

Abstract
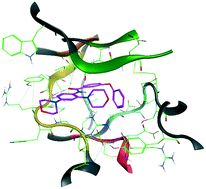
In silico virtual screening followed by in vitro biochemical, biophysical, and cellular screening resulted in the identification of distinctly different hTrkA kinase domain inhibitor scaffolds. X-ray structural analysis of representative inhibitors bound to hTrkA kinase domain defined the binding mode and rationalized the mechanism of action. Preliminary assessment of the sub-type selectivity against the closest hTrkB isoform, and early ADME guided the progression of select inhibitor leads in the screening cascade. The possibility of the actives sustaining to known hTrkA resistance mutations assessed in silico offers initial guidance into the required multiparametric lead optimization to arrive at a clinical candidate.
 Please wait while we load your content...
Please wait while we load your content...

