
Antiproliferative activities of tricyclic amides derived from β-caryophyllene via the Ritter reaction against MDA-MB-231 breast cancer cells†
XiXi
Xu
,
Ariane
Roseblade
,
Tristan
Rawling
and
Alison T.
Ung
 *
*
School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Broadway, NSW 2007, Australia. E-mail: Alison.Ung@uts.edu.au; Tel: +61 2 9514 1881
First published on 18th December 2019
Abstract
A library of novel tricyclic amides has been synthesised via the Ritter reaction from β-caryophyllene 1 and its monoepoxy derivative 4. The compounds were assessed for antiproliferative activities against the aggressive MDA-MB-231 breast cancer cell line. Of the synthesised compounds, eight were active. 3c and 6b were the most potent and inhibited proliferation with IC50 of 9.7 and 8.2 μM, respectively. Mechanistic studies revealed differences in their antiproliferative actions. 6b inhibited cell cycle progression and induced predominantly apoptotic cell death. In contrast, 3c did not affect cell cycle kinetics and favoured necrotic over apoptotic pathways. Screening against mammalian cells (VERO cells) indicates that 3c and 6b were more active towards MDA-MB-231 cells than noncancerous cells. Facile synthesis and biological results suggest that these caryophyllene derived amides are viable lead compounds for further development.
Introduction
Natural products are a well-established drug source and libraries of compounds developed from them can mimic or even exhibit better drug-like properties.1–4 These newly developed natural product-derived libraries of compounds can provide a new class of lead compounds for contemporary drug discovery.Breast cancer is the most common cancer amongst women and the second most common cancer overall with the majority of cases diagnosed in late stages.5
Breast cancers are commonly grouped into three subtypes that have different treatment strategies and patient outcomes. Hormone positive breast cancers are characterised by high expression of progesterone (PR) and estrogen receptors (ER) that drive tumour cell proliferation, and the development of targeted therapeutics such as the ER antagonist tamoxifen has achieved significant clinical success.6 Similarly, treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancers with targeted agents, for example, the monoclonal anti-HER2 antibody trastuzumab, has substantially improved disease-free and overall survival.6 The third subtype of breast cancer, triple negative breast cancers (TNBCs), lack ER, PR and HER2 expression and therefore do not respond to targeted agents. Non-targeted cytotoxic drugs such as paclitaxel can slow tumour progression; however, dose-limiting toxicities, multidrug resistance and the aggressive and metastatic nature of TNBCs limit therapeutic outcomes.6–8 Thus, there is a critical need to develop new agents to treat TNBCs.9–11
As part of our ongoing research in anti-cancer drug discovery, we use the Ritter reaction as a key reaction for the preparation of tricyclic amides as potential anti-cancer agents.12–15
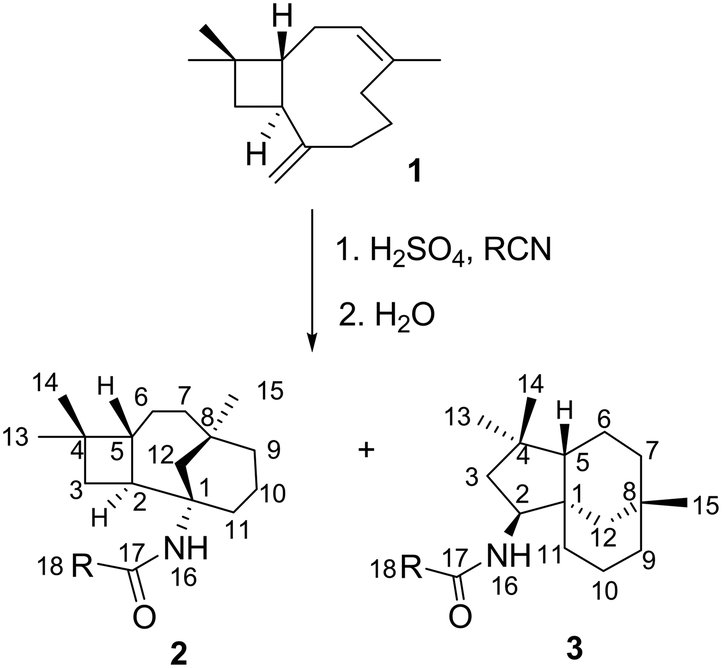
Transformations of β-caryophyllene 1 (Scheme 1) and its isomers, as well as their multistage rearrangements, are well documented via methods of acid-catalysed rearrangements16–18 and the nucleophilic addition of nitrile.19,20 Two major stereospecific tricyclic products were reported; moreover, they have been referred to as caryolane I (skeleton of 2) and clovane II (skeleton of 3), respectively. These core structures were elucidated by NMR spectroscopy and unequivocally confirmed by Robertson et al.21,22 using X-ray structural analysis. The stereochemistry of both structures was also well established.23
 | ||
| Scheme 1 General synthesis of caryophyllene derivatives via the Ritter reaction with various nitriles. | ||
β-Caryophyllene 1 is abundant in the clove tree (Eugenia caryophyllata) and exhibits anti-inflammatory, antibiotic, antioxidant, and anti-cancer activities.24,25 It is readily available commercially and therefore provides cost-effective access to natural product-like compounds that contain unusual trans-fused ring structures. In this study, we report the synthesis of novel tricyclic amides derived from β-caryophyllene 1 and its monoepoxide derivative 4via a single step Ritter reaction. The anticancer effects of these compounds were assessed against the aggressive MDA-MB-231 triple negative breast cancer cell line.
Results and discussion
Ritter reaction with β-caryophyllene 1 and various nitriles
Reactions of β-caryophyllene 1 with various nitriles under strong acidic conditions underwent a stereoselective Ritter reaction to afford optically active tricyclic amides 2a–i (caryolane I skeleton) and 3a–d (clovane II skeleton) (Scheme 1).19 The nitriles were chosen based on their reactivity and ability to provide a variety of substituents (Table 1) at the C-18 position, which can affect the biological activity of the respective compound. The structures of the novel compounds (2b–i and 3b–d) shown in Table 1 were established by 1D and 2D NMR spectroscopy. Their assignments were compared to their respective core structures to the NMR data of caryolane I (2a) and clovane II (3a) that were established in the literature.18–20,22| Entry | R | 2 (%) | 3 (%) |
|---|---|---|---|
| a | CH3 | 36 | 36 |
| b | Et | 55 | 28 |
| c | ClCH2 | 63 | 28 |
| d | Ph | 57 | 19 |
| e | n-But | 58 | |
| f | Cl-But | 59 | |
| g | Cl-Et | 29 | |
| h | H2NCH2 | 75 | |
| i | CH3S | 1 |
Similar chemical shifts and distinct splitting of H-2 and NH resonances proved the structural identities of the newly synthesised compounds (2b–i). It is worth noting that the 1H NMR spectra of all the caryolane (2a–i) skeleton compounds showed characteristic H-16 (δ 5.10 ppm), H-2 and H-11′ (multiplet, δ 2.00–2.50 ppm) resonances. The coupling constants of H-2 and H-11′ could not be measured due to overlapping of H-2 and H-11′ resonances, which was confirmed by HSQC NMR spectroscopy.
Cyclisation and Wagner–Meerwein rearrangements
As mentioned earlier, when β-caryophyllene 1 was reacted under strong acidic conditions, the sum of the products includes the four-membered tricyclic caryolanes 2a–i and the five-membered tricyclic clovanes 3a–d. The caryolanes and clovanes 3 arise from the two major conformations of β-caryophyllene 1, (ββ) and (αα), respectively (Scheme 2). These cyclisation and rearrangements are reversible and lead to the most stable isomers. The product ratio can also depend on the reactivity and the size of the nitriles used.18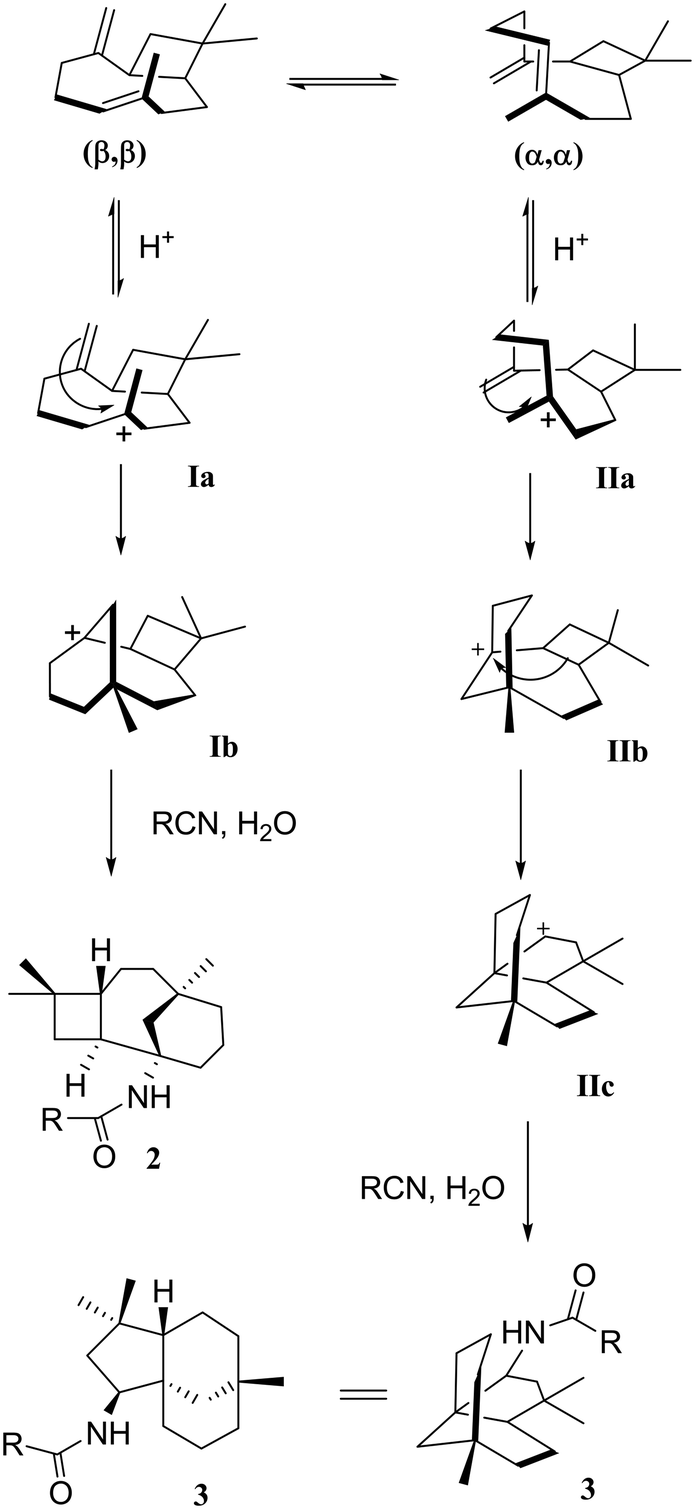 | ||
| Scheme 2 A suggested mechanism for the formation of caryolane 2 and clovane 3 chemical scaffolds. | ||
This selectivity was observed in the product ratios formed from the Ritter reaction of 1, where the caryolane skeleton (2) was the major product and was formed in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relative to the clovane skeleton (3). The observed selectivity can be explained by the reaction mechanism (Scheme 2). The internal bonds are more reactive than their corresponding external bonds because of the higher accumulation of sp2 electrons than that of the external bonds. The clovane skeleton 3 was formed via cyclisation on the lower face of the molecule after the expansion of the four-membered ring. Since the formation of the clovane skeleton 3 required an extra expansion step, it was, therefore, the kinetically unfavoured of the two skeletons.18,19
:1 relative to the clovane skeleton (3). The observed selectivity can be explained by the reaction mechanism (Scheme 2). The internal bonds are more reactive than their corresponding external bonds because of the higher accumulation of sp2 electrons than that of the external bonds. The clovane skeleton 3 was formed via cyclisation on the lower face of the molecule after the expansion of the four-membered ring. Since the formation of the clovane skeleton 3 required an extra expansion step, it was, therefore, the kinetically unfavoured of the two skeletons.18,19
Ritter reaction with β-caryophyllene monoepoxide 4 and various nitriles
The monoepoxide of caryophyllene 4 was prepared from 1 using a reported method.26 The Ritter reactions of 4 and various nitriles yielded alcohol–amide 5 and diamide structure 6, as shown in Scheme 3.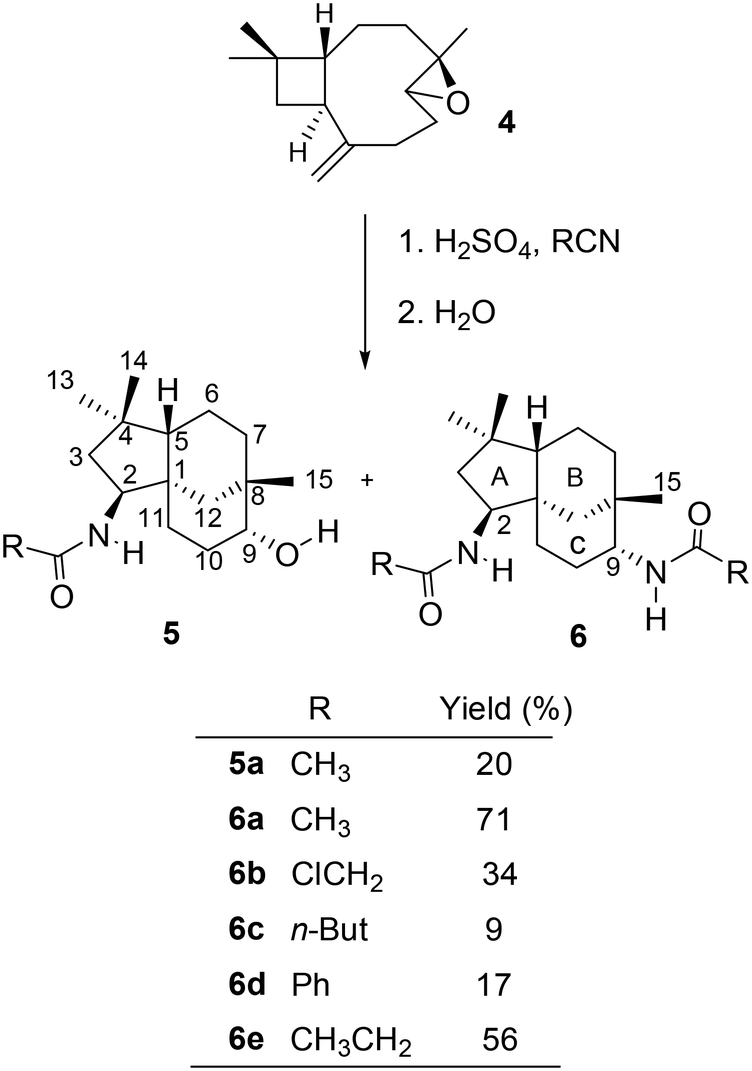 | ||
| Scheme 3 General synthesis of monoepoxide derivatives via the Ritter reaction with various nitriles. | ||
When the reactions were carried out for 30 minutes at 0 °C, it was found that only diamides 6a–e were formed. The ratio between the formation of compounds 5 and 6 is dependent on the duration of the reaction in an acidic medium as well as the size and nucleophilicity of the nitriles. Under strong acidic conditions, the hydroxy group in 5a would readily react to generate the carbocation at C-9, resulting in the formation of diamides 6b–e as the only isolated products (Scheme 3). In an attempt to favour the formation of alcohol–amide structure 5, reaction conditions such as time (1 min – 4 h and 24 h) and temperature (0 °C for 30 min then at room temperature) were varied. By using very short reaction times (4 minutes), we were able to isolate 5a in low yields. However, 4 minutes of reaction with other nitriles failed to yield other derivatives of 5.
The structures of the synthesised compounds 5a and 6a–e were confirmed by 1D and 2D-NMR experiments; their assignments were based on the spectral data from their corresponding mono-amides 3a–d. The stereochemistry of 5a was established by Nisnevich et al. on the reaction of 4 with CH3CN under the Ritter reaction.27 This was further supported by Pinto et al. when 4 reacted with CH3CN in the presence of BiBr3 under the Ritter reaction.28 In both cases, N-acylamino clovane-type compound 5a was obtained. The 1H NMR spectra for compounds 5a and 6a are analogous to each other, except H-9 resonances. The vicinal coupling constant for H-9 (doublet, δ 3.60 ppm, J = 9.00 Hz) for compound 6a indicates that it is in the equatorial conformation as the C ring exists in a chair conformation. Similar coupling constants were observed for all diamide compounds 6a–e. A 2D NOESY experiment was carried out for compound 6a to ascertain the relative stereochemistry at C-9. There were no cross-peaks observed between H-9 and H-15 protons which supported the (R)-configuration at C-9. The lowest-energy conformation of 6a was calculated using Spartan 10 molecular modelling software (WAVEFUNCTION, INC) (Fig. 1). The calculations were carried out using the setup menu in Spartan, with the specification of equilibrium conformation for ground states (Hartree–Fock 3-21G model at the AM1 level). The molecular models of 6a showed the distance between H-9 and H-5 to be 4.42 Å, which confirms the absence of their NOE interaction in 2D NOESY NMR.
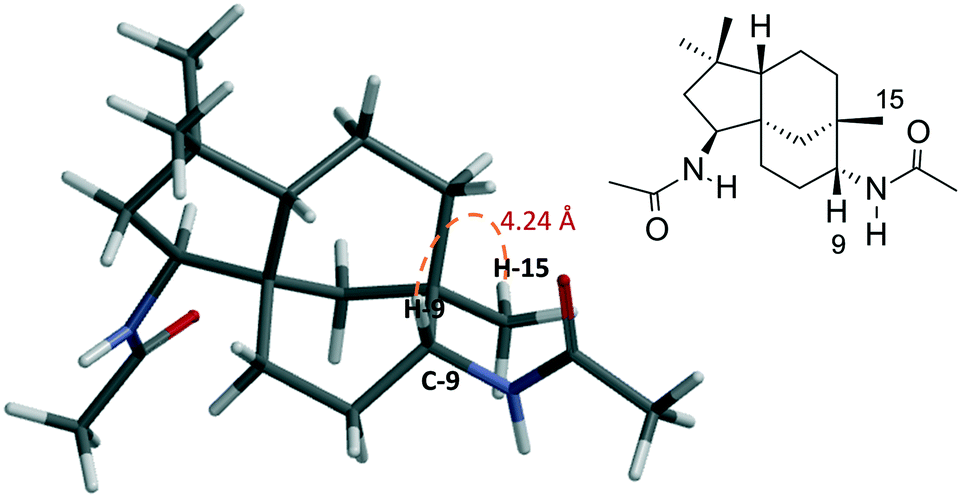 | ||
| Fig. 1 Molecular model of compound 6a (Hartree–Fock 3-21G model at the AM1 level) showing the distance (4.24 Å) between H-9 and H-15 protons. | ||
The relative stereochemistries at C-2 and C-9 are supported by the accessible sites of the carbocation intermediates 4b and 4c. The formation of 4b resulted from the trans-diaxial ring-opening of compound 4, as shown in Scheme 4. The addition of RCN to the most accessible face of 4b led to the formation of 5. Acid-catalysed dehydration of 5 gave 4c which further reacted with the second mole of RCN to give 6.
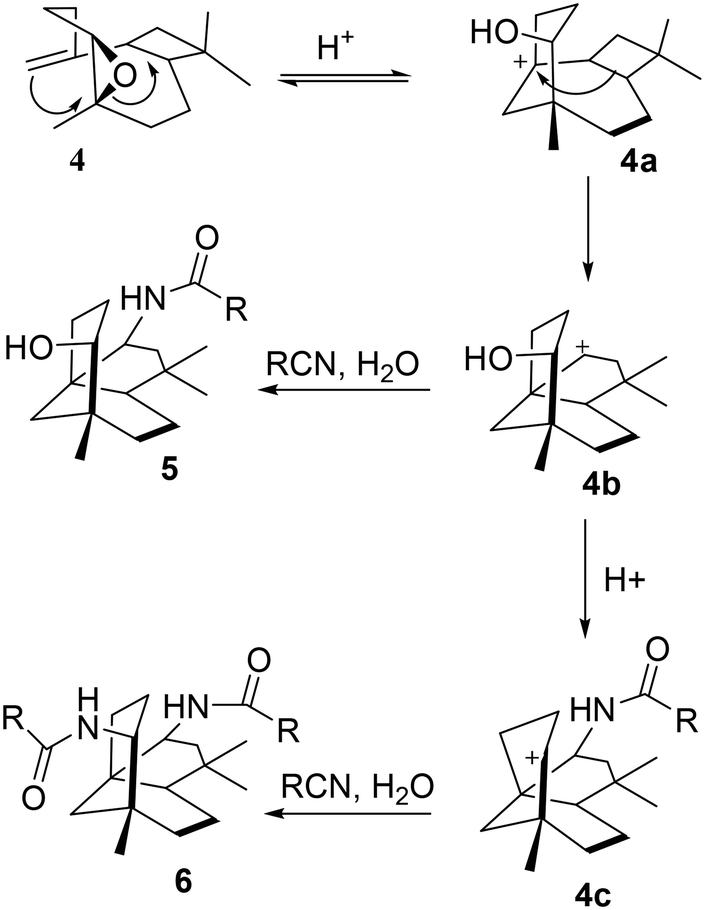 | ||
| Scheme 4 A suggested mechanism for the stereoselective formation of 5 and 6. | ||
Amide cleavage under mild acidic conditions
The chloroacetyl (ClCH2CO) group was readily cleaved from 2c, 3c and 6b under mild conditions with thiourea and acetic acid to afford primary amines 7, 8 and 9 in reasonable yields, 58, 65, and 33%, respectively (Scheme 5).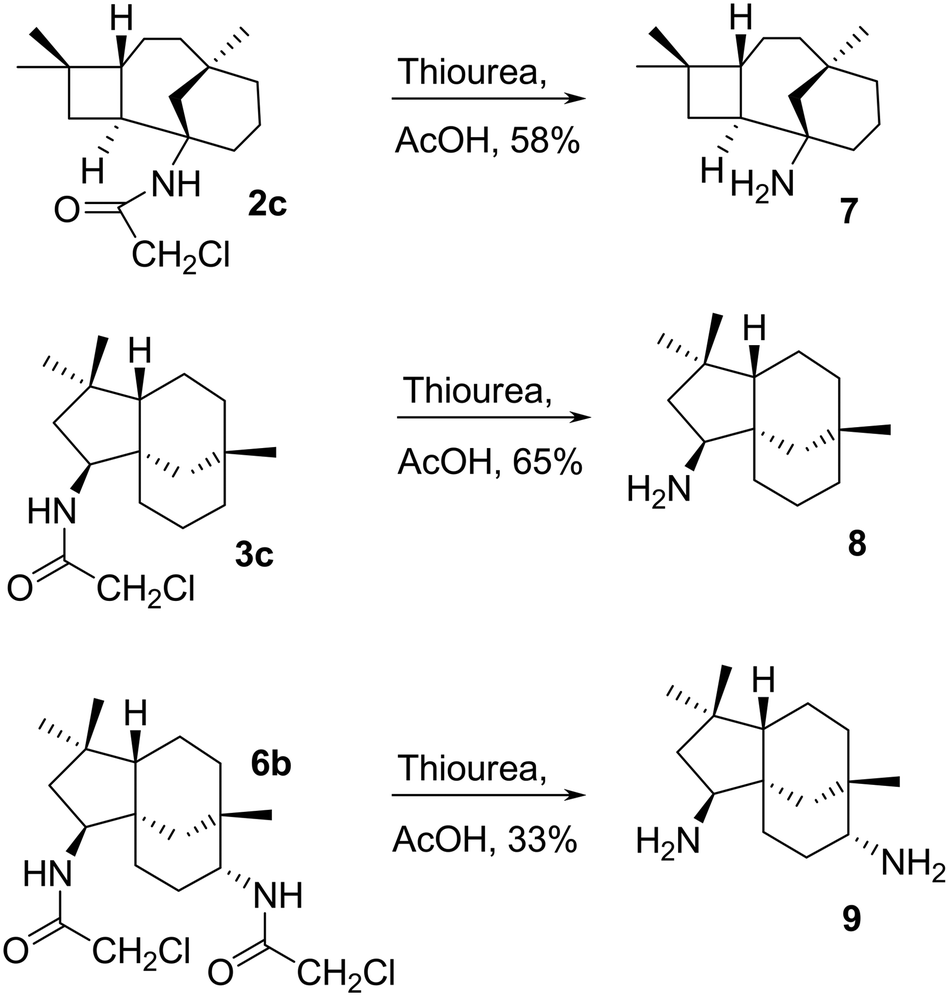 | ||
| Scheme 5 Amide cleavage of 2c, 3c and 6b under mild conditions to afford primary amines 7, 8 and 9. | ||
Amines 7 and 8 were further derivatised via reductive alkylation with various aldehydes, as shown in Scheme 6. Di-alkylated products 10a and 11a–11b were obtained (Scheme 6). The least hindered formaldehyde appeared to alkylate faster due to less steric hindrance, while the bulky butanal required a longer reaction time for the formation of di-alkylated products. The mono-alkylated product was observed in the mixture of the di-alkylated product. However, this could not be controlled by either stoichiometry or reaction time. Furthermore, it was challenging to isolate the mono-alkylated product from the di-alkylated product. Hence, it was decided to ultimately synthesise di-alkylated products. The yields of 10c and 11b highlight the significance of steric hindrance of the substituents at the C-1 position in compound 7 in comparison to that at the C-9 position of compound 8.
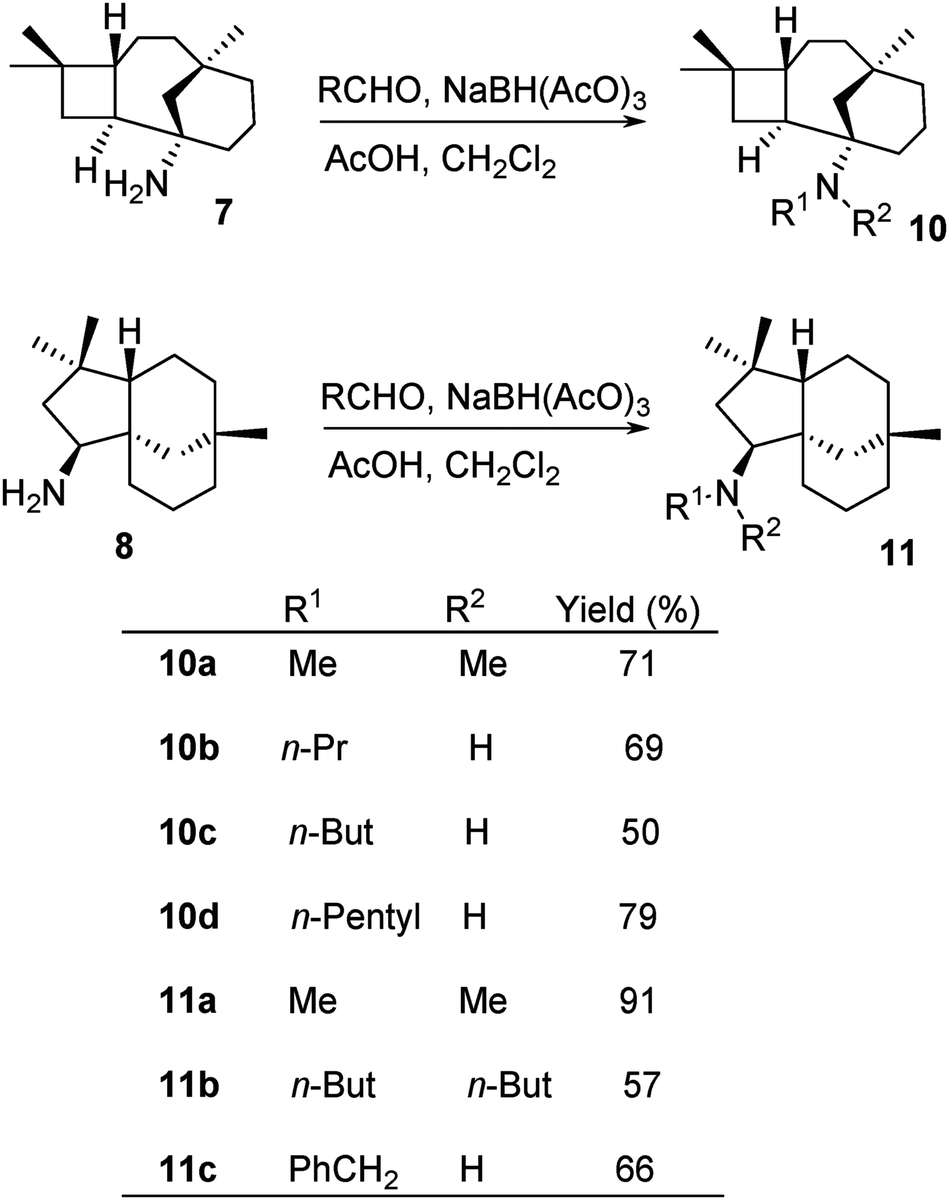 | ||
| Scheme 6 Reductive alkylation of 7 and 8. | ||
Biological evaluation
Antiproliferative activity in breast Cancer
The synthesised compounds were screened for anticancer activity against the aggressive triple negative MDA-MB-231 breast cancer cell line using the MTS cell viability assay (Fig. 2). Eight (2c, 2e, 3c, 6b, 7, 10b, 10c, and 11c) of the twenty-nine compounds were identified as active and significantly reduced cell viability at the concentrations tested (25 and 50 μM, 48 h). Compound 10d also appeared to reduce viability at 50 μM; however, the results were highly variable and not statistically significant. The variability was attributed to its poor aqueous solubility, and therefore, this compound was not selected for further evaluation. The activity of 2f could not be determined at either test concentration due to its poor solubility.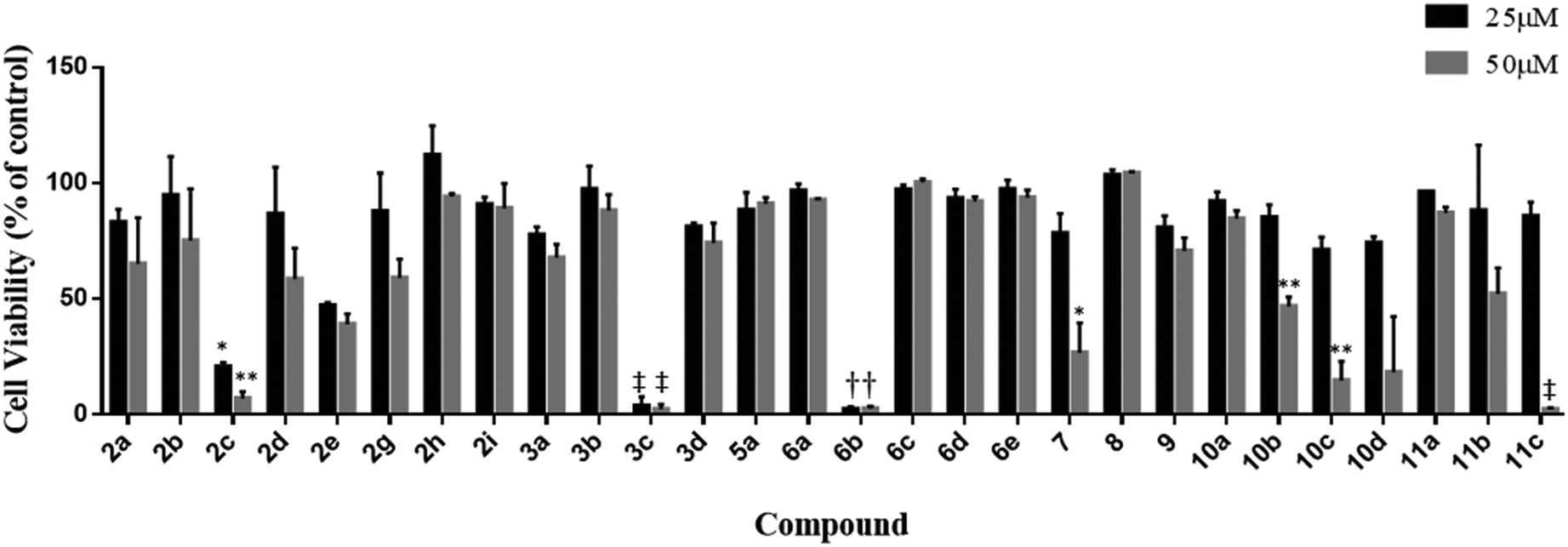 | ||
| Fig. 2 Effects of caryophyllene and monoepoxide derivatives on the viability of MDA-MB-231 breast cancer cells (48 hours, 25 and 50 μM). All data are the mean ± SEM from three separate experiments. Different from the DMSO-treated control: (*) p ≤ 0.05, (**) p ≤ 0.01, (†) p ≤ 0.001, (‡) p ≤ 0.0001. | ||
In follow up assays, the cytotoxicity of the active compounds was assessed over a wide concentration range, and the data were used to calculate IC50 values (Table 2). Compounds 2c, 2e, 7, 10b, 10c, and 11c displayed a moderate activity, with IC50 values above 25 μM. 3c and 6b were the most potent of the series and reduced cell viability with IC50 values in the low micromolar range. The dose–response curves for these compounds are shown in Fig. 3. 3c and 6b both contain an α-chloro amide group (ClCH3CONH), which indicates that the presence of this functional group improves the antiproliferative activity. Although this group has the potential to act as a reactive toxophore, we note that compound 2c, which also contains an α-chloro amide group, was significantly less active (IC50 = 21.4 ± 1.9 μM). This suggests that 2c, 3c, and 6b do not share a common mechanism of action that involves non-specific reactivity. This is further supported by the differences in the primary mechanisms of action for 3c and 6b (see the following section). Indeed, we have prepared alkaloid-like anticancer agents with this group that are selectively toxic towards cancer cells over non-cancerous cells.15
| Compound | IC50 (μM) |
|---|---|
| 2c | 21.4 ± 1.9 |
| 3c | 9.7 ± 2.6 |
| 2e | 55.3 ± 12.5 |
| 7 | 34.9 ± 2.2 |
| 10b | 45.9 ± 5.0 |
| 10c | 37.3 ± 1.1 |
| 11c | 23.6 ± 0.5 |
| 6b | 8.2 ± 0.5 |
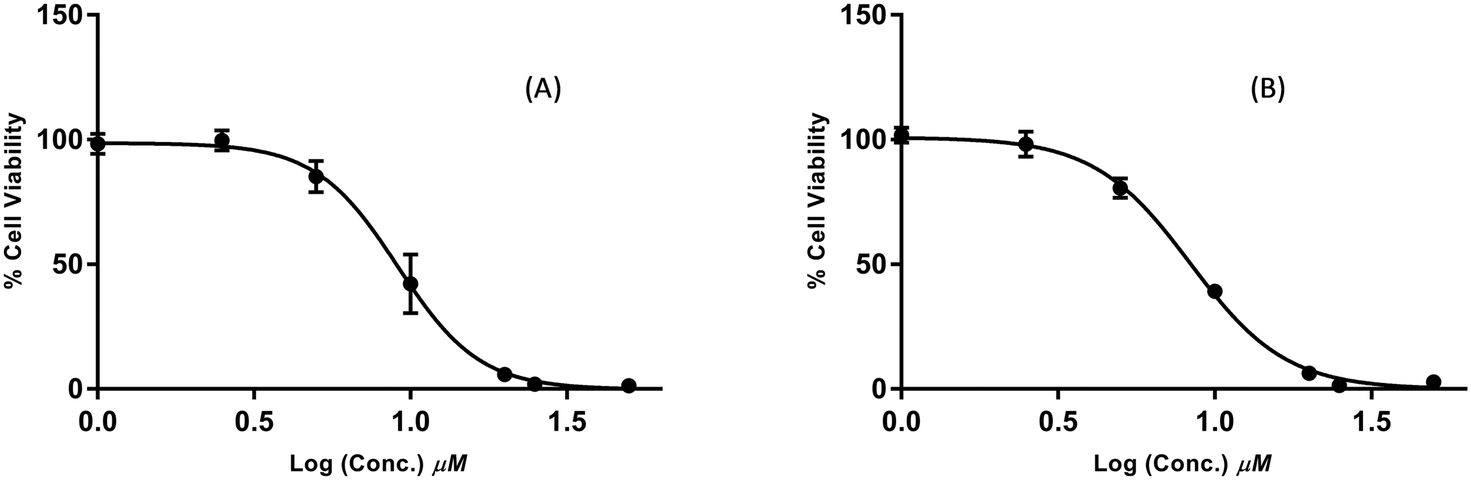 | ||
| Fig. 3 Dose–response curves of samples 3c (A) and 6b (B) on the proliferation of MDA-MB-231 breast cancer cells. | ||
Cytotoxicity of the active compounds against VERO cells
The selectivity of 3c and 6b towards cancer cells was assessed by determining the cytotoxicity of these compounds against the non-cancerous mammalian VERO cell line (African green monkey kidney cells).293c and 6b were found to exhibit cytotoxicity towards the VERO cells at 168 μM and 129 μM, respectively. In these assays, 3c and 6b reduced the VERO cell viability to 89 and 88% of the control, respectively. 3c and 6b reduced the MDA-MB-231 cell viability to similar extents, but at much lower concentrations (∼5 μM). These data also demonstrate that 3c and 6b are much more cytotoxic against MDA-MB-231 than the noncancerous mammalian cell line. We estimate 3c and 6b to be approximately 33 and 25-fold more toxic toward MDA-MB-231 cells than the non-cancerous cell line, respectively.Primary mechanism of action studies
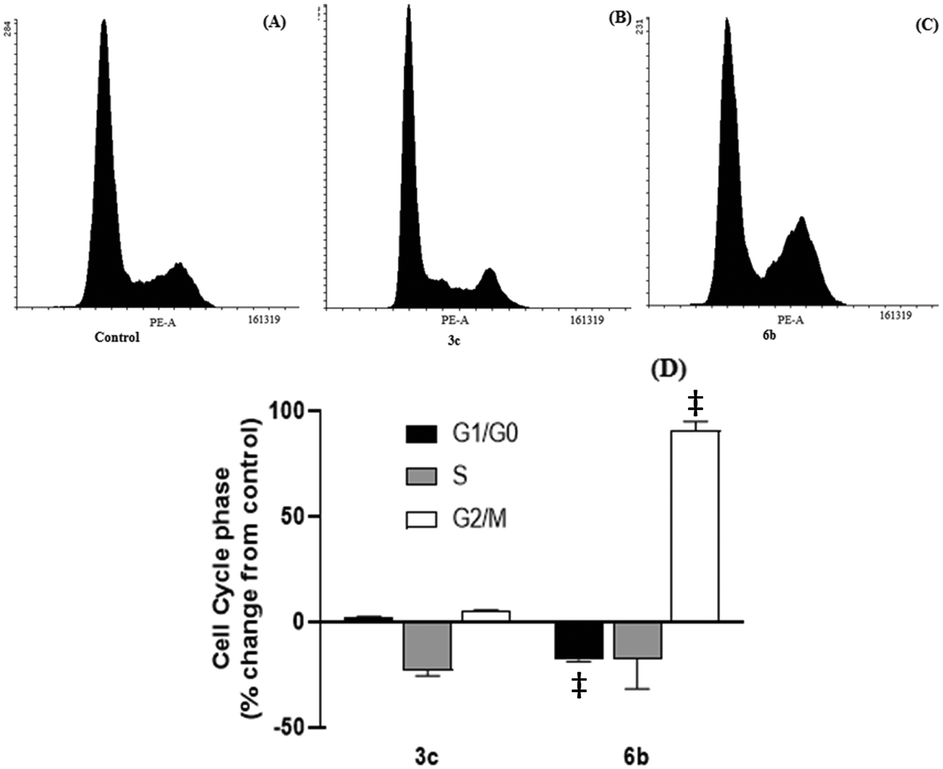 | ||
| Fig. 4 Effect of 3c and 6b on the cell cycle progression of the MDA-MB-231 cells. Cells were treated for 24 h with (A) DMSO (control), (B) 3c (10 μM), and (C) 6b (9 μM). Panel (D) shows the percentage change from the control. All data are the mean ± SEM from three separate experiments. Different from the DMSO-treated control: (‡) p < 0.0001. | ||
After 48 hour treatments, 3c and 6b (20 μM) decreased the population of unstained (live) cells relative to the control and increased the number of dual stained cells, consistent with extensive cell death (Fig. 5). The extent of cell death observed following 6b treatment was consistent with its IC50 determined from MTS assays. In the case of 3c, the population of live cells was greater than anticipated from the MTS data. This discrepancy may reflect the different endpoints measured in these assays. Both compounds increased the number of cells stained with either Annexin V or PI, indicating early apoptosis and necrosis, respectively, but to varying extents. 3c appears to kill cells predominantly through necrosis, as evidenced by intense PI staining. In contrast, higher Annexin V staining was observed in 6b-treated cells, indicating that 6b preferentially elicits apoptotic cell death.
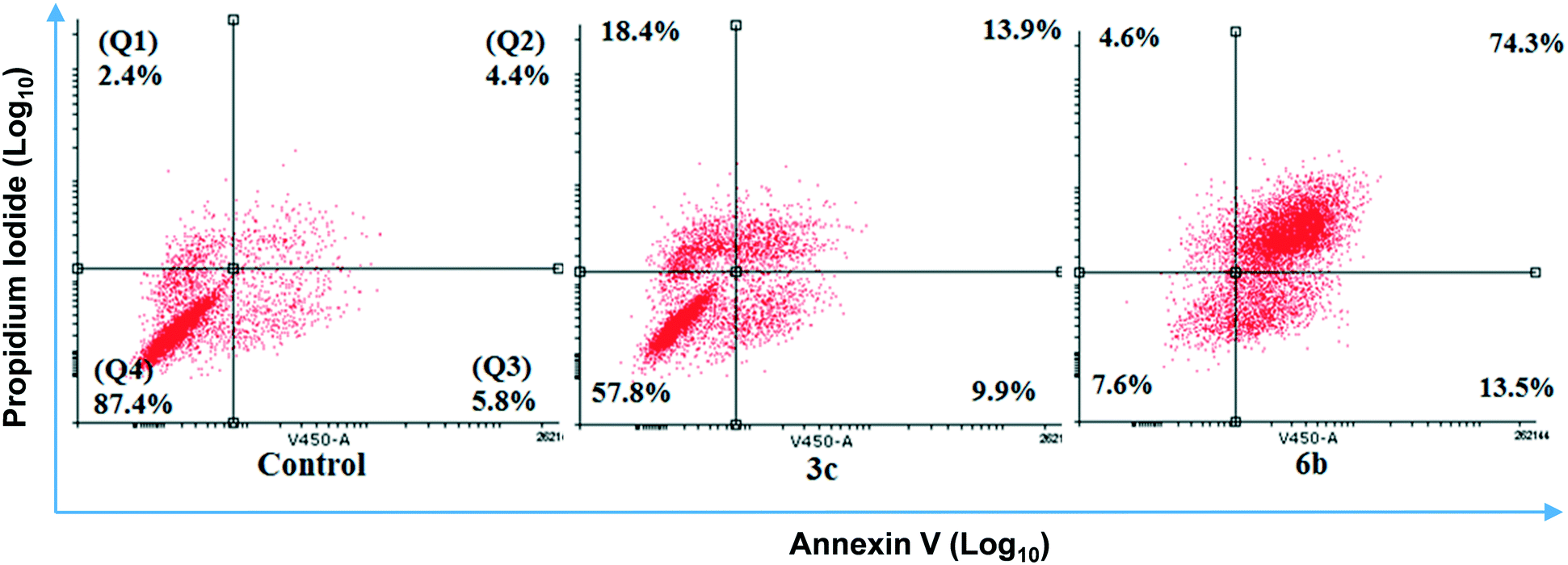 | ||
| Fig. 5 Effect of compounds 3c and 6b on the induction of apoptosis in MDA-MB-231 cells. Cells were treated with 20 μM 3c and 6b for 48 h and stained with Annexin V-FITC and PI, and the apoptotic effect was assessed by flow cytometry. Representative results are shown, with quadrants indicating the proportion of necrotic: Q1, late apoptotic: Q2, early apoptotic: Q3, and healthy cells: Q4. The images are representative of three independent experiments. | ||
Conclusions
A library of 29 tricyclic amides was successfully synthesised from β-caryophyllene 1 and its monoepoxide 4via the Ritter reaction. Eight active analogues were identified that reduced the viability of MDA-MB-231 cells. The most potent compounds in the series, 3c and 6b, inhibited proliferation with IC50 values of 9.7 and 8.2 μM, respectively, and mechanistic studies revealed differences in their antiproliferative actions. 6b inhibited cell cycle progression and induced predominantly apoptotic cell death. In contrast, 3c had no effect on cell cycle kinetics and favoured necrotic over apoptotic pathways.Screening against mammalian cells (VERO cells) indicates that the compounds were more active towards cancerous cells than noncancerous cells. In summary, the Ritter reaction was successfully used as a viable method for generating a chemically diverse library of β-caryophyllene-derived compounds that exhibit promising anticancer activities towards the MDA-MB-231 TBNC cell line.
Experimental section
General chemistry methods, synthetic procedures and analyses of compounds plus representative NMR spectra are available in the ESI.† Cell biology methods such as general reagents for cell culture, cell viability, flow cytometry assays, apoptosis/necrosis, cytotoxicity against VERO cells and statistical analysis can also be found in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We like to thank the University of Technology of Sydney for supporting this study. The Human MDA-MB-231 breast cancer cells were obtained as a gift from Prof. Michael Murray (University of Sydney).References
- V. Amirkia and M. Heinrich, Phytochem. Lett., 2014, 10, xlviii–liii CrossRef CAS.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
- G. M. Cragg and J. M. Pezzuto, Med. Princ. Pract., 2016, 25, 41–59 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- W. H. Organisation, World Cancer Report 2014, International Agency for Research on Cancer, France, 2014, pp. 362–369 Search PubMed.
- G. W. Sledge, E. P. Mamounas, G. N. Hortobagyi, H. J. Burstein, P. J. Goodwin and A. C. Wolff, J. Clin. Oncol., 2014, 32, 1979–1986 CrossRef CAS PubMed.
- Cancer in Australia 2016, ed. A. I. O. H. Welfare, Australian Institute of Health and Welfare, Canberra, 2016 Search PubMed.
- L. M. Heiser, A. Sadanandam, W.-L. Kuo, S. C. Benz, T. C. Goldstein, S. Ng, W. J. Gibb, N. J. Wang, S. Ziyad, F. Tong, N. Bayani, Z. Hu, J. I. Billig, A. Dueregger, S. Lewis, L. Jakkula, J. E. Korkola, S. Durinck, F. Pepin, Y. Guan, E. Purdom, P. Neuvial, H. Bengtsson, K. W. Wood, P. G. Smith, L. T. Vassilev, B. T. Hennessy, J. Greshock, K. E. Bachman, M. A. Hardwicke, J. W. Park, L. J. Marton, D. M. Wolf, E. A. Collisson, R. M. Neve, G. B. Mills, T. P. Speed, H. S. Feiler, R. F. Wooster, D. Haussler, J. M. Stuart, J. W. Gray and P. T. Spellman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2724–2729 CrossRef CAS PubMed.
- J. P. Gillet and M. M. Gottesman, Methods Mol. Biol., 2010, 596, 47–76 CrossRef CAS.
- B. C. Baguley, Mol. Biotechnol., 2010, 46, 308–316 CrossRef CAS PubMed.
- J. Stagg and B. Allard, Ther. Adv. Med. Oncol., 2013, 5, 169–181 CrossRef CAS PubMed.
- A. T. Ung, S. G. Williams, A. Angeloski, J. Ashmore, U. Kuzhiumparambil, M. Bhadbhade and R. Bishop, Monatsh. Chem., 2014, 145, 983–992 CrossRef CAS.
- A. T. Ung, A. N. West, M. J. A. Phillips and S. G. Williams, Monatsh. Chem., 2016, 10, 1737–1746 CrossRef.
- S. G. Williams, M. Bhadbhade, R. Bishop and A. T. Ung, Tetrahedron, 2017, 73, 116–128 CrossRef CAS.
- X. Xu, T. Rawling, A. Roseblade, R. Bishop and A. T. Ung, MedChemComm, 2017, 8, 2105–2114 RSC.
- G. G. Henderson, R. O. O. McCrone and J. M. Robertson, J. Chem. Soc., 1929, 1368–1372 RSC.
- (a) A. Aebi, D. H. R. Barton, A. W. Burgstahler and A. S. Lindsey, J. Chem. Soc., 1954, 4659–4665 RSC; (b) D. H. R. Barton and A. Nickon, J. Chem. Soc., 1954, 4665–4669 RSC.
- I. G. Collado, J. R. Hanson and A. J. Macias-Sanchez, Nat. Prod. Rep., 1998, 15, 187–204 RSC.
- O. Yarovaya, D. Korchagina, T. Rybalova, Y. Gatilov, M. Polovinka and V. Barkhash, Russ. J. Org. Chem., 2004, 40, 1593–1598 CrossRef CAS.
- (a) V. V. Fomenko, D. V. Korchagina, N. F. Salakhutdinov and V. A. Barkhash, Helv. Chim. Acta, 2001, 84, 3477–3487 CrossRef CAS; (b) R. J. Phipps, L. McMurray, S. Ritter, H. A. Duong and M. J. Gaunt, J. Am. Chem. Soc., 2012, 134, 10773–10776 CrossRef CAS PubMed.
- J. M. Robertson and G. Todd, J. Chem. Soc., 1955, 1254–1263 RSC.
- K. Gollnick, G. Schade, A. F. Cameron, C. Hannaway and J. M. Robertson, J. Chem. Soc. D, 1971, 46–46 RSC.
- A. Horeau and J. K. Sutherland, J. Chem. Soc. C, 1966, 247–248 RSC.
- E. J. Corey, R. B. Mitra and H. Uda, J. Am. Chem. Soc., 1965, 86, 485–492 CrossRef.
- J. Legault and A. Pichette, J. Pharm. Pharmacol., 2007, 59, 1643–1647 CrossRef CAS PubMed.
- G. I. Bombarda, E. M. Gaydou, J. Smadja and R. Faure, Bull. Soc. Chim. Fr., 1995, 132, 836–842 Search PubMed.
- G. Nisnevich, D. Korchagina, V. Makalskii, Z. V. Dubovenko and V. Barkhash, Zh. Org. Khim., 1993, 29, 524–541 CAS.
- R. M. A. Pinto, J. A. R. Salvador and C. Le Roux, Synlett, 2006, 2047–2050 CAS.
- Bioassay Laboratory, National Center for Genetic Engineering and Biotechnology (BIOTEC), 113 Thailand Science Park, Phahonyothin Road, Khlong Nueng, Khlong Luang, Pathum Thani 12120 Thailand, http://www.biotec.or.th/bioassay/index.php/bioassay-service, (accessed on 10/03/ 2017) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c9md00237e |
| This journal is © The Royal Society of Chemistry 2020 |