
Design of protein-based “turn on” molecular probes for intracellular bond cleavage
Michelle R. Sorkin†
,
Joshua A. Walker†,
Francis Ledesma,
Nicole P. Torosian and
Christopher A. Alabi
 *
*
Robert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, 113 Ho Plaza, Ithaca, NY 14850, USA. E-mail: caa238@cornell.edu
First published on 26th November 2019
Abstract
The clinical success of antibody-drug conjugates and numerous other stimuli-responsive drug delivery systems motivates the need to develop molecular tools to quantitatively study their intracellular processing. To this end, researchers have developed fluorescent-based probes utilizing Förster resonance energy transfer (FRET). Quenched probes have the potential to eliminate fluorescence bleed through, which is common in FRET-based systems. However, the hydrophobicity of many broad-spectrum fluorescence quenchers can complicate the design of protein-based molecular probes. In this work, we investigate the potential for 2,4-dinitroaniline (2,4-DNA) to serve as a hydrophilic fluorescence quencher. A support-free synthesis of oligothioetheramide (oligoTEA) linkers was developed and applied to the design of quenched fluorescence probes. These quenched probes were based on intramolecular static quenching of boron dipyrromethene (BODIPY)-FL by 2,4-DNA. Probes containing a reduction-sensitive disulfide bond and a protease-sensitive valine–citrulline-PABC linker were synthesized using a model protein – human transferrin. Within HeLa cells, the apparent degradation rate of the disulfide bond was greater than the valine–citrulline-PABC linker. This work establishes a versatile method for synthesizing multifunctional crosslinkers and identifies 2,4-DNA as an effective fluorescence quencher for protein-based bioconjuates.
 Christopher Alabi | Christopher Alabi received his B.S. in Chemistry and B.E. in Chemical Engineering from New York University and Stevens Institute of Technology in 2004. He obtained a graduate degree at Caltech with Dr. Mark Davis and was an NIH Postdoctoral Fellow with Dr. Langer and Anderson from 2009–2013. Chris Alabi joined Cornell thereafter as an Assistant Professor in Chemical Engineering. In 2019, He was promoted to Associate Professor with Indefinite Tenure. Research in the Alabi lab seeks to understand how the sequence of a macromolecule affects its properties with an eye towards engineering sustainable materials and biomolecular therapeutics. |
Design, System, ApplicationUnderstanding how degradable cross-linkers respond to intracellular biological stimuli is a critical component of any successful drug delivery system. With the recent clinical success of antibody drug conjugates and numerous stimuli-responsive drug delivery systems in development, molecular tools are needed to quantitatively study their intracellular processing. In this article, we developed a support-free synthetic strategy and applied it to the design of quenched fluorescent probes that can be used to directly monitor intracellular bond cleavage kinetics. Probes containing a reduction-sensitive disulfide bond and a protease-sensitive valine–citrulline dipeptide linker were synthesized and conjugated to human transferrin. We show in HeLa cells that disulfide bond degradation occurs more rapidly than the widely used valine–citrulline dipeptide linker. Limitations of this work include the effect of cargo with different physicochemical properties on the bond cleavage kinetics. This limitation can ultimately be overcome by incorporating the cargo directly into the probe linker at one of the pendant positions. Nevertheless, this approach provides insight into intracellular processing and highlights the utility of multifunctional crosslinkers for creating protein-based bioconjugate probes. |
Introduction
Targeted drug delivery aims to selectively localize therapeutics to a specific site within the body. To ensure stability in the extracellular milieu and release at the desired intracellular site, a variety of nanoparticle1 and conjugate-based2 drug delivery systems have been designed to respond to intracellular stimuli. Antibody-drug conjugates (ADCs) are a class of conjugate-based drug delivery systems that combine the antigen specificity of antibodies with the chemotherapeutic potential of small molecule drugs. ADCs are a proven therapeutic platform with five FDA-approved compounds and many more in the clinical pipeline.2 Of these five compounds, four contain stimuli-responsive chemistry.3–6The clinical success of antibody-drug conjugates has motivated researchers to quantitatively study intracellular processing.7 Particular attention has been paid to studying the intracellular degradation of the reduction-sensitive disulfide bond8 and protease-sensitive valine–citrulline-PABC linker9 as these bonds are present in four of the five clinically approved ADCs. Many studies have relied on Förster resonance energy transfer (FRET)-based probes for studying intracellular bond degradation.8–10
Fluorescence quenched probes that use “dark quenchers” with no inherent fluorescent signal hold potential advantages over FRET-based systems with two fluorophores. Namely, quenched systems exist in a dark state, which eliminates the bleed through of fluorescence signal and crosstalk between channels that is commonly observed in FRET-based systems.11 However, designing a quenched fluorescence probe using “dark quenchers” is not without challenges. Dark quenchers largely operate via static quenching. That is, the fluorophore and quencher interact, usually via hydrophobic or π–π interactions, to form a non-fluorescent dimer. The hydrophobicity of most quenchers, which renders them efficient at fluorescent quenching, poses a significant challenge to their use in the biological milieu. Excessive hydrophobicity can lead to challenging reaction conditions between the chemical crosslinker and protein carrier. Further, conjugation of hydrophobic cargos has been shown to destabilize protein carriers.12
The static quencher 2,4-dinitroaniline (2,4-DNA), which is relatively hydrophilic, has the potential to circumvent these design challenges. Intramolecular quenching via 2,4-DNA has been reported for dyes across the blue to red spectrum. This has been leveraged to develop model protease substrates,13 visualize enzymatic processing of lipids,14 and image intracellular RNA dynamics.15 However, to the best of our knowledge, static quenching via 2,4-DNA has not been reported within the context of a protein-based bioconjugate. Therefore, we investigated the feasibility of utilizing static quenching via 2,4-DNA for the design of protein-based “turn on” molecular probes for intracellular bond cleavage.
The intracellular probe scaffold for bond cleavage must also remain stable in the harsh environment of the endosome. To achieve biostability, we turned to oligothioetheramides (oligoTEAs), a class of sequence-defined biomacromolecules developed by our lab five years ago.16 Much like peptide-based crosslinkers, oligoTEAs enable the linear incorporation of multiple functional groups. However, in contrast to peptide-based crosslinkers the oligoTEA backbone is abiotic and is therefore stable in the biological milieu.17 Further, oligoTEA synthesis is compatible with a diverse set of dithiol backbone monomers. These monomers can be used to mimic the flexibility and hydrophilicity of PEG-based crosslinkers or to incorporate biologically inert, chemically triggered cleavage sites.18
We recently reported a support-free methodology for oligoTEA synthesis that combined the backbone and pendant group functionalities into a single monomer unit.19 This greatly improved the efficiency of synthesizing short, hydrophilic oligoTEA-based crosslinkers. However, it requires multi-step monomer synthesis. For certain backbone and pendant group functionalities this can be challenging. In this work, we present an updated support-free methodology for oligoTEA synthesis. This approach simplifies monomer synthesis by utilizing sequential, one-pot thiol-Michael and thiol–ene reactions.
We demonstrate the utility of this synthetic methodology by synthesizing three heteromultifunctional chemical crosslinkers containing four independently addressable chemical handles. This synthesis was then applied to the design of stimuli-responsive quenched fluorescence crosslinkers containing boron dipyrromethene (BODIPY)-FL and the static quencher 2,4-DNA. Intracellular processing of crosslinkers containing a disulfide bond or a valine–citrulline-PABC linker was evaluated using HeLa cells and a model carrier protein – human transferrin. These studies represent the first demonstration of 2,4-DNA-mediated fluorescence quenching within the context of a protein-based bioconjugate. We demonstrate that dequenching of these systems can be used to detect intracellular bond degradation. Further, we find that relative to the valine–citrulline-PABC linker, the disulfide bond is degraded with a faster apparent rate within the transferrin-mediated endocytic pathway.
Results and discussion
Support-free synthesis of multifunctional oligoTEAs
We envisioned a support-free strategy to synthesize oligoTEAs containing up to three pendant functional groups (Fig. 1A). The first pendant group is incorporated through a starting material containing both a BOC-protected secondary amine and a terminal thiol. The second pendant group is installed through sequential, one-pot thiol-Michael and thiol–ene reactions using an N-allylacrylamide monomer and a monoacetylated dithiol co-monomer. Treatment of this intermediate with ammonia was used to liberate the terminal thiol. A second synthetic cycle using an N-allylacrylamide monomer and an unprotected dithiol was used to install the third functional group. RP-HPLC was used to achieve stepwise purification. Each oligomer synthesized in this manner contains a BOC-protected secondary amine and a terminal thiol.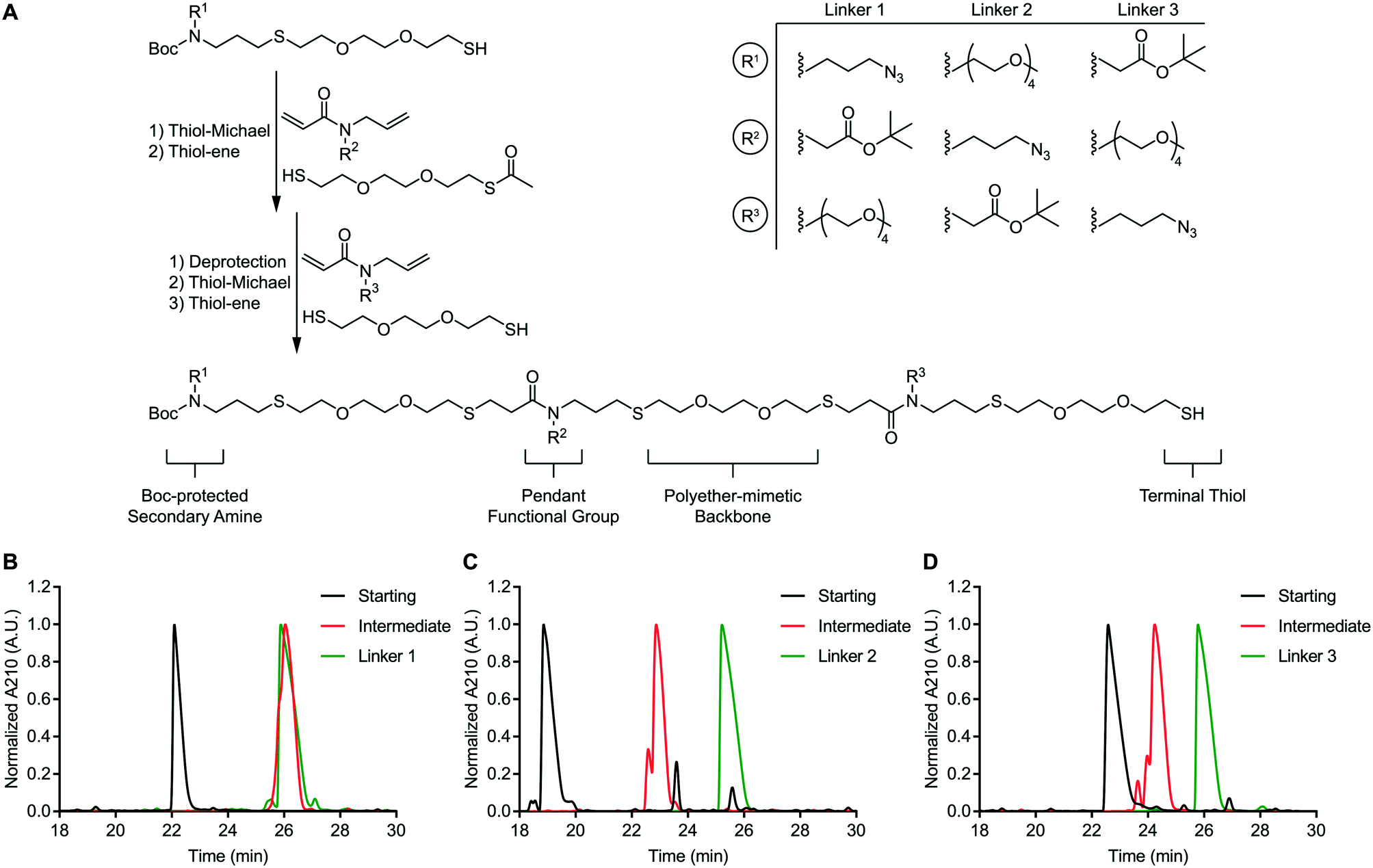 | ||
| Fig. 1 Support-free synthesis of oligoTEAs. A) Overview of support-free synthesis of oligoTEAs and design of multifunctional, sequence-defined crosslinkers. Linkers 1–3 are notated based on the placement of the pendant tert-butyl-protected carboxylic acid in positions 1–3 respectively. B) Analytical RP-HPLC of linker 1 synthesis. C) Analytical RP-HPLC of linker 2 synthesis. D) Analytical RP-HPLC of linker 3 synthesis. | ||
This approach was used to synthesize three constitutionally isomeric oligomers. The backbone of these oligomers was comprised of (2,2′)-ethylenedioxy-diethanethiol to mimic the polyether nature of polyethylene glycol (PEG)-based crosslinkers. These oligomers contained diverse pendant functionalities; an azide, a tert-butyl-protected carboxylic acid, and a four-unit PEG chain. The pendant PEG chain serves as a handle to mask the hydrophobicity of attached cargos and thereby improve conjugate stability. This effect has been shown to improve the efficacy of antibody-drug conjugates.20,21 The azide and carboxylic acid side chains are compatible with commonly used azide-alkyne “click” chemistry and amide coupling reactions, respectively. The secondary amine can be derivatized via activated esters or aza-Michael addition. Finally, the terminal thiol can undergo a thiol-Michael addition or thiol–disulfide exchange.22 In total, these oligomers possess four independently addressable chemical handles. Therefore, these oligomers represent a highly versatile class of multifunctional chemical crosslinkers.
These oligomers (Fig. S48–S50), henceforth referred to as linkers 1–3, were analysed via RP-HPLC after each synthetic step (Fig. 1B–D). RP-HPLC confirmed that each starting material, intermediate oligomer, and final linker was isolated in high purity. The purity of the linkers 1–3 was further confirmed via nuclear magnetic resonance (NMR) spectroscopy (Fig. S51A–C). These data speak to the robustness of our support-free methodology.
“Turn on” molecular probes for intracellular bond degradation
With proof-of-concept in hand, we applied this methodology to design molecular probes for intracellular bond degradation. We adopted an approach based on intramolecular fluorescence quenching of BODIPY-FL via 2,4-DNA. To demonstrate the versatility of our approach, we investigated two prominent stimuli-responsive chemistries; the disulfide bond and the valine–citrulline-PABC linker.2To create quenched crosslinkers, two additional multifunctional chemical crosslinkers were synthesized. Linker 4 (Fig. S52) was created using a single synthetic cycle as described above. The starting material contained a BOC-protected primary amine and a hydrophilic L-dithiothreitol (L-DTT) dithiol monomer. A thiol-Michael addition was carried out between this starting material and an N-allylacrylamide monomer functionalized with a hydrophilic, azide-modified two-unit PEG chain. Linker 4 was subsequently capped with a monoacetylated (2,2′)-ethylenedioxy-diethanethiol co-monomer. Linker 5 (Fig. S53) was synthesized in the same manner except that capping was performed with 2-mercaptoacetic acid. The primary amine, terminal thiol, and terminal carboxylic acid functional groups provide sites for further derivatization. Meanwhile, the pendant azide functionality provides a handle for bioconjugation via copper-free “click” chemistry.
The reduction-sensitive linker (Fig. 2A), was synthesized by derivatizing linker 4 with BODIPY-FL-NHS ester and performing thiol–disulfide exchange with a pyridyl disulfide-functionalized version of 2,4-DNA (Fig. S57). The protease-sensitive linker (Fig. 2A), was synthesized by derivatizing linker 5 with BODIPY-FL NHS ester and performing a carbodiimide coupling of the terminal carboxylic acid with a valine–citrulline-PABC-functionalized version of 2,4-DNA (Fig. S58). A non-cleavable control linker (Fig. 2A), was synthesized by derivatizing linker 4 with BODIPY-FL-NHS ester and performing a thiol-Michael addition with a maleimide-functionalized version of 2,4-DNA (Fig. S59). These fluorescent linkers will henceforth be referred to as the disulfide, dipeptide, and control linkers respectively. Each quenched fluorescence linker was analysed via RP-HPLC. This analysis indicated that each linker was isolated in high purity (Fig. 2B).
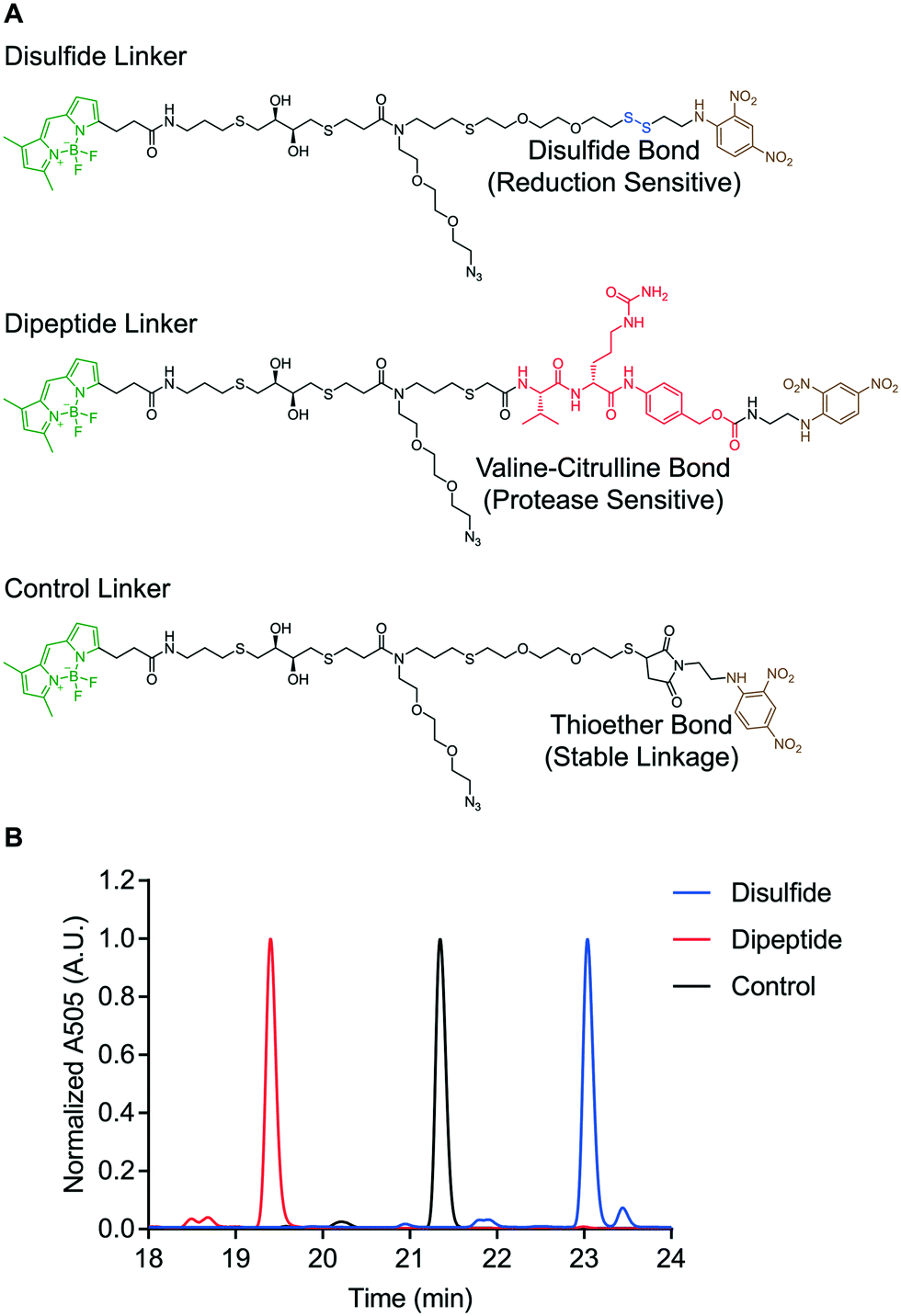 | ||
| Fig. 2 Design of “turn on” molecular probes. A) Structure of disulfide, dipeptide, and control linkers. B) RP-HPLC of quenched fluorescence linkers. | ||
Each linker was then conjugated to a model carrier protein – human transferrin. Transferrin was first modified via coupling a strained dibenzocylcooctyne (DBCO) functional group to exposed lysine residues via NHS ester chemistry (Fig. 3). Each linker was then conjugated to DBCO-modified transferrin via copper-free “click” chemistry as described previously.18 The resulting compounds are henceforth referred to as the disulfide, dipeptide, and control probes. Having demonstrated successful linker synthesis and synthesized the resulting bioconjugates, we sought to validate their functional properties.
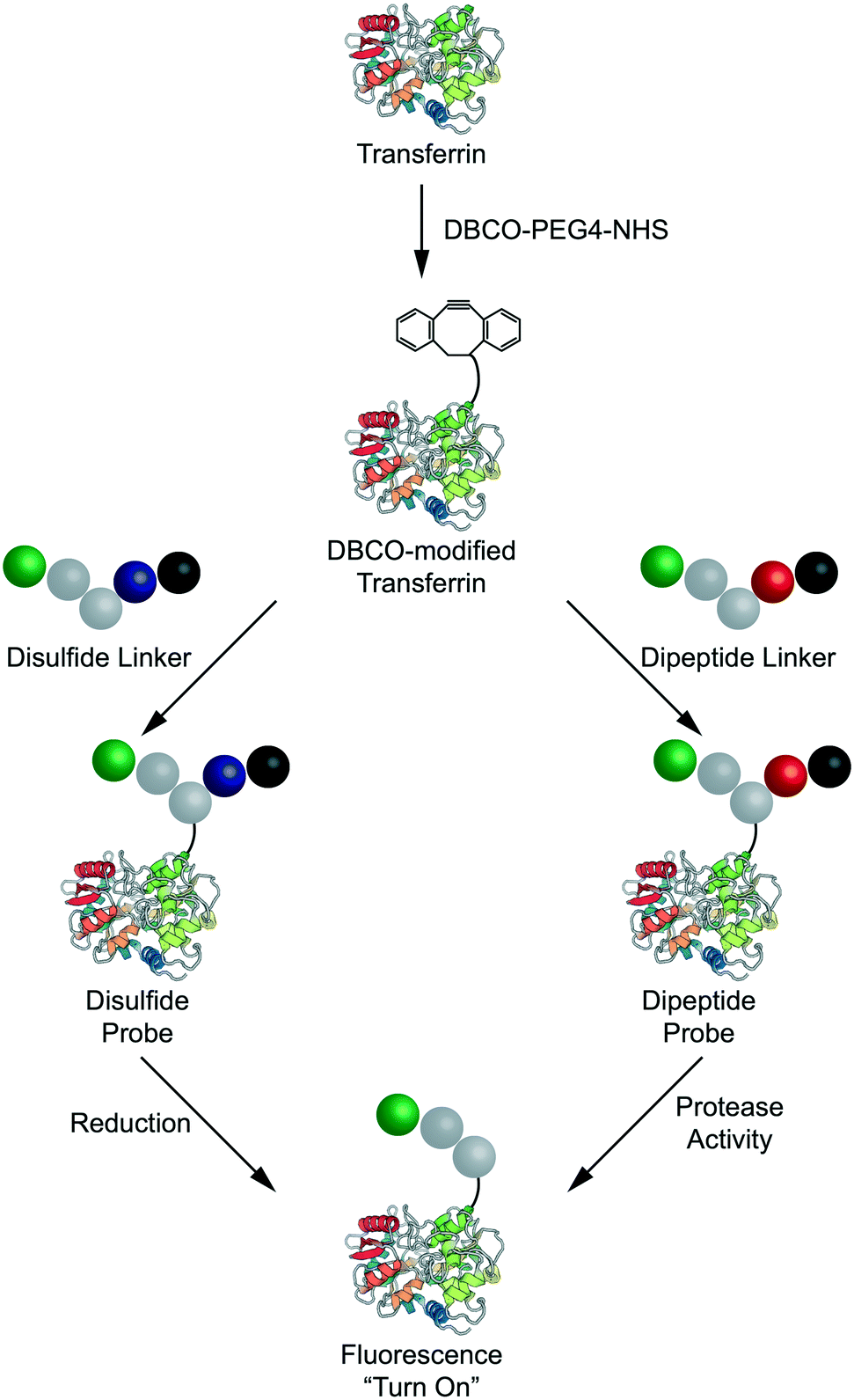 | ||
| Fig. 3 Design and synthesis of transferrin-based probes for intracellular bond degradation. | ||
Detection of intracellular disulfide bond reduction
A cell-free reduction assay was used to evaluate the efficiency of static quenching by 2,4-DNA within the disulfide linker (Fig. 4A). The disulfide linker was treated with excess of either DL-dithiothreitol (DL-DTT) or the tripeptide glutathione (GSH). BODIPY-FL-specific fluorescence was then monitored for up to 24 hours at room temperature. Compared to the control linker, treatment with both GSH and DL-DTT resulted in an increase in fluorescence. Reduction by DL-DTT occurred within 2 hours while reduction by GSH was seemingly incomplete after 24 hours. This result is in line with the high reducing potential of DL-DTT.23,24 Treatment with DL-DTT also resulted in a greater than 100-fold increase in fluorescence intensity. This indicates that static quenching via 2,4-DNA is efficient within the disulfide linker. Maleimide-conjugated payloads can exchange with albumin in vivo.25 However, this data suggests that the maleimide-conjugated linker used in this study is stable relative to the disulfide linker.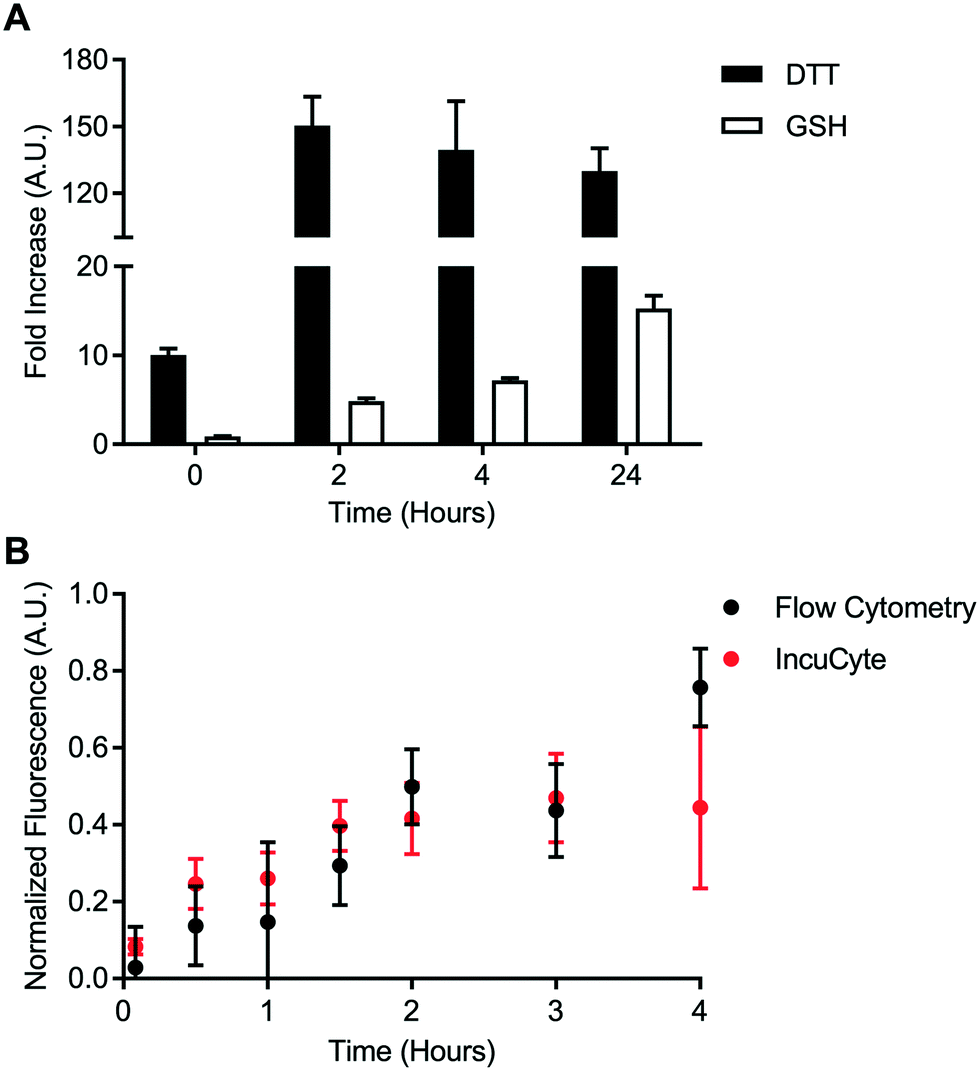 | ||
| Fig. 4 Intracellular disulfide bond reduction. A) Cell-free assay of disulfide bond reduction viaDL-dithiothreitol (DTT) and glutathione (GSH). Signal is represented as the fold increase in fluorescence upon treatment with reducing agent relative to the fold increase observed for the control linker. Error bars represent standard error of the mean. B) Intracellular bond degradation in HeLa cells evaluated via flow cytometry and IncuCyte. Data represents mean and standard error of the mean. | ||
Intracellular disulfide bond degradation was evaluated using the transferrin-based disulfide probe. HeLa cells were used due to their high expression of the transferrin receptor. HeLa cells were incubated with the disulfide probe for up to 4 hours. Fluorescence signal was quantified kinetically using two techniques: flow cytometry and IncuCyte ZOOM quantitative live cell imaging. Fluorescence dequenching was reported relative to the non-cleavable control probe and an “always on” BODIPY-FL-modified transferrin (Fig. 4B). This analysis revealed that the disulfide bond is processed within approximately 4 hours in the transferrin-mediated endocytic pathway. Additionally, measurements made via flow cytometry were in good agreement with data from IncuCyte ZOOM. This result is important for cleavable chemistries which degrade on a longer time scale since flow cytometry-based measurements may become tedious or unfeasible. Finally, representative images from IncuCyte ZOOM analysis show that the disulfide probe and control probe can be distinguished visually (Fig. S60). These data indicate that static quenching via 2,4-DNA can be used to detect intracellular bond degradation.
Detection of intracellular protease activity
A cell-free cleavage assay was used to validate the function of the dipeptide linker and the dipeptide probe. Both linker and probe were treated with cathepsin B and incubated at 40 °C. BODIPY-FL-specific fluorescence was then monitored for up to 12 hours (Fig. 5A). Within 4 hours, the dipeptide linker exhibited nearly complete dequenching. The kinetics of bond cleavage were slower for the dipeptide probe, which exhibited complete dequenching after approximately 8 hours. This could be explained by steric hindrance of the valine–citrulline-PABC linker when placed within the context of the carrier protein. Nevertheless, cathepsin B was able to act on both the dipeptide linker and the dipeptide probe.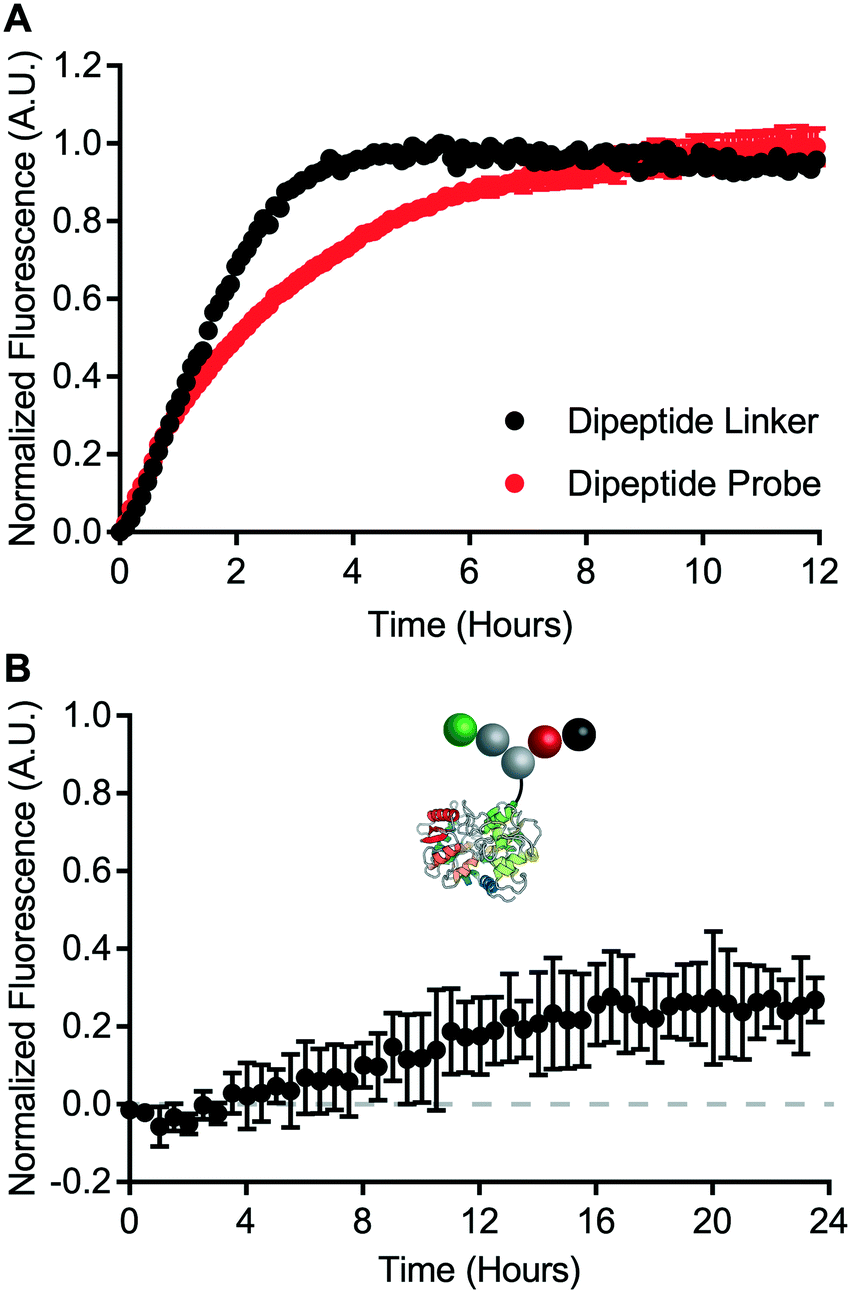 | ||
| Fig. 5 Intracellular degradation of valine–citrulline-PABC linker. A) Cell-free assay of dipeptide linker and dipeptide probe cleavage via cathepsin B. Error bars represent standard error of the mean. B) Intracellular bond degradation in HeLa cells measured via IncuCyte ZOOM. Data represents mean and standard error of the mean. | ||
Intracellular degradation of the valine–citrulline-PABC linker was evaluated using HeLa cells – a model transferrin receptor-positive cell line. Intracellular degradation of protease-sensitive linkers has been reported to take on the order of 12 hours or more.9 Therefore, dipeptide probe degradation was evaluated via IncuCyte ZOOM live cell imaging (Fig. 5B). HeLa cells were incubated with the dipeptide probe for 24 hours and fluorescence dequenching was monitored. Fluorescence dequenching was reported relative to the non-cleavable control probe and an “always on” BODIPY-FL-modified transferrin. This analysis revealed that the valine–citrulline-PABC linker is processed slowly within the transferrin receptor-mediated endocytic pathway. Detectable signal was not observed until approximately 12 hours. After 24 hours, fluorescence dequenching had only reached approximately 25% of the maximum defined by the “always on” control. This contrasts with the disulfide probe, which demonstrated complete degradation within 4 hours (Fig. 4B).
Conclusions
In conclusion, we have developed a support-free methodology for the synthesis of oligoTEAs. We demonstrated the versatility of this strategy by synthesizing three chemical crosslinkers containing four independently addressable chemical handles. We then applied this synthesis to the design of “turn on” molecular probes for intracellular bond degradation. Two stimuli-responsive chemistries were investigated; the disulfide bond and the valine–citrulline-PABC linker. Bond degradation was reported by intramolecular quenching of BODIPY-FL by 2,4-DNA. A model protein – human transferrin was used as the protein carrier. Static quenching via 2,4-DNA was found to be effective within the context of a protein-based bioconjugate. Intracellular disulfide bond reduction was found to occur within 4 hours. In comparison, valine–citrulline-PABC linker degradation occurred slowly with incomplete dequenching observed after 24 hours. Notably, this result was in contrast with a cell-free assay for valine–citrulline-PABC linker degradation, which demonstrated efficient cleavage of the dipeptide probe. These results establish the versatility of our support-free synthesis methodology. Further, these data support static quenching via 2,4-DNA as a potential alternative to bulky, hydrophobic broad-spectrum quenchers. Finally, our findings underscore the value of studying bond degradation within live cells. Taken together, this work provides important insights into the design of multifunctional bioconjugates and protein-based molecular probes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by funds from Cornell University and the Nancy and Peter Meinig Investigator Fellowship. MRS acknowledges the Fleming fellowship for financial support. JAW acknowledges the NSF GRFP (DGE-1650441) for financial support. Equipment used to perform this research was funded in part by the NSF MRI (CHE-1531632).Notes and references
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- B. Shor, H.-P. Gerber and P. Sapra, J. Controlled Release, 2014, 67, 107–116 Search PubMed.
- J. Baron and E. S. Wang, Expert Rev. Clin. Pharmacol., 2018, 11, 549–559 CrossRef CAS PubMed.
- D. Dornan, F. Bennett, Y. Chen, M. Dennis, D. Eaton, K. Elkins, D. French, M. A. T. Go, A. Jack, J. R. Junutula, H. Koeppen, J. Lau, J. McBride, A. Rawstron, X. Shi, N. Yu, S.-F. Yu, P. Yue, B. Zheng, A. Ebens and A. G. Polson, Blood, 2009, 114, 2721–2729 CAS.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- K. F. Maass, C. Kulkarni, A. M. Betts and K. D. Wittrup, AAPS J., 2016, 18, 635–646 CrossRef CAS PubMed.
- M. R. Sorkin, J. A. Walker, S. R. Kabaria, N. P. Torosian and C. A. Alabi, Cell Chem. Biol., 2019, 26, 1–9 CrossRef PubMed.
- B.-C. Lee, C. Chalouni, S. Doll, S. C. Nalle, M. Darwish, S. P. Tsai, K. R. Kozak, G. Del-Rosario, S.-F. Yu, H. Erickson and R. Vandlen, Bioconjugate Chem., 2018, 29, 2468–2477 CrossRef CAS PubMed.
- J. Yang, H. Chen, I. R. Vlahov, J.-X. Cheng and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13872–13877 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science and Business Media, New York, NY, USA, 3rd edn, 2013 Search PubMed.
- N. S. Beckley, K. P. Lazzareschi, H.-W. Chih, V. K. Sharma and H. L. Flores, Bioconjugate Chem., 2013, 24, 1674–1683 CrossRef CAS PubMed.
- M. Poreba, A. Szalek, W. Rut, P. Kasperkiewicz, I. Rutkowska-Wlodarczyk, S. J. Snipas, Y. Itoh, D. Turk, B. Turk, C. M. Overall, L. Kaczmarek, G. S. Salvesen and M. Drag, Sci. Rep., 2017, 7, 43135 CrossRef CAS PubMed.
- H. S. Hendrickson, E. K. Hendrickson, I. D. Johnson and S. A. Farber, Anal. Biochem., 1999, 276, 27–35 CrossRef CAS PubMed.
- A. Arora, M. Sunbul and A. Jäschke, Nucleic Acids Res., 2015, 43, e144 Search PubMed.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS PubMed.
- M. Porel, D. N. Thornlow, C. M. Artim and C. A. Alabi, ACS Chem. Biol., 2017, 12(3), 715–723 CrossRef CAS PubMed.
- M. R. Sorkin, J. A. Walker, J. S. Brown and C. A. Alabi, Bioconjugate Chem., 2017, 28, 907–912 CrossRef CAS PubMed.
- J. A. Walker, M. R. Sorkin, F. Ledesma, S. R. Kabaria, R. M. Barfield, D. Rabuka and C. A. Alabi, Bioconjugate Chem., 2019, 30(11), 2982–2988 CrossRef CAS PubMed.
- J. W. Buecheler, M. Winzer, J. Tonillo, C. Weber and H. Gieseler, Mol. Pharmaceutics, 2018, 15, 2656–2664 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier, 3rd edn, 2013 Search PubMed.
- P. K. Pullela, T. Chiku, M. J. Carvan and D. S. Sem, Anal. Biochem., 2006, 352, 265–273 CrossRef CAS PubMed.
- W. J. Lees and G. M. Whitesides, J. Org. Chem., 1993, 58, 642 CrossRef CAS.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
Footnote |
| † M. R. S. and J. A. W. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |