
Fast processing of highly crosslinked, low-viscosity vitrimers†
Christian
Taplan
 ,
Marc
Guerre
*,
Johan M.
Winne
* and
Filip E.
Du Prez
*
,
Marc
Guerre
*,
Johan M.
Winne
* and
Filip E.
Du Prez
*
Polymer Chemistry Research Group and Laboratory for Organic Synthesis, Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4-bis, Ghent B-9000, Belgium. E-mail: Marc.Guerre@UGent.be; Johan.Winne@UGent.be; Filip.DuPrez@UGent.be
First published on 23rd August 2019
Abstract
Here we describe a rational approach to go beyond the current processability limits of vitrimer materials, with a demonstration of low-viscosity fast processing of highly crosslinked permanent networks. Vitrimers are a recently introduced class of polymer networks with a unique glass-like viscoelastic behavior, in which bond exchange reactions govern the macroscopic material flow. The restricted chain mobility, only enabled by chemical exchanges, typically limits the use of continuous processing techniques, such as extrusion or injection moulding. Herein, we outline a straightforward materials design approach, taking into account both the effect of minor additives on the chemistry of bond rearrangement as well as the macromolecular architecture of the vitrimeric network. These combined effects are demonstrated to work in an additive fashion, culminating in stress relaxation times below 1 s at 150 °C. The observed rapid bond exchanges in permanent networks result in an unprecedented control of the polymer material behavior, where the material flow is still dominated by chemical exchanges, but only marginally limited by the chemical exchange rate, overcoming the challenges encountered so far in continuous processing of highly crosslinked vitrimeric systems.
New conceptsVitrimers are permanent macromolecular networks that undergo stress relaxation through bond exchanges. Because the viscous flow of a polymer network is limited by the rate of these chemical reactions, the (re)processing of vitrimers has been limited to compression molding, related to the long times (>10 seconds) it generally takes to reach chemical equilibrium. The presented work demonstrates a rational vitrimer material design, encompassing the exact macromolecular architecture, i.e., distribution of reactive moieties, and controlling the exchange chemistry in a straightforward manner to achieve a breakthrough in polymer network processing of highly crosslinked vitrimers by means of extrusion. This result stands in contrast to current existing vitrimers, as it is achieved without the need of reactive extrusion and works in a temperature range suitable for organic materials. The main principles underlying the rational design of these vitrimers is demonstrated for vinylogous urethane vitrimers, but should also be applicable to other vitrimer chemistries, pointing towards a general concept to achieve continuous processing of permanently crosslinked polymer networks. |
Introduction
Material science has evolved over the past few decades from designing tougher and more durable materials towards tailored degradability, processability and ultimately recyclability. This new particular public awareness originates from an ongoing process of reducing the environmental impact of synthetic materials by for instance extending their life-cycle or targeting second-life applications in a circular economy.1,2 However, when considering highly crosslinked materials, such as those used in composites for lightweight applications, or rubbers as found in tires or conveyor belts, recyclability is profoundly limited. Therefore, over the past decades, the scientific community has explored the application of reversible crosslinking points in thermoset-like materials, via dynamic covalent bonds (so-called covalent adaptable networks, CANs),3–5 or via non-covalent interactions such as hydrogen bonds,6 host/guest interactions7 or the design of thermoplastic elastomers.8 In CANs, the dynamic character is achieved by applying different stimuli, e.g., light,9 pH, heat,10 upon which an either dissociative or associative reaction mechanism is triggered. Importantly, in such materials, the viscoelastic properties or the rate of macroscopic flow is dictated by the underlying mechanism involved in the dynamic exchange reactions. For instance, a dissociative mechanism can be characterized by a net loss of crosslinking density, implying a loss of network integrity and resulting in a sudden macroscopic flow after the application of a stimulus. On the other side, an associative mechanism-based dynamic network keeps the crosslinking density intact because a new covalent bond needs to be formed either simultaneously or prior to breaking an existing covalent bond.11 Pioneered by Leibler and co-workers in 2011,12 such associative pathways have been successfully introduced into non-decrosslinking, permanent but at the same time dynamic networks based on a zinc catalyzed transesterification mechanism, which was demonstrated to undergo gradual topological rearrangements upon heating. Due to their peculiar viscoelastic behavior, i.e., gradual decrease of the viscosity, a behavior previously observed only in inorganic vitreous silica, this new generation of CAN materials were introduced as vitrimers.Ever since, vitrimers gained much attention and are an emerging new class of polymer materials.13–22 This interest arises from their outstanding properties, which provide a constant crosslinking density throughout the (re)processing, maintaining the chemical integrity of the polymer network and preventing dissolution. However, until now the technology transfer of these revolutionary materials from academic research to industrial manufacturing is limited by the industrial processability requirements. One reason is that the temperatures that would theoretically be required to achieve a sufficiently fast processing are beyond the thermal stability limits of most organic materials. These temperature limits prevent common continuous processing techniques like extrusion, or injection molding. In most cases described so far, the processability is limited to compression moulding.11 Recently, however, Leibler and co-workers presented a promising approach, where extrudable reinforced vitrimeric thermoplastics were designed via reactive extrusion.23 The inclusion of dynamic crosslinks in these materials markedly increased solvent resistance compared to non-modified materials, whereas the permanent crosslinks did not excessively slow down the processability due to their dynamic nature. In related studies, this reactive vitrimer extrusion was also applied in order to create more densely crosslinked materials based on semi-crystalline poly(butylene terephthalate).24 However, because of the highly crosslinked nature of the finally obtained material, further processing following the reactive extrusion step was again limited to compression molding.
Herein, we describe an approach that enables to overcome current processing limitations, set by highly crosslinked systems, by rational design and control of the bond exchange rates. For the first time, highly crosslinked vitrimeric materials, in this study based on poly(propylene glycol), a commonly used diol for elastomer applications was created to achieve a characteristic relaxation time well below one second. Further, we demonstrate that this design leads to a crosslinked material, which can be extruded in its fully cured form, while maintaining its material and rheological properties.
Results and discussion
As the macroscopic flow of a vitrimer is governed by the underlying exchange reaction, we were stimulated to design a vitrimer material in which the respective dynamic exchange is occurring so quickly that it does not hinder this flow. Therefore, the aim here was to develop a strategy that enables future vitrimer materials to be tailored regarding their processing and application potential. Bearing this in mind, the effect of the chemical nature of the crosslinking on the exchange kinetics rates was first investigated. In a vitrimer, composed of A- and B-type monomers (e.g., A/B standing for alcohols/carboxylic-acids,16,25 or amines/aldehydes,15,26,27 or amines/acetoacetates),20,28–30 it can be speculated that the dynamic character, and inherently the resulting exchange kinetics, of the formed AB bond (e.g., ester, imine, or vinylogous urethane respectively) is not exclusively governed by the dynamic exchanges but can further be influenced by the macromolecular architecture. Similar observations were made in dissociatively Diels–Alder based dynamic networks.31 Indeed, by definition, the viscoelastic properties of vitrimers are mainly chemistry-dependent. A potential underlying effect originating from the structure from which a vitrimeric macromolecular network was built, on the exchange reactions has not been explicitly considered so far in vitrimer design.To preliminarily evaluate the effect of macromolecular architecture, we designed on the one hand elastic A3 (trifunctional nucleophilic crosslinker) and on the other hand B3 (trifunctional electrophilic crosslinker) type vitrimers with a very similar overall composition (Fig. 1). The choice for the dynamic vinylogous urethane chemistry is based on our earlier research efforts with vitrimers in which the robust exchange reactions are based on the reaction of amines with vinylogous urethane groups, readily derived from amines and acetoacetate groups (β-ketoesters).20 The A3-type vitrimer was prepared based on a poly(propylene glycol) bis-acetoacetate (PPG400-AA), easily synthesized according to earlier reported procedures (see ESI†), crosslinked with tris(2-aminoethyl)amine (TREN), and compared to poly(propylene glycol) bis-amine (Jeffamine D-400, PPG-NH2) crosslinked with a tris-acetoacetate moiety (B3-type) (Fig. 1b). In both networks, the composition was chosen in such a way that the fraction of pendent amines (available nucleophiles essential for the associative dynamic character) remained constant (5 mol%). When investigating both types of materials with respect to stress relaxation, the characteristic relaxation time (τ*) differed quite significantly with values around 680 s for the A3-type structures and around 7000 s for the B3-type ones at 150 °C (Fig. 1c).
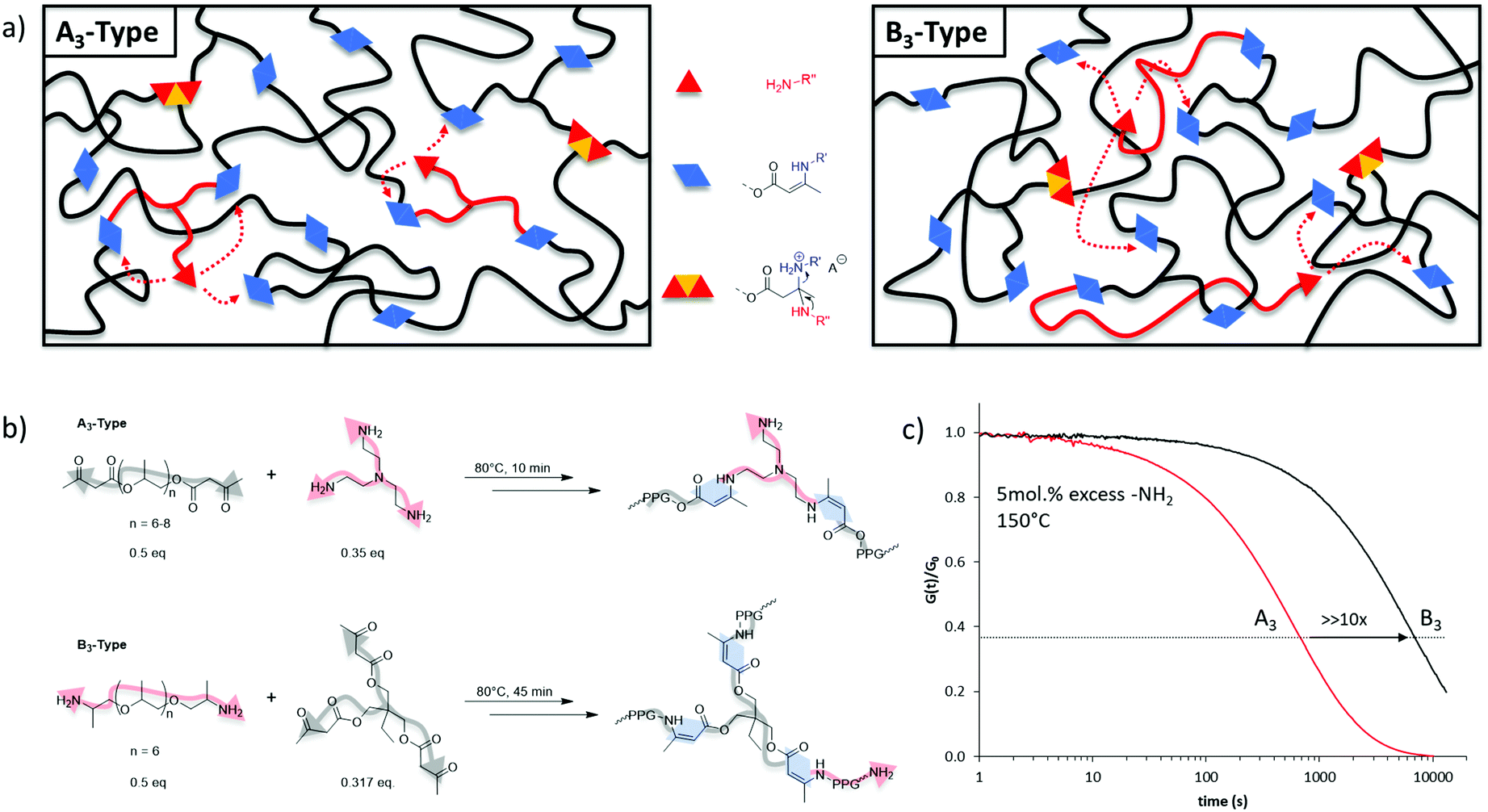 | ||
| Fig. 1 Different vitrimer networks based on trifunctional nucleophilic (A3-type) or trifunctional electrophilic (B3-type) crosslinkers. (a) Schematic representation of the two vitrimer materials, (b) chemical nature of the network and (c) the resulting characteristic relaxation times. | ||
By using a similar molecular weight, matrix and exchange chemistry, the remarkable difference in observed stress relaxation rates are not easy to explain but may be linked to chain length and the statistical distribution of the pendent primary amines in the network, originating from pendent amines, and crosslinking points throughout the macromolecular matrix of the created networks. When a nucleophilic crosslinker is used (A3-type, TREN), amine nucleophiles are likely to be situated close to two VU moieties and close to a tertiary amine (Fig. 1). Hence, in this A3-type vitrimer the statistical likelihood of a productive exchange can be enhanced by a number of effects that bring together the reaction partners, thus affecting the exchange rate, but not the activation energy. In contrast, the exchange reactions in B3-type vitrimers operate only via an amino-terminated long-chain nucleophile that will be more readily associated with the macromonomer backbone, and can thus be statistically less likely to undergo a productive exchange via the same effects. The tenfold difference observed in stress relaxation experiments clearly indicates that the crosslinkers’ chemical nature can significantly affect the exchange kinetics. Thus, in addition to the molecular mechanism of the underlying chemical exchange, more parameters that can possibly govern the dynamic material properties of vitrimers should be taken into account. Among these parameters, the precise nature and exact architecture of the polymer matrix require a close attention within the design and study of such type of materials.
The next aim of this study was to further tune the exchange kinetics for speeding up the vitrimer's processability. Thus, the A3-type molecular architecture was applied throughout the following investigation in order to push the processing limitation of densely crosslinked networks. Therefore, additional mechanistic design principles were investigated on behalf of vinylogous urethane exchange chemistry. Since an addition/elimination pathway is the underlying mechanism for the dynamic exchange reaction, the nucleophiles’ concentration (amines) dependence was investigated. In this context, several A3-type vitrimers were prepared following the same procedure as displayed in Fig. 1, but the proportion of pendent primary amines was screened, ranging from 5 mol% (N5) to 40 mol% (N40) with respect to the formed vinylogous urethane moieties. Each of the network composition led to a complete consumption of the acetoacetate groups, as shown by ATR-FTIR (Fig. S4 and S5, ESI†) in which the increase of pendent amines can be qualitatively monitored in the range of 3000 cm−1. Further TGA analyses showed a nearly similar thermal stability for both systems ranging from 260 °C to 280 °C, regardless the amount of primary amines (Fig. S7, ESI†). To compare the network formation, soluble fraction tests have been conducted in THF. As expected, a higher concentration of pendent amines causes also an increase of the respective soluble fraction (see Table 1, last column), ranging from 1.6 ± 0.3 wt% for the N5 vitrimers to 20 ± 0.2 wt% for the N40 ones. Although N40 is expected to possess a weaker network structure due to the high concentration of dangling units and reduced crosslinking density, with a soluble fraction of 20%, it can still be considered as a decent dynamically crosslinked system.
| Material | –NH2 (mol%) | pTsOH (mol%) |
τ
160°C*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (s) a (s) |
E a(I)b (kJ mol−1) | E a(II)b (kJ mol−1) | T V (°C) | T TGA-95% (°C) | Swel. rat.e (wt%) | Sol. frac.e (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| a Characteristic relaxation time (τ*) at 160 °C obtained from stress relaxation experiments. b Activation energies (Ea(I) and Ea(II) for the iminium and Michael-addition pathway respectively) obtained from Arrhenius plots. c Topology freezing temperature (Tv) calculated by extrapolation for a viscosity of 1012 Pa s. d TGA onset-temperatures after 5% weight-loss (TTGA-95%). e Swelling ratio and soluble fraction in THF or acetone (*) obtained from four samples measurement at r.t. for 24 h. | |||||||||
| N5 | 5 | — | 103 | 69 ± 1 | 250 ± 20 | 46 | 260 | 202 ± 5 | 1.6 ± 0.3 |
| N5H0.3 | 0.3 | 56 | 99 ± 3 | — | 48 | 270 | 225 ± 4 | 1.5 ± 0.2 | |
| N5H1.5 | 1.5 | 11 | 86 ± 1 | — | 16 | 245 | 272 ± 8 | 4.1 ± 1.1 | |
| N5H6 | 6 | 0.9 | 78 ± 2 | — | −17 | 230 | 316 ± 5 | 3.6 ± 0.6 | |
| N10 | 10 | — | 41 | 75 ± 1 | 180 ± 22 | 37 | 275 | 233 ± 12 | 1.4 ± 0.8 |
| N10H3 | 3 | 2.2 | 80 ± 2 | — | ±0 | 235 | 356 ± 10 | 6.8 ± 0.7 | |
| N15 | 15 | — | 16 | 83 ± 1 | 160 ± 10 | 32 | 280 | 262 ± 17 | 2.6 ± 0.3 |
| N15H4.5 | 4.5 | 0.8 | 78 ± 1 | — | −12 | 235 | 172 ± 7* | 4.8 ± 0.5* | |
| N20 | 20 | — | 10 | 102 ± 3 | 130 ± 12 | 42 | 265 | 280 ± 5 | 4.2 ± 1.4 |
| N20H1.5 | 1.5 | 1.0 | 78 ± 0.5 | — | −8 | 230 | 161 ± 4* | 3.6 ± 0.5* | |
| N20H6 | 6 | 0.3 | 70 ± 1 | — | −17 | 235 | 210 ± 2* | 8.3 ± 0.7* | |
| N40 | 40 | — | 2.0 | 76 ± 1 | — | −9 | 260 | 716 ± 10 | 20 ± 0.2 |
When looking at the rheological behavior of these vitrimers, it can be found that increasing the amount of pendent amine reduces the resulting characteristic relaxation time significantly (Table 1). Moreover, it appears that it also affects the recently described dual-temperature response for vinylogous urethane networks,32 which originates from two competing exchange pathways for the same dynamic chemistry platform, leading to a remarkable different activation energy from an iminium-type reaction (Ea(I)) and a Michael-addition originating (Ea(II)). Indeed, the increased presence of protic species provided by readily available amines, quenched the aprotic Michael-type pathway and pushes the temperature dependency towards the protic-iminium pathway (Fig. 2a). This results in a linear Arrhenius plot (regression grade R2 of 0.9986) with an activation energy of 76 kJ mol−1 (Fig. 2b, left). This swift transition towards the protic-iminium intermediate was also previously reported in the context of the addition of Brønsted acids.28
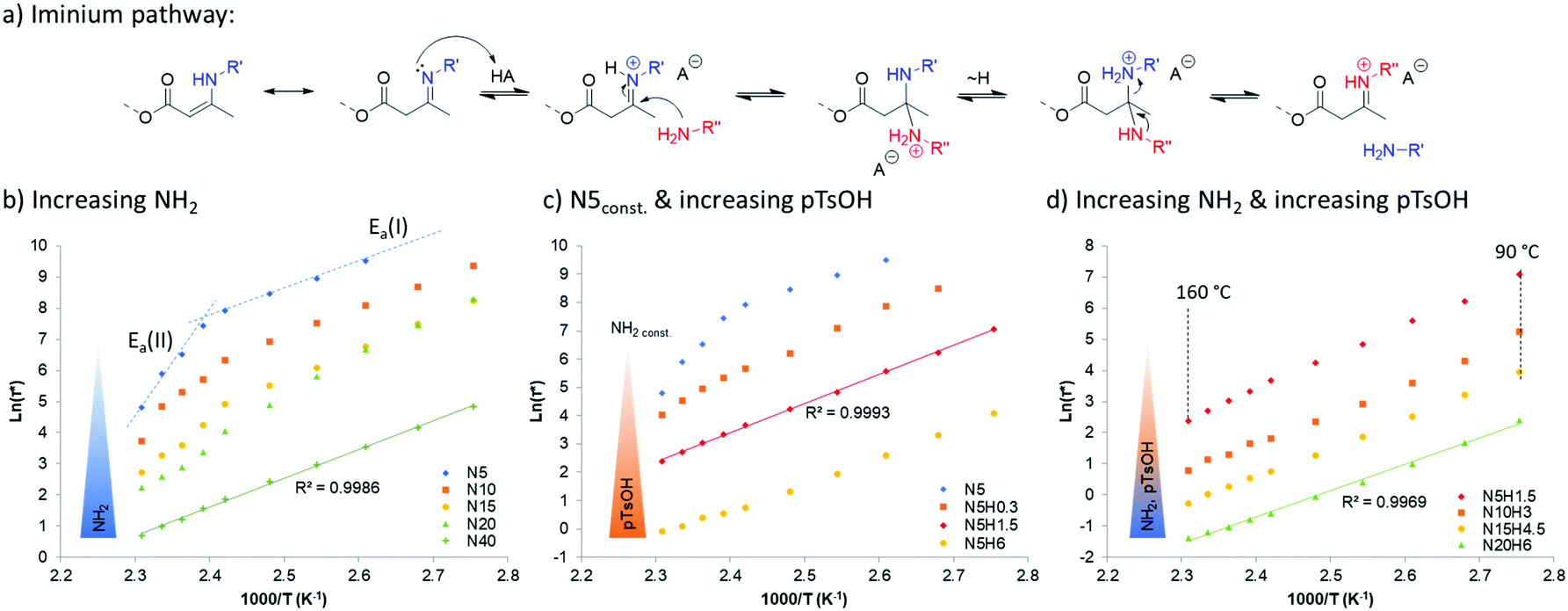 | ||
| Fig. 2 Description of the iminium pathway (a). Arrhenius plots obtained after stress relaxation experiments of PPG400 vinylogous urethane vitrimers with varying amine content N5–N40 (b), with constant amine content N5 and varying pTsOH content H0.3–H6 (c), and with variation of both amine and pTsOH content (d) over a temperature range from 90 °C to 160 °C. | ||
Since the final aim of this study is to push the actual processing boundaries of vitrimers, it is important to accelerate the exchange rates. When taking into account that the kinetically favored mechanism over the investigated temperature range follows the iminium pathway, one can predict that increasing proton mobility or accessibility can help reaching this target. Thus, several vitrimers were prepared with different amounts of para-toluene sulfonic acid. Indeed, an addition of only 0.3 mol% of non-volatile and temperature stable para-toluene sulfonic acid (pTsOH) (H0.3) on a material containing 5 mol% of pendent amines, already reduces drastically the attributed aprotic pathway (Michael-addition), as can be observed from the obtained Arrhenius-plots that show no dual temperature response anymore (Fig. 2c, N5H0.3). Further addition of pTsOH up to 6 mol%, while keeping the amount of pendent amines constant, enabled a remarkable acceleration of the exchange reaction. Indeed, for the first time in vitrimer research, characteristic relaxation times below 1 s could be obtained, shown for a material composed of only 5 mol% of pendent amines and incorporating 6 mol% of pTsOH (N5H6) that relaxes the stress of a 1% deformation in approximately 0.9 s at 160 °C. Combining both parameters, being an increase of the available nucleophilic species in an A3-type network with an increase of available externally provided protons, enabled us to further decrease the measured relaxation times down to 0.3 s (N20H6, Fig. 2d). Yet, the materials exhibit the characteristic linear behavior over the entire measured temperature range from 90 °C to 160 °C (regression grade of R(N20H6)2 = 0.9969) and a resulting activation energy of 70 kJ mol−1, which is in good agreement with the reported values for the iminium pathway.28 Because of this low relaxation time, which could in principle lower the accuracy of stress relaxation measurements, additional frequency sweep experiments were performed (Fig. 3).
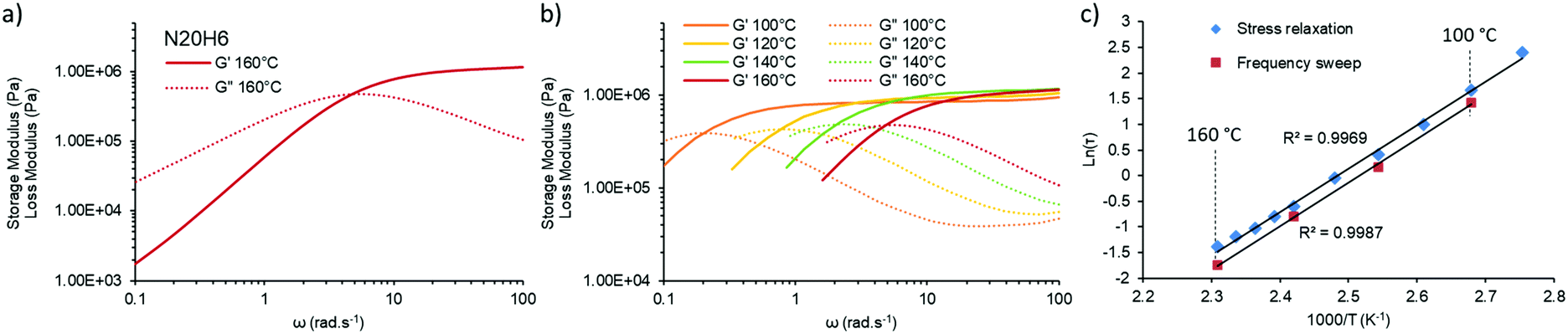 | ||
| Fig. 3 Frequency sweep measurements of vitrimer N20H6 at various temperatures (left) and the comparative plot of the respective Arrhenius plots, confirming the same temperature dependency obtained from stress-relaxation and frequency sweep experiments. | ||
A similar characteristic relaxation time at the crossover at approximately 5 rad s−1 (Fig. 3a), which calculates to a relaxation time of ca. 0.2 s at 160 °C and which is in good agreement with the value obtained via stress relaxation of approximately 0.3 s. When measured over different temperatures in the range of 100 °C to 160 °C the crossover shifts to lower frequencies with lower temperatures, while maintaining its storage modulus at high frequency confirming the associative nature of our underlying chemical mechanism (Fig. 3b).33 This information can be transformed into an Arrhenius-plot and it confirms the same temperature dependency (Fig. 3c) with a derived activation energy of 70 ± 1 kJ mol−1 on the one hand and therefore also the reliability of the performed stress relaxation experiments on the other.
To better evaluate the impact of this extremely fast relaxation with regard to the viscoelastic properties and processability, temperature sweep experiments with a constant angular frequency of 1 rad s−1 were performed on among others N5, N5H6, N20, and N20H6 vitrimer compositions (Fig. S10, ESI†). By monitoring the storage (G′) and the loss modulus (G′′) of these materials, it was observed that G′′ of N5 exhibits a slight increase in the early temperature range while the faster N20 composition displays a constant increase. When adding pTsOH (N5H6), it was possible to see a close crossover of G′ and G′′ at the point where the relaxation rate is around 1 s (Fig. S10, ESI†) at the respective temperature. Hence, a simultaneous increase of the amount of pendent amines and pTsOH content was intended to see if a crossover of a covalently crosslinked material can be achieved in a reasonable frequency range. Indeed, when looking at N5H1.5, N10H3 and N5H6 compositions, maintaining an anticipated crossover and thus a solid-liquid transition can be achieved and tuned accordingly (Fig. S10 and S14, ESI†). Going further with a combination of 15 mol% pendent amines with 4.5 mol% of pTsOH (N15H4.5) and 20 mol% amines with 6 mol% pTsOH (N20H6) resulted in a crossover at around 146 °C and 128 °C, respectively (Fig. S14, ESI†). Thus, with these two compositions, the corresponding networks exhibited predominantly a liquid behavior, correlating to a relaxation time of 1 s at temperatures far below degradation temperatures, opening the possibility for continuous processing (vide infra). In addition, this behavior was demonstrated to be fully reversible over at least three cycles as shown in Fig. S10 (ESI†) (N20H6), excluding the presence of significant side reactions.
However, when soluble fraction tests were performed in THF (24 h), the swollen networks of N15H4.5 and N20H6 were very weak, giving diffuse gel-like samples, so neither values for swelling ratio nor soluble fraction could be obtained in THF. On the other hand, since network N15 and N20 exhibited a low soluble fraction of 3 to 5 wt% (Table 1), the network formation of N15H4.5, N20H6 was assumed to have taken place. This hypothesis was verified by additional tests in a good swelling solvent that was also expected to reduce the reactivity of the free amines to prevent extreme swelling to a fragile network or organogel. For instance, when performing swelling tests in acetone, an excellent solvent for PPG that can additionally reduce the reactivity of the pendent amines due to an in situ imine/enamine formation,34 the network expansion as a result of swelling stress is reduced. Consequently, N15H4.5 and N20H6 exhibited measurable swelling ratios and soluble fractions in acetone, which were in line with the values of the aforementioned polymer networks without added catalysts (Table 1). These results indicate the fact that vitrimers, when they are exchanging bonds very rapidly, can show limitations to their inherent solvent resistance to the point where they can lose sample integrity, even when the network structure stays intact.
Additionally, TGA-analysis of these vitrimer networks (N15H4.5 and N20H6) showed that the thermal stability was only slightly affected (230 °C to 245 °C, Fig. S15 and S16, ESI†) compared to networks without acidic additives (260 °C to 280 °C). These values are well beyond the generally applied processing temperatures. Furthermore, isothermal TGA analyses was performed at 100 °C and 150 °C for N5 and N20H6. Interestingly, the latter showed merely less than 1% weight loss after two hours at 150 °C (Fig. S17, ESI†). Hence, we can reasonably conclude that this unprecedented temperature dependent behavior is not caused by thermal degradation of the network.
Next, we aimed for the extrusion of those carefully designed vitrimers showing rapid covalent adaptable fluid topology in order to obtain the first extruded, densely crosslinked covalent network. It should be emphasized that they remain fully crosslinked before, during and after processing, and that reactive extrusion is not required. Therefore, N20H6 was synthesized and dried thoroughly under vacuum for 48 h at 60 °C. A melt-flow-index (MFI) set-up was used to estimate the flow of this material at 170 °C and was determined to be 0.75 g per ten minutes. As expected according to the temperature sweep experiment mentioned before, this vitrimer exhibited a viscoelastic liquid behavior enabling sufficiently rapid macroscopic flow to obtain long cylindrical rods after the MFI measurement. Next, a double-twinscrew extruder was preheated to 150 °C for 10 min and fed with dried vitrimer N20H6 at a low shear rate of 4 rpm (Fig. 4a). While a shear rate of 1 rpm resulted in a smooth rod (Fig. 4b), higher rotation rates (2 and 4 rpm) resulted in helical wires, as can be seen in the video provided in the supporting information, which is ascribed to elastic memory effects of the material from the extruding screws.35
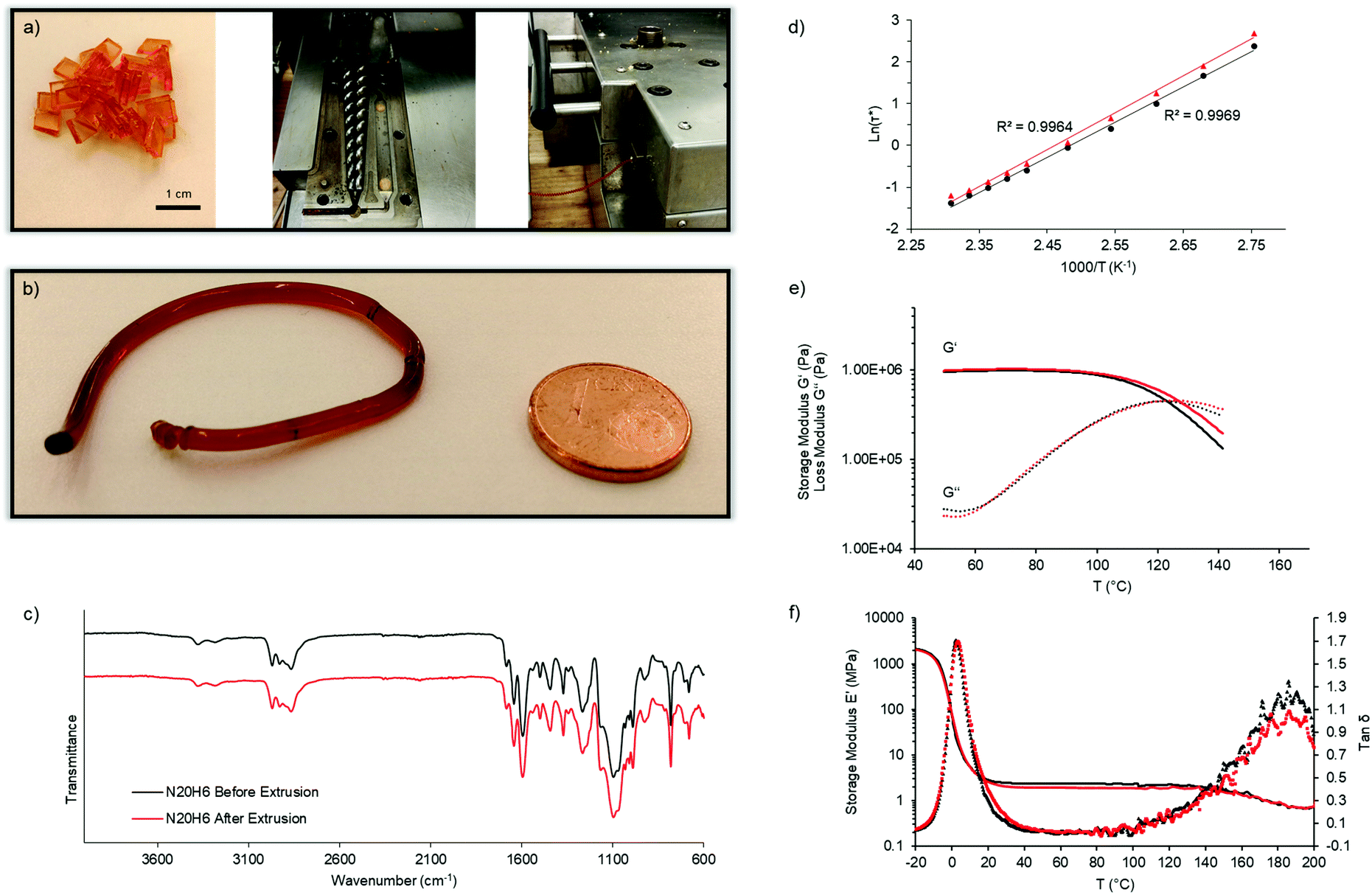 | ||
| Fig. 4 Extrusion and recyclability assessment of vitrimer N20H6. Elastic vitrimer pellets, and double-twinscrew extruder setup (a) used to create vitrimer extrudates (b) at 150 °C. Chemical and material integrity of the network is shown by ATR-FTIR (c), stress-relaxation and the corresponding Arrhenius plots (d), further temperature ramp showing shear-moduli (G′, G′′) (e) as well as Young-modulus (E′) with the respective tanδ (f), each before and after extrusion. | ||
The chemical integrity after extrusion of the material was then confirmed using ATR-FTIR spectroscopy (Fig. 4c). Part of the extruded material was then compression-molded at 130 °C for 3 min in order to check if it kept its thermal and rheological properties after extrusion (Fig. 4d–f). The results showed that the material's rheological behavior remained constant, as can be seen by stress-relaxation experiments with a resulting iminium pathway related activation energy of 70 ± 1 kJ mol−1 before and 73 ± 2 kJ mol−1 after extrusion, and with a characteristic relaxation time of about 0.4 s at 150 °C (Fig. 4d and e). Lastly, the resulting dynamic mechanical analyses (DMA) displayed similar glass transition temperatures of the network (5 °C) as well as a decrease of the elastic plateau beyond 140 °C (Fig. 4f). Only the temperature ramp behavior exhibited a slight alteration with regard to a shift of the crossover by 4 K to a higher temperature, which could be ascribed to oxidation of few pendent amines, thus marginally altering the relaxation speed (Fig. 4e). Overall, the performed measurements proved the ability of highly crosslinked materials to undergo continuous processing.
Conclusion
Different vitrimer architectures, with the exemplified use of vinylogous urethane exchange chemistry, were synthesized and compared regarding their dynamic stress relaxation behavior, demonstrating significant differences based on the nature of the crosslinker. This led to the selection of an amine-functional crosslinker that results in a tenfold decrease of the relaxation times within a given polymer matrix. This enhanced stress-relaxation was then strategically further accelerated by increasing the amounts of nucleophilic moieties, without compromising network integrity. In connection to this acceleration, a suppression of the characteristic dual temperature response of vinylogous urethane vitrimers was observed, leading predominantly to the iminium pathway with an activation energy of around 70–80 kJ mol−1. In this context, the iminium driven mechanism was further supported by adding para-toluene sulfonic acid. The acidification of the polyether network resulted in a further controlled decrease of the relaxation times below 1 s. As a combined result of these effects, certain combination of proportion of pendent amines with catalytic pTsOH, resulted in a crossover of the shear storage- and loss- moduli at time scales and within a temperature window that are relevant for continuous processing techniques (around 130 °C at 1 rad s−1). These covalently crosslinked permanent networks thus exhibit a pronounced viscoelastic-liquid behavior, presenting the first example of extrusion processing that can be achieved with densely cross-linked vitrimers.We believe that the material design concepts demonstrated herein will open the door to an equally unprecedented range of material applications, ranging from solvent-free 3D-printed recyclable networks up to fibers and coatings. This could possibly lead to new research areas as well as industrial applications, thanks to the herein presented low viscosity vitrimer networks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. T. and M. G. acknowledge the Research Foundation-Flanders (FWO) for the PhD and Postdoctoral fellowships. The authors would like to thank Bernhard De Meyer, Bastiaan Dhanis, Mustafa Erkoç, and Prof. Dr Ludwig Cardon for the technical support and use of equipment, as well as Prof. Dr Ludwik Leibler, Dr Lucie Imbernon, Dr Nezha Badi, and Yann Spiesschaert for the fruitful discussions. F. D. P. thanks BOF-UGent for GOA-funding.References
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- R. F. M. Lange, M. Van Gurp and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3657–3670 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2014, 26, 3938–3942 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- A. Demongeot, R. Groote, H. Goossens, T. Hoeks, F. Tournilhac and L. Leibler, Macromolecules, 2017, 50, 6117–6127 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- T. Philip, N. Huagang, Z. Chengpu, Y. Kai, L. Samuel, J. Yinghua, Q. H. Jerry and Z. Wei, Adv. Mater., 2016, 28, 2904–2909 CrossRef.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- Y. Spiesschaert, M. Guerre, L. Imbernon, J. M. Winne and F. Du Prez, Polymer, 2019, 172, 239–246 CrossRef CAS.
- K. Pahnke, J. Brandt, G. Gryn'ova, P. Lindner, R. Schweins, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Chem. Sci., 2015, 6, 1061–1074 RSC.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- J. Hine, M. S. Cholod and W. K. Chess, J. Am. Chem. Soc., 1973, 95, 4270–4276 CrossRef CAS.
- J. F. Stevenson, in Comprehensive Polymer Science and Supplements, ed. G. Allen and J. C. Bevington, Pergamon, Amsterdam, 1989, pp. 303–354, DOI:10.1016/B978-0-08-096701-1.00213-5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh01062a |
| This journal is © The Royal Society of Chemistry 2020 |