopeptides were enriched by hydrophilic interaction chromatography (HILIC) using a HyperSep Retain AX resin (Thermo). The full details of the protocol are yet to be published (manuscript in preparation). The data in the analyzed file was acquired on a Fusion Lumos Tribrid mass spectrometer (Thermo) where three types of fragmentation were employed on each precursor: HCD measured in the Orbitrap; EThcD measured in the Orbitrap; and ion trap CID measured in the ion trap. The EThcD data was analyzed by the search engine; the HCD data was used for MS-Filter, and the CID data was ignored.
The N-linked data was searched in Batch-Tag against human entries from SwissProt downloaded on 25 July 2017, along with randomized concatenated sequences, with a total of 20201 sequences considered. A precursor mass tolerance of 8 ppm and fragment tolerance of 20 ppm were permitted. Peptides were permitted to be semi-tryptic. Carbamidomethylation was set as a fixed modification. In addition to methionine and tryptophan oxidation, pyro-glutamate formation from N-terminal glutamine and pyro-carbamidomethyl formation from N-terminal cysteine a total of 206 glycan compositions and potassium-adduct versions were considered, only on sites within the consensus motif N-X-S/T, where X is not a proline. Two searches were performed; the only difference being whether incorrect monoisotopic peak assignment (i.e. whether the assigned mass is the second isotope) was considered. Results were thresholded at a 1% FDR at the unique peptide/glycopeptide level.
MS-Filter analyses were performed with Instrument setting ESI-Q-hi-res, with ‘Match Charge’ selected. All searches required matching m/z 204.087 in addition to the Y ions calculated by input of the list of unmodified peptides identified from database search results. The O-linked data was queried for Y0 and Y1 peaks, whereas the N-linked data was searched for either just Y1 or Y1 and Y2 as described in the results. All searches allowed precursor and fragment mass tolerances of ±20 ppm. The number of most intense peaks considered for matching the m/z 204 peak and the relevant Y ions was varied as described in the results. The output from these searches are in Tables S1 and S2 (ESI†).
Results
Fig. 1 presents a flowchart of the strategy we developed, and Fig. 2 displays the new input interface. MS-Filter is software that was developed to filter peak list files based on the presence of defined mass peaks, mass losses, precursor mass and/or charge state ranges. We added in new features that would also allow it to search for Y ion masses. To do this we created a field where users can enter peptide sequences. These can optionally also contain modifications, such as carbamidomethyl cysteine or oxidized methionine residues. We then created tick boxes that allow the user to select which Y ions and charge states to search peak list files for, and a ‘Min Mod Matches’ which specifies how many of these ions are required to match. The ‘Matched YOY1’ option specifically requires both the Y0 and Y1 to be matched (rather than, for example, meeting a ‘Min Mod Matches’ threshold by matching the Y1 in two different charge states). Finally, we provide sets of glycans to consider when trying to translate observed mass shifts to glycan compositions. There are several other parameters that can be set by the user, including how many of the ‘n’ most intense peaks to consider when looking for the fragment mass/es of interest and mass tolerance for matching these peaks. For evaluating different parameters in this study, the two parameters we varied were the ‘Max MSMS Peaks’, and whether to require one or two Y ion peaks to be matched.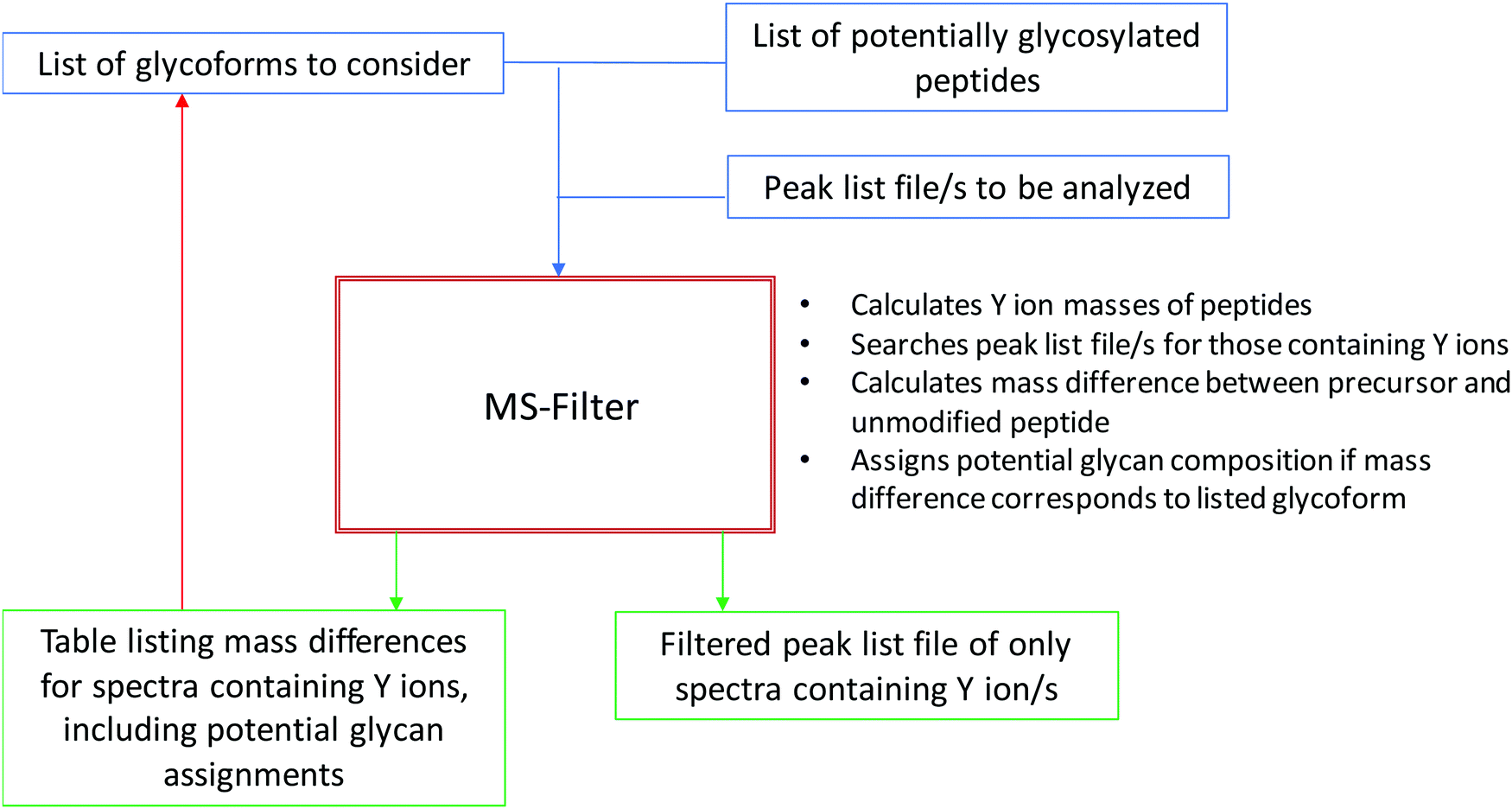 | ||
| Fig. 1 Flowchart of Y ion filtering process using MS-Filter. The input is peak list file/s and a list of candidate peptides. The user specifies parameters including how many Y ions need to be matched and at what mass tolerance, and what glycoforms to consider. The software outputs a new peak list file containing only those spectra that pass the filtering criteria, and also a table listing these spectra, the observed mass modification and potential glycan explanations for the masses. | ||
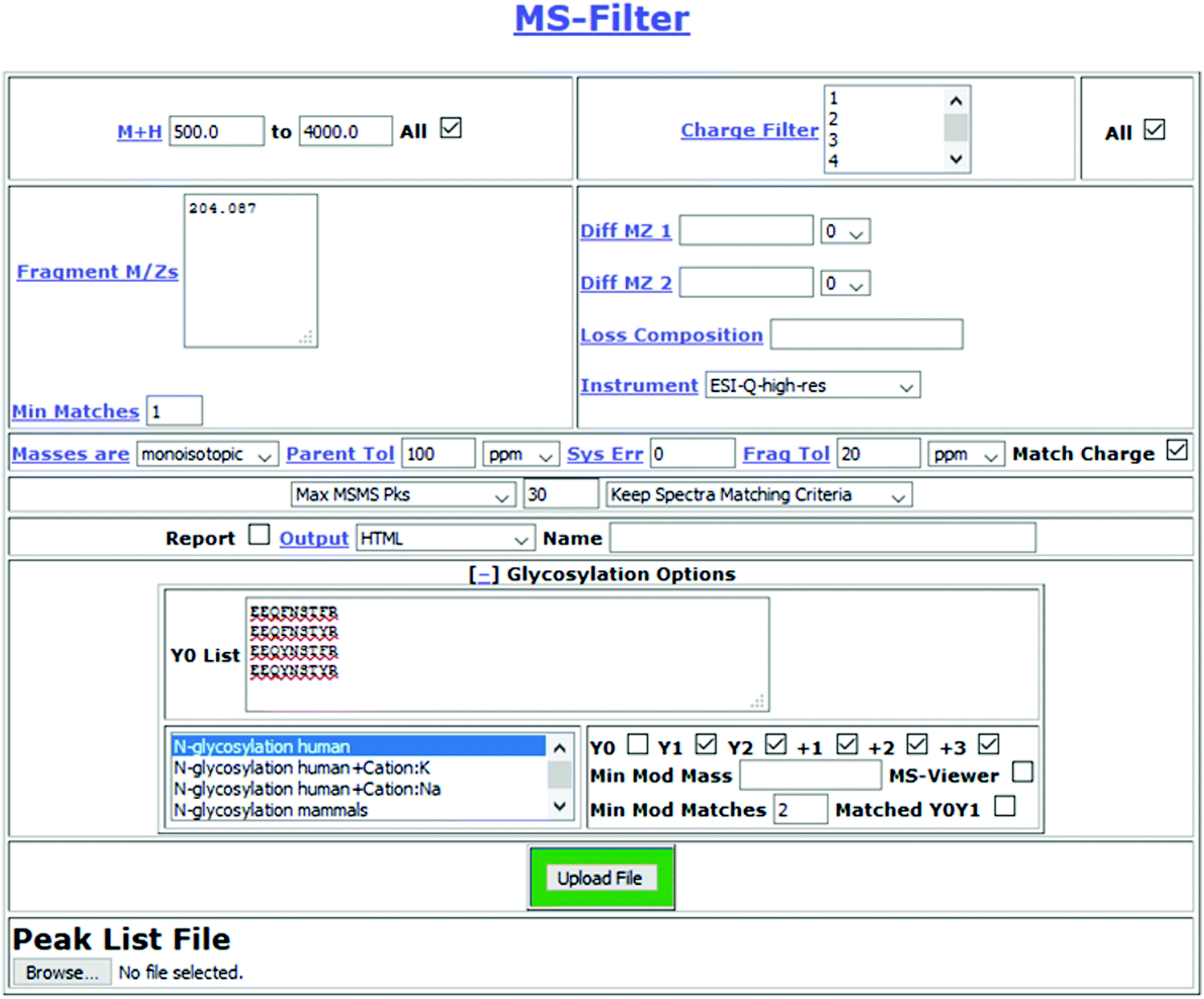 | ||
| Fig. 2 MS-Filter submission page. Features specific to glycosylation analysis are hidden as a default, but become visible after clicking on ‘[+] Glycosylation Options’. The input a list of candidate peptide sequences (optionally including non-glycosylation modifications), they select one or multiple sets of glycosylation sets to consider (from a list that is currently about twenty long), they specify which Y ion types to match and a minimum number to match. | ||
For evaluating the performance of Y ion filtering for analyzing an O-glycosylation data we used a single file from a large O-glycosylation dataset (60 files) derived from human urine that was produced by LWAC enrichment using WGA. Using Protein Prospector as the search engine, and considering the most common mucin-type structures, i.e. the asialo, mono- and disialo core-1, and the disialo core 2 oligosaccharides, 160 O-glycosylated peptides were identified in the entire dataset,5 but to simplify the evaluation we chose to focus only on the alphabetically first ten sequences. Of these, three were semi-tryptic peptides where a tryptic version was also observed, so these results represent seven glycosylation sites. In HCD O-linked glycopeptide spectra the Y0 and Y1 are generally very prominent. Hence, we evaluated requiring the HexNAc oxonium ion at m/z 204.087 and two Y ions (Y0 and Y1) to be present among the 5, 7, 10 or 15 most intense peaks. An initial MS-Filter analysis of this data indicated many mass modifications consistent with sodium salt adduct spectra, so we considered fully protonated and single sodium adduct glycan masses. A summary of the results is shown in Table 1. Considering the 15 most intense peaks returned 184 spectra, of which 60 produced a candidate glycopeptide assignment. At the other end of the scale, requiring the two Y ions to be among the five most intense peaks still reported a list of 54 spectra, the masses of 28 of which could be explained as potential glycoforms. The number of unique glycoforms assigned using only the top five peaks was only 14. This increased to 22 allowing for the sixth and seventh peak and went up to 29 with the top 10 and 33 considering the top 15. When using the top 10 or 15 peaks potential glycopeptide assignments for all ten considered peptide sequences were reported. Annotated spectra for the results using the top 10 peaks can be viewed through MS-Viewer14 using the search key yp7fdjrmu0.
| Filter (# peaks) | 15 | 10 | 7 | 5 |
| #Results | 184 | 143 | 95 | 54 |
| Suggested glycan spectra | 60 | 55 | 44 | 28 |
| Unique glycan assignments | 33 | 29 | 24 | 14 |
| Unique peptide sequences | 10 | 10 | 9 | 7 |
Spectrum 6780 was assigned in the top 10 and top 15 analyses only to the peptide ATAPTAQAPR + HexNAc2Hex2NeuAc2 + Cation:Na. Ions corresponding to Y0, Y0 + HexNAc, Y0 + HexNAcHex and Y0 + HexNAc2 were observed. However, several intense peaks including the base peak were observed between m/z 600–900 and were not immediately explained. These peaks correspond to doubly-charged Y ions with the sodium adduct still attached. The fully annotated spectrum is shown in Fig. 3. More Y ions can be matched to sodiated fragments than to protonated fragment ions.
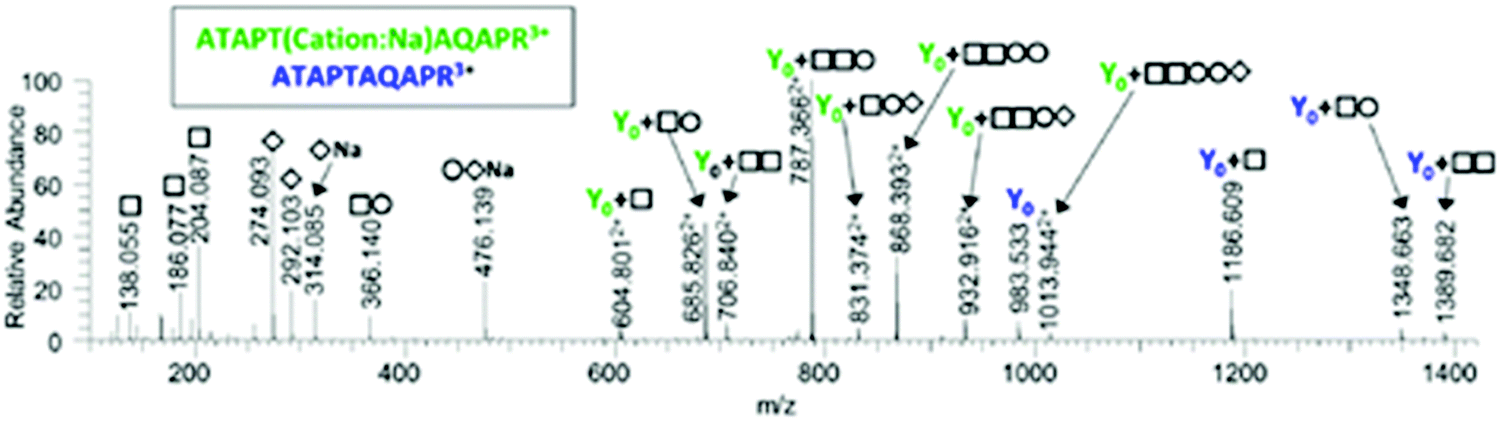 | ||
| Fig. 3 Annotated spectrum matched by MS-Filter to ATAPTAQAPR + HexNAc2Hex2NeuAc2 + Cation:Na. The sodium adduct Y ions are significantly more intense than the protonated fragments. | ||
Altogether 5 out of 145 spectra were solely assigned to sodium adducts of glycopeptides in the most permissive MS-Filter search. One of these spectra represented an unlikely glycan structure with weak Y0 and Y1 ions and no evidence for the presence of a sodium adduct (scan 11304 assigned to AVAVTLQSH + HexNAc3Hex3NeuAc6 + Na). Manual inspection of the other 4 spectra confirmed the glycan structures suggested by MS-Filter (scans 6780, 12028, 15872, 17651). However, lower relative abundance of protonated Y ions compared to sodiated Y ions meant these spectra were reported only when the more permissive filtering parameters were applied (Y0 and Y1 in the top 10 or 15 fragment ions). A similar experience was observed when analyzing the remaining datasets in this study (60 files): spectra assigned to peptides with metal ion adducts were disproportionately lost as the number of peaks considered was lowered.
We performed two database searches with the EThcD data of this single data file. When only the 3 most common mucin-type O-glycans (mono- and disialylated core-1 and disialylated core-2 glycans) were permitted 13 spectra were assigned to 8 out of the 10 peptides considered in the MS-Filter analysis, corresponding to 9 unique peptide + glycan combinations. Considering a ‘comprehensive’ O-glycan database 30 spectra were assigned: 9 of these corresponded to glycoforms already assigned in the ‘basic’ search, albeit the reported E-values were ∼10× higher; 21 PSMs were new but these represented only 7 of the peptide sequences used for MS-Filter analysis. These consisted of 13 unique peptide + glycan combinations. 9 out of 13 of these were reported by MS-Filter in the top 5 peak analysis; all 13 in the top 15 peak analysis. The 5 peak filtering reported five glycoforms not identified by the database search; the 15 peak analysis reported 20 more. Many of these are probably correct. For example, HexNAc2Hex2NeuAc4 was identified on all three cleavage forms of the peptide from Glycophorin-A. MS-Filter additionally matched HexNAc3Hex3NeuAc6 on AHEVSEISVRTVYPPEE (scan 15243, m/z 1196.7310, z = 4), which would correspond to an additional HexNAcHexNeuAc2 (one of the most common O-glycoforms on serum proteins), so this is probably evidence of an additional glycosylation site in the peptide.
For evaluating the performance of MS-Filter for analysis of complex N-linked glycosylation data we chose to use a dataset created for the Human Glycoproteomics Initiative study, where human serum was enriched for glycopeptides using HILIC chromatography, then the same sample was analyzed by six different combinations of fragmentation and measurement, including resonant excitation CID, beam-type CID (HCD), ETciD or EThcD, with fragments measured in either a quadrupole ion trap or orbitrap. In our submission to this study we analyzed the EThcD data using Protein Prospector and reported 574 non-redundant N-linked glycopeptides and 195 O-linked glycopeptides. For the evaluation of MS-Filter we chose to re-search the EThcD data only considering N-linked glycosylation, as we wanted to evaluate the value of Y1 and Y2 ions for this type of glycopeptides. We performed two searches; one allowing for incorrect monoisotopic peak assignment; the other not. The former search was designed to try to produce a longer list of potential glycopeptides and identified 260 glycosylated sequences for a total of 712 unique glycopeptides; the latter reported 248 glycosylated sequences but 751 unique glycopeptides, and was used for the subsequent comparison to the MS-Filter results (see below). These numbers are higher than those reported in our initial analysis, but the searches differ significantly due to not considering O-glycosylation in the newer searches. We previously reported a similar effect where increased glycopeptide IDs were reported when not considering O-glycosylation in a sample that contained both N- and O-glycosylation.15 We used the list of the 260 peptide sequences identified as the input list of peptides in MS-Filter for analyzing the HCD peak list file, where the only other modification permitted was fixed carbamidomethylation of cysteines. The HCD and EThcD were acquired in the same run and on an identical list of precursors, so there are corresponding HCD and EThcD spectra for every precursor fragmented, allowing comparison of results.
MS-Filter was used to return spectra containing a m/z 204.087 peak (HexNAc oxonium ion) and either just a Y1 or matching two peaks among different charges states of Y1 (peptide + HexNAc) or Y2 (peptide + HexNAc2). As, in our experience, these ions are typically not as intense as Y0 in O-linked data, these peaks had to match in either the 10, 20, 30 or 40 most intense peaks in the spectrum. In these analyses we chose not to allow for incorrect monoisotopic peak assignment, as we have found this leads to excessive levels of incorrect glycan assignments, so we compared all MS-Filter results to those from the database search that did not consider incorrect monoisotopic peak labeling. An initial MS-Filter search indicated prevalent potassium adducts, so these were also considered on all the glycans. A summary of the MS-Filter parameter analysis is presented in Table 2.
| Filter #peaks_# Y ions | 20_Y1 | 10_Y1 | 40_2 | 30_2 | 20_2 | 10_2 | 30_2_Cation:K |
|---|---|---|---|---|---|---|---|
| #Results | 3498 | 2474 | 1670 | 1351 | 827 | 405 | 1840 |
| #Spectra | 2674 | 2150 | 1466 | 1244 | 799 | 404 | 1568 |
| Suggested glycan spectra | 623 | 547 | 402 | 364 | 315 | 209 | 495 |
| Unique suggested glycans | 389 | 330 | 256 | 230 | 198 | 134 | 338 |
| Unique peptide sequences | 196 | 174 | 79 | 72 | 63 | 46 | 128 |
| Overlap with EThcD results | 138 | 120 | 84 | 77 | 67 | 49 | 111 |
There are 9776 HCD spectra in the file where HCD and EThcD spectra were acquired, of which 6464 contain a m/z 204.087 peak among the twenty most intense peaks. When the m/z 204 peak and Y1 ion for one of the 260 potential peptides was required to be present in the 20 most intense peaks in a spectrum 3498 results were returned, although these corresponded to only 2674 spectra; i.e. on average about a third of the spectra matched contained masses for two Y1 ions, indicating a significant level of false positive matches. Only 138 of these spectra correspond to glycopeptide identifications from the matching EThcD results. Reducing the number of peaks considered for matching the Y1 ion and m/z 204 to the 10 most intense peaks reduced the number of results to 2474, corresponding to 2150 spectra. Some of these spectra containing two Y ions could be products of mixture spectra, but there are still many false positives, caused by only requiring to match Y1 and considering a large number of peptides.
Requiring matching of two Y ions is a much more powerful filter. Needing two among the 40 most intense peaks to be either Y1 or Y2 led to 1670 spectra being kept, of which 402 (24%) corresponded to mass shifts consistent with a glycan. Reducing the number of peaks considered led to steady reductions in spectra but increases in the percentage of spectra whose mass could be postulated as a glycoform. At the highest stringency (matching two peaks among the 10 most intense) 52% (209 out of 405) of all matched spectra produced a glycoform assignment, but the number of candidate glycoforms halved compared to the 40 peak results. When requiring two peaks matched among the top 40, 84 out of the 256 postulated unique glycopeptides were identified in the corresponding EThcD database search results. These values dropped to 46 out of 134 when considering only the ten most intense peaks. In each case the overlap with the EThcD is only about a third of the results.
Visually comparing the lists of identified glycopeptides in the database search and from MS-Filter the standout difference is the percentage of identifications that contain a potassium salt adduct. Of the 835 glycopeptide spectra identifications from the database searching 406 (49%) are potassium adduct species, whereas for example in the search requiring two Y ion matches in the top 30 peaks only 14 out of 228 (6%) feature a salt adduct. This result suggests that HCD spectra of salt adduct species do not as consistently produce the expected prominent Y ions, which is consistent with the observation in the O-linked results as illustrated in Fig. 3, where sodiated Y ions were more intense and Y and oxonium ions were observed both protonated and sodiated. Hence, we tried an additional MS-Filter search where for each considered peptide we added an additional version with a fixed potassium adduct on the C-terminus, such that the Y ions searched for would be shifted by this mass. The results of this search, when requiring two Y ions to be matched among the top 30 peaks, are the final column in Table 2, and annotated spectra of these results can be viewed using MS-Viewer14 with the Search Key edorrpnirx. This search returned about 25% more results than the equivalent search without the potassium peptide modifications, which led to 110 more unique suggested glycans (338 instead of 228), and 44% higher overlap in results with the EThcD database search results (111 instead of 77). 42 out of the 338 unique glycopeptides were reported in MS-Filter with the expected Y ions and in a separate spectrum with the potassium adduct Y ions. There was only one spectrum that was returned by MS-Filter matching both the protonated and potassium-adduct Y ions; this is shown in Fig. S1 (ESI†). There were eight results where a glycopeptide was identified with a potassium adduct on both the peptide and glycan (i.e. double adduct species). Of these, for four of them there were alternative interpretations for the spectra, and none of them were convincing assignments.
Some of the most abundant glycopeptides in serum are those of the conserved N-glycosylation site in the core of IgG. These peptides have the sequence EEQxNSTxR, where ‘x’ can be either F or Y depending on the IgG isoform. In the search allowing for peptide potassium adducts and requiring two Y ions to be matched among the 30 most intense peaks MS-Filter returned 103 results to these peptides. MS-Filter suggested 22 defined unique glycopeptides, whereas the semi-tryptic database search reported 28 glycoforms from EThcD data, with an overlap of 17 glycopeptides. Hence, together they are reporting 33 glycoforms. Looking at the corresponding EThcD spectra for the extra assignments in the MS-Filter results, for all five the database search reported the corresponding glycopeptide as the top ranked identification, but the result was below the 1% FDR threshold.
Of the extra identifications from database searching, eight out of eleven were to potassium salt adducts of glycopeptides. Even though the search considered both regular Y ions and potassium adduct versions these glycopeptides were not identified. Looking at the corresponding HCD spectra for these identifications, six of the eight contained normal Y1 ions but of low intensity and most were weak spectra. The quality of these spectra suggests that the salt adduct glycopeptides do not fragment as readily upon collisional activation as the protonated equivalents, which has previously been observed for peptides.16 Fig. S2 (ESI†) shows the HCD spectrum corresponding to one of the precursors identified by EThcD. The EThcD spectrum was reported as either EEQYN(HexNAc4Hex5FucNeuAc)STYR or EEQFN(HexNAc4Hex6NeuAc)STYR; i.e. the data did not allow distinguishing between whether the extra oxygen was in the peptide (tyrosine contains an extra oxygen compared to phenylalanine) or in the glycan (fucose is a deoxyhexose). Although the HCD spectrum is extremely weak, it clearly contains a m/z 1392.59 peak, which corresponds to the Y1 ion of the peptide sequence containing two tyrosines. Therefore, the HCD clears up the ambiguity in the EThcD assignment.
46 of the 103 reported MS-Filter matches to the IgG glycopeptides corresponded to mass modifications above 3000 Da. Although a few of these can be assigned to tetraantennary complex glycans we believe that the majority of these results represent false positives. Several of these reported large modifications are due to incorrect precursor and charge state determination. For example, the precursor for scan 4285 was reported as m/z 1187.269 5+, whereas the actual precursor was m/z 1187.476 2+, which corresponds in mass to EEQFN(HexNAc2Hex5)STFR (see Fig. S3, ESI†).
Discussion
In this work we have developed new software that allows one to filter a peak list file for the presence of Y ions from glycopeptides of interest. The software suggests candidate glycopeptides based on calculation of the mass difference between the unmodified peptide and observed precursor ion. We show that the software is effective for analysis of HCD data from both O- and N-linked glycopeptide data, but the optimal parameters for each differ due to Y ions being more prominent in O-linked spectra than in N-linked glycopeptide data. For N-linked data we evaluated requiring matching of one or two Y ions and found that for complex mixture analysis matching one peak happens too frequently at random for it to be a reliable filter. Requiring two peak matches was much more appropriate, although there were still a lot of spectra that were matched but could not be described. Some of the larger of these reported modifications could be explained by errors in monoisotopic peak and charge state labeling, but there was still a significant false positive level. For this reason, such an analysis on its own is not reliable enough. However, we think it is a very effective approach to produce a focused list of candidate spectral assignments that could then be followed up on manually, and the ability to upload the results to MS-Viewer makes the further analysis easier. We have incorporated glycan Y ion and oxonium ion labeling into the Protein Prospector package to facilitate this type of analysis.When comparing MS-Filter results to those from database searching of corresponding EThcD data we discovered significant under-representation of spectra of metal ion adduct species, particularly in the N-linked data. Further investigation showed that fragmentation spectra of metal ion adduct glycopeptides regularly produce metal ion adduct Y ions. This was slightly surprising, as the increased prevalence of metal ion adducts of glycopeptides in comparison to unmodified peptides would imply that the metal ion associates with the glycan, and previous studies of sodiated glycopeptides have suggested that the metal ion normally binds to the glycan portion of the molecule.17 However, in our analyses we observe metal ions attached to the peptide, the glycan or both. When MS-Filter considered such mass-shifted Y ion series it led to a third more suggested glycopeptide identifications and much greater overlap with the corresponding search engine results. This indicates that if metal ion adducts are prevalent (which in our experience is relatively common in glycopeptide datasets), then metal ion adduct Y ions should be considered in order to get more comprehensive coverage. However, it should be noted that there were metal ion-adduct glycopeptides identified only by matching the protonated Y ions, so the propensity to form one or the other is presumably influenced by factors such as the peptide sequence or glycan type. It should also be recognized that metal ion adducts do not as readily fragment in HCD as the fully protonated equivalents, so even when considering both series of Y ions there will still be many metal ion adduct spectra that are not reported.
The overlap between MS-Filter results from the HCD data and database search results of the matched EThcD data was relatively low; only about a third of MS-Filter assignments were reported in the search results. This is a much lower overlap than between database search results of the two sets of spectra, where just over sixty percent of the HCD spectrum search results were in the EThcD results (not shown). The lower overlap is expected because the MS-Filter results are primarily driven by how well the glycan fragments, whereas the database search results are relying on peptide backbone fragmentation.
A more detailed analysis of the results matching IgG peptides showed that for relatively abundant glycopeptides in a complex mixture an increase in glycoforms can be achieved through the use of MS-Filter to find Y ions. The analysis of the IgG peptides also highlights other advantages and disadvantages of the Y ion filtering approach. On the one hand, using mass alone it is unable to differentiate between EEQFNSTYR and EEQYNSTFR, leading to multiple results to the same spectrum. However, the matching of the Y ions is able to distinguish between whether an extra oxygen is in the peptide (Y vs. F) or in the glycan (Hex vs. Fuc), as illustrated in Fig. S2 (ESI†). For exactly the same reason MS-Filter can clear up the ambiguity of some Met-containing glycopeptide assignments, pointing out whether the peptide or the glycan features the extra oxygen. It also has been reported that Met residues may get carbamidomethylated, and this may lead to glycoform misassignments.18 However, filtering for the unmodified peptide will not identify these MS/MS spectra as potential different glycoforms due to the mass-shifted Y ions.
The MS-Filter analysis of the O-linked data had a bigger impact on identifying reliable extra glycoforms than the N-linked data, where using the top 15 peaks there were 33 unique glycopeptides reported compared to only 13 in the database search. There are multiple factors that probably contributed to this. The diagnostic Y ions are typically relatively more intense in O-linked spectra, so are more consistently matched. However, the major factor is probably a lack of ‘sensitivity’ of the database searching; by considering semi-tryptic peptides due to protease activity in body fluids, and also considering a large number of glycoforms, a lot of the results drop below the confidence acceptance threshold. For N-linked glycosylation, where one can impose a glycosylation motif for modification, only a small subset of peptides are considered glycosylated. However, as no such motif exists for O-linked glycosylation, and serines and threonines are prevalent, the search expansion when considering O-glycosylation is dramatic, so results become statistically much less confident. One way to address this is through the use of a site database,15 but a comprehensive library of O-linked sites does not currently exist.
It should be noted that many of the O-glycoforms reported by MS-Filter are not the products of single glycosylation sites; they are combined compositions from multiple glycosylations within the peptide, which is common for O-glycosylation. Determining the number of glycans that add up to the composition observed and the sites they are attached to is a challenging process. It requires observation of peptide backbone fragment ions with the glycan remaining attached, so can only be achieved in ETxD data, and even then the desired fragments may not be present. MS-Filter does not help address this issue directly, but as it can quickly suggest glycan compositions and the presence of other unpredicted modifications such as metal adducts, and this information could be used for more targeted database searching considering permutations of glycans that sum to the observed modification. We think using MS-Filter to derive a list of glycoforms present is a more powerful approach than only targeting an expected list of glycoforms as neutral losses during database searching, which we have published doing previously19 and another more recent publication performed on a larger scale.20 Database searching of O-linked glycopeptide data allowing for large numbers of glycoforms and multiple modifications per peptide can be very slow, but in a targeted search, such as using a list of accession numbers of proteins identified, or restricting the glycans considered based on MS-Filter results, this type of analysis is relatively quick.
Many mass modifications were reported that do not correspond to glycan masses. Errors in monoisotopic peak labeling and charge state determination may lead to reporting incorrect mass differences. Glycopeptides, especially N-linked glycopeptides, are generally of quite high mass: the average mass of the glycopeptides identified by Protein Prospector in the HGI dataset search was 4678 Da (the range was from 2456 Da to 9092 Da). Hence, the monoisotopic peak can often be low intensity, so the second or even third isotope may be labeled. In a complex mixture there are also quite often overlapping isotope clusters and so the software will sometimes conflate peaks from different clusters to report an incorrect charge state (e.g. see Fig. S3, ESI†).
MS-Filter occasionally returned multiple glycopeptide suggestions for some masses, creating ambiguity. Calculating the mass accuracy measurement of the precursor with these potential glycoforms one could predict which glycoform is more likely. However, we allowed 20 ppm tolerance on the mass modification observed because the software is calculating the mass accuracy of the mass modification rather than the whole precursor species, so if for example the peptide and glycan both had similar masses, then the relative error on the mass modification measurement will be roughly double that on the precursor measurement.
In theory one could use MS-Filter on EThcD or ETcID data, as these produce Y ions. However, in our hands the observation of small Y ions in EThcD data of glycopeptides is inconsistent (larger Y ions are more common), and when observed it is not one of the most intense peaks. Hence, we think for most EThcD data there will be too many spectra that do not pass the filtering to make it worthwhile employing, although potentially if one used an unusually high HCD collision energy in the EThcD it could be useful.
The higher number of identifications from the database searching supports the notion that EThcD data is more information-rich than HCD data. For complex glycopeptide mixture analysis the results here suggest that Y ion filtering may be effective as a way to boost the confidence of borderline matched EThcD results. Indeed, for all of the extra identifications reported by MS-Filter for the IgG peptides in the N-linked data the search engine reported the identification as the top match, but the result did not pass the FDR threshold employed. MS-Filter may also be effective at identifying spectra where there are errors in the monoisotopic peak or charge state assignment.
It would be desirable if MS-Filter could attach some sort of score to a given result to flag which are better matches. We employed a threshold minimum number of Y ions to match, but some spectra matched more than the minimum; for example, in the N-linked data there were several spectra matching both Y1 and Y2 in two different charge states. These results were more likely to be explained with a mass shift of a glycan, but not all could be explained. A good scoring system would probably need to account for (relative) peak intensity of Y ions in addition to the number matched. One could also use glycan oxonium ions to support glycan assignments, but in enriched glycopeptide datasets this is problematic in our experience, as for normally intense oxonium ions such as those from NeuAc, they are regularly observed as background in spectra of non-sialylated peptides. However, a lack of observation of NeuAc-containing oxonium ions seems to be a reliable parameter for excluding sialylated assignments. A future development will be to try to match peptide backbone fragment ions to provide a score associated with the peptide sequence identification. This can already be indirectly done, as the output from MS-Filter can be uploaded to MS-Viewer (as was done for two of the sets of results in this study), and from there one can search individual spectra, but it would be preferable to automatically calculate these scores.
Availability
MS-Filter, MS-Viewer and all other programs in the Protein Prospector package are available on the web at http://prospector.ucsf.edu, and the package can also be downloaded and installed locally for free for academic use.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. J. C. and P. R. B. were supported by funding from the Adelson Medical Research Foundation. K. F. M., Zs. D. and A. P. were supported by the following grants: the Economic Development and Innovation Operative Programmes GINOP-2.3.2-15-2016-00001, and GINOP-2.3.2-15-2016-00020 from the Ministry for National Economy.References
- B. Domon and C. E. Costello, A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates, Glycoconjugate J., 1988, 5, 397–408 CrossRef CAS.
- S.-W. Wu, T.-H. Pu, R. Viner and K.-H. Khoo, Novel LC-MS2 product dependent parallel data acquisition function and data analysis workflow for sequencing and identification of intact glycopeptides, Anal. Chem., 2014, 86, 5478–5486 CrossRef CAS PubMed.
- J. Nilsson, Liquid chromatography-tandem mass spectrometry-based fragmentation analysis of glycopeptides, Glycoconjugate J., 2016, 33, 261–272 CrossRef CAS PubMed.
- K. F. Medzihradszky, K. Kaasik and R. J. Chalkley, Characterizing sialic acid variants at the glycopeptide level, Anal. Chem., 2015, 87, 3064–3071 CrossRef CAS PubMed.
- Z. Darula, Á. Pap and K. F. Medzihradszky, Extended Sialylated O-Glycan Repertoire of Human Urinary Glycoproteins Discovered and Characterized Using Electron-Transfer/Higher-Energy Collision Dissociation, J. Proteome Res., 2019, 18, 280–291 CAS.
- H. Yang, C. Yang and T. Sun, Characterization of glycopeptides using a stepped higher-energy C-trap dissociation approach on a hybrid quadrupole orbitrap, Rapid Commun. Mass Spectrom., 2018, 32, 1353–1362 CrossRef CAS PubMed.
- Q. Yu, B. Wang, Z. Chen, G. Urabe, M. S. Glover, X. Shi, L.-W. Guo, K. C. Kent and L. Li, Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization, J. Am. Soc. Mass Spectrom, 2017, 28, 1751–1764 CrossRef CAS PubMed.
- Y. Zhang, X. Xie, X. Zhao, F. Tian, J. Lv, W. Ying and X. Qian, Systems analysis of singly and multiply O-glycosylated peptides in the human serum glycoproteome via EThcD and HCD mass spectrometry, J. Proteomics, 2018, 170, 14–27 CrossRef CAS PubMed.
- Z. Chen, Q. Yu, L. Hao, F. Liu, J. Johnson, Z. Tian, W. J. Kao, W. Xu and L. Li, Site-specific characterization and quantitation of N-glycopeptides in PKM2 knockout breast cancer cells using DiLeu isobaric tags enabled by electron-transfer/higher-energy collision dissociation (EThcD), Analyst, 2018, 143, 2508–2519 RSC.
- B. L. Parker, M. Thaysen-Andersen, N. Solis, N. E. Scott, M. R. Larsen, M. E. Graham, N. H. Packer and S. J. Cordwell, Site-specific glycan-peptide analysis for determination of N-glycoproteome heterogeneity, J. Proteome Res., 2013, 12, 5791–5800 CrossRef CAS PubMed.
- M. Saraswat, S. Joenväära, L. Musante, H. Peltoniemi, H. Holthofer and R. Renkonen, N-linked (N-) Glycoproteomics of Urimary Exosomes*, Mol. Cell. Proteomics, 2015, 14, 263–276 CrossRef CAS PubMed.
- K. Cheng, R. Chen, D. Seebun, M. Ye, D. Figeys and H. Zou, Large-scale characterization of intact N-glycopeptides using an automated glycoproteomic method, J. Proteomics, 2014, 110, 145–154 CrossRef CAS PubMed.
- R. J. Chalkley, P. R. Baker, K. F. Medzihradszky, A. J. Lynn and A. L. Burlingame, In-depth analysis of tandem mass spectrometry data from disparate instrument types, Mol. Cell. Proteomics, 2008, 7, 2386–2398 CrossRef CAS PubMed.
- P. R. Baker and R. J. Chalkley, MS-viewer: a web-based spectral viewer for proteomics results, Mol. Cell. Proteomics, 2014, 13, 1392–1396 CrossRef CAS PubMed.
- R. J. Chalkley and P. R. Baker, Use of a glycosylation site database to improve glycopeptide identification from complex mixtures, Anal. Bioanal. Chem., 2017, 409, 571–577 CrossRef CAS PubMed.
- M. Hamdan and O. Curcuruto, Collision-induced dissociation of some protonated peptides with and without mass selection, Rapid Commun. Mass Spectrom., 1994, 8, 274–279 CrossRef CAS PubMed.
- R. R. Seipert, E. D. Dodds, B. H. Clowers, S. M. Beecroft, J. B. German and C. B. Lebrilla, Factors That Influence Fragmentation Behavior of N-Linked Glycopeptide Ions, Anal. Chem., 2008, 80, 3684–3692 CrossRef CAS PubMed.
- Z. Darula and K. F. Medzihradszky, Carbamidomethylation Side Reactions May Lead to Glycan Misassignments in Glycopeptide Analysis, Anal. Chem., 2015, 87, 6297–6302 CrossRef CAS PubMed.
- Z. Darula, R. J. Chalkley, P. Baker, A. L. Burlingame and K. F. Medzihradszky, Mass spectrometric analysis, automated identification and complete annotation of O-linked glycopeptides, Eur. J. Mass Spectrom., 2010, 16, 421–428 CrossRef CAS PubMed.
- J. Mao, X. You, H. Qin, C. Wang, L. Wang and M. Ye, A New Searching Strategy for the Identification of O-Linked Glycopeptides, Anal. Chem., 2019, 91, 3852–3859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mo00178f |
| This journal is © The Royal Society of Chemistry 2020 |