
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of a bivalent probe targeting the putative mu opioid receptor and chemokine receptor CXCR4 heterodimer†
Bethany A.
Reinecke
a,
Guifeng
Kang
a,
Yi
Zheng
a,
Samuel
Obeng
a,
Huijun
Zhang
b,
Dana E.
Selley
c,
Jing
An
b and
Yan
Zhang
 *a
*a
aDepartment of Medicinal Chemistry, Virginia Commonwealth University, 800 East Leigh Street, Richmond, VA 23298, USA. E-mail: yzhang2@vcu.edu; Fax: +1 804 828 7625; Tel: +1 804 828 0021
bDepartment of Medicine, Division of Infectious Diseases, School of Medicine, University of California San Diego, 9500 Gilman Drive, Stein Clinical Research Building, Suite 410, La Jolla, CA 92093, USA
cDepartment of Pharmacology and Toxicology, Virginia Commonwealth University, 410 North 12th Street, VA 23298, USA
First published on 19th December 2019
Abstract
Opioid abuse and HIV/AIDS have been defined as synergistic epidemics. Opioids can accelerate HIV replication in the immune system by up-regulating the expression of HIV co-receptor CXCR4. Several hypotheses have been suggested as the mechanism of CXCR4 modulation by opioids through their activation on the mu opioid receptor (MOR). One hypothesis is the putative heterodimerization of the MOR and CXCR4 as a mechanism of cross-talk and subsequent exacerbation of HIV replication. Bivalent chemical probes can be powerful molecular tools to characterize protein–protein interactions, and modulate the function related to such interactions. Herein we report the design and synthesis of a novel bivalent probe to explore the putative MOR–CXCR4 dimerization and its potential pharmacological role in enhancing HIV progression. The developed bivalent probe was designed with two distinct pharmacophores linked through a spacer. One pharmacophore (naltrexone) will interact with the MOR and the other (IT1t) with the CXCR4. The overall synthetic routes to prepare the bivalent probe and its corresponding monovalent controls were comprised of 18–22 steps with acceptable yields. Preliminary biological evaluation showed that the bivalent probe preserved binding affinity and functional activity at both respective receptors, supporting the initial molecular design.
Introduction
The opioid epidemic represents a significant public health crisis in the United States. In 2016, it was estimated that more than 2.1 million Americans had some form of opioid use disorder.1,2 As the opioid crisis persists, there has been increasing concern regarding how injection drug use might impact transmission of viral infections among the opioid misuse population.3 Opioid abuse increases the risk of spreading and contracting HIV due to transmission through needle sharing.4 In March 2018, it was reported that approximately 9% of all new HIV diagnoses in the United States were a result of intravenous drug use.5 Aside from increasing the likelihood of viral spread from injection, drug use can also heighten the probability of high-risk sexual behaviors that could lead to transmission of the virus.4Besides the risk connections between opioid use and HIV transmission, it was shown that opioids have the capacity to accelerate HIV replication and progression at the protein level.6–8 Opioid exposure was found to alter HIV replication in T cells in a time-dependent manner,6,9,10 promote the survival/viability of simian immunodeficiency-infected (SIV) T cells,11 disinhibit HIV latency in T cells,12 and increase plasma viral loads in SIV-infected macaques.13 Chronic morphine exposure was also shown to cause a dysregulation in peripheral immune function through a shift in T-cell dynamics in SIV-infected macaques,14–16 which included dysregulation of T regulatory cells (Tregs) and a subset of T helper cells (Th17).17
One of the pathways by which opioids may accelerate HIV replication is through up-regulation of the viral entry receptor CXCR4. Reports from the literature showed that both [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO, a selective mu-opioid (MOR) agonist) and morphine, the exogenous MOR agonist, increased CXCR4 expression in lymphoblasts and CD14 monocytes by more than three-fold.18–20 An increase in CXCR4 expression provides more entry sites for viral entry and thus an enhanced replication of HIV as evidenced by an increase in p24 antigen levels.
Studies reporting the ability for MOR agonists to up-regulate CXCR4 expression have supported functional cross-talk between these two receptors.19–22 One hypothesis is that MOR–CXCR4 crosstalk is mediated by the formation of MOR–CXCR4 heterodimers.22 This hypothesis has been supported by both studies that substantiated the ability of MOR and CXCR4 to form homodimers23–31 and heterodimers with other GPCRs,31–36 and the finding that the MOR and CXCR4 were co-expressed on immune cells.37–40 As demonstrated by previous studies, the use of bivalent ligands can be a powerful strategy to characterize GPCR dimerization/oligomerization.41–43 Therefore, bivalent chemical probes that are capable of interacting with both receptors simultaneously would be invaluable in facilitating the study of the putative MOR–CXCR4 heterodimer and its role in opioid accelerated HIV replication. Herein, we report the design, synthesis, and preliminary biological evaluation of the first bivalent ligand (1) (Fig. 1) targeting the putative MOR–CXCR4 heterodimer. This ligand may serve as a potential chemical probe to help reveal the underlying mechanism of MOR–CXCR4 heterodimer formation in opioid enhanced HIV replication.
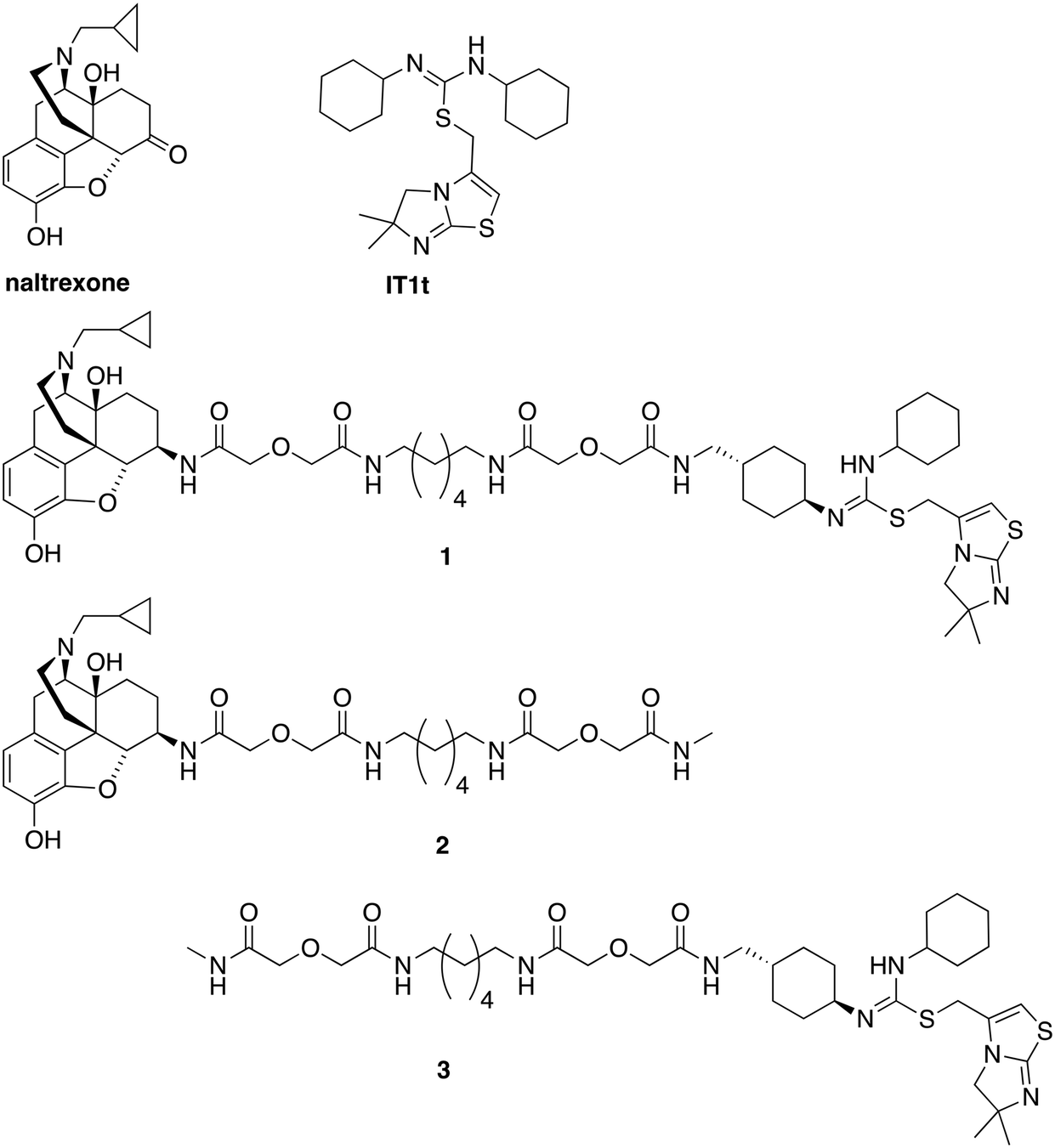 | ||
| Fig. 1 Chemical structures of naltrexone, IT1t, designed bivalent (1), and monovalent controls (2 and 3). | ||
Results and discussion
Bivalent ligand rational design
As this probe was intended to simultaneously block HIV-entry through interactions at the entry receptor CXCR4 as well as enhanced viral replication by opioids, our bivalent ligand is composed of MOR and CXCR4 antagonists linked through an appropriate spacer. Naltrexone (Fig. 1) was selected as the MOR antagonist due to its previous utilization in several published bivalent ligand series and dimerization studies related to the MOR,44–48 in addition to its clinical efficacy in treating opioid addiction.49–51 IT1t (Fig. 1) was selected as the CXCR4 antagonist moiety because of its well-established pharmacological profile52 and its successful employment in CXCR4 crystallographic structural studies.30 Additionally, both of these two ligands showed high affinity and reasonable selectivity toward the MOR and the CXCR4, respectively.52–54Based on previous bivalent ligand series reported from the literature, the attachment locus on naltrexone was chosen to be the C6-position after transforming the carbonyl group to the 6β-amino group.44,45,55–59 The attachment locus on IT1t was identified based on the available CXCR4–IT1t co-crystal structure (PDB: 3ODU).30 In the binding cavity it was observed that the cyclohexyl groups adjacent to the symmetric isothiourea moiety of IT1t were pointing upward and away from the TM bundle and toward the extracellular space. Due to the rotational C2 symmetry of IT1t, one of these cyclohexyl groups was chosen as the attachment site.
A number of previously reported studies indicated that a linker with a length of 16 to 22 atoms might be ideal for targeting GPCR dimers.32,44–48,60,61 Therefore, our first bivalent ligand (1) contained a spacer of 20 atoms as a starting point. The composition of the spacer, an alkyldiamine moiety and two flanking diglycolic units, was chosen based on previously studied GPCR bivalent ligands and ease of synthesis.62 For comparative purposes, monovalent ligands (2 and 3) (Fig. 1) were also designed as controls to study the potential influence of the spacer on receptor function.
Chemical syntheses
The retrosynthetic analysis of the designed bivalent probe revealed 3 major fragments, 6β-naltrexamine, the diacid spacer, and the CXCR4 pharmacophore aminomethyl-substituted IT1t (Scheme 1). 6β-Naltrexamine can be prepared from naltrexone according to the previously reported procedure.63 Nucleophilic reaction of 1,6-diaminohexane with diglycolic anhydride can afford the diacid spacer.64 The synthesis of aminomethyl-substituted IT1t can be split into 2 fragments according to the previously published synthetic route for IT1t. These fragments are 1-(4-(aminomethyl)cyclohexyl)-3-cyclohexylthiourea and 3-(chloromethyl)-5,6-dihydro-6,6-dimethylimidazo[2,1-b]thiazole. The 3-(chloromethyl)-5,6-dihydro-6,6-dimethylimidazo[2,1-b]thiazole can be readily prepared from the commercially available 4,4-dimethyl-imidazolidine-2-thione. The 1-(4-(aminomethyl)cyclohexyl)-3-cyclohexylthiourea was subsequently divided into 2 smaller fragments, Boc-protected 4-(aminomethyl)cyclohexan-1-amine and isothiocyanatocyclohexane. The isothiocyanatocyclohexane can be easily prepared using cyclohexylamine, and the Boc-protected 4-(aminomethyl)cyclohexan-1-amine fragment can be prepared from the commercially available trans-4 (aminomethyl)cyclohexanecarboxylic acid.65,66 Monovalent controls can be synthesized in a similar manner using 2-(2-(methylamino)-2-oxoethoxy)acetic acid as one of the linker building blocks instead of 2 units of diglycolic anhydride.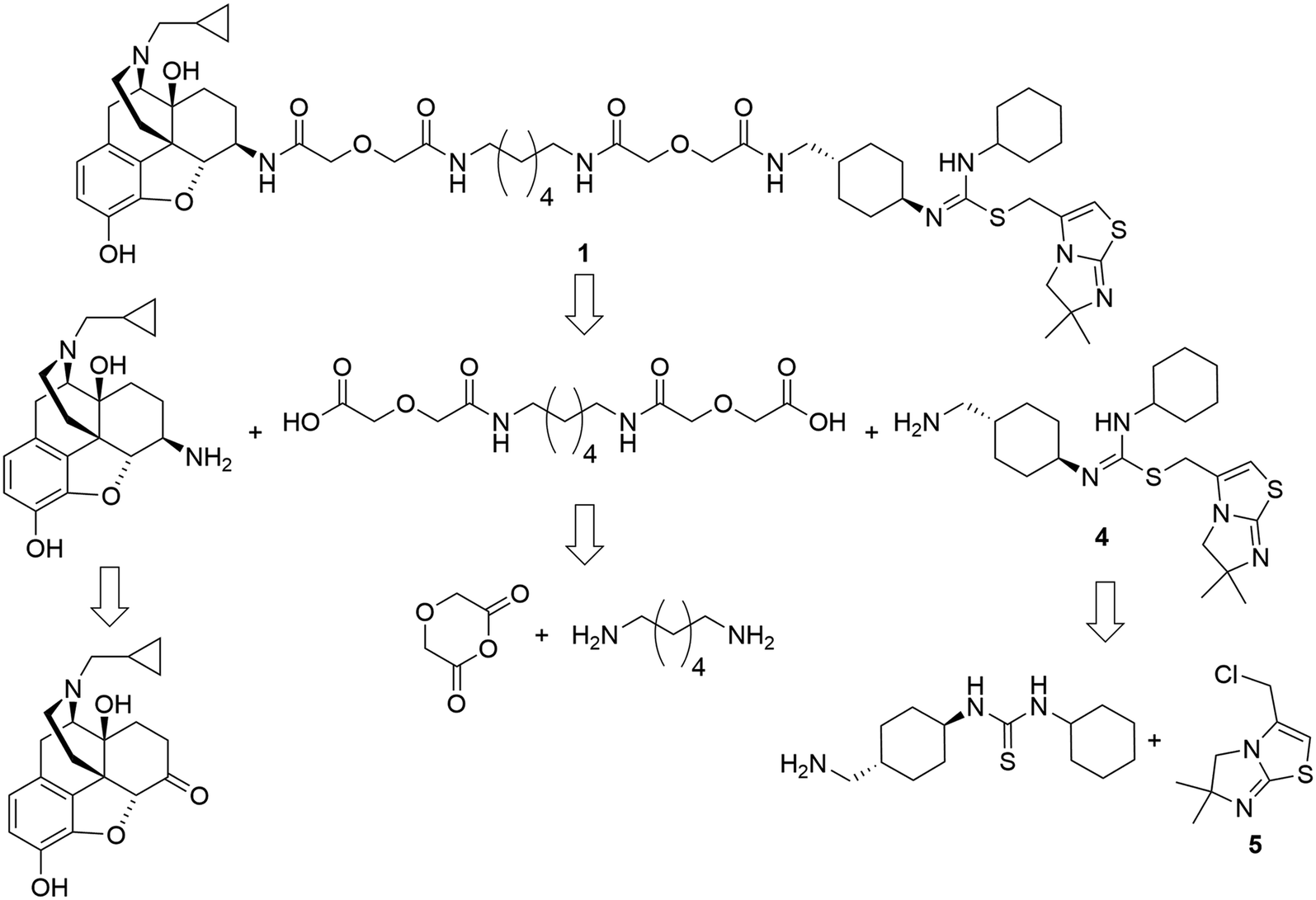 | ||
| Scheme 1 Retrosynthetic analysis of the designed bivalent ligand. | ||
The synthetic route for aminomethyl-substituted IT1t 4 is shown in Scheme 2. Intermediate 5 was prepared following a similar procedure as reported in the synthesis of IT1t.52trans-4-(Aminomethyl)cyclohexanecarboxylic acid was protected using Boc2O to give a moderate yield of 6. Intermediate 6 was then reacted with DPPA to generate an acyl azide. Successive Curtius rearrangement of the acyl azide produced the isocyanate intermediate which could then react with benzyl alcohol to afford 7.65,66 The Cbz protecting group was then removed via hydrogenation with Pd/C to produce 8. Compound 9 was synthesized by first forming the dithiocarbamate intermediate with CS2 and triethylamine, followed by tosyl chloride mediated decomposition to form the isothiocyanate 9 with a moderate yield.67 Isothiocyanate 9 was then reacted with 8 to produce the isothiourea 10. Compound 10 was then reacted with compound 5 in refluxing acetonitrile to produce 11. The final removal of the Boc protecting group was achieved to produce compound 4.
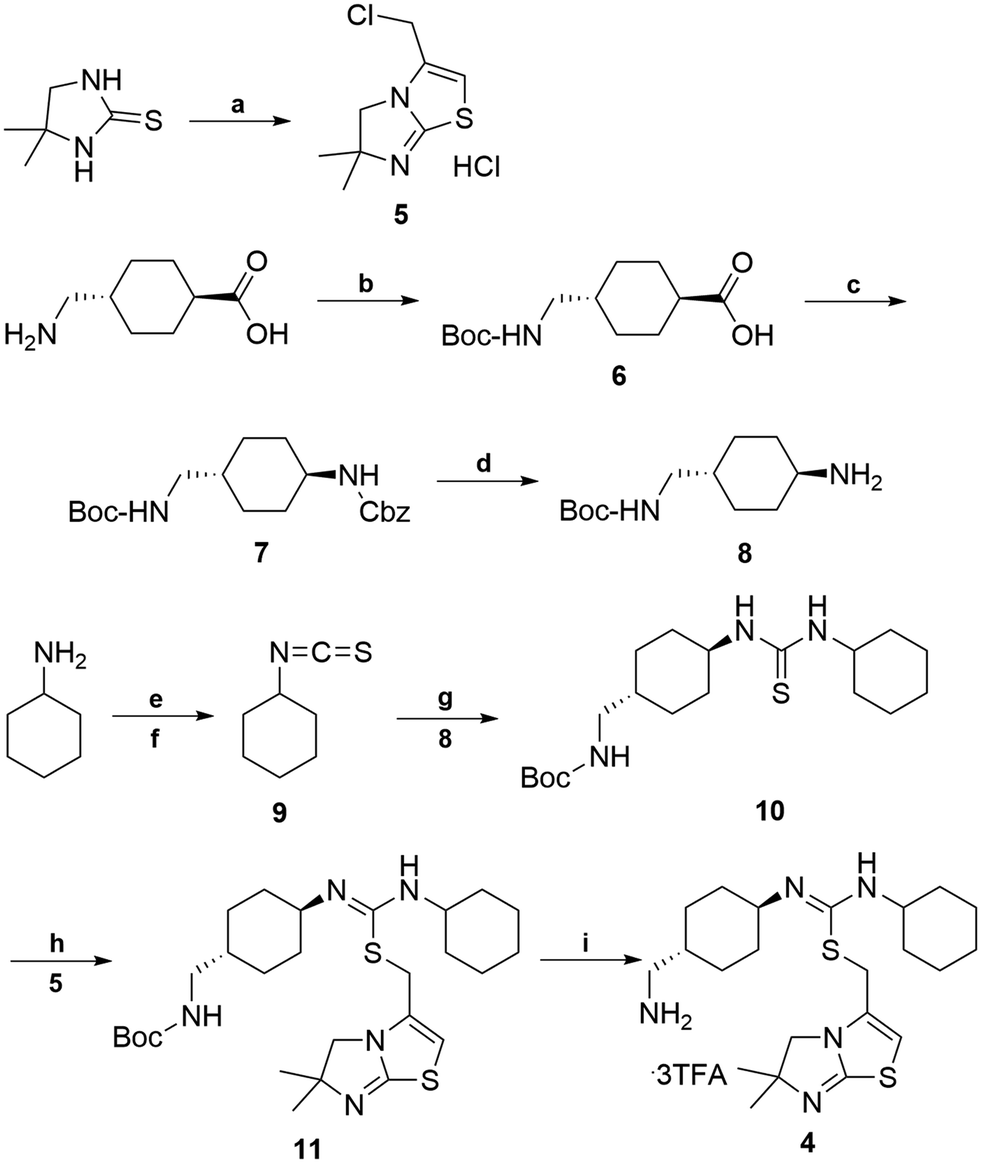 | ||
| Scheme 2 Synthesis of aminomethyl-substituted IT1t (4). Reagents and conditions: (a) 4,4-dimethyl-imidazoline-2-thione, 1,3 dichloroacetone, CH3CN, 1-methoxy-2-(2-methoxyethoxy)-ethane, 145 °C 50–60% (b) BOC2O, NaHCO3, 1,4-dioxane, −5 °C, 50–60% (c) DPPA, TEA, BnOH, 80 °C, 20–30% (d) Pd/C, MeOH, H2, 10 psi, rt, 90–95% (e) TEA, CS2, THF, 0 °C (f) TsCl, HCl, rt, 2 step 60–70% (g) CH2Cl2, 8, rt, 70–80% (h) CH3CN, reflux, 5, 50–60% (i) CH2Cl2, TFA, rt, 90–95%. | ||
The synthetic route for the bivalent ligand 1 is shown in Scheme 3. First, reaction of 1,6-diaminohexane with benzyl chloroformate on an ice-water bath generated the mono-Cbz protected intermediate 12 with a good yield.64 Compound 12 was then coupled to diglycolic anhydride to give 13.45 Compound 14 was prepared by coupling 6β-naltrexamine, obtained via previously reported procedures,63 with 13via the EDCI/HOBt coupling method. Deprotection of the Cbz group of 14 was achieved via hydrogenation with 10% Pd/C catalyst to give amine 15. Coupling of 15 again with diglycolic anhydride provided acid 16. Final coupling of 16 to the CXCR4 antagonist precursor 4via HOBt/EDCI coupling method produced the bivalent ligand 1.
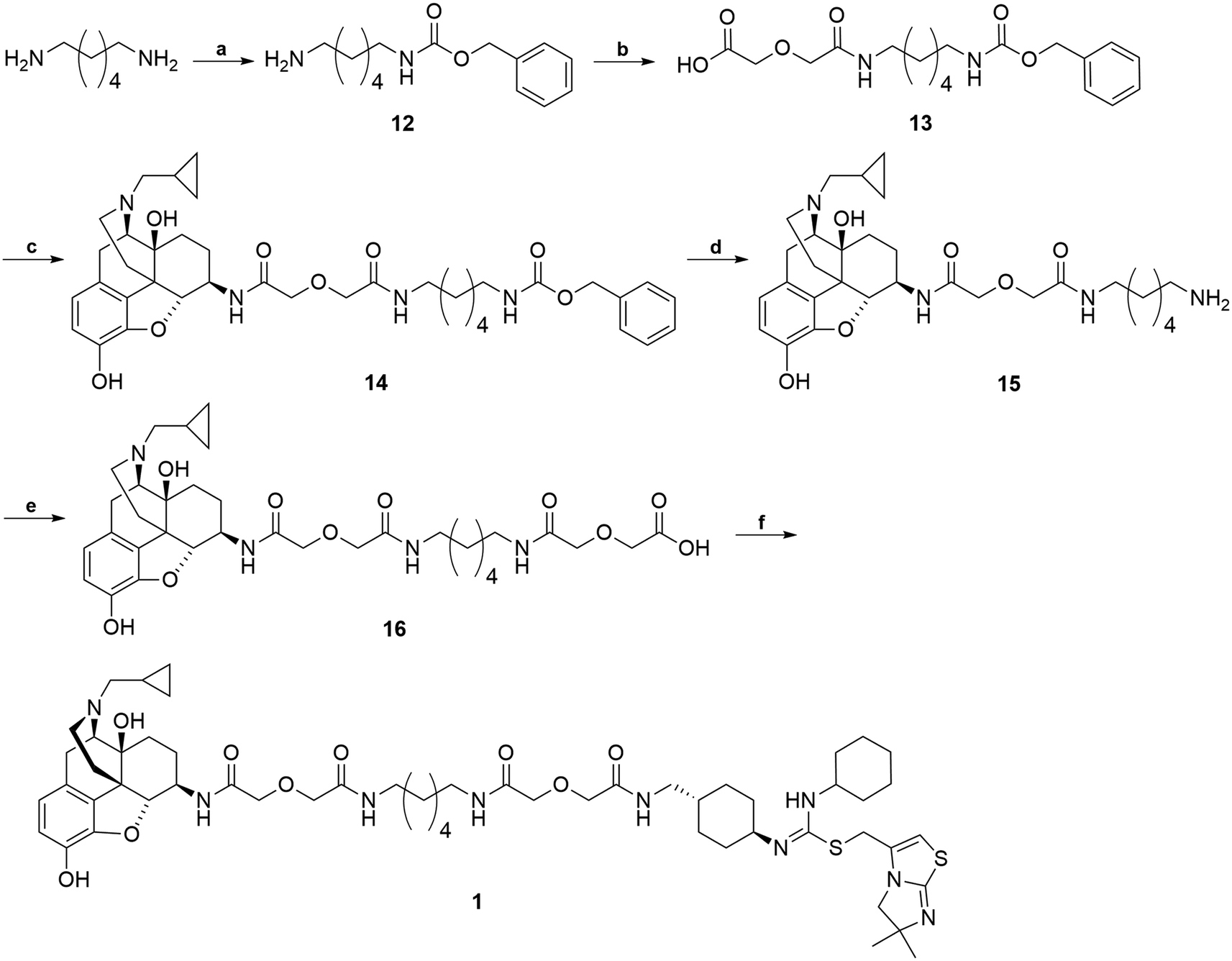 | ||
| Scheme 3 Synthesis of bivalent ligand (1). Reagents and conditions: (a) 1,6-diaminohexane, CbzCl, CH2Cl2, MeOH, 0 °C 70–80% (b) diglycolic anhydride, THF, rt, 50–70% (c) 6β-naltrexamine, EDCI, HOBt, TEA, DMF, 0 °C, 40–50% (d) H2, MeOH, Pd/C, 60 psi, rt, 90–95% (e) diglycolic anhydride, THF, rt, 85–95% (f) 4, EDCI, HOBt, TEA, DMF, 0 °C, 20–30%. | ||
Monovalent controls (2 and 3) were synthesized following a similar route outlined in Scheme 4.45 Methylamine was first condensed with diglycolic anhydride to produce intermediate 17. This intermediate was then coupled to 12via EDCI/HOBt coupling method to yield compound 18. The Cbz protection group was then removed via hydrogenation with Pd/C catalyst to produce 19. Compound 19 was then coupled to another molecule of diglycolic anhydride to yield 20. The final step was to couple either 6β-naltrexamine or the aminomethyl-substituted IT1t 4 with 20via EDCI/HOBt coupling reaction to produce the corresponding monovalent controls (2 and 3).
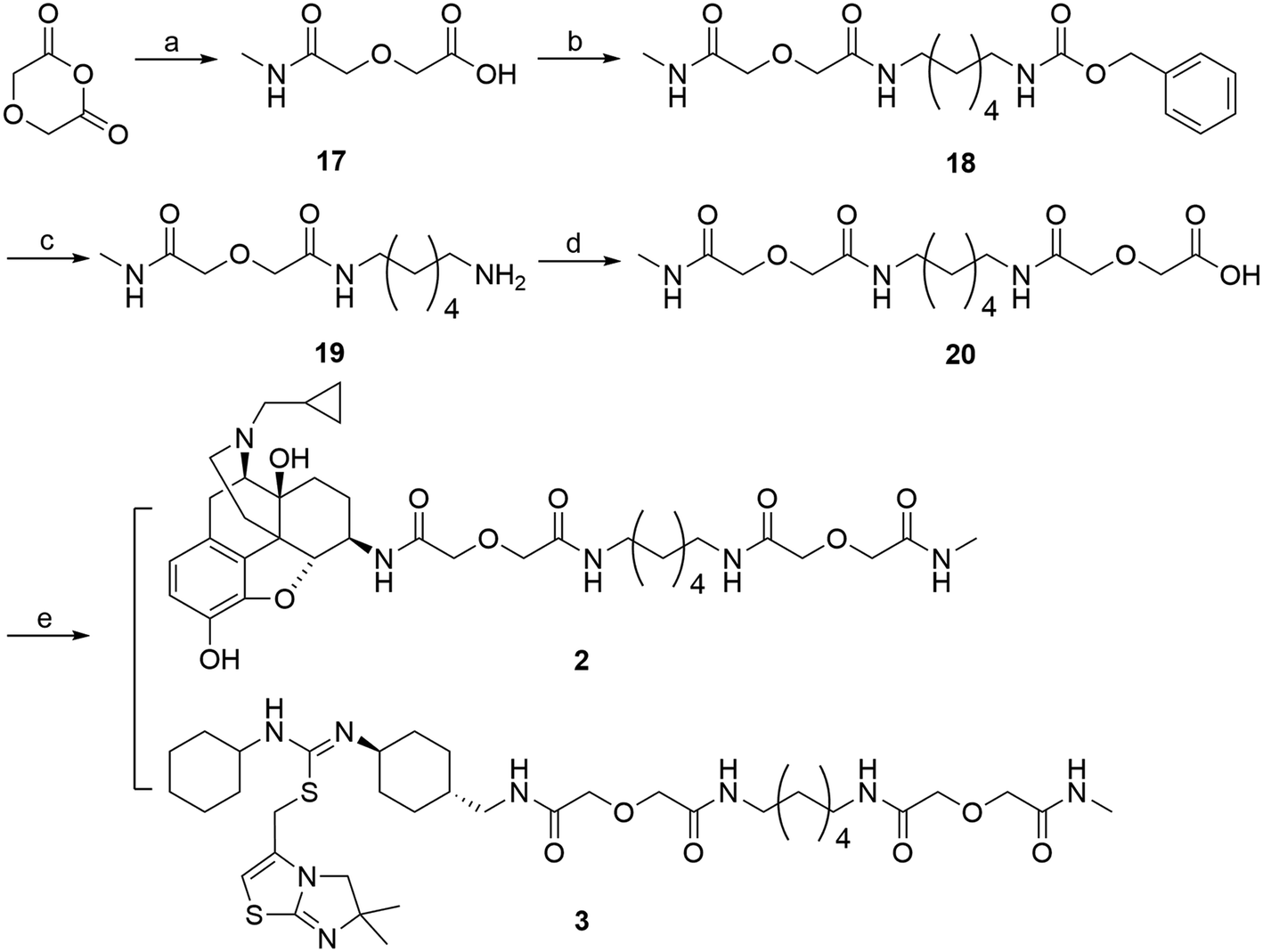 | ||
| Scheme 4 Synthesis of monovalent control compounds (2, 3). Reagents and conditions: (a) methylamine, diglycolic anhydride, THF, rt, 60-70% (b) 12, EDCI, HOBt, TEA, DMF, 0 °C, 40–50% (c) H2, Pd/C, MeOH, rt, 50–60% (d) diglycolic anhydride, THF, rt, 40–50% (e) 4 or 6β-naltrexamine, EDCI, HOBt, TEA, DMF, 0 °C, 20–40%. | ||
Biological evaluation
All three ligands were further characterized for their binding and functional activities on the corresponding receptors (Table 1). In radioligand binding assays with MOR-CHO cells, the bivalent ligand (1) showed to retain binding to the MOR with a Ki value of 25.4 ± 3.0 nM. The monovalent control maintained binding to the MOR with a Ki value of 2.0 ± 0.2 nM, similar to the parent pharmacophore naltrexone (Ki = 0.39 ± 0.04 nM), indicating that addition of the linker does not significantly impact receptor binding. In calcium mobilization assays with MOR-CHO cells, neither the bivalent ligand 1 nor monovalent control 2 showed apparent agonism. In antagonism studies, the monovalent control compound 2 inhibited DAMGO induced calcium flux with an IC50 value of 133.1 ± 15.5 nM, indicating that introduction of the linker does not lead to loss of receptor recognition on the MOR. Bivalent compound 1 inhibited DAMGO induced calcium flux with an IC50 value of 61.9 ± 11.3 nM. Although less potent than the parent pharmacophore naltrexone (8.9 ± 0.9 nM),58 both the bivalent and monovalent compounds were able to maintain functional antagonist activity at the MOR with nanomolar potency.| Compound | MOR binding Ki | CXCR4 binding IC50 | MOR activity IC50 | CXCR4 activity IC50 |
|---|---|---|---|---|
| Bivalent ligand (1) | 25.4 ± 3.0 nM | 17.2 ± 2.7 μM | 61.9 ± 11.3 nM | 3.3 ± 1.0 μM |
| Monovalent (2) | 2.0 ± 0.2 nM | N/A | 133.1 ± 15.5 nM | N/A |
| Monovalent (3) | N/A | 74.0 ± 21.0 μM | N/A | 78.3 ± 21.3 μM |
| Naltrexone | 0.39 ± 0.04 nM | N/A | 8.9 ± 0.9 nM | N/A |
| IT1t | N/A | 7.4 ± 3.8 nM | N/A | 32.6 ± 8.8 nM |
In antibody binding assays with CXCR4-CHO cells, the bivalent ligand (1) retained binding affinity to CXCR4 (IC50 = 17.2 ± 2.7 μM), however, this was substantially reduced as compared to the pharmacophore IT1t (IC50 = 7.4 ± 3.8 nM). Similarly, the monovalent control 3, also showed reduced binding affinity (IC50 = 74 ± 21 μM), indicating that the linker is likely interfering with binding to the CXCR4. In calcium mobilization assays with CXCR4-HOS cells,68,69 neither monovalent or bivalent compound showed any apparent agonism. Monovalent control compound 3 inhibited SDF-1 induced calcium flux with an IC50 value of 78.3 ± 21.3 μM. Bivalent compound 1 inhibited SDF-1 induced calcium flux with an IC50 value of 3.3 ± 1.0 μM. Again, although the relatively higher IC50 values for the bivalent ligand and monovalent controls as compared to IT1t (32.6 ± 8.8 nM) indicate apparent interference caused by the linker which is consistent with the antibody binding data, this preliminary biological evaluation supported our molecular design in that the bivalent probe was still able to maintain reasonable functional activity at the CXCR4.
Conclusions
A novel bivalent probe was designed and synthesized targeting the putative heterodimerization of the MOR and the HIV-1 entry receptor CXCR4. The synthetic route for this compound and its corresponding monovalent controls were comprised of 18–22 steps and were achieved with reasonable yields. Preliminary biological evaluation of this probe showed that the ligand was able to preserve binding and antagonist activity at both the MOR and CXCR4, substantiating our molecular design. Further characterization of this probe in primary cell lines expressing both the MOR and CXCR4 are underway to study the capacity for the probe to inhibit viral invasion and replication. This study will serve as a platform to utilize bivalent probes targeting the putative MOR–CXCR4 heterodimer to better understand its potential role in opioid enhanced HIV-1 infection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HOS.CXCR-4 from Dr. Nathaniel Landau. We thank funding from the NIMH Translational Research in NeuroHIV and Mental Health Pilot Grant R25MH080661 and the NIH Institutional Training Grant 5T32DA007027 (B. A. R.), and the NIDA grant R01DA044855 (Y. Z.).Notes and references
- N. I. on D. Abuse, Overview, https://www.drugabuse.gov/publications/research-reports/medications-to-treat-opioid-addiction/overview, (accessed August 14, 2018).
- N. I. on D. Abuse, Opioid Overdose Crisis, https://www.drugabuse.gov/drugs-abuse/opioids/opioid-overdose-crisis, (accessed August 14, 2018).
- C. S. C. H. B. last updated: M. 30 and 2018, Alcohol and Drug Use and HIV Risk, https://www.hiv.gov/hiv-basics/hiv-prevention/reducing-risk-from-alcohol-and-drug-use/alcohol-and-drug-use-and-hiv-risk, (accessed August 14, 2018).
- Injection Drug Use|HIV Risk and Prevention|HIV/AIDS|CDC, https://www.cdc.gov/hiv/risk/idu.html, (accessed August 14, 2018).
- HIV in the United States|Statistics Overview|Statistics Center|HIV/AIDS|CDC, https://www.cdc.gov/hiv/statistics/overview/ataglance.html, (accessed August 14, 2018).
- A. Tahamtan, M. Tavakoli-Yaraki, T. Mokhtari-Azad, M. Teymoori-Rad, L. Bont, F. Shokri and V. Salimi, Front. Microbiol., 2016, 7, 970 CrossRef PubMed.
- X. Wang, T. Zhang and W.-Z. Ho, J. Neuroimmune Pharmacol., 2011, 6, 477–489 CrossRef PubMed.
- P. V. B. Reddy, S. Pilakka-Kanthikeel, S. K. Saxena, Z. Saiyed and M. P. N. Nair, Interactive Effects of Morphine on HIV Infection, https://www.hindawi.com/journals/art/2012/953678/, (accessed October 30, 2017) Search PubMed.
- P. K. Peterson, G. Gekker, S. Hu, G. Cabral and J. R. Lokensgard, J. Neuroimmunol., 2004, 147, 123–126 CrossRef CAS PubMed.
- X. Liang, R. Liu, C. Chen, F. Ji and T. Li, Transl. Perioper. Pain Med., 2016, 1, 5–13 Search PubMed.
- P.-F. Li, Y.-S. Hao, D.-A. Huang, X.-H. Liu, S.-L. Liu and G. Li, Toxicol. In Vitro, 2004, 18, 449–456 CrossRef CAS PubMed.
- X. Wang, N. Tan, S. D. Douglas, T. Zhang, Y.-J. Wang and W.-Z. Ho, J. Leukocyte Biol., 2005, 78, 772–776 CrossRef CAS PubMed.
- N. K. Vaidya, R. M. Ribeiro, A. S. Perelson and A. Kumar, PLoS Comput. Biol., 2016, 12, e1005127 CrossRef PubMed.
- S. M. Bokhari, R. Hegde, S. Callen, H. Yao, I. Adany, Q. Li, Z. Li, D. Pinson, H.-W. Yeh, P. D. Cheney and S. Buch, J. Neuroimmune Pharmacol., 2011, 6, 626–639 CrossRef PubMed.
- A. Banerjee, M. Strazza, B. Wigdahl, V. Pirrone, O. Meucci and M. R. Nonnemacher, J. NeuroVirol., 2011, 17, 291–302 CrossRef CAS PubMed.
- T. J. Rogers, J. Neuroimmune Pharmacol., 2012, 7, 722–724 CrossRef PubMed.
- W. D. Cornwell, M. G. Lewis, X. Fan, J. Rappaport and T. J. Rogers, J. Neuroimmunol., 2013, 265, 43 CrossRef CAS PubMed.
- C. Happel, A. D. Steele, M. J. Finley, M. A. Kutzler and T. J. Rogers, J. Leukocyte Biol., 2008, 83, 956–963 CrossRef CAS PubMed.
- J. P. Patel, R. Sengupta, G. Bardi, M. Z. Khan, A. Mullen-Przeworski and O. Meucci, J. NeuroVirol., 2006, 12, 492–500 CrossRef CAS PubMed.
- A. D. Steele, E. E. Henderson and T. J. Rogers, Virology, 2003, 309, 99–107 CrossRef CAS PubMed.
- S. Mélik Parsadaniantz, C. Rivat, W. Rostène and A. Réaux-Le Goazigo, Nat. Rev. Neurosci., 2015, 16, 69–78 CrossRef PubMed.
- P. J. Barnes, J. Clin. Invest., 2006, 116, 1210–1212 CrossRef CAS PubMed.
- C. Li-Wei, G. Can, Z. De-He, W. Qiang, X. Xue-Jun, C. Jie and C. Zhi-Qiang, Protein Pept. Lett., 2002, 9, 145–152 CrossRef PubMed.
- D. Rodríguez and H. Gutiérrez-de-Terán, Proteins, 2012, 80, 1919–1928 Search PubMed.
- J. Wang, L. He, C. A. Combs, G. Roderiquez and M. A. Norcross, Mol. Cancer Ther., 2006, 5, 2474–2483 CrossRef CAS PubMed.
- L. Pawig, C. Klasen, C. Weber, J. Bernhagen and H. Noels, Front. Immunol., 2015, 6, 429 Search PubMed.
- G. J. Babcock, M. Farzan and J. Sodroski, J. Biol. Chem., 2003, 278, 3378–3385 CrossRef CAS PubMed.
- A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, J. M. Mathiesen, R. K. Sunahara, L. Pardo, W. I. Weis, B. K. Kobilka and S. Granier, Nature, 2012, 485, 321–326 CrossRef CAS PubMed.
- L. Qin, I. Kufareva, L. G. Holden, C. Wang, Y. Zheng, C. Zhao, G. Fenalti, H. Wu, G. W. Han, V. Cherezov, R. Abagyan, R. C. Stevens and T. M. Handel, Science, 2015, 347, 1117–1122 CrossRef CAS PubMed.
- B. Wu, E. Y. T. Chien, C. D. Mol, G. Fenalti, W. Liu, V. Katritch, R. Abagyan, A. Brooun, P. Wells, F. C. Bi, D. J. Hamel, P. Kuhn, T. M. Handel, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1066–1071 CrossRef CAS PubMed.
- C. W.-S. Lee and I.-K. Ho, Cell, 2013, 2, 689–714 CrossRef PubMed.
- E. Akgün, M. I. Javed, M. M. Lunzer, M. D. Powers, Y. Y. Sham, Y. Watanabe and P. S. Portoghese, J. Med. Chem., 2015, 58, 8647–8657 CrossRef PubMed.
- A. Al-Aoukaty, T. J. Schall and A. A. Maghazachi, Blood, 1996, 87, 4255–4260 CrossRef CAS PubMed.
- C. Chen, J. Li, G. Bot, I. Szabo, T. J. Rogers and L.-Y. Liu-Chen, Eur. J. Pharmacol., 2004, 483, 175–186 CrossRef CAS PubMed.
- P.-Y. Law, L. J. Erickson-Herbrandson, Q. Q. Zha, J. Solberg, J. Chu, A. Sarre and H. H. Loh, J. Biol. Chem., 2005, 280, 11152–11164 CrossRef CAS PubMed.
- O. M. Pello, L. Martínez-Muñoz, V. Parrillas, A. Serrano, J. M. Rodríguez-Frade, M. J. Toro, P. Lucas, M. Monterrubio, C. Martínez-A and M. Mellado, Eur. J. Immunol., 2008, 38, 537–549 CrossRef CAS PubMed.
- M. Strazza, A. Banerjee, A. Alexaki, S. R. Passic, O. Meucci, V. Pirrone, B. Wigdahl and M. R. Nonnemacher, BMC Res. Notes, 2014, 7, 752 CrossRef PubMed.
- S. Heinisch, J. Palma and L. G. Kirby, Brain, Behav., Immun., 2011, 25, 360–372 CrossRef CAS PubMed.
- B. Nash and O. Meucci, Int. Rev. Neurobiol., 2014, 118, 105–128 CrossRef PubMed.
- J. W. Rodríguez, R. Hunter, M. Nair and E. Rios-Olivares, J. Immunol., 2007, 178, LB4 CrossRef PubMed.
- I. Berque-Bestel, F. Lezoualc'h and R. Jockers, Curr. Drug Discovery Technol., 2008, 5, 312–318 CrossRef CAS PubMed.
- M. Decker and J. Lehmann, Curr. Top. Med. Chem., 2007, 7, 347–353 CrossRef CAS PubMed.
- W. S. Messer, Curr. Pharm. Des., 2004, 10, 2015–2020 CrossRef CAS PubMed.
- S. Zhang, A. Yekkirala, Y. Tang and P. S. Portoghese, Bioorg. Med. Chem. Lett., 2009, 19, 6978–6980 CrossRef CAS PubMed.
- Y. Zheng, E. Akgün, K. G. Harikumar, J. Hopson, M. D. Powers, M. M. Lunzer, L. J. Miller and P. S. Portoghese, J. Med. Chem., 2009, 52, 247–258 CrossRef CAS PubMed.
- P. S. Portoghese, G. Ronsisvalle, D. L. Larson, C. B. Yim, L. M. Sayre and A. E. Takemori, Life Sci., 1982, 31, 1283–1286 CrossRef CAS PubMed.
- P. S. Portoghese, D. L. Larson, L. M. Sayre, C. B. Yim, G. Ronsisvalle, S. W. Tam and A. E. Takemori, J. Med. Chem., 1986, 29, 1855–1861 CrossRef CAS PubMed.
- P. S. Portoghese, J. Med. Chem., 2001, 44, 2259–2269 CrossRef CAS PubMed.
- S. D. Comer, M. A. Sullivan and G. K. Hulse, Expert Opin. Invest. Drugs, 2007, 16, 1285–1294 CrossRef CAS PubMed.
- Y. Adi, A. Juarez-Garcia, D. Wang, S. Jowett, E. Frew, E. Day, S. Bayliss, T. Roberts and A. Burls, Health Technol. Assess., 2007, 11(iii–iv), 1–85 Search PubMed.
- B. A. Johansson, M. Berglund and A. Lindgren, Addiction, 2006, 101, 491–503 CrossRef PubMed.
- G. Thoma, M. B. Streiff, J. Kovarik, F. Glickman, T. Wagner, C. Beerli and H.-G. Zerwes, J. Med. Chem., 2008, 51, 7915–7920 CrossRef CAS PubMed.
- L. Toll, I. P. Berzetei-Gurske, W. E. Polgar, S. R. Brandt, I. D. Adapa, L. Rodriguez, R. W. Schwartz, D. Haggart, A. O'Brien, A. White, J. M. Kennedy, K. Craymer, L. Farrington and J. S. Auh, NIDA Res. Monogr., 1998, 178, 440–466 CAS.
- E. Kelly, S. J. Mundell, A. Sava, A. L. Roth, A. Felici, K. Maltby, P. J. Nathan, E. T. Bullmore and G. Henderson, Psychopharmacology, 2015, 232, 305–314 CrossRef CAS PubMed.
- C. K. Arnatt and Y. Zhang, Curr. Top. Med. Chem., 2014, 14, 1606–1618 CrossRef CAS PubMed.
- C. K. Arnatt, B. A. Falls, Y. Yuan, T. J. Raborg, R. R. Masvekar, N. El-Hage, D. E. Selley, A. V. Nicola, P. E. Knapp, K. F. Hauser and Y. Zhang, Bioorg. Med. Chem., 2016, 24, 5969–5987 CrossRef CAS PubMed.
- Y. Yuan, C. K. Arnatt, G. Li, K. M. Haney, D. Ding, J. C. Jacob, D. E. Selley and Y. Zhang, Org. Biomol. Chem., 2012, 10, 2633–2646 RSC.
- Y. Yuan, C. K. Arnatt, N. El-Hage, S. M. Dever, J. C. Jacob, D. E. Selley, K. F. Hauser and Y. Zhang, MedChemComm, 2013, 4, 847–851 RSC.
- N. El-hage, S. M. Dever, E. M. Podhaizer, C. K. Arnatt, Y. Zhang and K. F. Hauser, AIDS, 2013, 27, 2181–2190 CrossRef CAS PubMed.
- D. J. Daniels, A. Kulkarni, Z. Xie, R. G. Bhushan and P. S. Portoghese, J. Med. Chem., 2005, 48, 1713–1716 CrossRef CAS PubMed.
- D. J. Daniels, N. R. Lenard, C. L. Etienne, P.-Y. Law, S. C. Roerig and P. S. Portoghese, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 19208–19213 CrossRef CAS PubMed.
- F. Li, J. Liu, G. S. Jas, J. Zhang, G. Qin, J. Xing, C. Cotes, H. Zhao, X. Wang, L. A. Diaz, Z.-Z. Shi, D. Y. Lee, K. C. P. Li and Z. Li, Bioconjugate Chem., 2010, 21, 270–278 CrossRef CAS PubMed.
- L. M. Sayre and P. S. Portoghese, J. Org. Chem., 1980, 45, 3366–3368 CrossRef CAS.
- J. S. Squire, A. Sutti, G. Durand, X. A. Conlan and L. C. Henderson, New J. Chem., 2013, 37, 1895–1905 RSC.
- C.-F. Yen, J. Yuan and C.-H. Richard King, Lyophilization Formulation, US2010/0120719A1, 2015.
- M. Packiarajan, M. R. Marzabadi, M. Desai, Y. Lu, S. A. Noble, W. C. Wong, V. Jubian, G. Chandrasena, T. D. Wolinsky, H. Zhong, M. W. Walker, O. Wiborg and K. Andersen, Bioorg. Med. Chem. Lett., 2011, 21, 5436–5441 CrossRef CAS PubMed.
- J. E. Hodgkins and M. G. Ettlinger, J. Org. Chem., 1956, 21, 404–405 CrossRef CAS.
- H. Deng, R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. D. Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman and N. R. Landau, Nature, 1996, 381, 661–666 CrossRef CAS PubMed.
- N. R. Landau and D. R. Littman, J. Virol., 1992, 66, 5110–5113 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, including chemical synthesis procedures, biological screening methodologies, and spectral data, are in this section. See DOI: 10.1039/c9md00433e |
| This journal is © The Royal Society of Chemistry 2020 |