DOI:
10.1039/C9MD00473D
(Research Article)
RSC Med. Chem., 2020,
11, 148-154
Discovery and characterization of a novel peptide inhibitor against influenza neuraminidase
Received
4th October 2019
, Accepted 13th December 2019
First published on 22nd January 2020
Abstract
Neuraminidase, an abundant glycoprotein on the influenza virus surface, plays crucial roles in virus replication. Targeting neuraminidase could be a splendid way for the prevention of the spread of influenza infections. Herein, we have identified an octapeptide (errKPAQP) from a synthesized peptide library, originating from mimicking the binding pocket of oseltamivir in neuraminidase, as a potent peptide neuraminidase inhibitor. The docking-based virtual studies showed that errKPAQP exhibited a strong binding affinity (a docking score of −20.03) and nanomolar affinity (11 nM) to influenza neuraminidase, and can inhibit neuraminidase activity at a concentration as low as 4.25 μM, leading to effective protection of MDCK cells from influenza virus-induced death and replication. Furthermore, errKPAQP presented low hemolytic activity, minimal cytotoxicity, and good pharmacokinetic characteristics, which are imperative for an anti-influenza drug. Importantly, errKPAQP was capable of reducing influenza virus-induced inflammation, the serious damage to the lung tissues, and mortality rates in infected mice, indicating that it could protect against the lethal challenge of influenza viruses in vivo. Therefore, we have developed a novel neuraminidase peptide inhibitor with advantageous biological properties and high inhibitory activity towards neuraminidase, and it can serve as a promising anti-influenza drug.
Introduction
Influenza is an acute respiratory infection caused by influenza viruses, and its epidemic outbreak and spread are one of the most frightening causes of pandemics across the world, leading to a great number of deaths and hospitalizations.1 The most outstanding characteristic of influenza viruses is their high potential for mutation, leading to great difficulty in the treatment of infections. Therefore, anti-influenza therapeutics with new targets and action mechanisms are greatly needed to overcome newly emerging viruses and constantly mutating strains.2
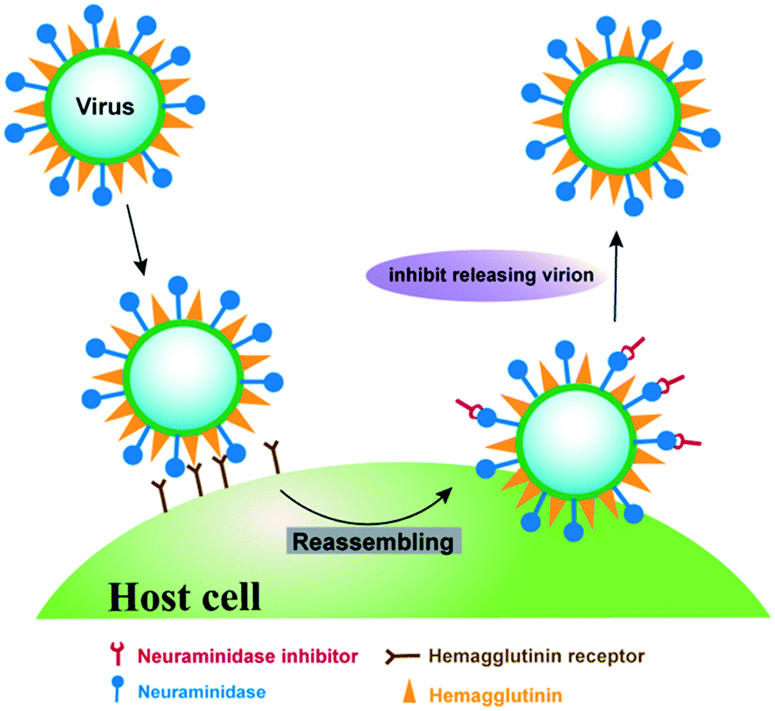
The current anti-influenza drugs majorly target membrane glycoproteins haemagglutinin and neuraminidase as well as the matrix-2 protein, which are critical for viral infections.3 Among these glycoproteins, neuraminidase is a surface glycoside hydrolase enzyme that selectively catalyzes the cleavage of the terminal sialic acid residues, and plays a critical role in the release and spread of progeny virions (Fig. 1), making it an excellent drug target for the prevention of influenza infections.4 Although several neuraminidase inhibitors have been approved for the treatment of influenza in clinics, including oseltamivir, zanamivir, peramivir and laninamivir,5 neuraminidase subtype-specific mutations frequently emerge at different rates in influenza virus, leading to serious drug resistance.6 For example, oseltamivir resistant strain H275Y, in which histidine 275 mutates to tyrosine and disrupts the hydrophobic binding pocket of oseltamivir, has a 300-fold lower binding affinity compared to the wide type strain.7 In another influenza mutant H274Y, tyrosine mutation is close to the hydrophobic binding site of oseltamivir, leading to a three orders of magnitude loss of the drug efficacy.8 Therefore, discovery of novel neuraminidase inhibitors is greatly needed for enhancing anti-influenza treatment in clinics.
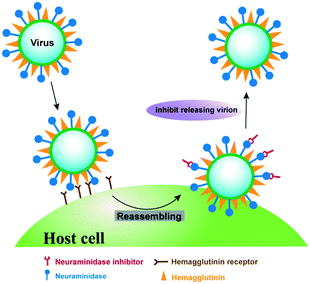 |
| | Fig. 1 The effect of neuraminidase inhibitor on preventing and treating influenza. Neuraminidase inhibitor is capable of blocking the enzyme active site of neuraminidase, thereby preventing virus release from the host cell. | |
Peptide drugs represent a unique class of pharmaceutical compounds, which have played a lot of notable roles in fundamental research9 and medical practice.10 In comparison with other chemical drugs, peptide drugs have been recognized as a promising alternative to biologics due to their high selectivity, efficacy and safety. Consequently, there is an increased interest in the development of peptide drugs, and approximately 140 peptide drugs have been approved in clinics.11 For example, cyclic peptide drugs such as oritavancin (bacteria), pasireotide (tumor) and peginesatide (anemia) show great success as therapeutics in multiple disciplines due to their high affinity and selectivity for targets, as well as good biocompatibility. In addition, peptides are easily synthesized by automated chemical synthesis, and can generate a large combinatory library for screening new drug candidates. Peptides have multiple functional moieties, such as amino, carboxyl and thiol groups, and are easy to modify, handle, and decorate to enhance their application potential, which are important properties for therapeutics. Most importantly, natural amino acids, as the starting materials for the synthesis of peptides, are widely accessible, allowing for the synthesis of a large amount of active pharmaceutical ingredients.12 Given their attractive synthetic feasibility, peptides present an ideal type of drug candidate that can closely mimic natural pathways.
Herein, we have identified an octapeptide (errKPAQP) from a synthesized peptide library, originating from mimicking the binding pocket of oseltamivir in neuraminidase. errKPAQP presented high binding affinity to influenza neuraminidase. Systemic evaluation of the inhibitory abilities against neuraminidase confirmed that errKPAQP could significantly inhibit neuraminidase activity in vitro. Furthermore, biocompatibility tests demonstrated that errKPAQP exhibited low hemolytic activity, minimal cytotoxicity, and good pharmacokinetic characteristics, which are imperative for an anti-influenza drug. Particularly, errKPAQP was capable of reducing influenza virus-induced cell death and minimizing mortality rates in infected mice, indicating that it could be a valid peptide inhibitor for the treatment of influenza.
Results and discussion
Design of neuraminidase inhibitors
By mimicking the binding pocket of oseltamivir in the influenza neuraminidase, a synthesized library of 1000 peptides was created, composed of natural L-amino acids (capital letters) and unnatural D-amino acids (lowercase letters) which were introduced in the peptide inhibitors for enhancing biostability.13 The docking scores, including van der Waals energy, electrostatic energy and desolvation energy, were employed to estimate the binding affinities of peptides against neuraminidase.14 Twenty peptides were identified as the potent neuraminidase inhibitors (Table 1), and peptides P1, P2, P4, P5, P8, P9, P13, P14, P16 and P18 with high docking scores were selected for further in vitro and in vivo validation.
Table 1 Sequences and docking scores of peptides
| Code |
Peptide sequence |
Docking score |
|
P1
|
GHrKPAQP |
−19.09 |
|
P2
|
errKPAQP |
−20.03 |
|
P3
|
MWRVHPKP |
−13.43 |
|
P4
|
eeEKPAQP |
−18.27 |
|
P5
|
efrTMVHPK |
−16.97 |
|
P6
|
hsFHHKPAQA |
−11.81 |
|
P7
|
ehdPKTAHQ |
−11.61 |
|
P8
|
edRKHAQA |
−19.20 |
|
P9
|
kEFHKTAQP |
−18.44 |
|
P10
|
GRRSKPAQP |
−15.48 |
|
P11
|
SfnARLPR |
−14.31 |
|
P12
|
vIsKHAQA |
−16.02 |
|
P13
|
frrRPLRA |
−17.57 |
|
P14
|
DdhKTAHQ |
−16.91 |
|
P15
|
VRKHHKPAQA |
−10.91 |
|
P16
|
GwRKPAQP |
−17.83 |
|
P17
|
iEERTMHH |
−15.34 |
|
P18
|
wKrLHHKPA |
−16.06 |
|
P19
|
wrSPTMVH |
−13.46 |
|
P20
|
yErKPAQP |
−15.60 |
Estimations of antiviral binding affinity of peptides with neuraminidase
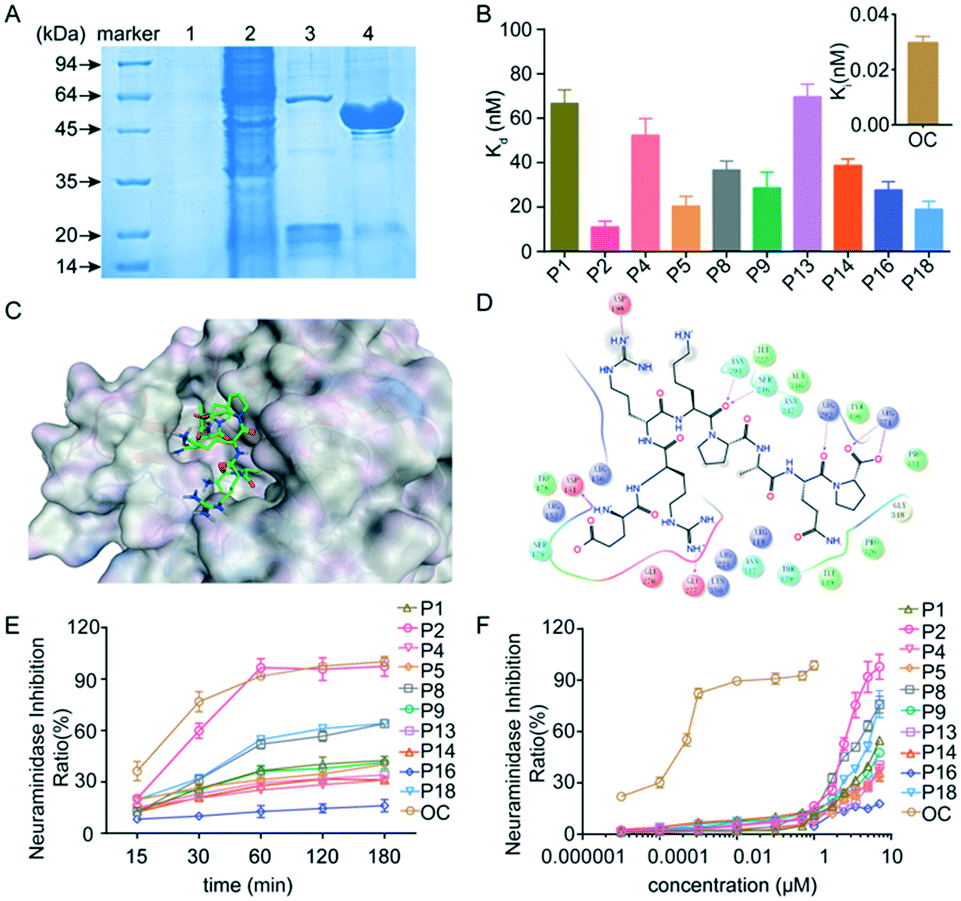
The purity of neuraminidase is more than 95% after purification (Fig. 2A), which was used for the following experiment. The dissociation constant (Kd) is an equilibrium constant for the dissociation of a complex into its components, and is widely used in biochemistry and pharmacology to measure the tendency of the dissociation of a substrate from an enzyme. The dissociation constant provides essential information about the affinity between ligands and their target proteins.15 Therefore, we have systematically measured the Kd of 20 peptides binding to neuraminidase. Fig. 2B shows that seven peptides, namely P2, P5, P8, P9, P14, P16 and P18, exhibited high affinities towards neuraminidase with Kd values of 11.0, 20.3, 36.6, 28.6, 38.6, 27.7 and 19 nM, respectively. Particularly, peptide P2 (errKPAQP) showed a high binding affinity to neuraminidase, indicating that peptide P2 could be a potent inhibitor candidate for treating influenza virus.
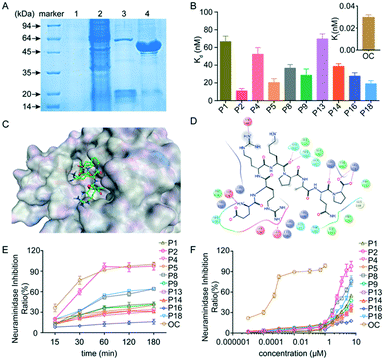 |
| | Fig. 2 Neuraminidase peptide inhibitors and their inhibitory effects. (A) The preparation of neuraminidase. Lane 1: the precipitate of sonicated cell lysate. Lane 2: the soluble fraction of sonicated cell lysate. Lane 3: the soluble protein from the washing step. Lane 4: purified protein. (B) The dissociation constants (Kd) between peptides or oseltamivir carboxylate (OC) and neuraminidase. (C) 3D structural analysis of peptide P2 to neuraminidase. (D) 2D structural analysis of cyclic peptide P2 binding to neuraminidase. (E) The neuraminidase inhibition ratio of peptide (25 μM) and OC (0.1 nM) in different times. (F) The inhibitory activity of peptides and OC against neuraminidase. Data were expressed as mean ± SD, n = 6. | |
Structural analysis of neuraminidase–ligand interactions
To investigate the binding affinity between peptide P2 and neuraminidase, their complex interactions were systematically analyzed using molecular dynamics, followed by post-processing based on the three-dimensional reference interaction site model (3D-RISM), which is a method for representing a molecular solvation structure in terms of 3D maps of solvent site densities around a solute macromolecule or a supramolecular assembly of arbitrary shape.16 The 2D and 3D images show that peptide P2 is docked into the binding cavity, and has strong electrostatic and hydrogen-bonding interactions with neuraminidase. Two D-arginines form strong hydrogen bonds with Asp198 and Glu277 of neuraminidase, while two prolines form strong hydrogen bonds with Asn294, Ser246, Arg292 and Arg371,17 indicating its strong electrostatic and hydrogen-binding interactions with neuraminidase.
Neuraminidase inhibitory assay
We further performed additional experiments to investigate the ability of peptides to inhibit influenza viral neuraminidase activity. The neuraminidase employed for experimental studies was prepared as described by Mayron and Rafelson,18 and the 4-MU-NANA based enzymatic assay was selected to assess the neuraminidase activity. In order to assess the chemical kinetics of inhibition of enzymatic reactions, peptide P2 (errKPAQP) (25 μM) was cultured with neuraminidase in the presence of 4-MU-NANA for 180 minutes, and the fluorescence intensity was measured at different time points. Fig. 2E shows that the inhibition of neuraminidase activity was fully achieved at 60 min. In addition, all peptides were tested to evaluate their inhibitory activity, and the results were expressed in terms of half-maximal inhibitory concentration (IC50) values, which were obtained from the initial incubation responses of test peptides in the presence of 4-MU-NANA. Peptides P1, P2, P8, P9 and P18 showed high inhibition activity with IC50 values less than 25 μM, indicating that these peptides were potential antiviral candidates against influenza, and particularly, peptide P2 exhibited a neuraminidase inhibitory effect with an IC50 value of 4.25 μM (Fig. 2F), which is comparable to previously reported influenza inhibitors.19
In vitro inhibitory effects of peptide neuraminidase inhibitors against influenza infection
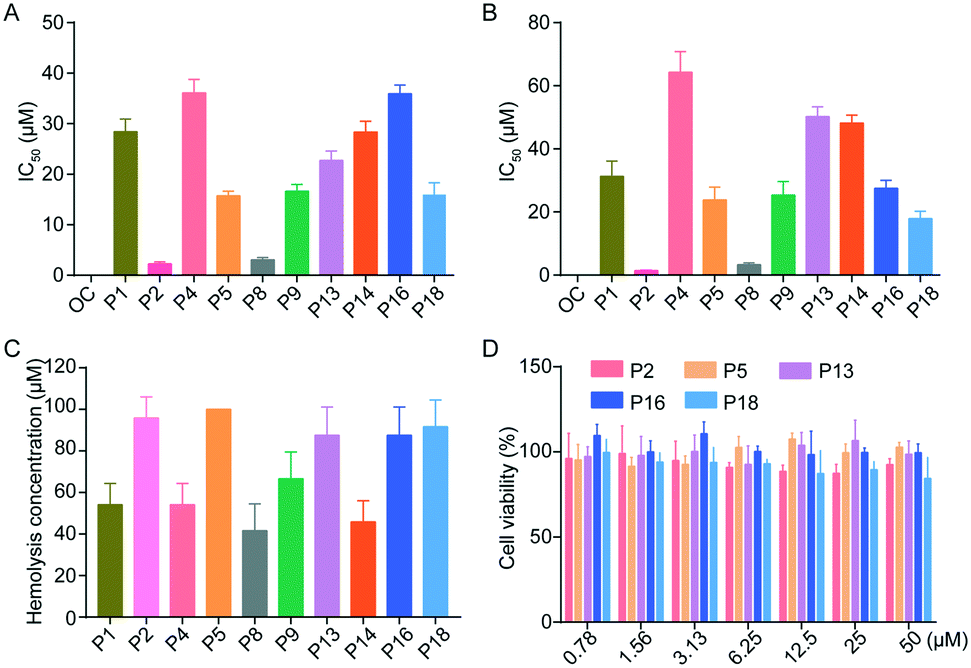
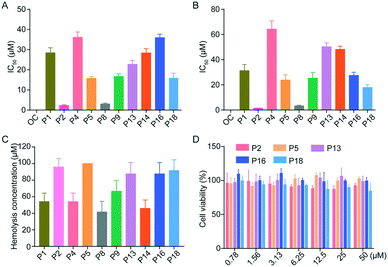
Cytopathic effect (CPE) assay was performed to confirm the antiviral efficacy of peptide inhibitors on H1N1 and H5N1. The inhibitory activity of all peptides was expressed in terms of half-maximal inhibitory concentration (IC50) values. Among all peptides, P2, P8 and P18 showed high neuraminidase inhibitory activity against H1N1 and H5N1. For example, peptides P2, P8 and P18 effectively inhibited H1N1 with IC50 values of 2.26, 3.08 and 15.99 μM, respectively, as well as H5N1 with IC50 values of 1.46, 3.32 and 17.89 μM, respectively (Fig. 3A and B and Table 2). In comparison with oseltamivir carboxylate, which has an IC50 value of 11.26 and 5.60 nM for H1N1 and H5N1, respectively, peptide inhibitors exhibit compromising inhibitory activity. However, peptide inhibitors, as natural substrates, have been recognized as a promising alternative drug resource due to their good selectivity, efficacy and biosafety, and can overcome the challenges of developing new drugs, such as poor cell permeability, low bioavailability and high production cost,11 indicating that novel peptide inhibitors may be potential drug candidates for treating influenza.
 |
| | Fig. 3
In vitro studies of peptides. (A) Antiviral activity of peptides against influenza H1N1, which was expressed in terms of IC50. (B) Antiviral activity of peptides against influenza H5N1, which was expressed in terms of IC50. (C) The hemolytic concentrations of peptides against red blood cells. (D) The cytotoxicity of peptides in HEK 293T cells. Data were expressed as mean ± SD, n = 6. OC: oseltamivir carboxylate. | |
Table 2 IC50 values of peptides against H1N1 and H5N1
|
|
H1N1 (mM) |
H5N1 (mM) |
|
OC: oseltamivir carboxylate. IC50 values of OC against H1N1 and H5N1 were 11.26 ± 2.79 nM and 5.60 ± 1.65 nM, respectively. OC: oseltamivir carboxylate.
|
| OCa |
11.26 ± 2.79 |
5.60 ± 1.65 |
|
P1
|
28.44 ± 2.51 |
31.28 ± 4.87 |
|
P2
|
2.26 ± 0.40 |
1.46 ± 0.12 |
|
P4
|
36.10 ± 2.66 |
64.29 ± 6.52 |
|
P5
|
15.73 ± 0.90 |
23.83 ± 4.09 |
|
P8
|
3.08 ± 0.44 |
3.32 ± 0.57 |
|
P9
|
16.65 ± 1.34 |
25.35 ± 0.57 |
|
P13
|
22.71 ± 1.87 |
50.20 ± 3.17 |
|
P14
|
28.36 ± 2.01 |
48.16 ± 2.56 |
|
P16
|
35.95 ± 2.11 |
27.49 ± 2.58 |
|
P18
|
15.99 ± 2.33 |
17.89 ± 2.35 |
Hemolysis and cytotoxicity studies of peptides
In order to ensure the biosafety of peptides, we investigated their hemolytic activity and cytotoxicity. Fig. 3C and D show that peptides had low hemolytic activity against red blood cells with HC50 (50% hemolytic concentration) ranging from 25 to 100 μM. Among them, peptides P2, P5, P13, P16 and P18 exhibited no hemolysis at a concentration of 100 μM, and no significant cytotoxicity was observed in normal HEK 293T cells even at a high dose of 50 μM, indicating that they have good biosafety.
In vitro stability of peptides in serum
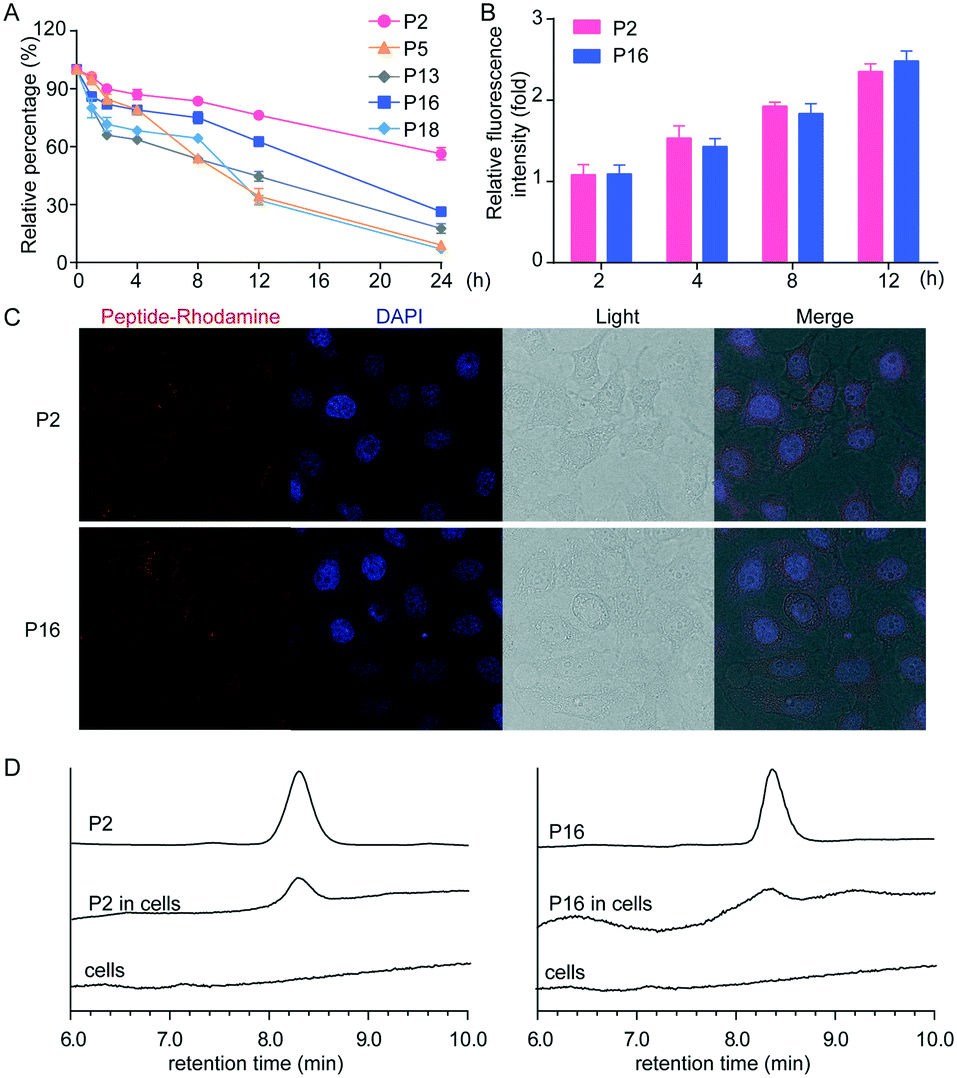
To investigate the translational potential of the peptides identified in this study, we performed in vitro stability analyses. The in vitro stability of peptides was assessed in human plasma, and peptides were incubated with the plasma at 37 °C, measured by LC. The percentages of the remaining peptides were plotted against the corresponding time points, and linear regression gave a straight line (shown in Fig. 4A). Most of the peptides did not show any degradation over 8 h (Fig. 4A). Particularly, peptides P2 and P16 were stable in the plasma even for 18 h, indicating that peptides containing specific unnatural amino acids endow them with resistance to proteolytic cleavage.
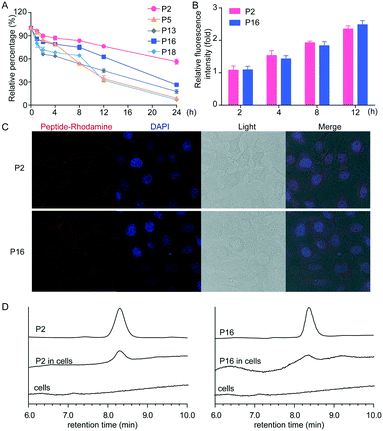 |
| | Fig. 4 Pharmacokinetic properties of peptides. (A) HPLC analysis of the stability of peptides in serum. (B) Flow cytometry analysis of the cellular uptake of peptides. (C) Confocal microscopy analysis of cellular uptake of peptides. (D) HPLC analysis of intracellular peptides. The pictures were magnified 200×. Data were expressed as mean ± SD, n = 6. | |
Cellular uptake of peptides
The key obstacle for the development of new antiviral agents is the challenges of delivery to infected cells in vivo, greatly decreasing the inhibitory activity of potential antiviral agents. Therefore, we performed extra experiments to investigate the cellular uptake of peptides P2 and P16 due to their high stability in serum. Rhodamine labeled peptides were synthesized, and their uptake in HEK 293T cells was measured by flow cytometry. As shown in Fig. 4B, all peptides were rapidly internalized into cells, and accumulated within cells, endowing them with the ability to effectively inhibit the influenza virus. In addition, the uptake of peptides in HEK 293T cells was further confirmed by confocal microscopy and HPLC (Fig. 4C and D), and the two peptides exhibited similar uptake rates and intracellular accumulation.
Peptides effectively prevent influenza H1N1 infections in vivo
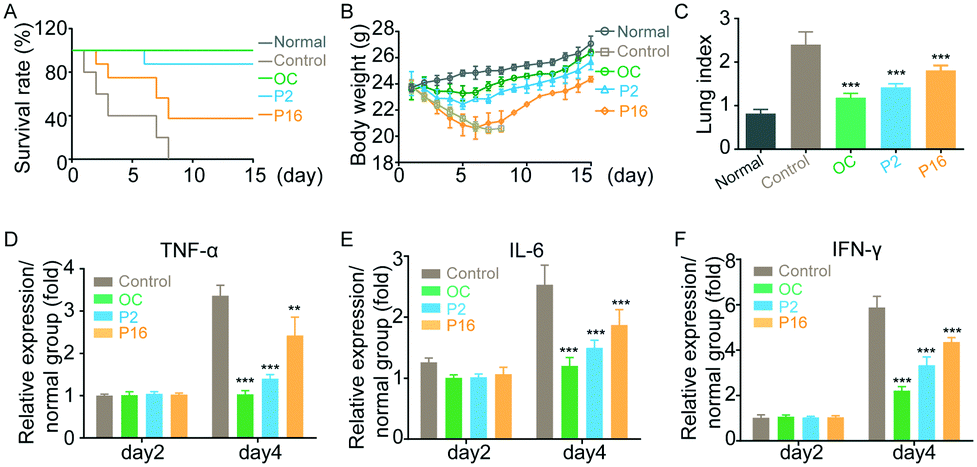
To further evaluate the antiviral efficacy of peptides in vivo, mice were inoculated with influenza virus (H1N1) for 4 h, followed by administration of either peptides (1 mg kg−1 per day) or oseltamivir carboxylate (50 mg kg−1 per day) for seven consecutive days. The mice in the control group were administrated with PBS. As shown in Fig. 5A, peptide P2 and oseltamivir carboxylate demonstrated similar anti-influenza activity, and generated mouse survival rates of 90% and 100%, respectively, while the mice in the control group died after 8-day post-infection. Although influenza infections significantly induced weight loss in the peptide-treated mice after initial infection, the mice body weights recovered rapidly 6 days after peptide treatment (Fig. 5B). In addition, the mice were sacrificed 15 days post-treatment, and the structure of the lung in peptide-treated mice appeared to be returning to normal (Fig. 5C), indicating that peptides show potential for use as a therapeutic for influenza virus infection in vivo.
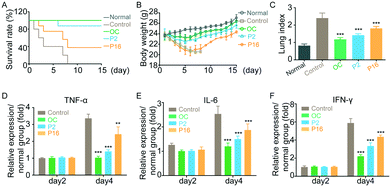 |
| | Fig. 5
In vivo anti-influenza activity of peptides. (A) The survival rate of infected mice treated with peptides. (B) Average body weight of mice over the 15-day treatment. (C) Lung index of mice. (D and E) The levels of inflammatory factors secreted from infected mice. (F) The level of IFN-γ secreted from infected mice. OC: oseltamivir carboxylate. Results are expressed in terms of mean ± SD (n = 6), **p < 0.01 and ***p < 0.001 compared to the normal group by Student's t test. | |
Peptides prevent the inflammatory response triggered by influenza virus
Normally, the robust inflammatory response that follows influenza infection is important for the control of virus proliferation but is also associated with lung damage,20 morbidity and death.21 Therefore, a growing number of studies suggest that the regulation of inflammation in infected mice may improve disease outcome without causing death. We performed the experiments to determine the inflammatory markers in mice after treatment. The mice serum was collected on day 2 and day 4 post-inoculation, and the level of cytokines TNF-α, IL-6 and IFN-γ22 was evaluated using ELISA (shown in Fig. 5D–F). In comparison with healthy mice, the levels of cytokines in infected mice increased. However, peptide treatment could significantly decrease the expression level of TNF-α, IL-6 and IFN-γ, indicating that peptides could improve the treatment outcome without affecting the ability of the host to deal with infection.
Conclusions
In summary, we have developed a novel neuraminidase peptide inhibitor (errKPAQP), which is capable of effectively inhibiting neuraminidase enzymatic activity in vitro and protecting mice from influenza infection in vivo. Therefore, errKPAQP is a new type of promising antiviral drug candidate against H1N1 influenza virus.
Experimental
Materials and instruments
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. Oseltamivir carboxylate was purchased from MedChemExpress. Monolayers of Madin–Darby canine kidney (MDCK) cells and human embryonic kidney cells transformed with large T antigen (HEK 293T) were obtained from ATCC (Shanghai, China). H1N1 (A/Puerto Rico/8/1934) and H5N1 (A/Vietnam/1203/2004) were kindly provided by Wuhan Institute of Virology, CAS. A microplate reader (TECAN, 200 pro, USA) was used for measuring cell viabilities. Confocal laser scanning microscopy (CLSM) (Zeiss, L710, Germany) was used for cell imaging.
All animal experiments were performed in accordance with the National Institute of Health Guidelines under the protocols approved by the ethics committee at the Affiliated Drum Tower Hospital of Nanjing University Medical School.
Virtual screening of neuraminidase inhibitors
A small peptide library was formed by imputing natural L-amino acids and unnatural D-amino acids to mimic the binding pocket of oseltamivir in neuraminidase. The crystal structure of H1N1 neuraminidase in complex with oseltamivir (PDB ID: 3TI6) was used for virtual screening neuraminidase inhibitors.23 Flexible docking was performed using MOEdock, and the Triangle Matcher algorithm and London dG score were utilized as the placement method and rescoring method, respectively.
Synthesis and characterization of peptides
A series of linear peptides varying in length and terminal-capping moieties were synthesized by standard Fmoc solid-phase peptide synthesis protocols using an ABI 433A automated peptide synthesizer, and purified by reverse-phase high-performance liquid chromatography. The molecular masses of the purified peptides (more than 98% purity) were measured by liquid chromatography/mass spectroscopy.
Construction and purification of neuraminidase
The DNA fragment of neuraminidase was constructed and synthesized by Sangon Biotech (China). The recombinant plasmid was transformed into Escherichia coli (E. coli, BL21, DE) competent cells, which were cultured in Luria Bertani (LB) medium at 37 °C for 4 h. Isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.4 mM) was then added into the medium, and the cells were cultured for another 5 hours, followed by centrifugation to harvest E. coli. The E. coli pellets were re-suspended in phosphate buffered saline (PBS, pH 7.4), and sonicated at 4 °C for 20 min to disrupt bacterial cells. The sonicated samples were centrifuged, and supernatants were collected and purified by Ni-Sepharose 6 fast flow affinity chromatography. Proteins were dialyzed in PBS to remove imidazole at 4 °C for 24 h. The neuraminidase was collected and analyzed using 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Binding affinity assays
Enzyme-linked immunosorbent assay (ELISA) was used to identify peptide binding affinity to neuraminidase. The purified neuraminidase was immobilized on the bottom of the enzyme strip, and bovine serum albumin (BSA) was used to block the strip. The neuraminidase strip was added into a 96 well plate, was incubated with different concentrations of peptides conjugated with biotin at 37 °C for 2 h, and then was washed with PBS three times to remove free peptides. Streptavidin conjugated with horseradish peroxidase (HRP) was added and incubated at 37 °C for 1 h. After being washed five times, the HRP substrates were added and incubated at 37 °C for 15 min, followed by measuring the optical density at 450 nm. The concentrations of peptides were defined by the mean absorbance values subtracting from the background intensities. The resulting absorbance value of the peptides was further used to calculate the equilibrium dissociation constant (Kd), which was obtained using the Prism software to fit a plot of the mean absorbance value of the specific binding intensities (Y) versus the peptide concentrations.24 Inhibition constants (Ki) of oseltamivir carboxylate were obtained by measuring the neuraminidase activity in the presence of 0.0002–50 nM oseltamivir carboxylate and 125 μM MUNANA.25
The neuraminidase inhibition assays
The neuraminidase inhibition activity was tested using the synthetic sialylated substrate 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (4-MU-NANA). Different concentrations of peptides were mixed with appropriate H1N1 viral dilutions and were incubated at 37 °C for 1 h. After adding 4-MU-NANA, the reaction mixtures were incubated at 37 °C for 18 h. The amount of free sialic acids liberated from 4-MU-NANA by the viral neuraminidase was measured. The fluorescence intensity of the released product 4-methylumbelliferone was measured (Ex 355/Em 460). Therefore, the inhibitory effect of peptides on the influenza virus neuraminidase is determined. The concentration of the peptides that is required to reduce 50% of the neuraminidase activity is given as the IC50 value.26
The cytopathic effect inhibition assay
MDCK cells (80% confluent) were infected with 0.01 multiplicity of infection (MOI) of influenza virus (H1N1 or H5N1) for 1 h at 37 °C. The viral inoculum was removed, and cells were washed twice with PBS. Different concentrations of peptides were mixed with 1 μg ml−1 1-(tosylamido-2-phenyl) ethylchloromethylketone (TPCK)-treated trypsin in serum-free minimum Eagle's medium (MEM), and were added into the cells. Oseltamivir carboxylate was used as a control. After incubating at 37 °C for 48 h, the cell viability was evaluated using the CCK-8 kit. The concentration that inhibits 50% of virus-induced cytopathic effect was defined as IC50.
Cell viability assay
HEK 293T cells were seeded in a 96-well plate (5000 cells per well) and incubated in Dulbecco's modified Eagle media (DMEM, Invitrogen) containing 10% FBS. Different concentrations of peptides (0.78 to 50 μM) were added into each well for 48 h, and cell proliferation and viability were evaluated using the CCK-8 kit.
Hemolysis assay
Fibrinogen-free rabbit blood was prepared and mixed with 0.9% saline, and the mixture was suspended for 1 min. The mixture was then centrifuged for 15 min at 1000–1500 rpm, and 2% red blood cell suspension was collected for the test assay. Peptide solutions and 2% erythrocyte suspension were mixed in a 96-well plate. After incubation at 37 °C for 60 min, the cell mixtures were centrifuged, and OD600 values of the supernatants were measured to check hemolysis activity.
Peptides serum stability
DMEM supplemented with 25% (v/v) human serum was allocated into a centrifuge tube, and the mixture was incubated at 37 °C for 15 min, followed by adding peptides. The reaction supernatant was then analyzed by using RP-HPLC.
The uptake of peptides into mammalian cells
HEK 293T cells were seeded into a confocal dish, and were then incubated with rhodamine labelled peptides (2 μM). At different time points (2, 4, 8 and 12 h), the cells were washed with PBS twice, fixed with 4% paraformaldehyde, and stained with DAPI (KeyGEN BioTECH). Stained cells were imaged using a laser scanning confocal microscope and were subjected to FACS analysis.
HEK 293T cells were seeded into a confocal dish and were then incubated with peptides P2 or P16 (2 μM) for 12 h, and the cells were washed with PBS twice. The cells were lysed, the proteins were precipitated and the peptides were analyzed by HPLC. HPLC column: VP-ODS C18 column, 150 × 4.6 mm, 5 μm. Flow rate: 1 mL min−1. Solvent: solvent A: 0.1% trifluoroacetic acid in H2O, solvent B: 0.1% trifluoroacetic acid in 80% acetonitrile + 20% H2O. Gradient: 14–34% solvent B from 0–20 min.
Anti-influenza activity of peptides in vivo
Mice (six-week-old male Balb/c mice) were randomly divided into the normal group, control group, and test group. Mice were anaesthetized with ethyl ether and were inoculated intranasally with 10× LD50 (50% lethal dose) of H1N1. Four hours after inoculation, mice were intravenously injected with either peptide inhibitors (1 mg kg−1 per day) or oseltamivir carboxylate (50 mg kg−1 per day) once a day for seven days. The body weight and mortality rates of mice were monitored for 15 consecutive days. The fifth day after influenza infection, the mice were weighed, and their lung tissues were extracted and washed with PBS, dried with gauze, and then weighed. The lung index was calculated as follows: lung index = lung weight/body weight × 100%.
Analysis of inflammatory factors in vivo
The lung homogenate was used to analyze the lung cytokine expression using mouse cytokine ELISA kits, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interferon-γ (IFN-γ) (CUSABIO). The content of TNF-α, IL-6 and IFN-γ was assessed according to the manufacturer's protocol. Absorbance at 450 nm was measured using a spectrophotometer.
Statistical analysis
Data are expressed as mean ± SD (n = 6 or more). All data were analyzed using GraphPad Prism 6 software. One-way ANOVA was used to compare each parameter. *Significance was established when P < 0.05, **P < 0.01, and ***P < 0.001.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The works were supported by the National Natural Science Foundation of China (Grant No. 21472090), Thousand Talents Program for Young Researchers, Shuangchuang Program of Jiangsu Province, Fundamental Research Funds for Central Universities Nanjing University and Scientific Research Foundation of Graduate School of Nanjing University (Grant No. 2017ZDL04).
Notes and references
- J. S. Peiris, L. L. Poon and Y. Guan, Science, 2012, 335(6073), 1173 CrossRef CAS PubMed.
- S. Barik, BMC Med., 2012, 10(1), 104 CrossRef PubMed.
- A. Loregian, B. Mercorelli, G. Nannetti, C. Compagnin and G. Palù, Cell. Mol. Life Sci., 2014, 71(19), 3659 CrossRef CAS PubMed.
- A. Jagadesh, A. A. Salam, P. P. Mudgal and G. Arunkumar, Arch. Virol., 2016, 161(8), 2087 CrossRef CAS PubMed.
- Y. Shobugawa, R. Saito, I. Sato, T. Kawashima, C. Dapat, I. C. Dapat, H. Kondo, Y. Suzuki, K. Saito and H. Suzuki, J. Infect. Chemother., 2012, 18(6), 858 CrossRef CAS PubMed.
- J. L. Mckimm-Breschkin, Influenza Other Respir. Viruses, 2013, 7(Suppl. 1), 25 CrossRef CAS PubMed; Y. Q. Chen, T. J. Wohlbold, N. Y. Zheng, M. Huang and P. C. Wilson, Cell, 2018, 173(2), 417 CrossRef PubMed.
- P. J. Collins, L. F. Haire, Y. P. Lin, J. Liu, R. J. Russell, P. A. Walker, S. R. Martin, R. S. Danniels, V. Gregory, J. J. Skehel, S. J. Gamblin and A. J. Hay, Vaccine, 2009, 27(45), 6317 CrossRef CAS PubMed.
- C. J. Woods, M. Malaisree, B. Long, S. Mcintosh-Smith and A. J. Mulholland, Biochemistry, 2013, 52(45), 8150 CrossRef CAS PubMed.
- G. Shantanu, Biophys. J., 2017, 112(3), 380A Search PubMed; A. T. Tucker, S. P. Leonard, C. D. Dubois, G. A. Knauf, A. L. Cunningham, C. O. Wilke, M. S. Trent and B. W. Davies, Cell, 2018, 172(3), 618 CrossRef CAS PubMed; B. Hussain, F. G. Robert, B. W. Russell and C. W. William, Biophys. J., 2011, 100(3), 216a Search PubMed; K. Singh and B. Chow, ACS Infect. Dis., 2016, 2(7), 471 CrossRef PubMed; Q. Chen and Y. Guo, ACS Infect. Dis., 2016, 2, 187 CrossRef PubMed.
- C. Li, N. Gao, Q. Xue, N. Ma, Y. Hu, J. Zhang, B. Chen and Y. Hou, Biotechnol. Lett., 2017, 39(Suppl 1), 1 Search PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20(1), 122 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24 CrossRef CAS PubMed.
- J. Liu, J. Liu, L. Chu, Y. Zhang, H. Xu, D. Kong, Z. Yang, C. Yang and D. Ding, ACS Appl. Mater. Interfaces, 2014, 6(8), 5558 CrossRef CAS PubMed; L. Zhong, Z. Xiao and Z. Shu, Macromol. Biosci., 2010, 8(8), 785 Search PubMed.
- C. Zhang, B. Tang, Q. Wang and L. Lai, Proteins: Struct., Funct., Bioinf., 2015, 82(10), 2472 CrossRef PubMed.
- L. Jaquillard, F. Saab, F. Schoentgen and M. Cadene, J. Am. Soc. Mass Spectrom., 2012, 23(5), 908 CrossRef CAS PubMed.
- J. P. Yesudas, N. Blinov, S. K. Dew and A. Kovalenko, J. Mol. Liq., 2015, 201, 68 CrossRef CAS.
- V. Mishra, S. Kashyap and Y. Hasija, J. Taibah Univ. Sci., 2015, 9(1), 20 CrossRef.
- L. W. Mayron, B. Robert, R. J. Winzler and M. E. Rafelson, Arch. Biochem. Biophys., 1961, 92(3), 475 CrossRef CAS PubMed.
- B. S. Hwang, M. S. Lee, S. W. Lee, I. K. Lee, G. S. Seo, H. J. Choi and B. S. Yun, Mycobiology, 2014, 42(2), 189 CrossRef CAS PubMed.
- S. Herold, C. Becker, K. M. Ridge and G. R. S. Budinger, Eur. Respir. J., 2015, 45(5), 1463 CrossRef CAS PubMed.
- J. Dushoff, J. B. Plotkin, C. Viboud, D. J. D. Earn and L. Simonsen, Am. J. Epidemiol., 2006, 163(2), 181 CrossRef CAS PubMed; C. Y. Cheung, L. L. Poon, A. S. Lau, W. Luk, Y. L. Lau, K. F. Shortridge, S. Gordon, Y. Guan and J. S. Peiris, Lancet, 2002, 360, 1831 CrossRef; J. R. Tisoncik, M. J. Korth, C. P. Simmons, J. Farrar, T. R. Martin and G. K. Michael, Microbiol. Mol. Biol. Rev., 2012, 76, 16 CrossRef PubMed.
- L. Kaiser, R. S. Fritz, S. E. Straus, L. Gubareva and F. G. Hayden, J. Med. Virol., 2010, 64(3), 262 CrossRef PubMed; B. Liu, D. Meng, T. Wei, S. Zhang, Y. Hu and M. Wang, PLoS One, 2014, 9(6), e100109 CrossRef PubMed; X. Shi, W. Zhou, H. Huang, H. Zhu, P. Zhou, H. Zhu and J. Dian, Crit. Care, 2013, 17, R301 CrossRef PubMed.
- C. J. Vavricka, Q. Li, Y. Wu, J. Qi, M. Wang, Y. Liu, F. Gao, J. Liu, E. Feng, J. He, J. Wang, H. Liu, H. Jiang and G. F. Gao, PLoS Pathog., 2011, 7(10), e1002249 CrossRef CAS PubMed.
- L. Y. Hung, C. H. Wang, Y. J. Che, C. Y. Fu, H. Y. Chang, K. Wang and G. B. Lee, Sci. Rep., 2015, 5, 10326 CrossRef PubMed.
- S. Yongkiettrakul, T. Nivitchanyong, S. Pannengpetch, A. Wanitchang, A. Jongkaewwattana and P. Srimanote, Virus Res., 2013, 175(2), 128 CrossRef CAS PubMed.
- S. K. Leang and A. C. Hurt, J. Visualized Exp., 2017,(122), e55570 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab