
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide directed phthalocyanine–gold nanoparticles for selective photodynamic therapy of EGFR overexpressing cancers†
Zoë Rachael
Goddard
 a,
Andrew Michael
Beekman
a,
Marco M. D.
Cominetti
a,
Maria A.
O'Connell
a,
Isabelle
Chambrier
b,
Michael J.
Cook
b,
María J.
Marín
b,
David A.
Russell
b and
Mark
Searcey
*a
a,
Andrew Michael
Beekman
a,
Marco M. D.
Cominetti
a,
Maria A.
O'Connell
a,
Isabelle
Chambrier
b,
Michael J.
Cook
b,
María J.
Marín
b,
David A.
Russell
b and
Mark
Searcey
*a
aSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: M.Searcey@uea.ac.uk
bSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
First published on 19th November 2020
Abstract
Gold nanoparticles, covalently functionalised with the photosensitiser C11Pc and PEG, were actively targeted towards epidermal growth factor receptor overexpressing cancers using the peptide FITC-βAAEYLRK. Selective phototoxicity was observed at nanomolar concentrations with minimal dark toxicity.
Photodynamic therapy (PDT) for cancer involves the combination of a photosensitiser and light to convert molecular triplet oxygen (3O2) to cytotoxic singlet oxygen (1O2) within cancerous cells, which ultimately leads to cell death. Ideal photosensitisers for PDT are isomerically pure, have good stability, a high molar extinction coefficient between 650–800 nm, a high quantum yield for singlet oxygen, have no dark toxicity, interact preferentially with cancer cells and clear relatively quickly from normal tissue.1,2 Most reported photosensitisers are hydrophobic aromatics3 as they fulfil many of these criteria, although the delivery of these photosensitisers intravenously is particularly challenging. One approach to overcoming the issues of photosensitiser hydrophobicity is the use of a delivery vehicle such as gold nanoparticles (AuNPs).2,4,5
AuNPs can be considered to be ideal drug carriers as they are multivalent, chemically inert and minimally toxic.6 It has previously been demonstrated that the functionalisation of ca. 4 nm AuNPs with a mixed monolayer of the highly hydrophobic zinc phthalocyanine C11Pc and poly(ethylene glycol) (PEG) allows for the delivery of the active photosensitiser in aqueous media.7 Antibodies and lectins have been covalently attached to these nanosystems to selectively deliver them to HER2 and Tf-antigen positive cell lines respectively.7–9
While proteins are strong candidates for the targeted delivery of AuNPs due to their extremely high selectivity towards their target, the sensitivity of proteins towards pH, temperature and organic solvents can lead to loss of activity upon sample preparation. Alongside high production costs, heterogeneity of samples can result in poor reproducibility. Recently, the targeting ability of peptides has increased in popularity10–15 as these chemically synthesised molecules can overcome many of the disadvantages that proteins present, while still maintaining selectivity towards the desired target.
The epidermal growth factor receptor (EGFR) is an attractive target for cancer therapeutics as it is overexpressed in a wide range of epithelial cancers, including lung,16 ovarian,17 colorectal18 and head and neck19 carcinomas. EGFR has been previously selected as a target for peptide-directed AuNPs for PDT. Here, the authors conjugated the EGFR targeting peptide GE11 (YHWYGYTPQNVI) to PEGylated AuNPs to selectively deliver the photosensitiser Pc4 to glioblastoma.20,21 In these systems the Pc4 is adsorbed to the ca. 5 nm gold core. These AuNPs were not internalised upon binding, and were thought to show selective phototoxicity through increased interaction with the EGFR, via desorption of Pc4 from the AuNPs.20 However, in a similar system targeting prostate specific membrane antigen, significant off-target phototoxicity was observed, likely due to the non-covalent attachment of the photosensitiser.22
While GE11 has been explored as a targeting ligand, several other EGFR specific peptides have been reported in the literature, including D4 (LARLLT)23 and AEYLR.24 These shorter peptides are appealing due to their ease of synthesis, while maintaining good selectivity for the EGFR. Of these, AEYLR is of particular interest as it shows higher binding affinity towards EGFR than D4,24 which in turn has better uptake characteristics than GE11 as the hydrophobicity of GE11 results in the aggregation of conjugates.25 AEYLR has been used to selectively deliver liposomes26 and chitosan microparticles27 towards EGFR expressing cancers. Here, we describe for the first time the use of the superior EGFR targeting peptide AEYLR for the selective delivery of gold nanoparticles to EGFR overexpressing cancer cells. In this work, we functionalised ca. 4 nm AuNPs with a self-assembled mixed monolayer of the photosensitiser C11Pc, PEG and targeting peptide. The covalent attachment of the photosensitiser removes the possibility of non-specific phototoxicity due to desorption, resulting in a more selective nanosystem.
The EGFR targeting peptide AEYLR was modified with a lysine residue at the C-terminus to allow for site selective amide bond formation to the PEG (HS-PEG-COOH), and with a FITC-βAla at the N-terminus, allowing for visualisation of this peptide, giving the directing peptide FITC-βAAEYLRK (Fig. 1). This peptide was synthesised by solid phase synthesis and purified by RP-HPLC.
 | ||
| Fig. 1 The structure of the peptide FITC-βAAEYLRK, highlighting the Lys (blue), βAla (pink) and FITC (green). | ||
Flow cytometry was used to confirm the expression level of EGFR on A549 pulmonary adenocarcinoma (EGFR overexpressing, ATCC no. CCL-185) and HEK293 embryonic kidney (basal level EGFR, ATCC no. CRL-1573) cell lines. Upon incubation with an EGFR antibody, A549 cells showed a 31-fold increase in fluorescence, compared to a 6.7-fold increase for HEK293 cells (Fig. S3, ESI†). To determine that the modifications made to the peptide did not affect its selectivity towards EGFR overexpressing cell lines, the selective uptake of this modified peptide was confirmed by fluorescence microscopy. Both cell lines were incubated for 1 h with the peptide, with only the A549 cells showing significant fluorescence, and therefore peptide binding (Fig. 2). Fluorescein was also incubated with both cell lines under the same conditions to ensure that this increased fluorescence observed in A549 cells was indeed due to binding of the peptide sequence, and not due to an interaction of the large FITC tag. The A549 cells did not show any binding of fluorescein (Fig. S4, ESI†), suggesting the uptake is due to the targeting of the peptide sequence itself. The peptide was also assessed for mitogenic activity in both A549 and HEK293 cells and no agonistic nor antagonistic effects were observed (Fig. S5, ESI†).
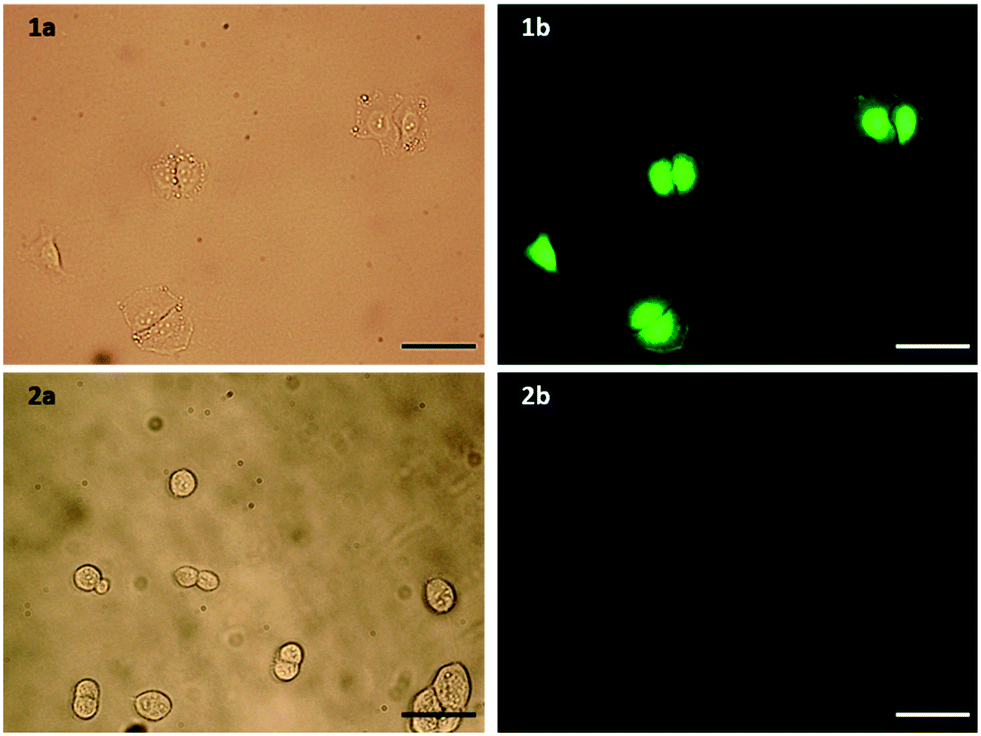 | ||
| Fig. 2 a) Bright field and b) fluorescence microscopy images of 1) A549 and 2) control HEK293 cells incubated with FITC-βAAEYLRK. Scale bar = 100 μm. | ||
Antibodies are commonly conjugated onto AuNPs post-synthesis through EDC/NHS mediated amide bond formation.28,29 These conjugations often give a poor yield due to competing hydrolysis reactions. A key benefit of peptides over antibodies is their significantly higher tolerability towards organic solvents, meaning that FITC-βAAEYLRK could be conjugated onto PEG prior to the synthesis of the AuNPs. Tighter control over the concentration of targeting moiety on the AuNP surface was possible by conjugation of peptide to PEG pre-AuNP synthesis. The peptide was conjugated onto HS-PEG-COOH via an amide linkage by stirring overnight with HATU and DIPEA. The resulting mixture was evaporated to dryness and used directly without purification for the synthesis of AuNPs. Due to the polydispersity of the PEG, it was difficult to purify at this stage but due to the efficiency of HATU for amide bond formation and the excess of both peptide and HATU, the reaction was assumed to go to completion. This was further indicated as the peptide–PEG showed solubility in THF, whereas the peptide itself was completely insoluble. Peptide–C11Pc–PEG–AuNPs (Fig. 3a) were synthesised following the one-pot protocol reported by Russell and co-workers to synthesise ca. 4 nm AuNPs.7,30,31 The resulting AuNPs were characterised by UV-vis and fluorescence spectroscopy (Fig. 3b and S6, ESI†). The presence of both C11Pc and FITC could be confirmed by their characteristic absorptions at ca. 645/696 nm and 495 nm respectively (Fig. 3b). The solubility of the nanoparticles in aqueous media confirmed that conjugation of C11Pc onto AuNPs in a mixed monolayer with PEG was successful as the photosensitiser itself is highly hydrophobic. For this initial study, peptide–PEG and non-conjugated PEG were added to the reaction mixture in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio as overcrowding of the AuNP surface with targeting ligand has been shown to reduce targeting efficiency.32 Comparison of the concentration of C11Pc and FITC on the surface of these AuNPs, estimated from the UV-vis spectrum, suggested that there were approximately 51 photosensitisers and 36 peptide moieties per AuNP (see ESI†). These AuNPs were seen to be below 5 nm in size due to the lack of a significant surface plasmon absorbance band in the UV-vis spectrum.33 The conjugation of FITC-βAAEYLRK to PEG prior to the synthesis of the AuNPs allows for a much higher level of conjugation, highlighted by previous reports that estimated only six targeting antibodies were conjugated to C11Pc–PEG–AuNPs using EDC/NHS post-synthesis.9
:50 ratio as overcrowding of the AuNP surface with targeting ligand has been shown to reduce targeting efficiency.32 Comparison of the concentration of C11Pc and FITC on the surface of these AuNPs, estimated from the UV-vis spectrum, suggested that there were approximately 51 photosensitisers and 36 peptide moieties per AuNP (see ESI†). These AuNPs were seen to be below 5 nm in size due to the lack of a significant surface plasmon absorbance band in the UV-vis spectrum.33 The conjugation of FITC-βAAEYLRK to PEG prior to the synthesis of the AuNPs allows for a much higher level of conjugation, highlighted by previous reports that estimated only six targeting antibodies were conjugated to C11Pc–PEG–AuNPs using EDC/NHS post-synthesis.9
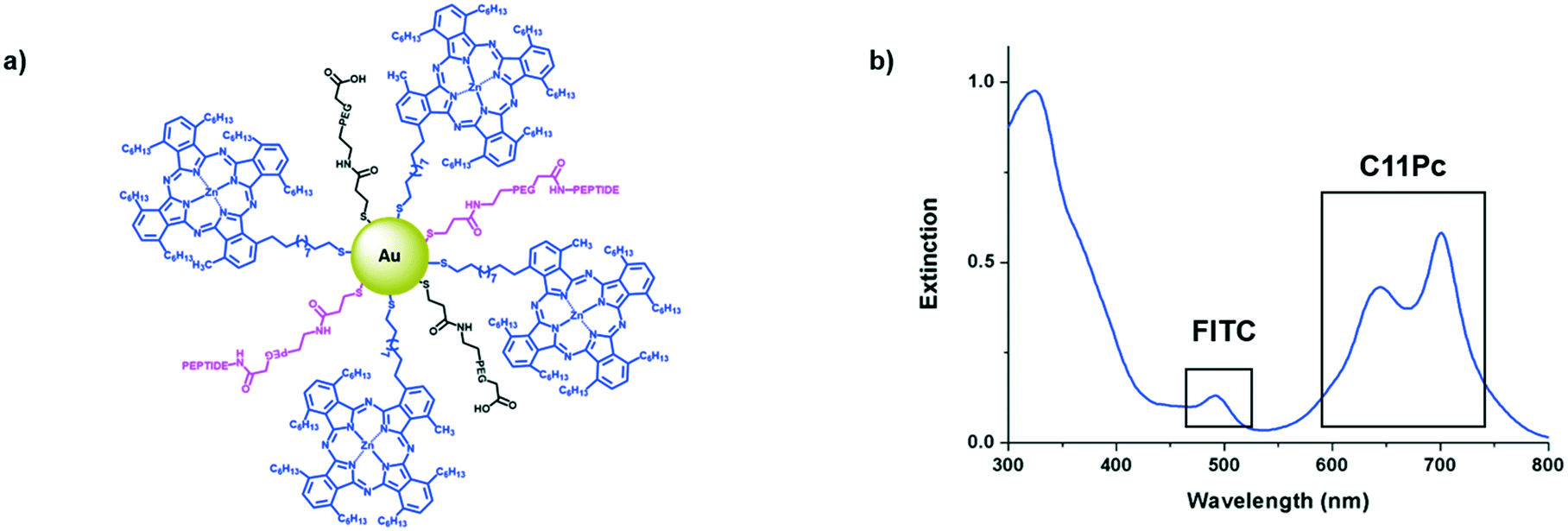 | ||
| Fig. 3 a) A schematic representation of the peptide–C11Pc–PEG–AuNPs with 50:50 PEG:peptide–PEG on the surface and b) the UV-vis extinction spectrum of peptide–C11Pc–PEG–AuNPs in PBS, showing the presence of FITC at 495 nm and the characteristic double peak of C11Pc at 645 nm and 696 nm. | ||
The ability of the synthesised nanoconjugates to produce singlet oxygen was assessed using the singlet oxygen probe 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABMA). A control of PEG–AuNPs was used to ensure that the recorded photobleaching was due to the photosensitiser and not the PEG functionalised gold core. While singlet oxygen lifetime is very short in PBS, significant singlet oxygen production was observed for the peptide–C11Pc–PEG–AuNPs (Fig. S7, ESI†). This was slightly reduced compared to that of non-targeted C11Pc–PEG–AuNPs, but this can be explained by the presence of tyrosine in the peptide sequence, a known singlet oxygen quencher.34
Once the nanocarriers were confirmed to produce singlet oxygen, they were tested for their ability to induce phototoxicity in EGFR overexpressing cell lines. The peptide–C11Pc–PEG–AuNPs were incubated with A549 cells for 3 h in serum-free media, before the wells were washed with PBS to remove any AuNPs that had not been internalised. The cells were irradiated for six minutes at 633 nm with a 10 mW HeNe laser (light dose 10.5 J cm−2), then incubated for 48 h before a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was used to assess the cell viability. To account for dark toxicity, a second plate of cells was treated with AuNPs in the same way, but not irradiated. In each plate, staurosporine was used as a positive control for cytotoxicity. The peptide–C11Pc–PEG–AuNPs showed negligible dark toxicity in A549 cells at and below 200 nM C11Pc, while significant phototoxicity was observed; a 7% cell viability was observed upon irradiation with 200 nM peptide–C11Pc–PEG–AuNPs (Fig. 4a), the equivalent of 4 nM AuNPs, and comparable to that of antibody-directed AuNPs.9 The IC50 of peptide–C11Pc–PEG–AuNPs in A549 cells was found to be 105.8 nM C11Pc (91.68 nM, 121.5 nM 95% CI) upon irradiation and >250 nM without irradiation (Fig. S8, ESI†). A549 cells were also incubated with non-targeted C11Pc–PEG–AuNPs at the same concentrations, and no phototoxicity was observed (Fig. 4b), suggesting there is a targeted photodynamic response for the peptide-conjugated nanocarriers due to the presence of the EGFR targeting peptide. To further confirm this, HEK293 cells were also treated with peptide–C11Pc–PEG–AuNPs and C11Pc–PEG–AuNPs, with no statistical difference observed between irradiated and non-irradiated cells (Fig. S9, ESI†). Interestingly, while there were no indications of cell death upon visual examination, there was a decrease in proliferation upon treatment of HEK293 cells with peptide–C11Pc–PEG–AuNPs. The reason for this decrease in cell proliferation is still under investigation, but the lack of phototoxicity suggests there is a targeted photodynamic response towards EGFR overexpressing cell lines.
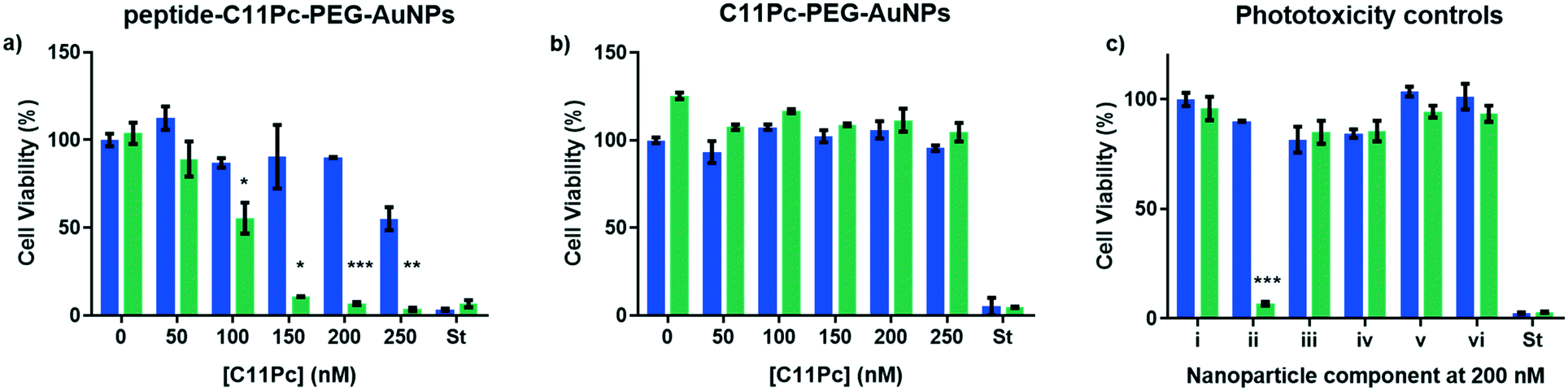 | ||
| Fig. 4 Cell viability of A549 cells treated with a) peptide–C11Pc–PEG–AuNPs, b) C11Pc–PEG–AuNPs or c) i) media, ii) peptide–C11Pc–PEG–AuNPs, iii) FITC-βAAEYLRK, iv) FITC–PEG–AuNPs, v) peptide–PEG–AuNPs and vi) FITC–C11Pc–PEG–AuNPs either with (green) or without (blue) irradiation. Where C11Pc was not present, cells were treated with 200 nM FITC. St = positive control of staurosporine. Error bars represent SE (n = 3). Statistically significant differences are indicated by * at P ≤ 0.05, ** at P ≤ 0.01, *** at P ≤ 0.001, obtained using a two-tailed Student's t-test. | ||
While significant photodynamic activity was observed in A549 cells after treatment with peptide–C11Pc–PEG–AuNPs, the ability of fluorescein to act as a photosensitiser35 led to the question of whether the photodynamic activity observed was due to the C11Pc alone, or a combination of C11Pc and FITC. The absorption of FITC is low at 633 nm36 and therefore it is unlikely to be responsible for this activity, however, to confirm this FITC–PEG–AuNPs and peptide–PEG–AuNPs were synthesised to determine if the FITC tag was responsible for any of the observed photodynamic activity. Control FITC–C11Pc–PEG–AuNPs were also synthesised to confirm whether the FITC tag was influencing the uptake of the nanocarriers, or whether this was due to the targeting ability of the peptide. The control AuNPs were characterised by UV-vis spectroscopy (Fig. S10, ESI†). As the peptide itself possesses both the targeting sequence and FITC, it was also tested for its ability to induce phototoxicity. A549 cells were incubated with these nanocarriers and the peptide for 3 h at 200 nM, a concentration shown to elicit a significant photodynamic response with peptide–C11Pc–PEG–AuNPs. The cells were irradiated as previously described and no cell death was observed for any of these control systems (Fig. 4c). These controls highlight that the phototoxicity observed for the peptide–C11Pc–PEG–AuNPs is due to the photocatalytic activity of the C11Pc. The lack of phototoxicity from FITC–C11Pc–PEG–AuNPs further confirms that the increased phototoxicity of peptide-conjugated AuNPs is due to the presence of the targeting peptide sequence itself, not the FITC tag.
In summary, we have demonstrated the viability of the peptide FITC-βAAEYLRK as a targeting ligand for the delivery of nanocarriers to EGFR overexpressing cell lines. We designed the modified peptide FITC-βAAEYLRK to allow for site selective conjugation onto PEG ligands. The FITC tag allows for quantification of the peptide on the surface of the AuNPs and provides an opportunity for intracellular imaging of these nanosystems. The designed peptide–PEG was used for the efficient synthesis of ca. 4 nm peptide–C11Pc–PEG–AuNPs in a one-pot method, with approximately 51 photosensitisers and 36 targeting ligands per nanoparticle. These nanocarriers are designed with the photosensitiser covalently bound to the core, avoiding any off target phototoxicity due to desorption of the photosensitiser. We demonstrated that these peptide–C11Pc–PEG–AuNPs produce singlet oxygen upon irradiation at 633 nm, and that selective phototoxicity can be observed in EGFR overexpressing A549 cells. These nanosystems displayed nanomolar potency in EGFR overexpressing A549 cells, with 200 nM C11Pc, the equivalent of 4 nM AuNPs, exhibiting 7% viability following irradiation and minimal dark toxicity. Finally, we demonstrated that the phototoxicity of these peptide–C11Pc–PEG–AuNPs is due to the dual effect of the targeting of the peptide sequence and the photocatalytic activity of the C11Pc, with the FITC tag neither causing phototoxicity nor encouraging uptake of the AuNPs. These results, and the relative tolerability of peptides towards organic solvents, highlight peptides as a feasible alternative to antibodies for the delivery of photosensitiser-baring nanoparticles to cancers, with phototoxicity observed to be comparable to that of antibody-directed C11Pc–PEG–AuNPs.9
The authors gratefully acknowledge the University of East Anglia, PDT Norfolk & the Anthony Long Trust for funding this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. B. Josefsen and R. W. Boyle, Met.-Based Drugs, 2008, 1–23 CrossRef CAS.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS.
- R. R. Allison, G. H. Downie, R. Cuenca, X.-H. Hu, C. J. Childs and C. H. Sibata, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS.
- P. García Calavia, G. Bruce, L. Pérez-García and D. A. Russell, Photochem. Photobiol. Sci., 2018, 17, 1534–1552 RSC.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- T. Stuchinskaya, M. Moreno, M. J. Cook, D. R. Edwards and D. A. Russell, Photochem. Photobiol. Sci., 2011, 10, 822–831 RSC.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 6158–6162 CrossRef CAS.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Photochem. Photobiol. Sci., 2015, 14, 737–747 RSC.
- E. Y. Xue, R. C. H. Wong, C. T. T. Wong, W.-P. Fong and D. K. P. Ng, RSC Adv., 2019, 9, 20652–20662 RSC.
- W. Li, S. Tan, Y. Xing, Q. Liu, S. Li, Q. Chen, M. Yu, F. Wang and Z. Hong, Mol. Pharmaceutics, 2018, 15, 1505–1514 CrossRef CAS.
- P. L. Bigliardi, B. Rout, A. Pant, V. Krishnan-Kutty, A. N. Eberle, R. Srinivas, B. A. Burkett and M. Bigliardi-Qi, Sci. Rep., 2017, 7, 15750 CrossRef.
- B. G. Ongarora, K. R. Fontenot, X. Hu, I. Sehgal, S. D. Satyanarayana-Jois and M. G. H. Vicente, J. Med. Chem., 2012, 55, 3725–3738 CrossRef CAS.
- O. C. Cartwright, A. M. Beekman, M. M. D. Cominetti, D. A. Russell and M. Searcey, Bioconjugate Chem., 2020, 31, 1745–1749 CrossRef CAS.
- Z. R. Goddard, M. J. Marín, D. A. Russell and M. Searcey, Chem. Soc. Rev. 10.1039/D0CS01121E.
- J. R. Grandis and J. C. Sok, Pharmacol. Ther., 2004, 102, 37–46 CrossRef CAS.
- Q. Sheng and J. Liu, Br. J. Cancer, 2011, 104, 1241–1245 CrossRef CAS.
- R. B. Cohen, Clin. Colorectal Cancer, 2003, 2, 246–251 CrossRef CAS.
- S. Kalyankrishna and J. R. Grandis, J. Clin. Oncol., 2006, 24, 2666–2672 CrossRef CAS.
- Y. Cheng, J. D. Meyers, R. S. Agnes, T. L. Doane, M. E. Kenney, A.-M. M. Broome, C. Burda and J. P. Basilion, Small, 2011, 7, 2301–2306 CrossRef CAS.
- J. D. Meyers, Y. Cheng, A. M. M. Broome, R. S. Agnes, M. D. Schluchter, S. Margevicius, X. Wang, M. E. Kenney, C. Burda and J. P. Basilion, Part. Part. Syst. Charact., 2015, 32, 448–457 CrossRef CAS.
- J. D. Mangadlao, X. Wang, C. McCleese, M. Escamilla, G. Ramamurthy, Z. Wang, M. Govande, J. P. Basilion and C. Burda, ACS Nano, 2018, 12, 3714–3725 CrossRef CAS.
- S. Song, D. Liu, J. Peng, H. Deng, Y. Guo, L. X. Xu, A. D. Miller and Y. Xu, FASEB J., 2009, 23, 1396–1404 CrossRef CAS.
- C. Y. Han, L. L. Yue, L. Y. Tai, L. Zhou, X. Y. Li, G. H. Xing, X. G. Yang, M. S. Sun and W. S. Pan, Int. J. Nanomed., 2013, 8, 1541–1549 Search PubMed.
- N. Zhao, T. M. Williams, Z. Zhou, F. R. Fronczek, M. Sibrian-Vazquez, S. D. Jois and M. G. H. Vicente, Bioconjugate Chem., 2017, 28, 1566–1579 CrossRef CAS.
- C. Y. Han, Y. Li, M. Sun, C. Liu, X. Ma, X. Yang, Y. Yuan and W. Pan, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 161–166 CrossRef CAS.
- I. Meerovich, D. D. Smith and A. K. Dash, J. Drug Delivery Sci. Technol., 2019, 54, 101288 CrossRef CAS.
- A. Popovtzer, A. Mizrachi, M. Motiei, D. Bragilovski, L. Lubimov, M. Levi, O. Hilly, I. Ben-Aharon and R. Popovtzer, Nanoscale, 2016, 8, 2678–2685 RSC.
- J. Spadavecchia, D. Movia, C. Moore, C. M. Maguire, H. Moustaoui, S. Casale, Y. Volkov and A. Prina-Mello, Int. J. Nanomed., 2016, 11, 791–822 CrossRef CAS.
- P. García Calavia, M. J. Marín, I. Chambrier, M. J. Cook and D. A. Russell, Photochem. Photobiol. Sci., 2018, 17, 281–289 RSC.
- P. García Calavia, I. Chambrier, M. J. Cook, A. H. Haines, R. A. Field and D. A. Russell, J. Colloid Interface Sci., 2018, 512, 249–259 CrossRef.
- N. A. Byzova, I. V. Safenkova, E. S. Slutskaya, A. V. Zherdev and B. B. Dzantiev, Bioconjugate Chem., 2017, 28, 2737–2746 CrossRef CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS.
- A. Michaeli and J. Feitelson, Photochem. Photobiol., 1994, 59, 284–289 CrossRef CAS.
- C. Balny and P. Douzou, Biochem. Biophys. Res. Commun., 1974, 56, 386–391 CrossRef CAS.
- R. Sjöback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7–L21 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00284d |
| This journal is © The Royal Society of Chemistry 2021 |