
Recombinant mucin-type proteins carrying LacdiNAc on different O-glycan core chains fail to support H. pylori binding†
Yolanda H.
Mthembu
 *a,
Chunsheng
Jin
b,
Médea
Padra
b,
Jining
Liu
a,
Johan Olofsson
Edlund
c,
Hanyue
Ma
d,
Janos
Padra
b,
Stefan
Oscarson
d,
Thomas
Borén
c,
Niclas G.
Karlsson
b,
Sara K.
Lindén
b and
Jan
Holgersson
a
*a,
Chunsheng
Jin
b,
Médea
Padra
b,
Jining
Liu
a,
Johan Olofsson
Edlund
c,
Hanyue
Ma
d,
Janos
Padra
b,
Stefan
Oscarson
d,
Thomas
Borén
c,
Niclas G.
Karlsson
b,
Sara K.
Lindén
b and
Jan
Holgersson
a
aDepartment of Laboratory Medicine, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, SE-41345 Gothenburg, Sweden
bDepartment of Medical Chemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, SE-40530 Gothenburg, Sweden
cDepartment of Medical Biochemistry and Biophysics, Umeå University, 901 87 Umeå, Sweden
dSchool of Chemistry Science Centre, Conway Institute of Biomolecular & Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland
First published on 30th March 2020
Abstract
The β4-N-acetylgalactosaminyltransferase 3 (B4GALNT3) transfers GalNAc in a β1,4-linkage to GlcNAc forming the LacdiNAc (LDN) determinant on oligosaccharides. The LacdiNAc-binding adhesin (LabA) has been suggested to mediate attachment of Helicobacter pylori to the gastric mucosa via binding to the LDN determinant. The O-glycan core chain specificity of B4GALNT3 is poorly defined. We investigated the specificity of B4GALNT3 on GlcNAc residues carried by O-glycan core 2, core 3 and extended core 1 precursors using transient transfection of CHO-K1 cells and a mucin-type immunoglobulin fusion protein as reporter protein. Binding of the LabA-positive H. pylori J99 and 26695 strains to mucin fusion proteins carrying the LDN determinant on different O-glycan core chains and human gastric mucins with and without LDN was assessed in a microtiter well-based binding assay, while the binding of 125I-LDN–BSA to various clinical H. pylori isolates was assessed in solution. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) and western blotting confirmed the requirement of a terminal GlcNAc for B4GALNT3 activity. B4GALNT3 added a β1,4-linked GalNAc to GlcNAc irrespective of whether the latter was carried by a core 2, core 3 or extended core 1 chain. No LDN-mediated adhesion of H. pylori strains 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 695 and J99 to LDN determinants on gastric mucins or a mucin-type fusion protein carrying core 2, 3 and extended core 1 O-glycans were detected in a microtiter well-based adhesion assay and no binding of a 125I-labelled LDN–BSA neoglycoconjugate to clinical H. pylori isolates was identified.
695 and J99 to LDN determinants on gastric mucins or a mucin-type fusion protein carrying core 2, 3 and extended core 1 O-glycans were detected in a microtiter well-based adhesion assay and no binding of a 125I-labelled LDN–BSA neoglycoconjugate to clinical H. pylori isolates was identified.
Introduction
Helicobacter pylori (H. pylori) is a Gram-negative bacterium1 causing gastritis, gastric and duodenal ulcers, and gastric cancer.2 Environmental factors and host-microbe characteristics influence susceptibility to H. pylori-induced disease. It infects half of the world's population often already in childhood.3H. pylori can survive in the acidic milieu of the stomach and can induce a chronic inflammation of the underlying gastric mucosa, which subsequently may lead to gastric carcinogenesis.4 It is estimated to be responsible for approximately 65% of all gastric cancers.5H. pylori carries carbohydrate-binding adhesins which enable them to bind to the epithelial cells of the gastric mucosa. The blood group antigen-binding adhesin (BabA) recognizes the Leb and H type-1 determinants,6 while the sialic acid-binding adhesin (SabA) recognizes sialylated oligosaccharides, for example sLex.7 A third, carbohydrate-binding adhesin, LabA, was recently described and was shown to bind the LacdiNAc (N,N′-diacetyllactosdiamine/GalNAcβ4GlcNAc, LDN) determinant.8 Highly glycosylated glycoproteins called mucins are covering the mucosal surfaces, both as the major component of the secreted mucus and as abundant components of the glycocalyx.9,10 Both secreted and membrane-bound gastric mucins can support attachment of H. pylori if glycan determinants that the H. pylori strains have adhesins for are present on the mucins.10 Binding of H. pylori to mucins that act as decoys can limit the access to determinants on the epithelial cells mediating binding.11,12LDN is a unique terminal structure in the outer chains of N- and O-glycans.13,14 LDN has been shown to be expressed at epithelial luminal surfaces of different organs like heart, colon, small intestines and salivary glands, and particularly on the epithelial surface of the stomach. It has also been identified by LC-MS/MS on MUC5AC, a secreted mucin present on the gastric mucosal lining.8,14 The presence of LacdiNAc was also examined among human gastric tissues and was expressed more deeply in pyloric glands while limited to the surface of the fundic mucosa and absent in cardiac glands.8 The LDN determinant has been suggested to regulate cancer stemness and invasiveness through modification of EGFR glycosylation and activity.15 The expression of LDN is high on colon cancer cells in vitro, and promotes increased tumor growth and metastasis in vivo.16 It is synthesized by β4-N-acetylgalactosaminyltransferase 3 (B4GALNT3) and 4 (B4GALNT4), which transfer GalNAc in a β1,4-linkage to GlcNAc forming the LDN determinant on N- and O-glycans.17,18 Their expression patterns are different; B4GALNT3 is predominantly expressed in stomach, colon and testis, whereas B4GALNT4 is predominantly expressed in ovary, fetal and adult brain, and fetal kidney and lung.17,18 The substrate specificity in vitro of B4GALNT3 and 4 was determined, and both enzymes were shown to prefer the O-glycan core 6 over core 2 and 3.17
Binding of H. pylori to gastric mucosa is mediated by the mucin O-GalNAc type glycans. The initiating step of O-GalNAc glycosylation is catalyzed by a large family of polypeptide GalNAc-transferases (ppGalNAc-Ts) and involves the addition of GalNAc to the hydroxyl group in serine or threonine.19 Three distinct regions are recognized in O-linked glycans and include the two or three innermost sugar residues nearest the peptide chain constituting the core region, the backbone region contributing to O-glycan chain length, and the terminal region with its bioactive determinants.20 The determinants are often sialylated, sulfated, acetylated and/or fucosylated.21 At least eight different core chain types, of which cores 1–4 are more common than the rare cores 5–8, have been identified in mammalian glycoproteins. All are based on the innermost α-GalNAc residue, which is further substituted at the C3, C6 or both positions.
The O-glycan core 1 is catalyzed by the ubiquitously expressed core 1 β1,3galactosyltransferase (B3GALT1), which adds galactose in a β1,3-linkage to the GalNAc residue.22 The core 2 structure is produced by the addition of an N-acetylglucosamine (GlcNAc) in a β1,6-linkage to the GalNAc of the core 1 structure; a reaction catalyzed by β1,6-N-acetylglucosaminyltransferases (GCNT1-3).23 The core 3 structure is generated upon addition of a GlcNAc in a β1,3-linkage to the innermost GalNAc. This step is catalyzed by a β3-N-acetylglucosaminyltransferase 6 (B3GNT6) and competes with the B3GALT1.24 A core 4 structure is generated when a GlcNAc residue is added in a β1,6-linkage to GalNAc of a core 3 structure by the GCNT2 enzyme.25 The core 3 and core 4 O-glycans are expressed in a more tissue-specific manner than the more abundant core 1 and core 2 structures. Besides structures based on core 1–3, structures based on extended core 1 (GlcNAcβ3Galβ3GalNAcα1-O-Ser/Thr) was investigated as to whether it could serve as a substrate for B4GALNT3 and, thus, carry LDN. Extended core 1 is generated by β3-N-acetylglucosaminyltransferase 3 (B3GNT3).26 As mentioned above, the core structures are then elongated, usually by repetitive Galβ4GlcNAc disaccharide (lactosamine) units, and terminally modified to generate complex O-linked glycans that are involved in a variety of biological processes.
A previous study showed that replacing fucosylated determinants with LDN determinants on gastric mucosa glycans prevented H. pylori binding,14 while others have reported a specific adhesion of H. pylori to LDN motifs located on MUC5AC mucins.8 To investigate this further, we engineered recombinant mucins carrying the LDN determinant on different core chains and assessed their capability to support adhesion of H. pylori strains claimed to carry the LabA adhesin. We show that B4GNT3 generated the LDN determinant on core 2, 3 and extended core 1 O-glycans, and that none of these structures supported binding of H. pylori in a LDN-dependent manner.
Materials and methods
Construction of expression plasmids
The B4GALNT3 cDNA in the expression vector pEZ-M67 was obtained from GeneCopoeia, Inc. (Rockville, MD, USA). The B4GALNT3 cDNA was amplified using QIAfilter Plasmid Mega Kit, (QIAGEN, Venlo, The Netherlands). Additional expression plasmids used to engineer the LDN determinant on different O-glycan core chains carried by the P-selectin glycoprotein ligand-1/mouse IgG2b (PSGL-1/mIgG2b) mucin-type fusion protein following expression in CHO-K1 cells were generated in the laboratory and are listed in Table 1.| Constructs | Glycosyltransferases | GENE | Selection drug | Ref. |
|---|---|---|---|---|
| PSGL-1/mIgG2b | Human PSGL-1 fused with the Fc fragment of mouse IgG2b | PSGL-1/mIgG2b | Puromycin | 51 |
| Extended C1 β3GnT3 | β-1,3-Galactosyl-O-glycosyl-glycoprotein β-1,3-N-acetylglucosaminyltransferase (EC 2.4.1.146) | B3GNT3 | Zeocin | 28 |
| C2 β6GnT1 | β-1,3-Galactosyl-O-glycosyl-glycoprotein β-1,6-N-acetylglucosaminyltransferase 1 (EC 2.4.1.102) | GCNT1 | Neomycin | 37 |
| C3 β3GnT6 | Acetylgalactosaminyl-O-glycosyl-glycoprotein β-1,3-N-acetylglucosaminyltransferase (EC 2.4.1.147) | B3GNT6 | Hygromycin | 52 |
| β4GnT3 | β1,4-N-Acetylgalactosaminyltransferase 3 | GALNT3 | Hygromycin | This study |
Glyco-engineering of CHO cells to reproduce GalNAcβ1,4GlcNAcβ1 (LacdiNAc; LDN) determinants on different O-glycan core chains of PSGL-1/mIgG2b
CHO-K1 cells (ATCC, Manassas, VA, USA) were seeded in 75 cm2 T-flasks (Nunc, Roskilde, Denmark) in Dulbecco's Modified Eagle's medium (DMEM, Lonza Group Ltd., Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS; Invitrogen AB, Stockholm, Sweden) and 100 units per mL penicillin and 100 μg mL−1 streptomycin (Invitrogen). The cells were maintained at 37 °C and 5.0% CO2 in a humidified incubator. CHO-K1 cells were transiently transfected 24 hours post-seeding at a cell confluency of 70–80%. The total concentration of plasmids encoding glycosyltransferases used for each transfection of 75 cm2 T-flask using the Lipofectamine 2000 Transfection Reagent Kit (Invitrogen AB) was 24 μg ensuring that equal concentrations were used (12 μg per plasmid for double transfections and 8 μg per plasmid for triple transfections.)A mucin-type fusion protein, PSGL-1/mIgG2b, with O-glycans carrying LDN determinants was generated. To achieve that, CHO cells in each 75 cm2 T-flask were transfected with plasmids encoding PSGL-1/mIgG2b and B4GALNT3 together with plasmids encoding glycosyltransferases supporting the generation of core 2, core 3 or extended core 1 O-glycans, respectively. Twenty-four hours post-transfection, the cell culture medium was changed to serum-free ProCHO-4 medium (Lonza, Basel, Switzerland) supplemented with 2 mM L-glutamine and 100 units per mL penicillin and 100 μg mL−1 streptomycin. The supernatants containing expressed proteins were collected 7–10 days post-incubation in serum-free medium, and cells were removed by centrifugation at 5000 × g, 30 min.
Purification of secreted recombinant PSGL-1/mIgG2b carrying the LDN determinant
For SDS–PAGE/western blotting and LC-MS/MS, PSGL-1/mIgG2b was purified from clarified supernatants on goat anti-mIgG agarose beads (Sigma-Aldrich, St. Louis, Missouri, USA; 60 μL slurry per 10 mL supernatant) by rolling head over tail at 4 °C overnight. The beads with captured fusion proteins were washed three times in PBS, resuspended in 4× SDS sample buffer without reducing agent (Invitrogen AB), and incubated at 95 °C for 5 min for protein denaturation.For H. pylori binding studies, purification procedures were carried out using columns (150 cm h−1, HR 16/10 column, 5 cm bed height) containing 4% cross-linked agarose Protein A Sepharose CL-4B (GE Healthcare, Chicago, IL, USA). The clarified supernatants from CHO cells were sterile-filtered using 0.22 μm polyether sulfone filter (Nalgene, Thermo Fisher Scientific, San Jose, CA) before loading onto the Protein A Sepharose CL-4B column (GE Healthcare), which was pre-equilibrated with five column volumes (CV) of phosphate-buffered saline (PBS). The supernatant was run at a flow rate of 1.5 mL min−1, and then washed with five column volumes of 0.15 M sodium chloride before eluting with 0.1 M sodium citrate, pH 3.0. Post elution, the fractions were neutralized with 800 μL mL−1 of 1 M Tris–HCl, pH 9.0 before dialysis (12–14 kDa cutoff) against MilliQ water at 4 °C. After dialysis, the samples were frozen, lyophilized, reconstituted with water until H. pylori LabA binding studies.
Quantification of PSGL-1/mIgG2b by enzyme-linked immunosorbent assay
A sandwich ELISA method was used for the quantitative measurement of PSGL-1/mIgG2b concentrations in supernatants. The 96-well ELISA plates (Costar 3590; Corning Inc., Corning city, NY, USA) were coated with an affinity-purified polyclonal goat anti-mIgG Fc-specific antibody (Sigma-Aldrich) at a concentration of 10 μg mL−1 in 50 mM carbonate buffer, pH 9.6, at 4 °C overnight. The plates were blocked with 1% (w/v) bovine serum albumin (BSA) in PBS to minimize non-specific binding. This buffer was also used as antibody dilution buffer. The plates were sealed and incubated at room temperature for 2 hours.Supernatants were incubated in the 96-well plate in triplicates for 2 hours before incubating with a peroxidase-conjugated anti-mIgG Fc-specific antibody (Sigma-Aldrich) diluted 1:2000 for 2 hours at room temperature. PBS containing 0.05% (v/v) Tween-20 (Sigma-Aldrich) and 0.15 M sodium chloride was used as the wash buffer between incubations. A bound peroxidase-conjugated antibody was visualized with 3,3′,5,5′-tetramethylbenzidine dihydrochloride (Sigma-Aldrich) as a substrate. The color reaction was stopped by the addition of 2 M H2SO4 and the optical density of the samples was read at a wavelength of 450 nm in a microplate reader (Synergy 2, Biotek Instruments Inc., Winooski, USA). The PSGL-1/mIgG2b concentration was estimated using a dilution series of purified mIgG2b (AbD Serotec, Oxford, UK) in blocking buffer as an internal standard.
SDS-PAGE and western blot analysis
NuPAGE 3–8% Tris–acetate gels and Tris–acetate SDS running buffer (Invitrogen AB) were used to separate goat anti-mIgG agarose bead-purified samples under non-reducing conditions. A precision protein standard (Hi-Mark, Invitrogen AB) was used as a molecular weight marker before running the gel at 120 volts for 2 hours.Separated proteins were electrophoretically blotted onto nitrocellulose membranes (Invitrogen AB) using the iBlot apparatus (Invitrogen AB). PBS containing 0.2% Tween-20 (PBS-T) and 3% BSA was used as the blocking buffer and as the diluent solution for antibodies and lectins. HRP-conjugated polyclonal goat anti-mouse IgG Fc (1:10000 dilution; Sigma Aldrich) was used to detect the Fc portion of PSGL-1/mIgG2b, whereas a mouse anti-human CD162 was used to detect the N-terminal part of PSGL-1 (1:1000 dilution; BD PharmMingen, San Diego, CA, USA) together with a HRP-conjugated polyclonal goat anti-mouse IgG (Fab-specific, 1:10000 dilution; Sigma Aldrich) used as secondary antibody. An anti-LDN (anti-LDN; mouse IgG, diluted 1:500–1:1000) antibody was used to detect LDN determinants27 together with HRP-conjugated polyclonal goat anti-mouse IgG (Fab-specific, 1:10000 dilution; Sigma Aldrich) used as secondary antibodies. All membranes stained with antibodies were incubated at room temperature for 1 h, and then washed three times with PBS-T after each incubation. Bound antibody and lectins were visualized by chemiluminescence using the ECL kit according to the manufacturer's instructions (GE Healthcare, Uppsala, Sweden).
Chemical release of O-linked glycans from purified PSGL-1/mIgG2b carrying the LDN determinant
O-Linked glycans were released by β-elimination and analyzed as native structures by LC-MS/MS analysis as described previously.14,28 Briefly, PSGL-1/mIgG2b purified on goat anti-mIgG agarose beads (60 μL slurry, 45 mL supernatant) were separated on SDS–PAGE (3–8% NuPAGE, Invitrogen AB) under non-reducing conditions. Separated proteins were electrophoretically blotted onto a PVDF membrane (0.45 μm, Millipore, Billerica, MA) using a semi-dry method. Protein bands were visualized by direct blue 71 (Sigma-Aldrich) staining (0.008% in 10% acetic acid and 40% ethanol, w/v). Bands containing recombinant proteins were excised and subjected to reductive β-elimination in a solution of 0.5 M NaBH4 in 50 mM NaOH and incubated overnight at 50 °C. The reaction was stopped by adding acetic acid. Released glycans were desalted on AG50WX8 cation exchange beads (Bio-Rad) and packed on top of a C18 Zip-tip (Millipore, Bedford, MA, USA). Samples were eluted with water and dried in a SpeedVac concentrator. Repeated evaporation steps with 1% acetic acid in methanol were performed until borate complexes were removed. Dried glycans were dissolved in water before analysis by LC-MS/MS.Liquid chromatography – mass spectrometry analyses
LC-MS/MS was performed on a porous graphitized carbon LC column coupled to an LTQ ion trap instrument (Thermo Fischer Scientific, San Jose, CA) as described previously.29 The column (10 cm × 250 μm i.d.) was packed with 5 μm porous graphitized carbon particles (Thermo Fischer Scientific). Glycans were separated using a gradient of 0–40% acetonitrile in 10 mM ammonium bicarbonate buffer over 40 min at a flow rate of 10 μL min−1. Eluted glycans were detected in negative mode by a full scan (m/z 380–2000) using an electrospray voltage of 3.5 kV, a capillary voltage of −33.0 V, and a capillary temperature of 300 °C. Air was used as sheath gas and the scanned mass range was defined according to the expected mass of the specific structure to be analyzed.The data were processed using the Xcalibur software (version 2.0.7, Thermo Fischer Scientific). Interpretation and annotations of MSn spectra were done manually. The biosynthesis of O-glycans was assumed to follow the classical pathways in CHO-K1 cells.28 The chain elongation was expected to be mediated by the addition of N-acetyllactosamine units. The terminal HexNAc2 determinants were presumed to be LDN. The annotated structures were submitted to the Unicarb-DB database (https://unicarb-dr.biomedicine.gu.se/references/378) and will be included in the next release. For comparisons between samples of the relative glycan abundance, the area under the curve (AUC) of each structure was normalized to the total AUC in the ion chromatogram and expressed as a percentage.
Bacterial strains and culture
H. pylori strains J99 and 26695 were cultured on Brucella agar plates (Brucella Medium Base, Oxoid; Basingstoke, Hampshire, England) supplemented with 10% citrated bovine blood (Svenska Labfab, Ljusne, Sweden), 1% IsoVitox (Oxoid), 4mg L−1 amphotericin B, 10mg L−1 vancomycin and 5mg L−1 trimethoprim in microaerophilic conditions (5% O2 and 15% CO2) at 37°C.
Isolation of genomic DNA from H. pylori J99 and 26695
Genomic DNAs from strains J99 and 26695 were purified using the DNeasy Blood & Tissue Kits (50) (QIAGEN, Venlo, The Netherlands). After isolation, the DNA concentration was measured using a spectrophotometer (Nanodrop 200 C, Thermo Fischer Scientific, San Jose, CA).Identification by PCR of labA genomic DNA sequences in H. pylori J99 and 26695
Isolated bacterial genomic DNA was used as template for PCR amplification (VetMAX™-Plus Multiplex One Step RT-PCR Kit; Thermo Fischer Scientific) of a LabA encoding sequence (GenBank accession number AAD05605.1) in the H. pylori J99 and 26695 strains.30 A 25 μL master mixture containing 50 ng of H. pylori genomic DNA, 2x multiplex buffer, a labA88 5′-GGCTATCAAATCGGTGAATCCGC-3′ forward and a labA340 5′-CTGCCGCATTCAAGGCTAAAAGCACG-3′ reverse primer, were used together with the Taq polymerase. PCR was performed on a 2720 Thermocycler (Applied Bio-systems; Foster City, CA, USA) using the following program: 95 °C denaturing, 63 °C annealing, 72 °C elongation, 33 cycles. PCR products were checked by electrophoresis (80 V, 40 min) on a 1.2% agarose gel in the presence of 0.5 g mL−1 of ethidium bromide (Sigma Aldrich, Steinheim, Germany) and illuminated under UV light.Identification of LabA in H. pylori strains J99 and 26695 by proteomic analyses
Proteomic analysis was performed at the Proteomics Core Facility at the Sahlgrenska Academy, University of Gothenburg. Cell pellets from H. pylori J99 and 26695 strains were homogenized in 2% sodium dodecyl sulfate (SDS) and 50 mM triethylammonium bicarbonate (TEAB) using lysis matrix D on a FastPrep-24 instrument (MP Biomedicals, Santa Ana, CA, USA). Protein concentration was determined using Pierce BCA Protein Assay Reagent (Thermo Fisher Scientific, San Jose, CA, USA), and 100 μg was digested with trypsin using a modified filter-aided sample preparation (FASP) protocol.31 Briefly, after reduction using 100 mM dithiothreitol at 60 °C for 30 min, samples were transferred onto 30 kDa MWCO Pall Nanosep centrifugal filters (Sigma-Aldrich) and washed repeatedly with 8 M urea. Alkylation was performed with 10 mM methyl methane thiosulfonate (MMTS) diluted in digestion buffer [1% sodium deoxycholate (SDC), 50 mM TEAB] for 30 min at room temperature. Trypsin (ratio 1:100, Pierce Trypsin Protease, MS Grade; Thermo Fisher Scientific) in digestion buffer was added and the samples were incubated at 37 °C overnight. A second incubation with trypsin for 3 hours ensured complete digestion of the proteins. Peptides were collected by centrifugation, and SDC was removed by acidification with 10% trifluoroacetic acid (TFA). The two different samples were purified using HiPPR Detergent Removal Resin (Thermo Scientific) according to manufacturer's instruction.
Peptide samples were analyzed on an QExactive HF mass spectrometer interfaced with Easy-nLC1200 liquid chromatography system (Thermo Fisher Scientific). Peptides were trapped on an Acclaim Pepmap 100 C18 trap column (100 μm × 2 cm, particle size 5 μm, Thermo Fisher Scientific) and separated on an in-house packed analytical column (75 μm × 350 mm, particle size 3 μm, Reprosil-Pur C18, Dr Maisch) using a gradient from 7% to 100% B over 90/120 min, at a flow of 300 nL min−1, where solvent A was 0.2% formic acid and solvent B was 80% acetonitrile in 0.2% formic acid. The instrument operated in data-dependent mode where the precursor ion mass spectra were acquired at a resolution of 60000, the 10 most intense ions were isolated in a 1.2 Da isolation window and fragmented using collision energy HCD settings at 28. MS2 spectra were recorded at a resolution of 30000 with charge states 2 to 4 selected for fragmentation and dynamic exclusion set to 20 s.
Data analysis was performed using Proteome Discoverer version 1.4 (Thermo Fisher Scientific) against the SwissProt databases for Helicobacter pylori J99 and 26695 strains, from Aug 2019 (Swiss Institute of Bioinformatics, Switzerland). Mascot 2.5 (Matrix Science) was used as a search engine with precursor mass tolerance of 5 ppm and fragment mass tolerance of 0.2 Da. Tryptic peptides were accepted with one missed cleavage. Variable modification of methionine oxidation and fixed cysteine alkylation was selected. The detected peptide threshold in the software was set to a significance level of Mascot 99%. The mass spectrometric proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD015288.
Glycan quantification using periodate oxidation and biotin conjugation
Samples were lyophilised after dialysis against two changes of distilled water before H. pylori LabA-binding studies were conducted. The glycan contents on coated PSGL-1/mIgG2b glycoforms and gastric mucin 1, 2 and 3 were determined by the detection of glycans as periodate-oxidizable structures using a microtiter-based assay. NUNC polysorb plates were coated with 100 μL sample, covered with parafilm before incubating overnight at 4 °C. Post-washing three times with DELFIA washing solution (5 mM Tris–HCl, 0.15 M NaCl, 0.005% Tween 20, 0.02% NaN3, pH 7.75), the glycans were periodate-oxidized by treatment with 100 μL 25 mM sodium metaperiodate in 0.1 M sodium acetate buffer, pH 5.5 for 20 min in room temperature. The plates were re-washed using Delfia and manually with PBS 0.05% Tween before blocking with DELFIA blocking solution (50 mM Tris–HCl, 0.15 M NaCl, 90 mM CaCl2, 4 mM EDTA, 0.02% NaN3, 6% sorbitol, 0.1% BSA, pH 7.75) for 1 h.After further washing steps, the samples were incubated for 1 h with 200 μL 2.5 mM biotin hydrazide solution in 10 mL 0.1 M sodium acetate buffer, pH 5.5, followed by another washing. Wells were incubated with 100 μL Europium-labeled streptavidin which was diluted 1:1000 in DELFIA assay buffer (50 mM Tris–HCl, 0.15 M NaCl, 20 mM DTPA, 0.01% Tween 20, 0.02% NaN3, 1.5% BSA, pH 7.75). The plates were washed further 1 h post-incubation and then incubated with 200 μL enhancement solution (0.05 M NaOH, 0.1 M ftalat, 0.1% Triton X-100, 50 mM TOPO, 15 mM b-NTA) on a shaker for 5 min. The mucin glycan values were measured using Wallac 1420 VICTOR2 plate reader with the Europium label protocol (PerkinElmer, Waltham, MA, USA). The concentrations were calculated from a standard curve of a fusion protein of MUC1. For the binding assay, all mucin samples were diluted in 4 M GuHCl to 4 μg mL−1 and coated on 96 well plates in 4 replicates.
H. pylori binding assay
To assess the binding of H. pylori to LDN-containing glycoconjugates and mucin samples in vitro, a previously described microtiter well-based assay was used.32 The mucins, the mucin-type fusion proteins (LDN carried by different O-glycan core structures on PSGL-1/mIgG2b) and the human serum albumin neoglycoprotein (Leb-HSA) purchased from Dextra (Reading, UK) were diluted in 4 M GuHCl to 4 μg mL−1 and coated in 96-well polysorb plates over night at 4 °C. The plates were washed 3 times with PBS containing 0.05% Tween using an ELISA plate washer before blocking for 1 hour with 1% blocking buffer (Blocking Reagent for ELISA (Roche) containing 0.05% Tween). Freshly harvested bacteria were washed twice in blocking buffer (2500 rpm, 5 min) and diluted in blocking buffer to an OD600 value of 0.01. The blocking buffer was discarded from the plates and the bacteria were added and incubated for 2 hours in a shaker at 120 rpm at 37 °C. The plates were washed 3 times and incubated for 1 hour in RT with rabbit anti-HP serum in a dilution of 1:1000. The plates were again washed 3 times before incubating in RT for 1 hour with horseradish peroxidase-conjugated donkey anti-rabbit IgG diluted 1:10000. TMB (3,3′,5,5′-tetramethylbenzidine) was used as peroxidase substrate and the wells were incubated with this reagent for 20 min. An equivalent amount of 0.5 M H2SO4 was used to stop the reaction, after which the plate was read in a microplate reader at 450 nm.
Binding of 125I-labeled LDN–BSA neoglycoconjugates to H. pylori strains J99 and 26695 as well as various H. pylori clinical isolates
For description and characterization of the LDN (16R)–BSA conjugate used in the H. pylori binding assay, see ESI.† The binding assay was performed as previously described by Aspholm et al.33 The LDN-BSA was labeled with 125I by the chloramine-T method. The labeling yielded about 40k CPM per ng of LacdiNAc. One ng radiolabeled conjugate was incubated in OD: 0.1 (A600) of bacterial suspension over night on a rocking table in PBS containing 0.5% (w/v) bovine serum albumin (BSA)/0.05% (v/v) Tween 20. After incubation and centrifugation, the pellet and supernatant were separated, and the CPM content was measured for each of the samples. The CPM in the H. pylori pellet was measured in a 2470 Wizard2 Automatic Gamma counter (PerkinElmer, Waltham, Massachusetts, USA). In total 17 strains were tested, including an E. coli control. The 17 H. pylori strains tested were: 17875/Leb, 17875/DM described in Mahdavi et al.,7 J166 (USA, from Jay Solnick), I9 (India34), J532, J519, J512 (Japan) and S865 (Spain) described in Aspholm et al.,35 26695/TB (from Kathryn Eaton), J99/TB (from Richard Peek), MC-337 (Mexico from Javier Torres), J532, J519, J512, Sv89, Sv41, Sv64 from Sweden described in Ilver et al.,36 as well as the J99/SL and 26695/SL strains also used in the binding assay performed in the microtiter plates assay described above. The strains selected for testing, besides from the J99 and 26695 strains, were selected from world-wide different populations.Statistical analyses
Statistical analyses were performed using SPSS statistics17.0 (SPSS Inc., IL, USA) and Graph Pad Prism 7.0 (GraphPad Software Inc.) software package. One-way ANOVA test was used when comparing 3 or more groups, and to ascertain that the multiple testing did not add to the chance of finding statistically significant differences, the Tukey's test was used. One-way ANOVA, Dunnett's multiple comparisons test was used to determine the statistical difference between the H. pylori binding signal and the background signal.Results
Generation of LDN determinants on PSGL-1/mIgG2b carrying different O-glycan core structures
PSGL-1/mIgG2b was expressed in CHO-K1 cells following transient co-transfection of plasmids encoding the LDN-generating enzyme, B4GALNT3, and the O-glycan core 2 (B6GNT1), core 3 (B3GNT6) or extended core 1 (B3GNT3) enzymes (Fig. 1 and Table 1). Fusion proteins were immunoaffinity-purified from the cell culture medium on anti-mouse IgG agarose beads and were analyzed by SDS–PAGE and western blot using antibodies specific for mIgG Fc (Fig. 2A), the N-terminal of PSGL-1 (Fig. 2B) and the LDN determinant (Fig. 2C). As reported before,37 the fusion protein had an apparent MW between 268 and 460 kDa under non-reducing conditions suggesting that the fusion protein is produced as a homodimer (Fig. 2A and B). Additional bands seen between the 117 and 268 kDa marker upon anti-mIgG Fc (Fig. 2A) and anti-PSGL-1 (Fig. 2B) antibody staining indicate the presence of PSGL-1/mIgG2b monomers and degradation products. Anti-LDN antibody staining (Fig. 2C) revealed strong staining of the fusion protein when the B4GALNT3 was co-expressed with the core 2 (lane 2) and the core 3 (lane 3) enzymes, while no or very weak staining of the fusion protein was seen when it was generated on extended core 1 (lane 4) or on core 1 O-glycans in wild-type CHO-K1 cells (lane 1). PSGL-1/mIgG2b expressed in CHO-K1 cells without co-expression of the B4GALNT3 enzyme was used as negative control (lane 5) and a lysate of porcine aortic valve tissue previously shown to contain glycoproteins carrying the LDN determinant (unpublished) was used as positive control (lane 6). To assess the purity of isolated fusion proteins, the presence of any additional glycosylated and non-glycosylated proteins of significant abundance was excluded following staining of the gels using a glycoprotein staining kit and the Ruby stain, respectively (Fig. 2D and E).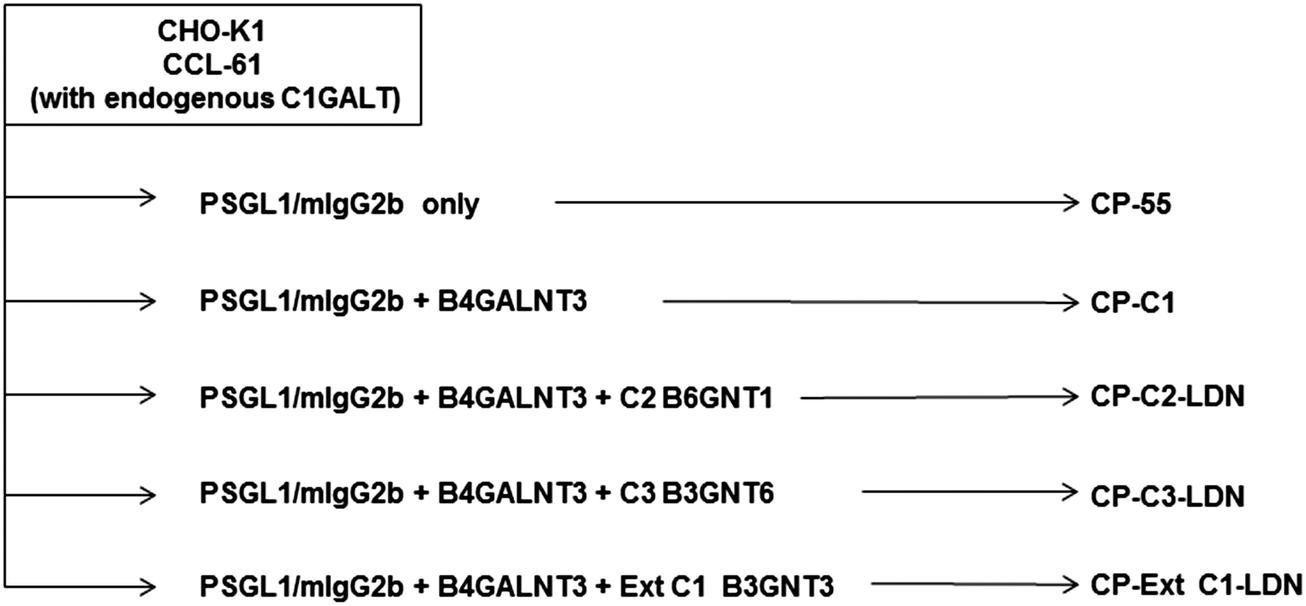 | ||
| Fig. 1 Transfection scheme. Glycosyltransferase cDNAs used to generate recombinant P-selectin glycoprotein ligand-1/mouse IgG2b fusion proteins carrying the LDN determinant on different O-glycan core chains by transient transfections in CHO-K1 cells. | ||
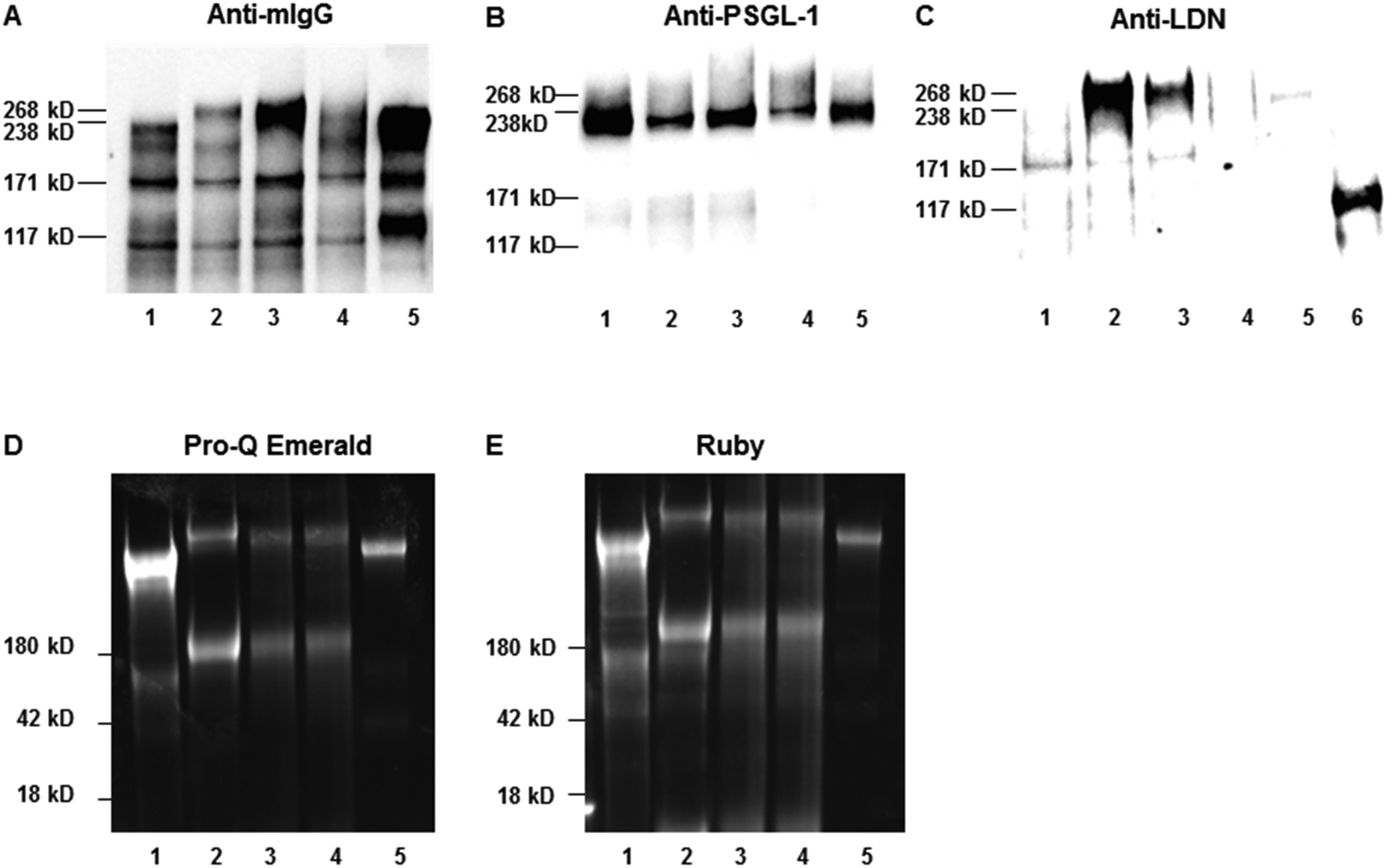 | ||
| Fig. 2 Western blot analysis of affinity-purified recombinant P-selectin glycoprotein ligand-1/mouse IgG2b (PSGL-1/mIgG2b) expressed in CHO-K1 cells following co-transfection with only the B4GALNT3 cDNA (lane 1; CP-C1), or together with GCNT1 generating core 2 (lane 2; CP-C2-LDN), B3GNT6 generating core 3 (lane 3; CP-C3-LDN) or B3GNT3 generating extended core 1 (lane 4; CP-ext C1-LDN) O-glycans. PSGL-1/mIgG2b carrying mono- and disialylated core 1 (lane 5; CP-55) O-glycans was used a as a negative control. A lysate of porcine aortic valve tissue containing glycoproteins carrying the LDN determinant was used as positive control (lane 6). Affinity-purified PSGL-1/mIgG2b (200 ng) were separated under non-reducing conditions, blotted onto nitrocellulose membranes and were stained in western blot experiments by anti-mouse IgG (mIgG-Fc) (panel A), anti-PSGL1 (panel B) and anti-LDN (panel C) antibodies. Glycosylation proteins were detected by Pro-Q Emerald (panel D) and Ruby staining (panel E). | ||
Liquid chromatography – mass spectrometry supports the presence of LDN determinants on core 2, core 3 and extended core 1 O-glycans
Recombinant PSGL-1/mIgG2b secreted into the medium of transiently transfected CHO-K1 cells was purified by affinity chromatography and subjected to β-elimination to release the O-linked glycans that were subsequently analysed by LC-MS/MS in negative ion mode. The LC-MS base peak chromatograms of O-glycans from PSGL-1/mIgG2b produced in CHO-K1 cells transiently transfected with the fusion protein cDNA and co-transfected with the B4GALNT3-encoding plasmid alone (Fig. 3A, CP-C1), or together with plasmids encoding the core 2, core 3 or extended core 1 enzymes are shown (Fig. 3B–D). As previously reported,37 PSGL-1/mIgG2b expressed in wild type CHO-K1 cells express predominantly mono- and disialylated core 1 O-glycans (675 and 966 in Fig. 3A and 675-1, 675-2, and 966 in Table 2) and lack B4GALNT3 precursor saccharides. Thus, the LDN determinant is not detected on O-glycans on PSGL-1/mIgG2b expressed in wild-type CHO-K1 cells upon co-transfection with the B4GALNT3 enzyme alone (Fig. 3A and Table 2).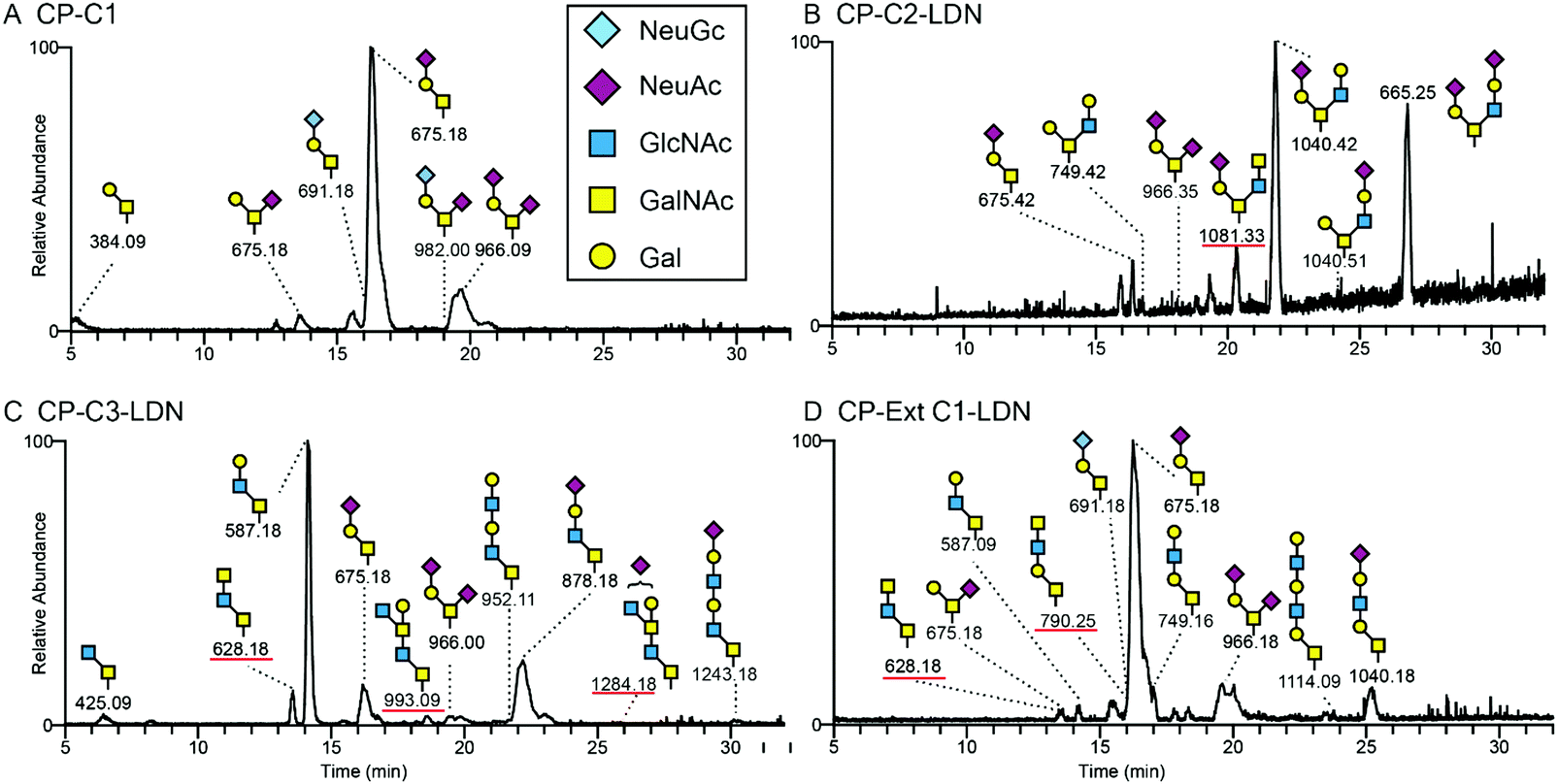 | ||
| Fig. 3 Liquid chromatography – mass spectrometry chromatograms of O-glycans released from recombinant P-selectin glycoprotein ligand-1/mouse IgG2b and based on core 1 (chromatogram A), core 2 (chromatogram B), core 3 (chromatogram C) or extended core 1 (chromatogram D) structures. (A) HexNAc–HexNAc sequence supporting the presence of the LDN determinant was detected on O-glycans based on the core 2 (B), core 3 (C) and extended core 1 structures (D). LC-MS of O-glycans released from PSGL-1/mIgG2b produced in CHO-K1 cells supporting expression of native core 1 structures only, did not reveal any HexNAc–HexNAc determinants suggesting that B4GALNT3 does not support the biosynthesis of LDN on core 1 O-glycans. The major structures are depicted using the Consortium for Functional Glycomics symbol nomenclature. | ||
| Cores | Observed mass [M − nH]n− | Composition | Putative structure | B4GALNT3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PSGL-1/m-IgG2b (Mock) | Endogenous C1 (in CHO-K1 cells) | Extended C1 β3GnT3 | C2 β6GnT1 | C3 β3GnT6 | |||||||||
| RT | RI | RT | RI | RT | RI | RT | RI | RT | RI | ||||
| Core 1 O-glycans | 384 | Hex1HexNAc1 | Galβ3GalNAcol | 5.2 | 26.4 | — | — | — | — | — | — | — | — |
| 675-1 | NeuAc1Hex1HexNAc1 | Galβ3(NeuAcα6)GalNAcol | 13.6 | 4.8 | 15.6 | 6.5 | 13.5 | 2.8 | — | — | 13.6 | 0.4 | |
| 675-2 | NeuAc1Hex1HexNAc1 | NeuAcα3Galβ3GalNAcol | 16.3 | 44.1 | 18.0 | 42.8 | 16.2 | 21.7 | 16.4 | 4.0 | 16.2 | 4.2 | |
| 691 | NeuGc1Hex1HexNAc1 | NeuGcα3Galβ3GalNAcol | 16.3 | 2.4 | 17.7 | 3.6 | 16.2 | 3.3 | — | — | — | — | |
| 749-2 | Hex2HexNAc2 | Galβ4GlcNAcβ3Galβ3GalNAcol | — | — | — | — | 17.0 | 8.3 | — | — | — | — | |
| 790-1 | Hex1HexNAc3 | GalNAcβ4GlcNAcβ3Galβ3GalNAcol | — | — | — | — | 16.0 | 4.5 | — | — | — | — | |
| 966 | NeuAc2Hex1HexNAc1 | NeuAcα3Galβ3(NeuAcα6)GalNAcol | 19.7 | 20.5 | 21.0 | 40.7 | 19.9 | 22.9 | 18.1 | 8.2 | 19.9 | 6.6 | |
| 982 | NeuGc1NeuAc1Hex1HexNAc1 | NeuGcα3Galβ3(NeuAcα6)GalNAcol | 19.0 | 1.8 | 20.7 | 6.4 | — | — | — | — | |||
| 1040-3 | NeuAc1Hex2HexNAc2 | NeuAcα3Galβ4GlcNAcβ3Galβ3GalNAcol | — | — | — | — | 25.2 | 5.1 | — | — | |||
| 1114 | Hex3HexNAc3 | Galβ4GlcNAcβ3Galβ4GlcNAcβ3Galβ3GalNAcol | — | — | — | — | 24.0 | 11.1 | — | — | — | — | |
| Core 2 O-glycans | 749-1 | Hex2HexNAc2 | Galβ3(Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | 16.7 | 19.1 | — | — | ||
| 1040-1 | NeuAc1Hex2HexNAc2 | NeuAcα3Galβ3(Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 21.8 | 12.7 | — | — | |
| 1040-2 | NeuAc1Hex2HexNAc2 | Galβ3(NeuAcα3Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 24.2 | 6.0 | — | — | |
| 1081 | NeuAc1Hex1HexNAc3 | NeuAcα3Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 20.4 | 8.5 | — | — | |
| 1331 | NeuAc2Hex2HexNAc2 | NeuAcα3Galβ3(NeuAcα3Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 26.8 | 21.5 | — | — | |
| 1347 | NeuAc1NeuGc1Hex2HexNAc2 | NeuGc + NeuAc + Galβ3(Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 26.5 | 13.4 | — | — | |
| 1696 | NeuAc2Hex3HexNAc3 | NeuAcα3Galβ3(NeuAcα3Galβ4GlcNAcβ3Galβ4GlcNAcβ6)GalNAcol | — | — | — | — | — | — | 31.4 | 6.6 | — | — | |
| Core 3 O-glycans | 425 | HexNAc2 | GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 6.4 | 9.7 |
| 587 | Hex1HexNAc2 | Galβ4GlcNAcβ3GalNAcol | — | — | — | — | 14.2 | 8.7 | — | — | 14.1 | 47.2 | |
| 628 | HexNAc3 | GalNAcβ4GlcNAcβ3GalNAcol | — | — | — | — | 13.6 | 4.5 | — | — | 13.6 | 5.0 | |
| 790-2 | Hex1HexNAc3 | GlcNAcβ3Galβ4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 19.0 | 2.4 | |
| 878 | NeuAc1Hex1HexNAc2 | NeuAcα3Galβ4GlcNAcβ3GalNAcol | — | — | — | — | 22.22 | 7.2 | — | — | 22.2 | 9.0 | |
| 894 | NeuGc1Hex1HexNAc2 | NeuGcα3Galβ4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 21.7 | 1.4 | |
| 952 | Hex2HexNAc3 | Galβ4GlcNAcβ3Galβ4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 21.9 | 3.0 | |
| 993 | Hex1HexNAc4 | GlcNAcβ3(Galβ4)GalNAcβ4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 18.6 | 6.5 | |
| 1243 | NeuAc1Hex2HexNAc3 | NeuAcα3Galβ4GlcNAcβ3Galβ4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 30.1 | 2.4 | |
| 1284 | NeuAc1Hex1HexNAc4 | NeuAc + GlcNAcβ3(Galβ4)GalNAcβ1-4GlcNAcβ3GalNAcol | — | — | — | — | — | — | — | — | 25.8 | 2.3 | |
The base peak chromatogram of O-glycans released from PSGL-1/mIgG2b expressed in CHO-K1 cells co-transfected with the B4GALNT3 and core 2 (B6GNT1) enzyme cDNAs contained one peak with a composition of NeuAc1Hex1HexNAc3 (Fig. 3B; [M − H]− ion of m/z 1081), the MS/MS spectrum of which suggested the presence of a structure tentatively assigned as NeuAcα3Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol (Fig. 4B). The peak accounted for 9% of the total O-glycan repertoire released from this fusion protein (Fig. 3B and Table 2). The predominant O-glycans were mainly core 2 O-glycans (79%), as well as mono- and disialylated core 1 O-glycans (12%).
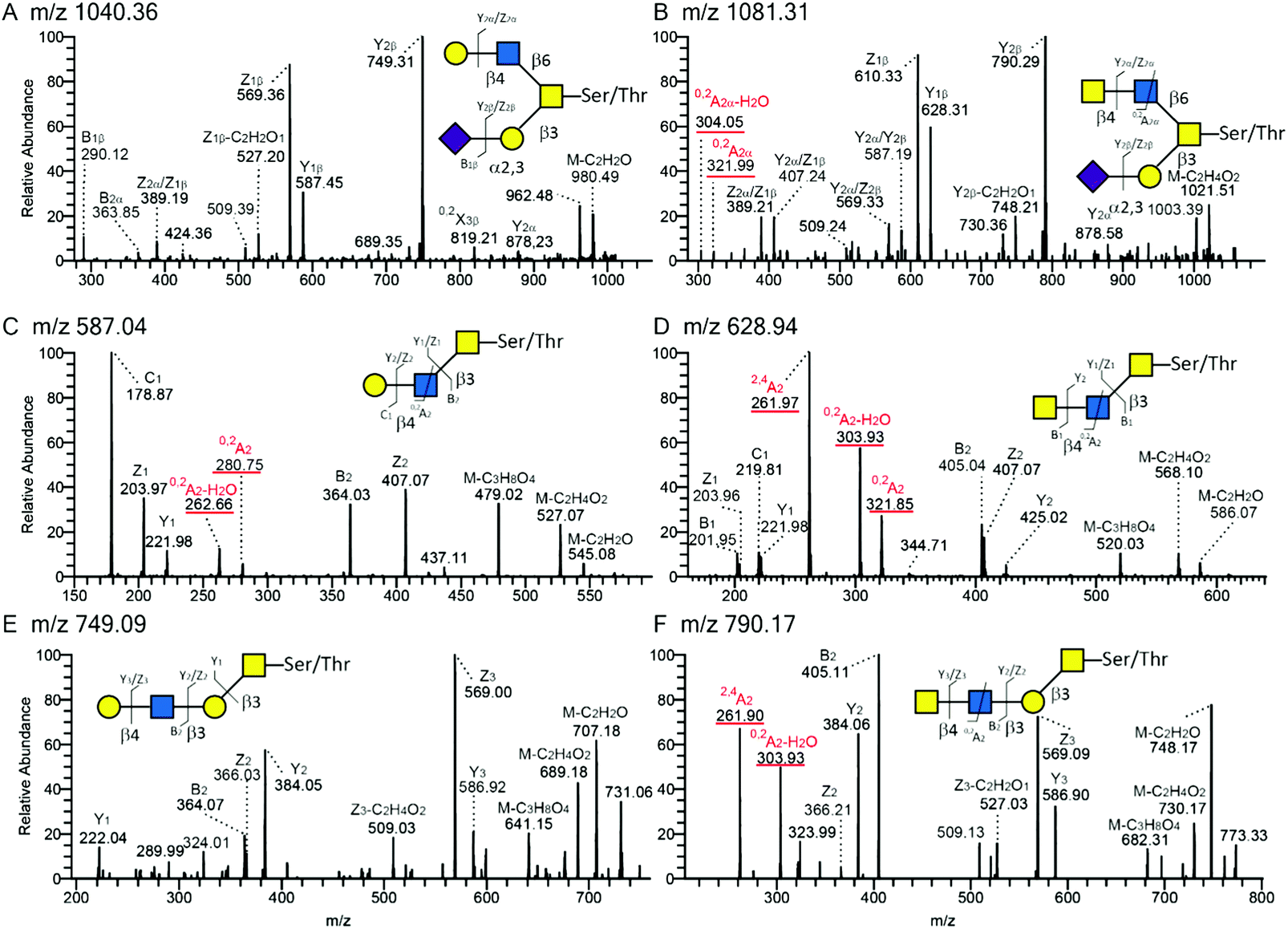 | ||
| Fig. 4 Tandem mass spectra (MS/MS) of O-glycans released from recombinant P-selectin glycoprotein ligand-1 carrying core 2 (panel A and B; [M − H]− ions of m/z 1040.36 and 1081.31, respectively), core 3 (panel C and D; [M − H]− ions of m/z 587.04 and 628.94) or extended core 1 (panel E and F; [M − H]− ions of m/z 749.09 and 790.17) and with terminal Hex–HexNAc (i.e. LacNAc); (A, C and E) or HexNAc–HexNAc (i.e. LDN; B, D and F) sequences. The MS/MS spectra are annotated according to the nomenclature suggested by Domon and Costello.50 | ||
LC-MS/MS of O-glycans of PSGL-1/mIgG2b expressed together with B4GALNT3 and the core 3 (B3GNT6) enzyme cDNA indicated three peaks containing possible LDN sequences (Fig. 3C). The first peak at m/z 628 has a composition of HexNAc3. The other two structures with Hex1HexNAc4 ([M − H]− ion of m/z 993) and NeuNAc1Hex1HexNAc4 ([M − H]− ion of m/z 1284) compositions account for 7% and 2% of the total O-glycan repertoires, respectively. The first one ([M − H]− ion at m/z 628) had the same retention time as the one found on PSGL-1/mIgG2b with extended core 1 O-glycans (Fig. 3D) and constituted 5% of the total O-glycans.
The LC-MS chromatogram of O-glycans of PSGL-1/mIgG2b co-expressed with B4GALNT3 and the extended core 1 enzyme (B3GNT3) suggested the presence of two peaks ([M − H]− ions of m/z 628 and 790; Fig. 3D), which have a composition HexNAc3 and Hex1HexNAc3, respectively, and accounting for up to 9% of the fusion protein O-glycans produced in these cells. Consistent with our previous investigation28 core 3 O-glycans ([M − H] ions of m/z 587, 628 and 878 in Table 2) accounted for up to 20% of the total O-glycans, while approximately one third (29%) of the O-glycans contained the extended core 1 structure.
All annotated MS/MS spectra were listed in Table 2 and deposited in UniCarb-DR (https://unicarb-dr.biomedicine.gu.se/references/378). The presence of terminal β1,4-linked GalNAc was inferred by the diagnostic fragmentation ions arising from a cross-ring cleavage of GlcNAc (0,2AGlcNAc–H2O and 0,2AGlcNAc or m/z 304 and 322).29,38 For example, in comparison with terminal LacNAc with such ions at m/z 263 and 281 (Fig. 4A, C and E), terminal LDN yielded fragmentation ions at m/z 304 and 322 (Fig. 4B, D and F). As for neutral O-glycans, another cross-ring cleavage of GlcNAc (2,4AGlcNAc) was observed at m/z 262 (Fig. 4D and F) and a B2 ion was seen at m/z 405 (Fig. 4D and F), respectively. Based on this, base peaks at m/z 628, 790 and 1081 were assigned as GalNAcβ4GlcNAcβ3GalNAcol, GalNAcβ4GlcNAcβ3Galβ3GalNAcol, and NeuAcα3Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol. Two [M − H]-ions at m/z 993 and 1284 were assigned as LDN-like structures: GlcNAcβ3(Galβ4)GalNAcβ4GlcNAcβ3GalNAcol and NeuNAc + GlcNAcβ3(Galβ4)GalNAcβ4GlcNAcβ3GalNAcol, respectively (MS/MS spectra not shown). An overexpressed B3GNT6 may explain the generation of the latter two structures. Taken together, these LC-MS/MS analyses support the presence of terminal LDN determinants on core 2, core 3 and extended core 1 O-glycans following co-transfection of the B4GALNT3 cDNA.
Detection of LabA in H. pylori strains J99 and 26695
The HP0025 gene in strain 26695 belongs to the large Hop family of H. pylori outer membrane protein (OMP) genes and encodes the HopD (OMP-2) protein,39 renamed as LabA.8 To confirm the presence of LabA in the H. pylori strains J99 and 26695 used in the binding experiments, we used liquid chromatography – mass spectrometry analyses of trypsin-digested protein lysates of each strain and identified 7 peptides in J99 and 15 peptides in strain 26695 matching the Omp2 sequences from strain J99 (UniProtKB – Q9ZN38)40 and 26695 (UniProtKB – O24870),41 respectively (Table 3). The mass spectrometric proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD015288.| H. pylori strain | Gene name | Uniprot accession(s) | Protein sequence | Observed mass MH+ [Da] |
|---|---|---|---|---|
| J99 (ATCC 700824) | jhp_0021 | Q9ZN38 | IVAANAQNQR | 1084.6 |
| LGEHYDSITAAISSLPDAQSLQNVVSK | 2843.4 | |||
| QSADPNAINNAR | 1270.6 | |||
| NAAIAVEQSK | 1030.6 | |||
| GIQDLSDSYER | 1282.6 | |||
| DSIAHFGDQAER | 1345.6 | |||
| AYQIIQK | 863.5 | |||
| 26695 (ATCC 700392) | HP_0025 | O24870 | GTPSGTVTSNTWGAGcAYVGETVTNLK | 2717.3 |
| KLGEHYDSITAALSSLPDAQSLQNVVSK | 2971.5 | |||
| LNNLLTSYSALNTLIR | 1806.0 | |||
| AFGASGQDIPALSDTK | 1577.8 | |||
| NLAYTLANFSGQYK | 1589.8 | |||
| IVTANAQNQHNLDTGK | 1723.9 | |||
| NLAYTLANFSGQYKK | 1717.9 | |||
| DANFAQSMFANAR | 1442.6 | |||
| NAEIAVEQSK | 1088.6 | |||
| TNPNSPQGIQDNYYIDSNIHSQVQSR | 2975.4 | |||
| VSNNAQELLK | 1115.6 | |||
| GIQDLSDSYER | 1282.6 | |||
| AFNPYKDANFAQSMFANAR | 2163.0 | |||
| DSLGVcHEK | 1033.4 | |||
| ELNFEIK | 892.5 |
Using forward and reverse primers based on the LabA sequence of the H. pylori J99 strain (gene bank accession no: AE001439) and PCR, LabA amplicons of the expected size of 252 bp were identified in both the J99 and 26695 strain (Fig. 5). These results confirm the presence of LabA in the J99 and 26695 H. pylori strains used in our binding experiments.
 | ||
| Fig. 5 Agarose gel electrophoresis of amplicons (252 bp) obtained following PCR amplification of a LabA gene sequence in genomic DNA isolated from H pylori strains J99 (lane 2) and 26695 (lane 3). A 100 bp DNA ladder was used for fragment size determination (lane 1). | ||
Absence of H. pylori binding to LDN-containing glycoconjugates
Binding assays were carried out to investigate the binding of H. pylori strains J99 and 26695 to LDN presented on different O-glycan core structures carried by mucin-type fusion proteins. Both strains carry the LDN-binding LabA adhesin and strain J99 carries in addition the BabA and SabA adhesins that mediate binding to Leb and sialyl-Lea/x, respectively. Using Tukey's multiple comparisons test there was no statistically significant difference in the binding of strain J99 to PSGL-1/IgG2b carrying core 1 O-glycans without LDN and core 2, core 3 and extended core 1 O-glycans with terminal LDN (p > 0.05, Fig. 6A). In the name designation, C stands for CHO-K1, P for PSGL-1/mIgG2b, then followed by the core structure carrying the desired carbohydrate determinant.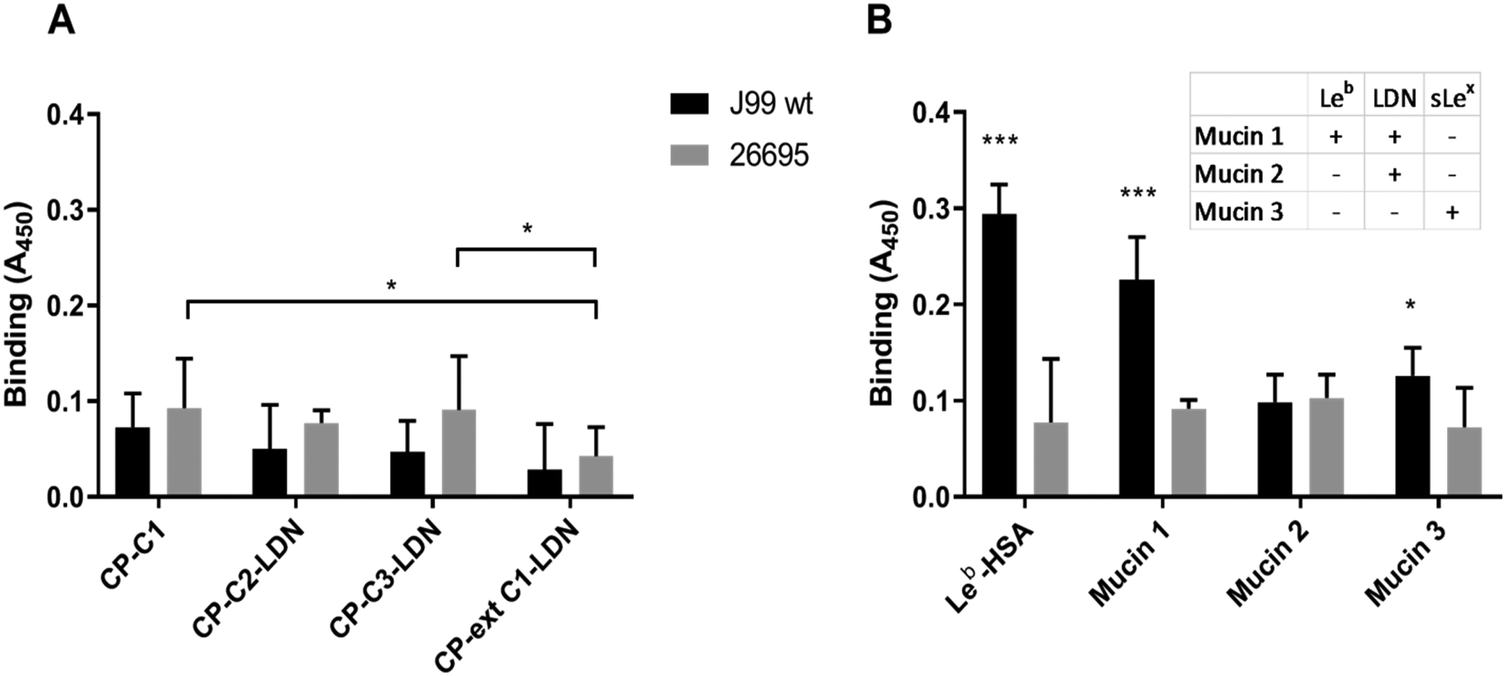 | ||
| Fig. 6 Binding of H. pylori to LDN-carrying mucin-type fusion proteins and purified gastric mucins. (A) Binding of H. pylori strains J99 and 26695 to LDN-carrying mucin-type fusion proteins. The values are presented after subtracting the background signal from each binding value. The non-specific background signal of the assay (i.e. binding signal from wells with no coated glycoconjugates) has been subtracted before plotting the binding data. This background signal had an average of 0.2. *Symbols represent the statistical difference between the binding to CP-ext C1-LDN and binding to CP-C1 and CP-C3–LDN (*p ≤ 0.05, One-way ANOVA, Tukey's multiple comparisons test). (B) Binding of H. pylori strains J99 and 26695 to two LDN-positive (human gastric mucin 1 and mucin 2b) and one LDN-negative purified monkey gastric mucin 3x. *Symbols represent the statistical difference between the binding signal and the background signal (*p ≤ 0.05, ***p ≤ 0.001, One-way ANOVA, Dunnett's multiple comparisons test). | ||
Strain 26695 bound with higher avidity to CP-C1 (p < 0.05) and CP-C3-LDN (p < 0.05) than to CP-Ext C1-LDN (Fig. 6A, grey bars). The fact that the H. pylori 26695 strain bound the CP-C1 fusion protein lacking LDN determinants better than CP-Ext C1-LDN carrying LDN determinants suggests that this H. pylori strain does not show specific binding to the LDN structure (Fig. 6A, grey bars). Furthermore, there appeared to be no relation between the level of binding of H. pylori 26695 and the anti-LDN antibody to the different fusion proteins carrying the LDN determinant when comparing the staining intensity with the anti-LDN antibody to the amount of bound H. pylori 26695 (compare Fig. 2C with Fig. 6A, grey bars).
To study the binding ability of H. pylori to natural mucins that carry the LDN glycan structure, we carried out binding assays using LDN-positive and LDN-negative purified gastric mucin samples (Fig. 6B). Leb conjugated to HSA was included in the assay as positive control. Strain J99 showed strong binding to the Leb–HSA glycoconjugate (p < 0.001) whereas strain 26695 did not bind to this structure (p > 0.05; Fig. 6B). Strain 26695 did not bind to any of the tested mucin samples. Human gastric mucin 1 contains Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol and human gastric mucin 2 contains Fucα2Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol (Table S1, ESI†). Strain J99 bound to human gastric mucin 1 that was positive for both LDN and Leb but it did not bind to human gastric mucin 2 that was positive for LDN only. Strain J99 also bound to the LDN-negative monkey gastric mucin 3 sample that carries the sLex structure but not the LDN and Leb determinants. Thus, neither for the native mucins, nor for the mucin-type fusion proteins any relation between level or presence of LDN and binding with these LabA positive strains was detected, whereas BabA- and SabA-dependent binding to their respective glycans was identified.
Binding of the 125I-labelled LDN–BSA conjugate to various strains of H. pylori in suspension
A series of world-wide H. pylori strains were assessed with regards to their binding to a 125I-labelled LDN–BSA conjugate. None of the tested 17 H. pylori strains (including the J99 and 26695, labelled with SL), bound more than 0.1% of the LDN-conjugate (Fig. 7A). The I9 and Sv64 strains provided a detectable signal, but below 1% of the 125I-LDN-conjugate added to the assay was bound and hence, the results are considered negative for binding. For comparison, 95% of the Leb conjugate binds to the 17875/Leb reference strain, while only 0.4% of the sLex conjugate do so. 0.14% of the LDN conjugate and 0.11% of the Leb conjugate were bound to the BabA minus (DM) strain. The background binding was slightly higher for the sLex conjugate compared to the Leb and LDN conjugates, possibly because of the charged sialic acid residues.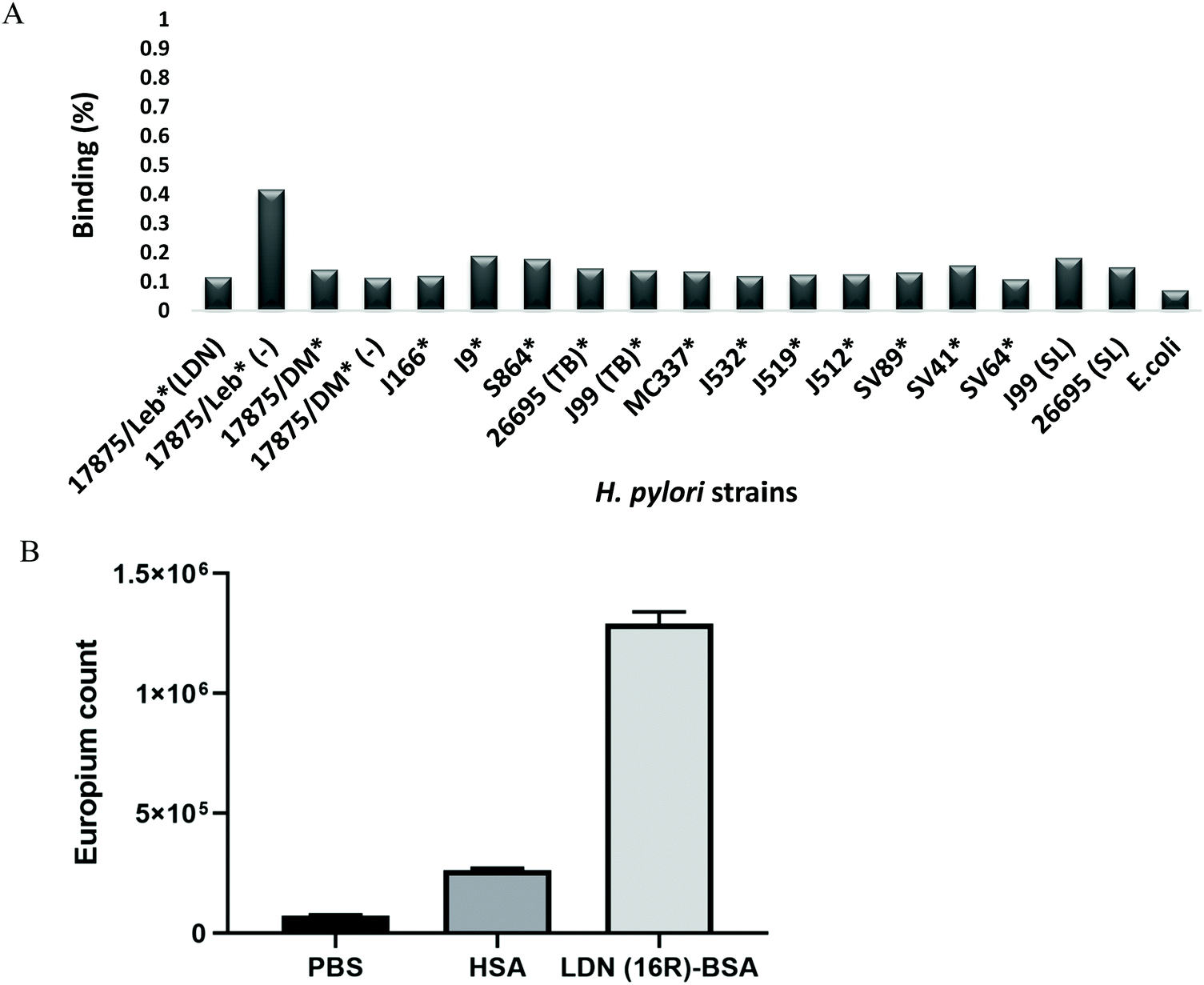 | ||
| Fig. 7 (A) Binding of H. pylori in suspension to a soluble 125I-labeled LDN–BSA neoglycoconjugate was measured by separating the pellet and supernatant after incubating the LDN conjugate with the bacteria overnight, and measuring the ratio of CPM in the pellet compared to total CPM count per sample. Binding is expressed as percentage of the total amount of 125I conjugate added to each of the bacterial suspensions. Data are presented as mean percentage of maximal radioactivity. (B) Verification of LDN on the LDN–BSA in an ELISA using Galectin-3. Binding of Galectin-3 to empty wells (PBS) and wells coated with HSA were used as controls. | ||
That LDN–BSA carried functional LDN determinants was verified by the selective binding of a recombinant human Galectin-3 to the LDN–BSA using an ELISA-type assay (Fig. 7B). Galectin-3 did not show any binding to non-coated wells (PBS) or wells coated with HSA. Galectin-3 binding to LDN(16R)–BSA showed a significant binding with p < 0.05 using the Anova test.
Discussion
To investigate the B4GALNT3 specificity in intact mucin molecule, we transiently transfected CHO-K1 cells with plasmids encoding a mucin-type fusion protein, PSGL-1/mIgG2b, which was used as a reporter protein to assess generated O-glycan phenotypes. By co-expressing glycosyltransferases responsible for core 2 (GCNT1), core 3 (B3GNT6) or extended core 1 (B3GNT3) O-glycans, the ability of B4GALNT3 to utilize these different core chains as precursor chains for LDN generation was investigated. Both western blot and LC-MS/MS experiments did not identify the LDN determinant on the fusion protein carrying only sialylated core 1 O-glycans, which is the predominant O-glycan core chain in wild type CHO-K1 cells.28 This is also in line with previous studies claiming β4GalNAcT activity in CHO cells even though no LDN sequences were detected on CHO glycoproteins.42,43 In contrast, the LDN determinant, or a HexNAc-HexNAc sequence in case of LC-MS/MS, was identified on core 2, core 3 and extended core 1 O-glycans upon transfection. All these core chains contain a terminal β-linked GlcNAc that could serve as a substrate for B4GALNT3 and the addition of a terminal β1,4-linked GalNAc. Our western blot results generated with the LDN antibody showed that core 2 and 3 chain O-glycans carried the LDN determinant, while staining of PSGL-1/mIgG2b carrying extended core 1 chains was very weak if there at all. However, LC-MS/MS of O-glycans released from the fusion protein generated in the presence of glycosyltransferases supporting core 2, 3 and extended core 1 biosynthesis identified glycans with terminal HexNAc–HexNAc sequences suggesting the presence of the LDN determinant on all these core chains.Based on the anti-LDN staining intensity, the fusion protein with core 2 O-glycans appeared to carry more LDN determinants than did PSGL-1/mIgG2b with core 3 and extended core 1 chains. There are four potential explanations for this finding; (i) the core 2 enzyme, GCNT1, generates more precursor chains for the B4GALNT3 enzyme; (ii) B4GALNT3 prefers core 2 over core 3 and extended core 1 chains; (iii) core 3 and extended core 1 chains are more efficiently β1,4-galactosylated than core 2 chains; and/or (iv) the anti-LDN antibody binds better to the LDN determinant situated on core 2 than on core 3 and extended core 1 chains. The fact that the staining of LDN on extended core 1 chains is very weak despite detected HexNAc–HexNAc determinants by LC-MS, suggests that the anti-LDN antibody recognizes the LDN determinant in a core chain-dependent manner. B4GALNT3 may have co-evolved to utilize core 2 O-glycans better than the other core chains because this O-glycan chain type has been reported to be the most dominant in human stomach.21
Based on experiments in which free LDN saccharides were shown to inhibit binding of several H. pylori strains (B128, 26695, J99, G27, P12 and B38) to gastric mucosa samples, the presence in those strains of a novel LDN-binding adhesin was suggested.8 A LDN-binding protein, the LabA adhesin, was subsequently purified by SDS–PAGE from lysates of the B128 strain and sequenced by mass spectrometry.8 To further characterize the LDN-binding specificity of the LabA adhesin, including a potential O-glycan core chain-dependent binding, we generated recombinant mucin-type fusion proteins carrying well-defined O-glycan repertoires including O-glycans based on core 1, core 2, core 3 or extended core 1 structures. In a microtiter well-based binding assay in which H. pylori strains 26695 and J99 were assessed for binding to coated recombinant mucin-type fusion proteins, purified gastric mucins or neoglycoproteins, we did not observe a binding that correlated to the level of LDN expression on the proteins. This conclusion is drawn because PSGL-1/mIgG2b lacking LDN determinants and carrying mainly mono- and disialyl core 1 O-glycans (CP-C1), bound 26695 H. pylori better than PSGL-1/mIgG2b carrying LDN on extended core 1 chains (CP-ext C1-LDN). Also, PSGL-1/mIgG2b carrying LDN on core 2 O-glycans (CP-C2-LDN) and staining the strongest with anti-LDN antibodies, did not bind 26695 bacteria significantly more than any the other mucin-type fusion proteins. In addition, there was no correlation between adhesion of the 26695 strain to native gastric mucins and the presence of the LDN determinant on those mucins. The J99 strain bound Leb-HSA and mucin 1 known to carry the Leb determinant (Table S1, ESI†), while strain 26695 did not. The inability of strain 26695 to bind Leb determinants was expected since this strain has the babA gene but does not express the BabA protein.44 Further, we failed to show binding of a LDN-BSA conjugate to any of 17 H. pylori strains/isolates in suspension, including the 26695 and J99 strains used in the solid phase, microtiter well-based adhesion assay and shown to carry the LDN adhesin. In contrast to the results of Rossez and coworkers,8 we could not verify a LDN-mediated adhesion of H. pylori strain 26695, despite the relative abundance of LDN was similar among the LDN positive human gastric mucins in the present study as among the mucins analysed by Rossez et al. The reason for this discrepancy is not obvious but may be related to the assay system used; inhibition of adhesion to tissue sections by means of free oligosaccharides or blocking antibodies vs. a microtiter well-based adhesion assay using recombinant mucin-type proteins or purified gastric mucins as substrates and an adhesion assay in solution using a LDN-conjugate. Further, the fine structure of LDN O-glycans represented in the two assay systems may differ, at least regarding the recombinant PSGL-1/mIgG2b. At least two LDN-containing oligosaccharides were identified in human gastric mucin, Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol and Fucα2Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol,14 whereas the predominant LDN-containing core 2 O-glycan on CP-C2-LDN was NeuNAcα3Galβ3(GalNAcβ4GlcNAcβ6)GalNAcol. Whether the sialic acid residue on the C3 branch of this core 2 oligosaccharide may prevent binding to the LDN determinant on the C6 branch is not known. Nevertheless, the gastric mucins used in the adhesion assay, and which did not support binding either, carried the LDN O-glycans described by Kenny et al.14 and verified by Rossez et al.8 making this explanation less likely. Further studies are needed to verify the binding specificity of LDN-binding H. pylori strains. Previous studies have demonstrated that the BabA-mediated adhesion to fucosylated structures is the dominating binding mode to gastric mucins from non-cancerous individuals.45–47 In Sweden, approximately 75% of H. pylori strains express BabA and bind to ABO blood group antigens in gastric mucosa.35 The great majority of H. pylori from patients with overt gastric disease, such as duodenal ulcer and cancer, are “triple-positive” because they have the full set of BabA, VacA and CagA, i.e the major established virulence markers.48 When taken together this suggests that H. pylori adherence to gastric mucosa, is a multi-step process, with initial binding and attachment to high affinity/avidity binding sites such as the ABO antigens. This initial binding could then proceed to a more targeted interaction to certain cells and structures found in more inflamed and/or dysplastic cells.49 The role of Lab A in such a multistep binding process is currently not known.
Conclusion
We have shown that B4GALNT3 can efficiently transfer GalNAc to GlcNAc-terminated O-glycans based on the core 2, core 3 and extended core 1 structures, while a mucin-type fusion protein carrying only core 1 O-glycans could not be modified to carry LDN. Further, we did not detect any LDN-mediated adhesion of H. pylori strains 26695 and J99 in a microtiter well-based adhesion assay. Similarly, no binding was found of a world-wide series of H. pylori strains to a soluble LDN-conjugate. Further studies are warranted to verify that LDN constitutes a significant binding site for H. pylori adhesion to the gastric mucosa and mucins, and in addition, how LDN binding relates to the LabA protein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Professor Richard D. Cummings (Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School) for the anti LDN antibody and Prof. Lothar Elling, Laboratory for Biomaterials, Institute for Biotechnology and Helmholtz-Institute for Biomedical Engineering, RWTH Aachen University for the LDN (16R-BSA). Proteomic analysis was performed by Carina Sihlbom at the Proteomics Core Facility of Sahlgrenska Academy, University of Gothenburg. This work was supported by the Swedish state under an agreement between the Swedish government and the county council, the ALF-agreement [ALFGBG-725381] to J. H., the Swedish Cancer Society (S. K. L. and T. B.), the Swedish Research Council (T. B.) and the RR Julin foundation to S. K. L. Y. M. was supported by a Marie Curie Fellowship from the European Union FP7 GastricGlycoExplorer ITN [316929] to J. H., S. K. L. and N. G. K. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- B. J. Marshall and J. R. Warren, Lancet, 1984, 1, 1311–1315 CrossRef CAS
.
- J. C. Atherton, Annu. Rev. Pathol.: Mech. Dis., 2006, 1, 63–96 CrossRef CAS PubMed
- C. Imrie, M. Rowland, B. Bourke and B. Drumm, Pediatrics, 2001, 107, 373–380 CrossRef CAS PubMed
- P. C. Konturek, S. J. Konturek and T. Brzozowski, J. Physiol. Pharmacol., 2009, 60, 3–21 CAS
- M. M. Walker, L. Teare and C. McNulty, Postgrad. Med. J., 2008, 84, 169 CrossRef PubMed
- T. Boren, P. Falk, K. A. Roth, G. Larson and S. Normark, Science, 1993, 262, 1892–1895 CrossRef CAS PubMed
- J. Mahdavi, B. Sonden, M. Hurtig, F. O. Olfat, L. Forsberg, N. Roche, J. Angstrom, T. Larsson, S. Teneberg, K. A. Karlsson, S. Altraja, T. Wadstrom, D. Kersulyte, D. E. Berg, A. Dubois, C. Petersson, K. E. Magnusson, T. Norberg, F. Lindh, B. B. Lundskog, A. Arnqvist, L. Hammarstrom and T. Boren, Science, 2002, 297, 573–578 CrossRef CAS PubMed
- Y. Rossez, P. Gosset, I. G. Boneca, A. Magalhaes, C. Ecobichon, C. A. Reis, C. Cieniewski-Bernard, M. Joncquel Chevalier Curt, R. Leonard, E. Maes, B. Sperandio, C. Slomianny, P. J. Sansonetti, J. C. Michalski and C. Robbe-Masselot, J. Infect. Dis., 2014, 210, 1286–1295 CrossRef CAS PubMed
- M. A. McGuckin, S. K. Linden, P. Sutton and T. H. Florin, Nat. Rev. Microbiol., 2011, 9, 265–278 CrossRef CAS PubMed
- S. Linden, H. Nordman, J. Hedenbro, M. Hurtig, T. Boren and I. Carlstedt, Gastroenterology, 2002, 123, 1923–1930 CrossRef CAS PubMed
- S. K. Linden, Y. H. Sheng, A. L. Every, K. M. Miles, E. C. Skoog, T. H. Florin, P. Sutton and M. A. McGuckin, PLoS Pathog., 2009, 5, e1000617 CrossRef PubMed
- S. Linden, C. Semino-Mora, H. Liu, J. Rick and A. Dubois, Helicobacter, 2010, 15, 251–258 CrossRef PubMed
- A. Dell, S. Chalabi, R. L. Easton, S. M. Haslam, M. Sutton-Smith, M. S. Patankar, F. Lattanzio, M. Panico, H. R. Morris and G. F. Clark, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15631–15636 CrossRef CAS PubMed
- D. T. Kenny, E. C. Skoog, S. K. Linden, W. B. Struwe, P. M. Rudd and N. G. Karlsson, Glycobiology, 2012, 22, 1077–1085 CrossRef CAS PubMed
- M. I. Che, J. Huang, J. S. Hung, Y. C. Lin, M. J. Huang, H. S. Lai, W. M. Hsu, J. T. Liang and M. C. Huang, Oncotarget, 2014, 5, 3673–3684 CrossRef PubMed
- J. Huang, J. T. Liang, H. C. Huang, T. L. Shen, H. Y. Chen, N. Y. Lin, M. I. Che, W. C. Lin and M. C. Huang, Mol. Cancer Res., 2007, 5, 543–552 CrossRef CAS PubMed
- M. Gotoh, T. Sato, K. Kiyohara, A. Kameyama, N. Kikuchi, Y. D. Kwon, Y. Ishizuka, T. Iwai, H. Nakanishi and H. Narimatsu, FEBS Lett., 2004, 562, 134–140 CrossRef CAS PubMed
- T. Sato, M. Gotoh, K. Kiyohara, A. Kameyama, T. Kubota, N. Kikuchi, Y. Ishizuka, H. Iwasaki, A. Togayachi, T. Kudo, T. Ohkura, H. Nakanishi and H. Narimatsu, J. Biol. Chem., 2003, 278, 47534–47544 CrossRef CAS PubMed
- E. P. Bennett, U. Mandel, H. Clausen, T. A. Gerken, T. A. Fritz and L. A. Tabak, Glycobiology, 2012, 22, 736–756 CrossRef CAS PubMed
- F. G. Hanisch, D. Bonar, N. Schloerer and H. Schroten, J. Biol. Chem., 2014, 289, 27363–27375 CrossRef CAS PubMed
- C. Jin, D. T. Kenny, E. C. Skoog, M. Padra, B. Adamczyk, V. Vitizeva, A. Thorell, V. Venkatakrishnan, S. K. Linden and N. G. Karlsson, Mol. Cell. Proteomics, 2017, 16, 743–758 CrossRef CAS PubMed
- T. Ju, K. Brewer, A. D'Souza, R. D. Cummings and W. M. Canfield, J. Biol. Chem., 2002, 277, 178–186 CrossRef CAS PubMed
- M. F. Bierhuizen and M. Fukuda, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9326–9330 CrossRef CAS PubMed
- T. Iwai, N. Inaba, A. Naundorf, Y. Zhang, M. Gotoh, H. Iwasaki, T. Kudo, A. Togayachi, Y. Ishizuka, H. Nakanishi and H. Narimatsu, J. Biol. Chem., 2002, 277, 12802–12809 CrossRef CAS PubMed
- J. C. Yeh, E. Ong and M. Fukuda, J. Biol. Chem., 1999, 274, 3215–3221 CrossRef CAS PubMed
- J. C. Yeh, N. Hiraoka, B. Petryniak, J. Nakayama, L. G. Ellies, D. Rabuka, O. Hindsgaul, J. D. Marth, J. B. Lowe and M. Fukuda, Cell, 2001, 105, 957–969 CrossRef CAS PubMed
- A. K. Nyame, A. M. Leppanen, R. DeBose-Boyd and R. D. Cummings, Glycobiology, 1999, 9, 1029–1035 CrossRef CAS PubMed
- J. Liu, C. Jin, R. M. Cherian, N. G. Karlsson and J. Holgersson, J. Biotechnol., 2015, 199, 77–89 CrossRef CAS PubMed
- N. G. Karlsson, B. L. Schulz and N. H. Packer, J. Am. Soc. Mass Spectrom, 2004, 15, 659–672 CrossRef CAS PubMed
- V. Paraskevopoulou, V. G. Artiaga, R. Rowlinson, G. S. Winkler, P. Gellert, S. Stolnik, R. Overman and F. H. Falcone, Protein Expression Purif., 2019, 163, 105446 CrossRef CAS PubMed
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef CAS PubMed
- E. C. Skoog, M. Padra, A. Aberg, P. Gideonsson, I. Obi, M. P. Quintana-Hayashi, A. Arnqvist and S. K. Linden, Sci. Rep., 2017, 7, 40656 CrossRef CAS PubMed
-
M. Aspholm, A. Kalia, S. Ruhl, S. Schedin, A. Arnqvist, S. Lindén, R. Sjöström, M. Gerhard, C. Semino-Mora, A. Dubois, M. Unemo, D. Danielsson, S. Teneberg, W. K. Lee, D. E. Berg and T. Borén, Methods Enzymol., Academic Press, 2006, vol. 417, pp. 293–339 Search PubMed
- J. A. Bugaytsova, O. Bjornham, Y. A. Chernov, P. Gideonsson, S. Henriksson, M. Mendez, R. Sjostrom, J. Mahdavi, A. Shevtsova, D. Ilver, K. Moonens, M. P. Quintana-Hayashi, R. Moskalenko, C. Aisenbrey, G. Bylund, A. Schmidt, A. Aberg, K. Brannstrom, V. Koniger, S. Vikstrom, L. Rakhimova, A. Hofer, J. Ogren, H. Liu, M. D. Goldman, J. M. Whitmire, J. Aden, J. Younson, C. G. Kelly, R. H. Gilman, A. Chowdhury, A. K. Mukhopadhyay, G. B. Nair, K. S. Papadakos, B. Martinez-Gonzalez, D. N. Sgouras, L. Engstrand, M. Unemo, D. Danielsson, S. Suerbaum, S. Oscarson, L. A. Morozova-Roche, A. Olofsson, G. Grobner, J. Holgersson, A. Esberg, N. Stromberg, M. Landstrom, A. M. Eldridge, B. A. Chromy, L. M. Hansen, J. V. Solnick, S. K. Lindén, R. Haas, A. Dubois, D. S. Merrell, S. Schedin, H. Remaut, A. Arnqvist, D. E. Berg and T. Boren, Cell Host Microbe, 2017, 21, 376–389 CrossRef CAS PubMed
- M. Aspholm-Hurtig, G. Dailide, M. Lahmann, A. Kalia, D. Ilver, N. Roche, S. Vikström, R. Sjöström, S. Lindén, A. Bäckström, C. Lundberg, A. Arnqvist, J. Mahdavi, U. J. Nilsson, B. Velapatiño, R. H. Gilman, M. Gerhard, T. Alarcon, M. López-Brea, T. Nakazawa, J. G. Fox, P. Correa, M. G. Dominguez-Bello, G. I. Perez-Perez, M. J. Blaser, S. Normark, I. Carlstedt, S. Oscarson, S. Teneberg, D. E. Berg and T. Borén, Science, 2004, 305, 519 CrossRef CAS PubMed
- D. Ilver, A. Arnqvist, J. Ogren, I. M. Frick, D. Kersulyte, E. T. Incecik, D. E. Berg, A. Covacci, L. Engstrand and T. Borén, Science, 1998, 279, 373 CrossRef CAS PubMed
- J. Liu, A. Gustafsson, M. E. Breimer, A. Kussak and J. Holgersson, Glycobiology, 2005, 15, 571–583 CrossRef CAS PubMed
- N. Karlsson, B. Schulz and N. Packer, J. Am. Soc. Mass Spectrom, 2004, 15, 659–672 CrossRef CAS PubMed
- R. A. Alm, J. Bina, B. M. Andrews, P. Doig, R. E. Hancock and T. J. Trust, Infect. Immun., 2000, 68, 4155–4168 CrossRef CAS PubMed
- R. A. Alm, L.-S. L. Ling, D. T. Moir, B. L. King, E. D. Brown, P. C. Doig, D. R. Smith, B. Noonan, B. C. Guild, B. L. deJonge, G. Carmel, P. J. Tummino, A. Caruso, M. Uria-Nickelsen, D. M. Mills, C. Ives, R. Gibson, D. Merberg, S. D. Mills, Q. Jiang, D. E. Taylor, G. F. Vovis and T. J. Trust, Nature, 1999, 397, 176–180 CrossRef PubMed
- J.-F. Tomb, O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. S. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser and J. C. Venter, Nature, 1997, 388, 539–547 CrossRef CAS PubMed
- K.-Y. Do, S. Do and R. D. Cummings, Glycobiology, 1997, 7, 183–194 CrossRef CAS PubMed
- C. H. Hokke, A. A. Bergwerff, G. W. K. Van Dedem, J. Van Oostrum, J. P. Kamerling and J. F. G. Vliegenthart, FEBS Lett., 1990, 275, 9–14 CrossRef CAS PubMed
- E. C. Skoog, M. Lindberg and S. K. Linden, Helicobacter, 2011, 16, 9–19 CrossRef CAS PubMed
- M. P. Quintana-Hayashi, R. Rocha, M. Padra, A. Thorell, C. Jin, N. G. Karlsson, M. Roxo-Rosa, M. Oleastro and S. K. Lindén, Virulence, 2018, 9, 1699–1717 CrossRef CAS PubMed
- S. Lindén, J. Mahdavi, C. Semino-Mora, C. Olsen, I. Carlstedt, T. Borén and A. Dubois, PLoS Pathog., 2008, 4, e2 CrossRef PubMed
- S. K. Lindén, C. Wickström, G. Lindell, K. Gilshenan and I. Carlstedt, Helicobacter, 2008, 13, 81–93 CrossRef PubMed
- M. Gerhard, N. Lehn, N. Neumayer, T. Borén, R. Rad, W. Schepp, S. Miehlke, M. Classen and C. Prinz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12778–12783 CrossRef CAS PubMed
- A. Javaheri, T. Kruse, K. Moonens, R. Mejias-Luque, A. Debraekeleer, C. I. Asche, N. Tegtmeyer, B. Kalali, N. C. Bach, S. A. Sieber, D. J. Hill, V. Koniger, C. R. Hauck, R. Moskalenko, R. Haas, D. H. Busch, E. Klaile, H. Slevogt, A. Schmidt, S. Backert, H. Remaut, B. B. Singer and M. Gerhard, Nat. Microbiol., 2016, 2, 16189 CrossRef CAS PubMed
- B. Domon and C. E. Costello, Biochemistry, 1988, 27, 1534–1543 CrossRef CAS PubMed
- J. Liu, Y. Qian and J. Holgersson, Transplantation, 1997, 63, 1673–1682 CrossRef CAS PubMed
- J. Lofling, M. Diswall, S. Eriksson, T. Boren, M. E. Breimer and J. Holgersson, Glycobiology, 2008, 18, 494–501 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mo00175a |
| This journal is © The Royal Society of Chemistry 2020 |