
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme degradable star polymethacrylate/silica hybrid inks for 3D printing of tissue scaffolds†
Anna
Li Volsi
 ab,
Francesca
Tallia
b,
Haffsah
Iqbal
b,
Theoni K.
Georgiou
*b and
Julian R.
Jones
*b
ab,
Francesca
Tallia
b,
Haffsah
Iqbal
b,
Theoni K.
Georgiou
*b and
Julian R.
Jones
*b
aDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
bDepartment of Materials, Imperial College London, London, SW7 2AZ, UK. E-mail: julian.r.jones@imperial.ac.uk; t.georgiou@imperial.ac.uk
First published on 20th November 2020
Abstract
There is unmet clinical need for scaffolds that can share load with the host tissue while biodegrading under the action of enzymes present at the site of implantation. The aim here was to create the first enzyme cleavable inorganic–organic hybrid “inks” that can be 3D printed as scaffolds for bone regeneration. Inorganic–organic hybrids are co-networks of inorganic and organic components. Although previous hybrids performed well under cyclic loads, there was little control over their degradation. Here we synthesised new hybrids able to degrade in response to endogenous tissue specific metallo proteinases (collagenases) that are involved in natural remodeling of bone. Three well-defined star polymers, of the monomer 3-(trimethoxysilyl)propyl methacrylate (TMSPMA) and of methyl methacrylate (MMA), of different architectures were prepared by RAFT polymerisation. The linear arms were connected together at an enzyme degradable core using a collagenase cleavable peptide sequence (GLY-PRO-LEU-GLY-PRO-LYS) modified with dimethacryloyl groups as a crosslinker for RAFT polymerisation. The effect of polymer architecture, i.e. the position of the TMSPMA groups on the polymers, on bonding between networks, mechanical properties, biodegradation rate and 3D printability, via direct ink writing, was investigated for the first time and was proven to be critical for all three properties. Specifically, hybrids made with star polymers with the TMSPMA close to the core exhibited the best mechanical properties, improved printability and a higher degradation rate.
Introduction
In the field of regenerative medicine, scaffolds are temporary templates with interconnected 3D pore architectures designed to promote cell migration and tissue ingrowth.1 There is an unmet clinical need for scaffolds that can share load with the host tissue while biodegrading under the action of enzymes present at the site of implantation. A hypothesis is that enzyme degradation, particularly by enzymes naturally responsible for tissue remodelling, will enable a more controlled and natural biodegradation profile compared to other degradation mechanisms, such as hydrolysis of synthetic polyesters, which are used as sutures. An example of unmet clinical need is bone regeneration, where bone graft scaffolds tend to be porous bioactive ceramics, such as synthetic hydroxyapatite (sHA),2 or putties containing bioactive ceramic particles such as sHA or bioactive glass.3While porous bioceramic scaffolds can be biodegradable, stimulate bone regeneration4–7 and can have suitable compressive strength,8 especially if 3D printed,9,10 they are too brittle and too stiff for regeneration of sites that are under cyclic loads. To overcome the brittle properties, conventional composites of bioactive glass particles and biodegradable polymer were produced.11 The most common polymers used are poly(lactide), poly(glycolic acid), poly(ε-caprolactone) and their copolymers.12 There has been limited clinical translation of conventional polymer composites implant devices, where inorganic particles are dispersed in a polymer matrix. Reasons for this are that the polymer covers the majority of the bioactive particles, masking their bioactivity and the polyester matrices degrade by autocatalytic hydrolysis, resulting in a dramatic loss of mechanical strength once degradation initiates.13
Inorganic/organic hybrid materials were developed to overcome the issue of composites not attaining true synergy of their component materials.14,15 Sol–gel hybrids consist of co-networks of inorganic and organic components and are often produced by introducing a polymer into the sol–gel process.13 The most common sol–gel hybrids have silica as the inorganic network, which is synthesised through hydrolysis of an alkoxide precursor such as tetraethylorthosilicate (TEOS) and the subsequent gelation of the silicate network through polycondensation of Si–OH bonds.16,17 Novak introduced “classes” of sol–gel hybrids.17 In Class I hybrids, the inorganic and organic co-networks interact through molecular entanglements only, while Class II hybrids also have covalent bonds between the networks by introducing a functionalised polymer into the sol–gel synthesis. The covalent bonding is critical for scaffold production because if it is not present, water will penetrate and cause rapid dissolution (separation) of the co-networks.18
Previously, Class II hybrids were produced by functionalizing natural polymers, such as gelatin19,20 and poly(γ-glutamic acid),18,21–23 using a coupling agent (3-glycidyloxypropyl)trimethoxysilane (GPTMS). The epoxy ring in the GPTMS was opened by nucleophilic attack of the –COOH groups on the polymers,24–26 under suitable conditions. However, batches of natural polymers are not reproducible and GPTMS reactions are difficult to control.26 Use of synthetic polymers can improve reproducibility, but the majority of publications on hybrids with synthetic polyesters have not reported suitable mechanical properties.12,27,28
One hybrid that did reach highly elasticity, including the ability to sustain cyclic load, combined with high compressive strength was the silica/polytetrahydrofuran/polycaprolactone hybrid,29 synthesised by in situ cationic ring-opening polymerisation (CROP) of tetrahydrofuran (THF) within the sol–gel process (Class IV hybrid). The hybrid sol was also 3D printed into a 3D grid-like scaffold. However, CROP is not a living polymerisation under these conditions, hence it does not allow control over the final molecular weight of PTHF; and the resulting tri-component hybrid had a slow degradation rate, which depended on the hydrolytic scission of PCL. New, designed-for-purpose synthetic polymers are needed that can deliver controlled degradation and mechanical strength to the hybrid, requiring a finely-controlled polymerisation method.
Chung et al. synthesised copolymers of methyl methacrylate (MMA) and 3-(trimethoxysilyl) propyl methacrylate (TMSPMA), crosslinked either with EGDMA or with disulphide based dimethacrylate (DMSDMA), to obtain different polymer architectures (e.g. branched or star copolymers).30–32 The TMSPMA provided bonding to the silicate network though Si–O–Si bond formation by condensation. While mechanical properties were promising, including the ability to take cyclic load, they were either not biodegradable or were only degradable by a reductive environment (by exposure to glutathione).
Here, we make a significant step change in controlled biodegradability by introducing enzymatic degradation to these polymers and hybrids. The aim was to create the first enzyme cleavable silica/polymer hybrid that can potentially be printed into a scaffold morphology and to investigate how the polymer architecture, including the detail of star polymer architecture, affects the physiochemical properties of the hybrids.
The most prominent advancements in polymer science have arisen from developments of controlled polymerisation techniques, e.g. reversible addition fragmentation chain transfer (RAFT).33–35 This method has allowed researchers to prepare materials with controlled molecular weight distributions, versatility in terms of block architecture, and functional end-groups, tuning physiochemical properties and as consequence their biological properties.36
Here, the new hybrids were designed to degrade in response to endogenous tissue specific metalloproteases (MMPs 1, 8, 13) that are involved in natural remodelling of bone.37 Three well-defined star polymers were prepared by RAFT polymerisation, using the monomers TMSPMA and MMA. The role of the TMSPMA is to form covalent bonds with the silicate network. This is the first time an enzyme degradable peptide core has been used in sol–gel hybrids. The effect of the star polymer architecture, i.e. the position of the TMSPMA groups on the star (AB block based versus BA based versus statistical star polymer), was investigated and this is the first study where the effect of star polymer architecture has been investigated in polymer/silica hybrids. The linear arms were connected via a collagenase sensitive peptide-based cross linker GLY-PRO-LEU-GLY-PRO-LYS chemically modified with methacryloyl chloride to introduce methacryloyl groups that act as cross-linkers via controlled radical polymerisation methods. An example of changes in architecture is the position of the TMSPMA groups relative to the peptide core. Arms of the stars were 90 mol% MMA and 10 mol% TMSPMA, but the TMSPMA could be anywhere along the arm, i.e. statistically distributed along the arm poly(MMA-stat-TMSPMA)-star or as a block at the outer end of the arm poly(TMSPMA-b-MMA)-star, or as a block next to the inner core poly(MMA-b-TMSPMA)-star (Fig. 1).
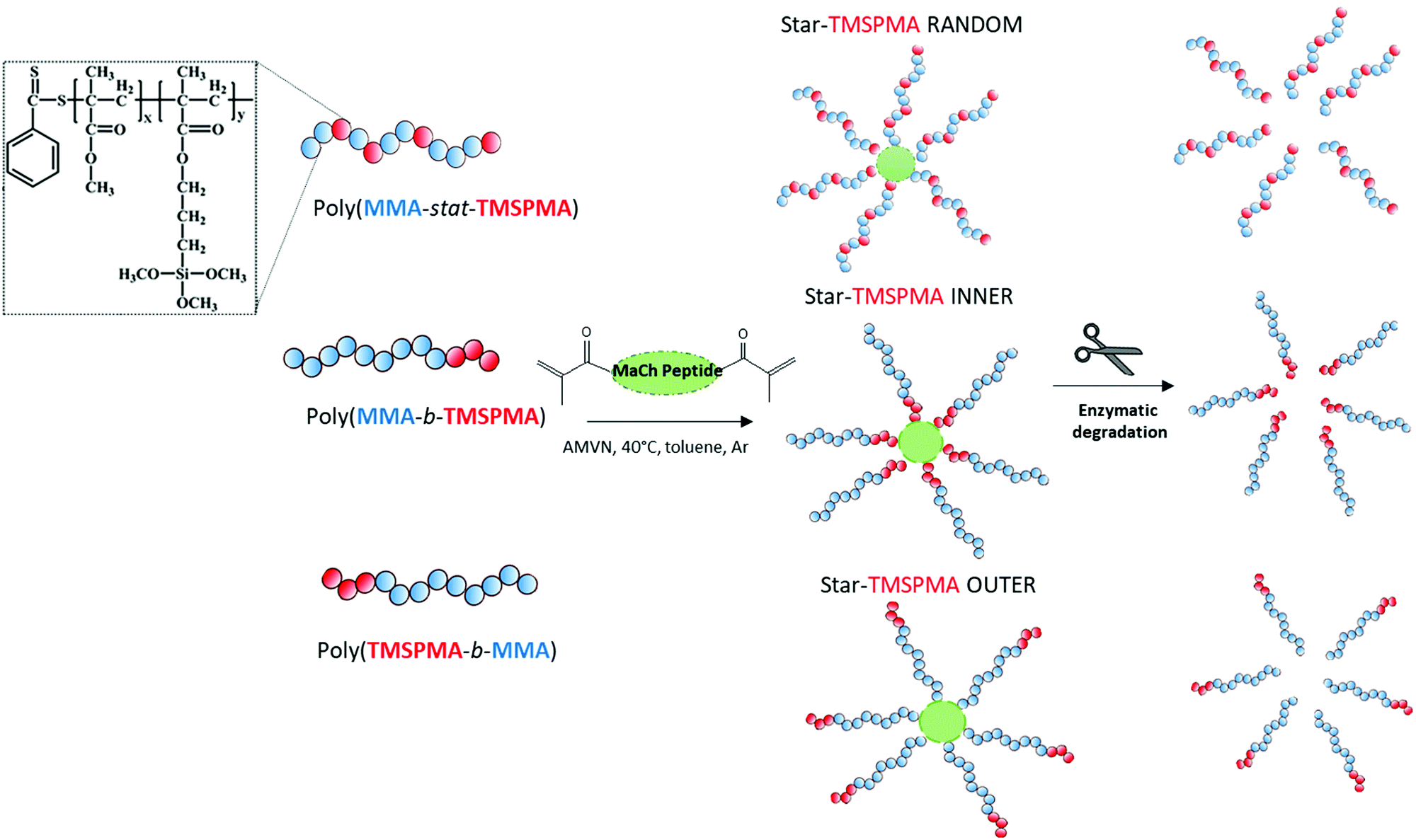 | ||
| Fig. 1 Schematic illustration of the poly(MMA-TMSPMA) star polymers synthesised with arms of three different architectures (random, inner and outer) cross-linked by MaCh-peptide core and cleavage by collagenase activity. | ||
Experimental section
Methyl methacrylate (MMA, 99%), 3-(trimethoxysilyl)propyl methacrylate (TMSPMA, 98%), 2-phenyl-2-propyl benzodithioate (CDB, 99%), 2,2-azobis(2-methylpropionitrile) (AIBN, initiator, 98%), methacryloyl chloride (>97%), triethylamine (TEA, 99%) 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH, free radical inhibitor, 99%), calcium hydride (CaH2, 95%), silica gel (60 Å, 70–230 mesh), basic alumina (Al2O3, 95%), n-hexane (95%), toluene (polymerisation solvent, 99%), tetrahydrofuran (THF, analytical and HPLC grade, 99.9%), deuterated chloroform (CDCl3, 99.8%), tetraethyl orthosilicate (TEOS, 98%), and hydrochloric acid solution (1 M HCl) were purchased from Sigma-Aldrich. GLY-PRO-LEU-GLY-PRO-LYS (peptide, >85%) was purchased from GenScript USA Inc. 2,2′-Azobis (4-methoxy-2,4-dimethylvaleronitrile) (AMVN, initiator) was purchased from Cayman Chemical, USA. Collagenase type I from Clostridium Hystocolicum was purchased from Alfa Aesar. Prior to polymerisation, MMA was passed through basic alumina columns to remove inhibitors and acidic impurities (TMSPMA was passed through neutral alumina due to its alkoxysilane group hydrolyzing). Then, they were stirred over CaH2 for 1 h to neutralize traces of moisture in the presence of DPPH. Finally, all the monomers were vacuum distilled prior to the polymerisation. AIBN and AMVN were recrystallised in ethanol. Toluene was dried in the presence of silica gel, which was heated up to 250 °C for 4 h prior to use. All the glassware was dried overnight at 120 °C and assembled hot under dynamic vacuum before use.Synthesis of dimethacryloyl peptide (MaCh-pep)
200 mg of peptide (0.352 mmol), Scheme S1 (ESI†), was dissolved in 500 μL of anhydrous DMSO and then diluted with anhydrous DCM (3 mL). Freshly distilled TEA (400 μL, 1.41 mmol) from CaH2 was added. Then, the solution was cooled to 0 °C using a water/ice-bath and methacryloyl chloride was added dropwise (200 μL, 2.11 mmol) (Scheme 1). The resulting mixture was heated up to 25 °C and then stirred for 24 h. Then the mixture was filtered to remove the triethyl ammonium salt. The resulting solution was purified through a basic alumina column to remove the unreacted methacryloyl chloride and then TEA was removed using a rotary evaporator. Then the product was precipitated in diethyl ether and further purified by washing 3 times in ether/dichloromethane 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v.
:1 v/v.
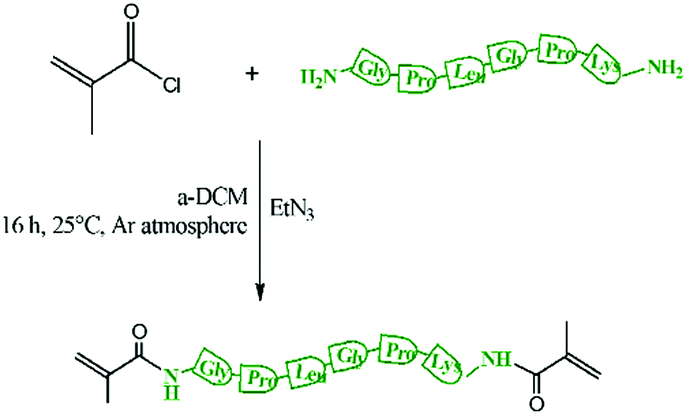 | ||
| Scheme 1 Synthesis of dimethacryolyl peptide (MaCh-peptide). | ||
Synthesis of poly(MMA-b-TMSPMA), poly(TMSPMA-b-MMA) and poly(MMA-stat-TMSPMA) linear arms
The linear arms, or the macro RAFT, of the star polymers were one of linear poly(MMA100-b-TMSPMA10), poly(TMSPMA10-b-MMA100) or poly(MMA100-co-TMSPMA10). Each type of arm was a copolymer with the same composition and similar molar mass (13 kg mol−1 < Mw < 15 kg mol−1) but different architecture in terms of TMSPMA position within the polymer chain (Fig. 1). For the synthesis of poly(MMA-b-TMSPMA), a Schlenk tube was loaded with AIBN (0.13 mmol, 0.02 g), CDB (0.25 mmol, 0.06 mL), MMA (0.06 mol, 6.50 mL), TMSPMA (6.72 mmol, 1.60 mL) with toluene as a solvent (20 wt%). The components of the Schlenk tube were subsequently degassed three times by freeze–vacuum–thaw cycle under argon atmosphere and were heated in an oil bath to 70 °C to initiate the polymerisation. The polymerisation was terminated after 6 h, corresponding to 50% conversion. After the polyMMA was synthesised, it was purified by precipitation in n-hexane and washed three times in the same solvent. Then a Schlenk tube was loaded with 2.5 g of dry polyMMA100 (0.25 mol), TMSPMA (6.72 mmol, 1.60 mL), (AIBN (0.13 mmol, 0.02 g)) and toluene as solvent (20 wt%). The components of the Schlenk tube were subsequently degassed three times by freeze–vacuum–thaw cycle under argon atmosphere, and were heated in an oil bath to 70 °C to initiate the polymerisation. The polymerisation was terminated after 2.5 h (50% conversion). After the linear macro RAFT poly(MMA-b-TMSPMA) was synthesised, it was purified by precipitation in n-hexane and washed with hexane and ethanol. For the synthesis of poly(TMSPMA-b-MMA), the same procedure and composition were used except from the order of the addition of monomers. The statistical linear macro RAFT poly(MMA-co-TMSPMA) was synthesised using the same composition as the previously synthesised linear polymers. Thus, AIBN (0.13 mmol, 0.02 g), CDB (0.25 mmol, 0.06 mL), MMA (0.06 mol, 6.50 mL), and TMSPMA (6.00 mmol, 1.45 mL) were used with toluene as a solvent. After the statistical macro RAFT was synthesised, similarly to the previous macro-RAFT agents, it was purified by precipitation in n-hexane.Star copolymers syntheses
Three star copolymers (Fig. 2) were synthesised by RAFT using the “arm-first” approach method. The arms, i.e. the linear macro RAFT, of the star polymers were of the same composition (90 mol% MMA and 10 mol% TMSPMA) and similar molar mass (13 g mol−1 < Mw < 15 g mol−1 as determined from GPC characterisation, Fig. 1) but different architecture (TMSPMA position). Thus, after the linear arms poly(MMA-b-TMSPMA), poly(TMSPMA-b-MMA) and poly(MMA-stat-TMSPMA), macro RAFT, were synthesised and purified, they were dissolved again in dry toluene in a Schlenk tube and AMVN (radical initiator) and the MaCh-peptide cross-linker were added. The molar ratios used were Macro RAFT/MACh-peptide/AMVN 8:1:0.3. The contents were degassed three times by the freeze–vacuum–thaw cycle under argon atmosphere and were heated in an oil bath to 40 °C. The polymerisation was kept for 24 h to obtain three star copolymers with different architecture. Specifically, the TMSPMA was either in the inner core of the star (using poly(MMA-b-TMSPMA), “inner”), in the outer part (using poly(TMSPMA-b-MMA), “outer”) or randomly distributed along the arms (using poly(MMA-co-TMSPMA), “random”), Fig. 1. Then, the polymer was precipitated in n-hexane and washed in ethanol. The precipitation was repeated 3 times for unreacted arms to be removed.
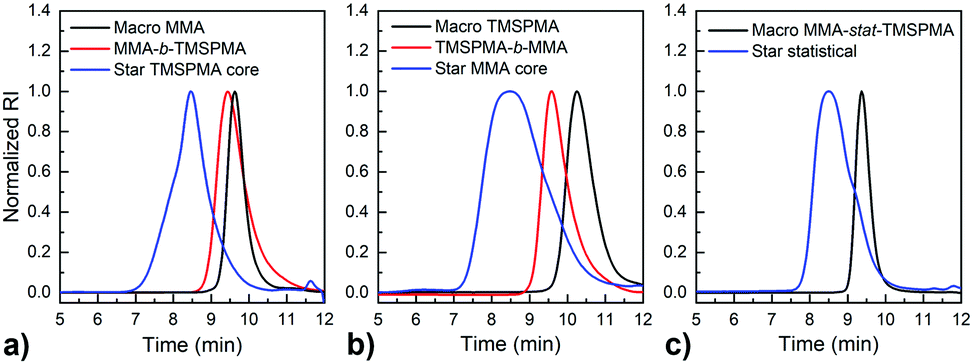 | ||
| Fig. 2 GPC traces of the three linear MMA–TMSPMA copolymers (used for the arms of the star polymers) compared to homopolymers and star homo-polymers crosslinked with dimethacryloyl peptide (blue): (a) poly(MMA-b-TMSPMA) compared to linear macro RAFT MMA (black) and TSPMA star; (b) poly(TMSPMA-b-MMA) compared to linear macro RAFT TMSPMA (black) and MMA star; (c) poly(MMA-stat-TMSPMA) compared to a poly(MMA-stat-TMSPMA) star polymer (blue). | ||
Polymer characterisation
The average molar masses (MMs) and dispersities (Đs) for all the macro RAFT, star polymers, and cleaved polymer fragments from degradation studies were determined by gel permeation chromatography (GPC). An Agilent, SECurity GPC system, with an Agilent Mixed D (PL gel 5 μm) column was used. All the copolymers were dissolved in THF and were filtered through 0.45 μm PTFE syringe filters. GPC eluent was THF, which was pumped with a flow rate of 1 mL min−1 by ‘1260 Iso’ isocratic pump. An Agilent 1260 RID detector was used to measure the refractive index signal. The calibration curve was based on PMMA standards with MMs of 2, 4, 8, 20, 50, 100 kg mol−1. Polymer structure and composition and the peptide structure were determined using proton nuclear magnetic resonance (1H NMR) spectroscopy. This was performed in deuterated CDCl3 using a 400 MHz Advance Bruker NMR spectrometer.Star polymer core cleavage by collagenase
For biodegradation studies of the polymers, the following degradation buffer was prepared: Collagenase Type I was dissolved (1 mg mL−1) in TRIS HCl buffer 0.1 M pH 7.4, which also contained CaCl2 5 mM (degradation buffer). 500 mg of poly(MMA-b-TMSPMA)-star, poly(TMSPMA-b-MMA)-star and poly(MMA-stat-TMSPMA)-star copolymers were placed in the solution. Degradation was carried out at 37 °C in orbital shaker. After 4 days, the copolymers were washed with distilled water, freeze-dried and dissolved in THF for gel permeation chromatography analysis.Hybrid syntheses
The three star copolymers were dissolved in THF (1 g mL−1). In a different pot, TEOS was hydrolyzed in the molar ratio of TEOS:water:HCl 1 M of 1:4:0.01. The amount of TEOS was added as such so the overall wt% of the hybrid to have 70 wt% organic and 30 wt% inorganic. After 1 h, when the TEOS was fully hydrolysed, the star copolymer solution was added to the hydrolysed TEOS and stirred for 10 minutes at room temperature. The mixture was then poured into a polytetrafluoroethylene (PTFE) cylindrical mold (∅ = 15 mm; height = 25 mm), and then it was sealed within a Nalgene polymethylpentene (PMP) container. The container was placed in 40 °C oven to age sealed for 3 days and then dried by slow opening of the lid for approximately 3 weeks.
Hybrid characterisation
The functional groups of the hybrids were examined by Fourier transform infrared spectroscopy (FTIR, NicoletiS10, Thermo Scientific) with an attenuated total reflectance module. 32 scans were averaged to yield 4 cm−1 resolution. The mechanical properties of the hybrid cylinders were investigated by uniaxial compression test to failure, using cylinders with height: diameter >1, with diameters of ∼10 mm and heights of 11–16 mm. The sample ends were ground with a sandpaper until they were flat and parallel. Compression testing was performed using Zwick/Roell instrument equipped with Zwick/Roell “testXpert II” software and 10 kN load cell set in displacement control with a compression speed of 0.1 mm min−1. Force (N) and displacement (mm) were measured during the test and corresponding stress and strain values were calculated from the sample dimensions as force/area (MPa) and displacement/initial height, respectively. Modulus of toughness was calculated by measuring the area underneath the compression stress–strain curve (where εc is unitless) using Origin Software (OriginLab Corporation, USA). Young's modulus was calculated as the slope of the tangent to the compression stress–strain curve (where εc is unitless) in the elastic region using Origin Software (OriginLab Corporation, USA). Yield point of the compression stress–strain curve was calculated as intercept between the tangent to the elastic region and the initial part of the plastic region (n = 3 for each hybrid material). Statistical analysis was performed using a one-way analysis of variance (ANOVA). A posthoc Tukey test with statistical significance evaluated at p < 0.05 was used as a comparison of means. Thermogravimetric analysis (TGA) was performed with NETZSCH STA 449C to confirm the organic content of the hybrid materials, equal to burnt-out mass percentage during the heating process. The hybrid samples were manually ground to a fine powder and were placed in a platinum crucible. The samples were heated up to 800 °C at 10 °C min−1 in continuously flowing air.Degradation studies of hybrids
For the evaluation of hybrid biodegradability, accurately weighed hybrids in bulk form (Mi) were introduced in the degradation buffer (Collagenase Type I dissolved (1 mg mL−1) in TRIS HCl buffer 0.1 M pH 7.4, with CaCl2 5 mM), and placed in 37 °C orbital shaker for two months. At each time point, the recovered hybrid samples were washed with distilled water and dried in the vacuum-oven. The weight was then measured and denoted Mf. The mass loss % was then calculated from the following equation.After two months, the polymer chains present in the medium were detected by GPC and 1H NMR spectroscopy as described previously.
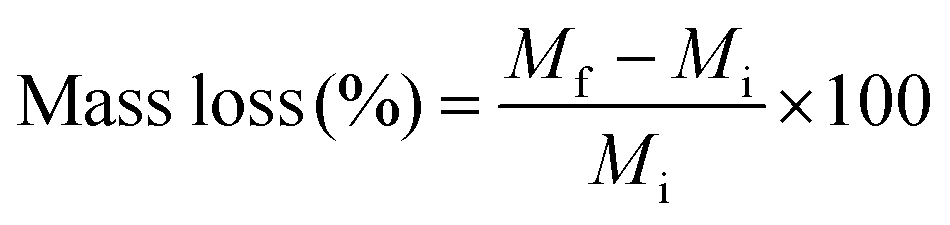
3D Extrusion printing
The hybrid sol of composition 70 wt% polymer 30 wt% silica was used for direct 3D extrusion printing without additional carriers or binders, following a similar protocol previously described by Tallia et al.29 The hybrid sol obtained by combining hydrolysed TEOS and polymer solution was stirred until a suitable viscosity was reached that allowed transfer into a 3 mL Luer-lock plastic syringe (VWR International, UK), equipped with a tapered tip for 3D printing (Nordson EFD, UK) with 200 μm nozzle diameter. The time required for gelation depended on the star polymer architecture, as discussed in the Results section. The syringe was then placed into a Direct Ink Writing machine (“Robocaster”, 3d Inks LLC, USA), connected to a computer equipped with the software “Robocad” (3d Inks LLC, USA), which controlled the printing of porous scaffolds following a pre-defined CAD file. The printing window, defined as the window of time after introduction of the polymer to the silica sol that the “ink” is printable, was determined through printing trials. If the ink was printed before a critical viscosity is reached, the structure collapsed, and printing too late means the ink gelled inside the syringe or nozzle. The three different hybrid inks were printed into 3D grid-like porous scaffolds, following an orthogonal lattice with aligned channels as previously described.29 The following printing parameters were used: conical nozzle with internal diameter of 200 μm, printing speed of 10 mm s−1, z-spacing (i.e. vertical displacement of the tip when moved to the next layer) of 0.22 mm, strut spacing (i.e. distance between the axis of adjacent rods in the same layer) of 0.60 mm. Cubic scaffolds with side dimension of 10.8 mm and height resulting from stacking of 20 layers were printed and, while still wet, were placed in PMP containers at 40 °C for ageing sealed for 3 days and drying over 7–10 days with gradual opening of the lid.SEM characterization
Scanning electron microscopy (SEM) analysis of the 3D printed scaffolds was carried out using a JEOL 6010LA, with an accelerating voltage of 20 kV; a working distance of 16 mm; and a spot size of 45 mm. Prior to imaging, samples were coated with a layer of gold in order to make them conductive. SEM images of the top surface of the scaffolds were analysed with ImageJ software to evaluate vertical channel size and strut size on the x–y plane (n ≥ 40)Results and discussion
Polymer characterisation
MMPs are key enzymes that regulate extracellular matrix remodelling. Here, the peptide sequence GLY-PRO-LEU-GLY-PRO-LYS, which is cleavable by MMP-1, 8 and 13 (collagenases), was used as enzyme cleavable core of three different star polymers. MMP-1, 8 and 13 were chosen as they are expressed by bone marrow cells (MSCs) and by osteoclasts29 and are strongly expressed in damaged bone.37 The bis-amino ended collagenase sensitive peptide sequence was chemically modified using a threefold molar excess of methacryloyl chloride respect to the end-amino groups of the peptide in presence of TEA (Scheme 1). The functionalisation with methacryloyl groups enabled the cross-linking of the peptide to our linear polymer arms via a controlled radical polymerisation method. After purification, the obtained dimethacryloyl peptide (MaCh-peptide) was characterised by 1H NMR, which confirmed the reaction occurred and the quantitative derivatisation of amine groups, by comparing the integrals of the peak at δ 5.5 and 6.0, ascribable to the vinyl protons of methacryloyl chloride, with the integral of the peaks related to protons at δ 0.8–0.85 assignable to the methyl (CH3)2 protons of Leucyine (Fig. S2 for further details, ESI†). The degree of derivatisation mol% in methyl methacrylate (DDMaCh%) was equal to 93.3 mol%. Three well-defined star polymers with arms of TMSPMA and MMA copolymers linked by peptide-based cross linker (MaCh-peptide) as an enzyme degradable core arms were synthesised. Three different arm architectures were successfully synthesised by RAFT polymerization.38 Three copolymers were used to make the stars with 90 mol% MMA and 10 mol% TMSPMA: one was a statistical star ((poly(MMA-stat-TMSPMA)-star), named TMSPMA random-star); two stars were made with block co-polmyer arms, poly(MMA-b-TMSPMA)-star and poly(TMSPMA-b-MMA)-star, named TMSPMA inner-star and TMSPMA outer-star respectively (Scheme 1). 1H-NMR analysis was used to characterize the structure and composition of the macro RAFT linear arms and the star polymers (Fig. S1, ESI†). The comparison of the peaks at 3.6 ppm (–O–CH3 group, a & b) and 0.67 ppm (Si–CH2–, c) confirmed that the molar ratios for all the copolymers were close to our targeted value (9 mol% of TMSPMA).GPC traces of the three linear arms and respective purified star polymers are shown in Fig. 2. The GPC traces show successful syntheses of the block copolymers poly(MMA-b-TMSPMA) and poly(TMSPMA-b-MMA), with no extra peaks present in the block copolymers traces (Fig. 2a and b), and overall a successful crosslinking of the linear arms using the collagenase sensitive dimethacryloyl peptide to obtain three star polymers with a peptide-based core (blue curve, at lower elution time) and different architecture. However, the shoulder-shaped peaks indicate that some linear macro RAFT were still present, not having yet interlinked to produce a star polymer. This was expected for the “arm-first” approach to produce star polymers after purification by sequential precipitations and washes.32Table 1 shows the molar masses (MMs) and the dispersity index (Đs) of all the copolymers. The aim was for all to have similar MMs. The Mns of all the linear arms were very similar (between 10 and 12 kg mol−1), and had satisfactorily narrow Đs ranging from 1.12 to 1.3. The Mns of the stars, following the crosslinking of the arms to the peptide core, were also similar (38.8 to 39.7 kg mol−1). To confirm that the star polymers can biodegrade by collagenase activity, the copolymers were introduced in a buffer containing collagenase Type I. The enzyme was hypothesised to cleave the star polymers down to smaller linear polymer fragments as schematically illustrated in Fig. 1, since collagenases recognize the sequence -R-Pro-/-X-Gly-Pro-R-, where X is most often a neutral amino acid.38Table 2 shows that the MMs of the poly(MMA-TMSPMA)-star copolymers decreased significantly after 4 days of incubation with collagenase Type I. Mn and Mw values of the three star polymers decreased by more than 2-fold, obtaining values near to the initial MM values of the linear arms, a size that can potentially excreted by kidney filtration, being lower than 30 kg mol−1 target MM for kidney filtration.
| Polymer name | M w (kg mol−1) | M n (kg mol−1) | Đ | TMPSMA molar% (from NMR) |
|---|---|---|---|---|
| poly(MMA-stat-TMSPMA) | 13.6 | 12.4 | 1.12 | 9 |
| poly(MMA-stat-TMSPMA)-star | 38.8 | 27.2 | 1.41 | 9 |
| poly(MMA-b-TMSPMA) | 15.0 | 11.5 | 1.30 | 9 |
| poly(MMA-b-TMSPMA)-star | 39.7 | 26.0 | 1.53 | 9 |
| poly(TMSPMA-b-MMA) | 13.8 | 10.4 | 1.33 | 9 |
| poly(TMSPMA-b-MMA)-star | 38.8 | 23.9 | 1.62 | 9 |
| Polymer name | M w (kg mol−1) | M n (kg mol−1) | Đ |
|---|---|---|---|
| Cleaved poly(MMA-stat-TMSPMA)-star | 15.8 | 9.22 | 1.71 |
| Cleaved poly(MMA-b-TMSPMA)-star | 14.5 | 8.65 | 1.68 |
| Cleaved poly(TMSPMA-b-MMA)-star | 14.1 | 7.78 | 1.81 |
GPC traces of the copolymers and their cleaved fragments are shown in Fig. 3. In each case, the cleaved stars displayed a significant shift of the peak to almost match that of linear arm. However, there was an unexpected trace in the lower elution time region (5–7 min) for the diblock arms. This was possibly due to the condensed alkoxysilane groups from TMSPMA in the block copolymers, since the polymer was exposed to water for 4 days, or could be attributed to the polymer linear arms connecting to the same double bond of the crosslinker core, as previously reported by Patrickios's group,39–41 Themistou's group42 and Chung et al.30
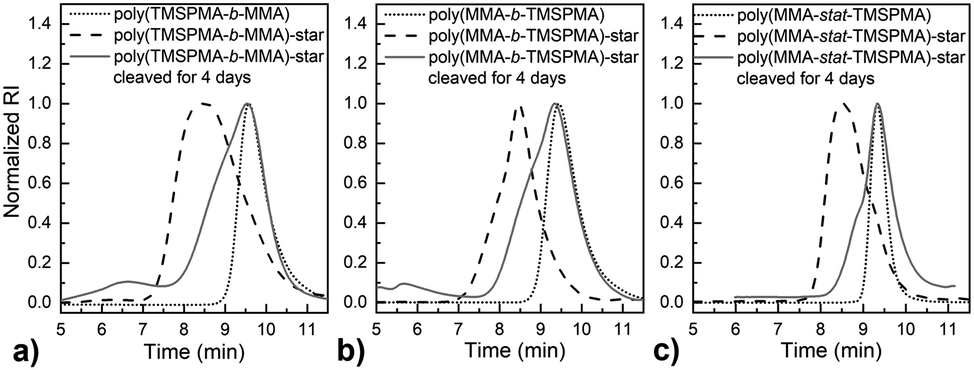 | ||
| Fig. 3 GPC traces of the star polymers of poly(MMA-TMSPMA) crosslinked by dimethacryloyl peptide, with the three arm architectures before (dash) and after (solid grey) peptide cleavage by exposure to collagenase for 4 days: (a) polyTMSPMA-b-MMA; (b) polyMMA-b-TMSPMA; and (c) polyMMA-stat-TMSPMA. The GPC traces for the linear arms (macro RAFT) prior to making stars are shown in dotted lines. | ||
Hybrid characterisation
Hybrids with the three enzyme cleavable star polymers with different architecture were successfully synthesised. Fig. 4a shows that crack-free transparent inorganic–organic Class II hybrid cylinders were fabricated. The transparency is an indication that co-networks formed well. The Class II hybrids were made with organic:inorganic ratio of 70:30 wt%, because from a previous study it was shown to be the only composition that allowed for the fabrication of tall crack-free monolith samples. Hybrids shrink during drying and here, the cylinders shrank approximately homogeneously by ∼65–80%, maintaining their cylindrical shape (Fig. 4a), with the hybrids made with arms of the “outer” architecture shrinking more than the others (∼76%), due to them having more sites for forming bonds with the silicate network (random was ∼65% and inner ∼68%). Thermal analysis was conducted to confirm the final inorganic:organic ratio (Fig. 4b), as the burn-out mass corresponds to the organic component in the hybrid material. All TGA traces of the hybrids confirmed that the composition of the hybrids was close to our targeted organic:inorganic composition of 70:30 wt% and showed thermal decomposition of the organic component starting at ≈342 °C.
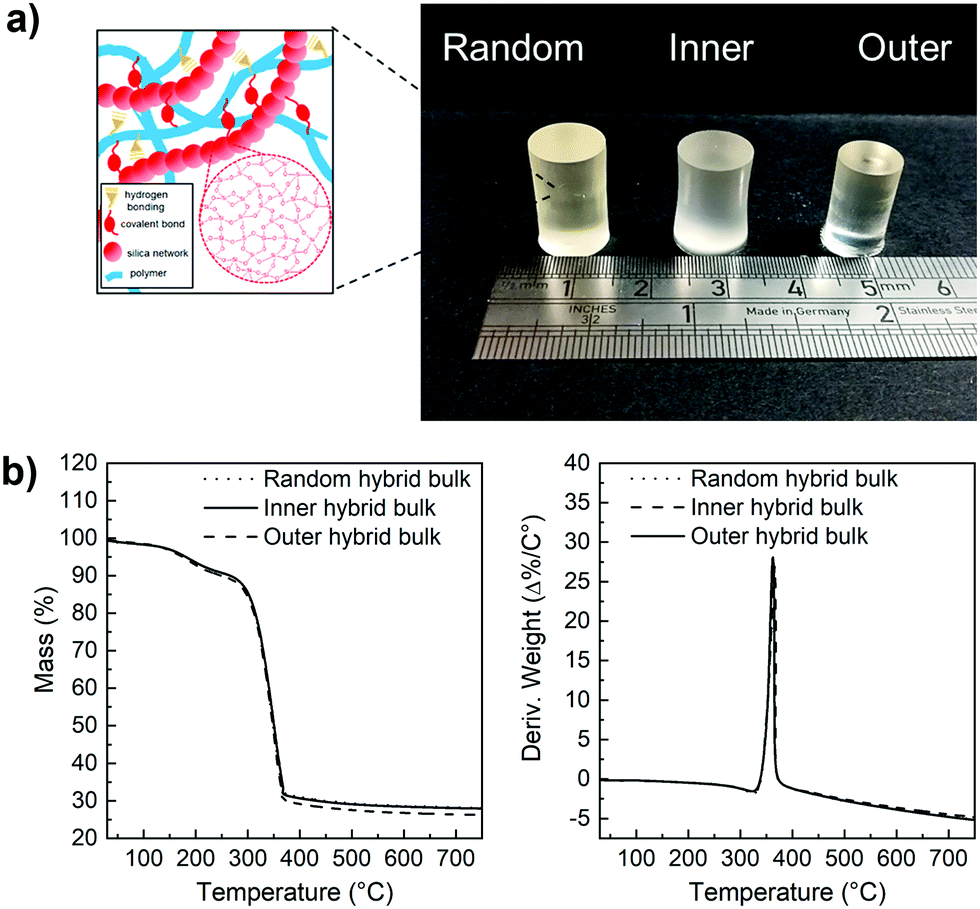 | ||
| Fig. 4 (a) Photographs of poly(MMA-TMSPMA)-star/silica hybrids, inset shows a schematic of the hybrid structure; (b) thermogravimetry (TGA, left) and differential scanning calorimetry (DSC, right) curves of the poly(MMA-TMSPMA)-star/silica hybrids. Random = poly(MMA-stat-TMSPMA)-star; Inner = poly(MMA-b-TMSPMA)-star; Outer = poly(TMSPMA-b-MMA)-star. | ||
The different architecture of the star significantly affected the gelation time of the sol–gel hybrids. When the TMSPMA block was in the outer part of the star (poly(TMSPMA-b-MMA)-star), it formed a chemically crosslinked gel in less than 5 minutes. When the TMSPMA was randomly distributed in the linear arms (poly(MMA-stat-TMSPMA)-star), gelation occurred in about 40 minutes, but when TMSPMA was in the inner part of the star (poly(MMA-b-TMSPMA)-star), it required an overnight period at 37 °C to gel. The different gelation times can be explained based on how accessible the methoxysilane groups were, that were able to form covalent bonds with silica network. Thus, the star architecture can be used to tailor the gelation time that could be critical for some applications, i.e. for the direct 3D printing from sol–gel. The chemical structure of the three different hybrids and copolymers was confirmed by FTIR analysis (Fig. 5). Vibration bands expected in methacrylate based copolymer were present in all three copolymer spectra: C–H bending (1485 cm−1) from the methacrylate backbone; C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1729 cm−1), C–O asymmetric stretching (1239 cm−1), and C–O–C symmetric stretching (1140 cm−1) of the ester group from the methacrylate moieties and Si–O–CH3 (1087 cm−1) alkoxysilane group from TMSPMA. Upon hybrid synthesis, stronger absorption bands characteristic of the condensed silica network arose, such as Si–O–Si asymmetric stretching while possessing the polymer absorption bands. FTIR spectra showed that the copolymers and silica network were both present in the hybrid system.
O stretching (1729 cm−1), C–O asymmetric stretching (1239 cm−1), and C–O–C symmetric stretching (1140 cm−1) of the ester group from the methacrylate moieties and Si–O–CH3 (1087 cm−1) alkoxysilane group from TMSPMA. Upon hybrid synthesis, stronger absorption bands characteristic of the condensed silica network arose, such as Si–O–Si asymmetric stretching while possessing the polymer absorption bands. FTIR spectra showed that the copolymers and silica network were both present in the hybrid system.
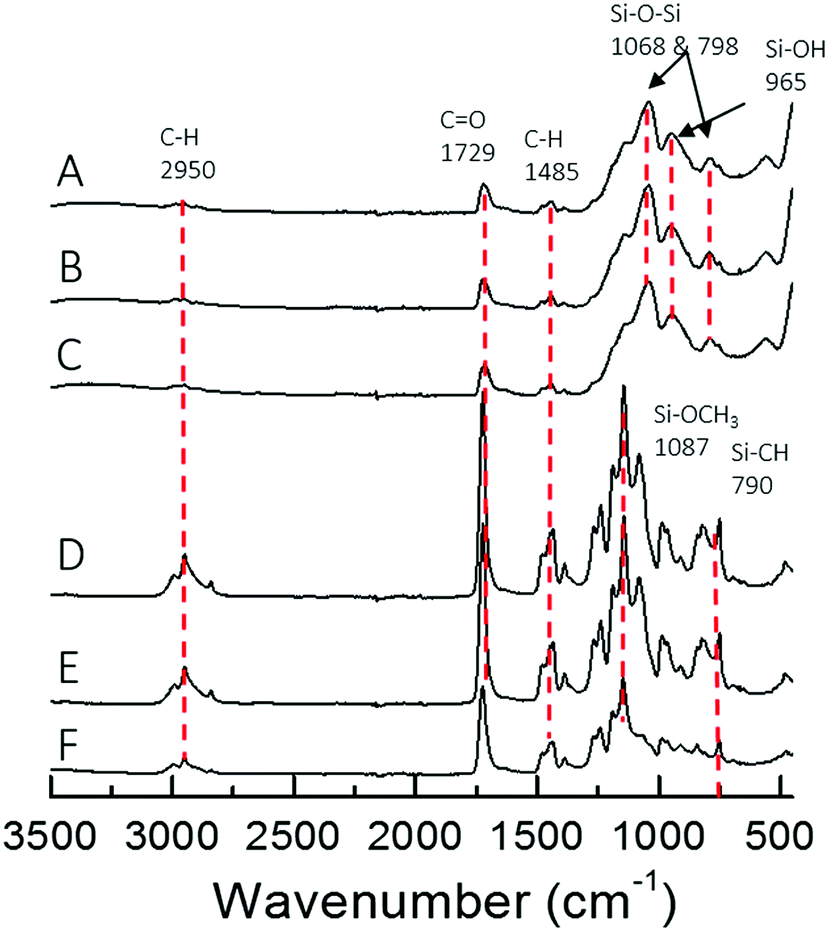 | ||
| Fig. 5 FTIR spectra of (A) poly(MMA-b-TMSPMA)-star/silica hybrid, (B) poly(TMSPMA-b-TMSPMA)-star/silica hybrid, (C) poly(MMA-stat-TMSPMA)-star/silica hybrid, and their respective star polymers (D, E and F). | ||
Previous poly(MMA-co-TMSPMA) hybrids, with the same percentage polymer, that had a disulfide based di-methacrylate core that could be degraded by glutathione, had a yield stress of 27 MPa at a strain of 4%.30 Here, a yield stress was in the range of 15–20 MPa at strain of ∼4%. Polymer architecture also affected the mechanical properties of the hybrids. Fig. 6 shows that all the three hybrids were characterised by an initial elastic deformation, similar for each of the hybrids (∼2–3% strain), followed by much larger plastic deformation regions with significant differences in terms of stress and strain at failure among the star architectures. There was no significant difference among the three hybrids in relation to Young's modulus, yield strain (∼4%) and stress (∼15–19 MPa) (Table 3). Stress and strain at failure, and modulus of toughness, for hybrids with TMSPMA blocks in the inner part of the hybrid were significantly higher than for hybrids with TMSPMA blocks randomly distributed or in the outer part of the star polymer, whereas there is no significant difference between outer and random (Table 3). The significantly lower values for hybrids with TMSPMA blocks randomly distributed or in the outer part of the star polymers can be related to a higher crosslinking density with the silica network, as the trimethoxy silane groups were more accessible. The higher crosslinking density leads to a more rigid structure and then higher brittleness of the materials (Table 3).
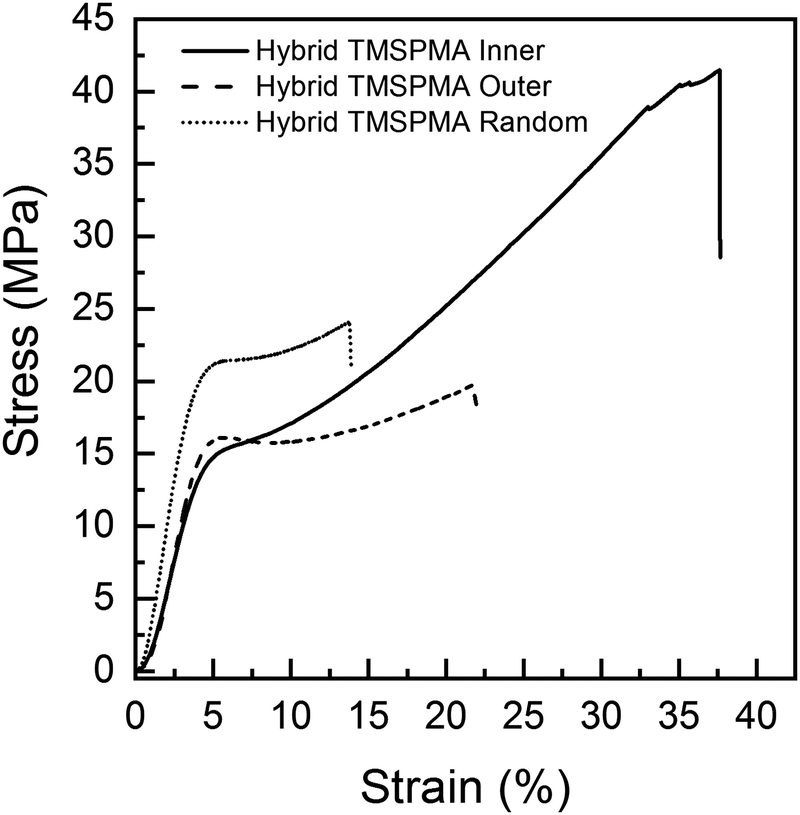 | ||
| Fig. 6 Representative uniaxial compression test curves of the poly(MMA-TMSPMA)-star/silica hybrid monoliths. Random = poly(MMA-stat-TMSPMA)-star; Inner = poly(MMA-b-TMSPMA)-star; Outer = poly(TMSPMA-b-MMA)-star. | ||
:inorganic 70:30 wt% composition tested in compression. Random = poly(MMA-stat-TMSPMA)-star; Inner = poly(MMA-b-TMSPMA)-star; Outer = poly(TMSPMA-b-MMA)-star. Values are reported as mean ± standard deviation
| Hybrid name | Yield stress (MPa) | Yield strain (%) | Young's modulus, E (MPa) | Stress at failure (MPa) | Strain at failure (%) | Modulus of toughness UT (MPa) |
|---|---|---|---|---|---|---|
| Hybrid-TMSPMA inner | 15.8 ± 3.9 | 4.0 ± 0.5 | 486 ± 68 | 45.7 ± 10.4 | 38.2 ± 2.6 | 10.4 ± 3.2 |
| Hybrid-TMSPMA random | 19.6 ± 1.5 | 4.2 ± 0.6 | 567 ± 121 | 24.1 ± 1.2 | 16.6 ± 2.8 | 3.0 ± 0.4 |
| Hybrid-TMSPMA outer | 15.2 ± 2.1 | 4.7 ± 0.7 | 458 ± 111 | 20.9 ± 2.9 | 27.8 ± 5.9 | 4.1 ± 0.8 |
Degradation studies of the hybrids were performed in TRIS buffer, containing collagenase Type I, at pH 7.4, to confirm the hypothesis that our new hybrids could degrade in response to endogenous tissue specific collagenases involved in natural remodelling. As shown in Fig. 7, the enzyme sensitive peptide-based core of the star polymers enabled a sustained degradation of the hybrids, which may allow the maintaining of strength and bioactive properties during cartilage healing. In particular, after two months’ immersion, the maximum of weight loss % was approximately 22% for the poly(MMA-b-TMSPMA)-star (inner)/silica hybrid, and only about 12% and 10% for the star polymers containing TMSPMA in the outer part and randomly distributed, respectively. Thus, the architecture of the star polymers significantly affected the degradation rate. This difference in degradation rate can be related to the fact that when TMSPMA is in the inner part of the star it is “hidden” and less accessible to form chemical bonds with silica network. This results in a higher degradation rate (less bonds need to be cleaved by water) which means a higher weight loss % as a function of time.
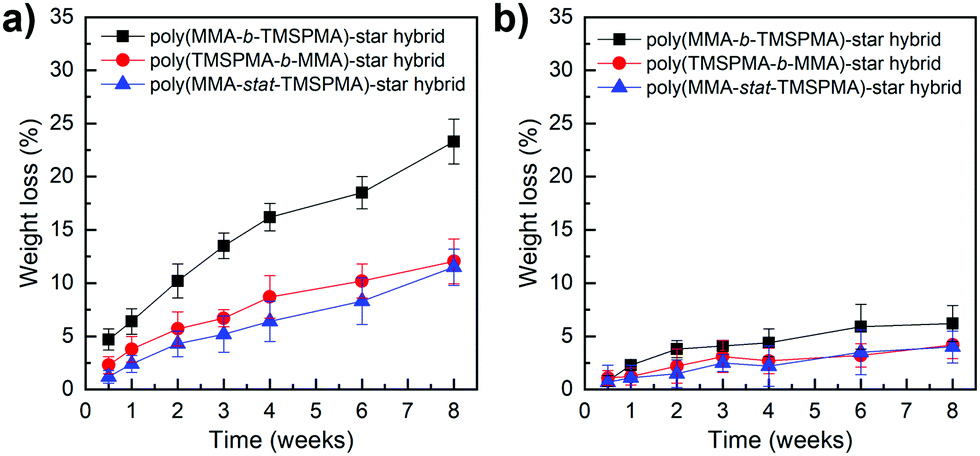 | ||
| Fig. 7 Weight loss % of poly(MMA-TMSPMA)-star/silica hybrids as a function of time: (a) with collagenase and (b) without collagenase. | ||
The same degradation studies were carried out without collagenase. It is worth noting that there was not a significant weight loss % without the presence of the enzyme (highest weight loss% equal to 4.4% after 1 month) suggesting that the degradation is stimulate/responsive to the presence of collagenase. To confirm that the weight loss was related to the loss in polymer after the activity of collagenase, the degradation medium was investigated by GPC and 1H NMR after 2 months of incubation. The GPC trace (Fig. 8, top graphs) showed a broad peak near to that of the linear arms (as measured prior to making the stars), indicating that the core of the star had degraded, leaving the low Mw arms. The MMs and dispersities of the cleaved hybrids are reported in Table 4. Finally, 1H NMR characterisation confirmed the presence of the polymer in the degradation medium (Fig. 8, bottom graphs). Cell viability studies were previously performed on hybrids with similar degradation products and they were found to be non-cytotoxic to the MC3T3 pre-osteoblast cell line.30
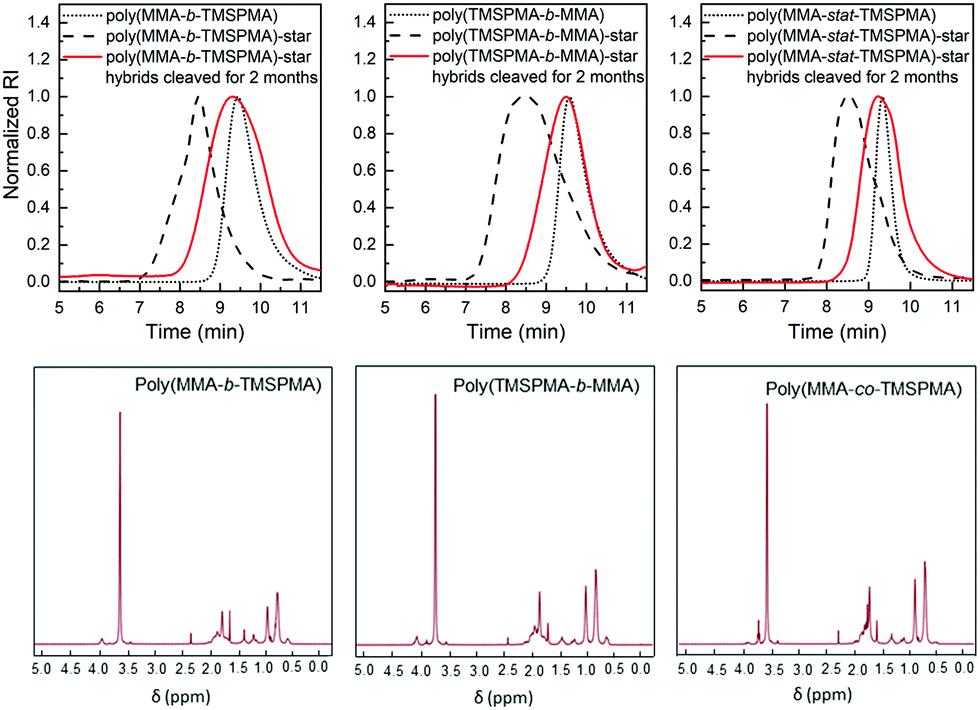 | ||
| Fig. 8 Characterisation of degradation products from three types of poly(MMA-TMSPMA)-star/silica hybrids after 2 months of incubation with collagenase (red lines): (top graphs) GPC traces, (bottom graphs) 1H NMR spectra. The GPC traces of the star polymers, prior to making hybrids, are shown as dashed lines; GPC traces of the linear arms (Macro RAFT) of the star polymers, prior to making stars, are shown as the dotted line. | ||
| Hybrid name | M w (kg mol−1) | M n (kg mol−1) | Đ |
|---|---|---|---|
| Cleaved poly(MMA-stat-TMSPMA)-star/hybrid | 16.25 | 8.86 | 1.83 |
| Cleaved poly(MMA-b-TMSPMA)-star/hybrid | 15.53 | 8.88 | 1.75 |
| Cleaved poly(TMSPMA-b-MMA)-star/hybrid | 16.09 | 9.4 | 1.71 |
The sol–gel process for hybrid synthesis allowed them to be used as an ‘‘ink’’ for 3D computer-controlled additive manufacturing.29,43 Direct ink write printing was used to fabricate porous scaffolds at room temperature, following a grid-like pattern with aligned 20 layers. Additive manufacturing, or 3D printing, is more advantageous over other methods, i.e. foaming19,20 and freeze casting,44 because it allows to tune independently the channel size and the mechanical properties by controlling the strut separation and the strut diameter in the design file. Gao et al. demonstrated the feasibility of the direct 3D printing of a Class I hybrid sol–gel ink of silica and gelatin.45 Tallia et al. were the first to 3D print covalently-bonded sol–gel silica/polytetrahydrofuran/poly-caprolactone hybrids with 200 μm channel size.29 Here, the printability of the three different star architecture of poly(MMA-TMSPMA)-star/silica hybrids was investigated. The hybrid inks were prepared by mixing the star-copolymer solution (0.7 g mL−1 in THF) to hydrolyzed TEOS to obtain a ratio of 70:30 wt% organic:inorganic component (i.e. same composition of the investigated monoliths). The polymer architecture had a significant effect on the printability of the inks. In particular, the poly(TMSPMA-b-MMA)-star (outer)/silica hybrid ink could not be 3D printed due to a too rapid gelation time (less than 5 minutes) that made the ink impossible to handle and to pour in the syringes. Conversely, the other two hybrid materials could be successfully extruded through the 200 μm nozzle without the need of additives, but with very different timing and printing window, i.e. the window of time during which the gel was fluid enough to flow smoothly through the nozzle and viscous enough to solidify and retain shape without collapsing.29 The poly(MMA-stat-TMSPMA)-star (random)/silica hybrid ink was printed within one hour of the transfer into the syringe. The syringe loaded with the poly(MMA-b-TMSPMA)-star (inner)/silica hybrid ink was instead only printable after 24 h at 37 °C, after which the printing window was ∼2 h. Within the printing window, the poly(MMA-stat-TMSPMA)-star (random)/silica hybrid and poly(MMA-b-TMSPMA)-star (inner)/silica hybrids could be 3D printed into hybrid scaffolds with well inter-connected porous structure (Fig. 9). However, the poly(MMA-b-TMSPMA)-star (inner)/silica hybrid ink showed a better printability (i.e. more controllable increase of viscosity that determines a better extrusion of the filament) than the poly(MMA-stat-TMSPMA)-star (random)/silica hybrid due to a more controllable gelation. The slower gelation time of the poly(MMA-b-TMSPMA)-star (inner)/silica architecture led to a gradual increase in viscosity of the ink which led to a manageable direct extrusion printing, whereas the poly(MMA-stat-TMSPMA)-star (random)/silica hybrid showed a too fast gelation after the ink was transferred into the syringe, which resulted in a variation of the extruded filament with increasing layers (i.e. the ink viscosity increased layer-by-layer causing a reduction of the filament diameter and more irregular struts). This is very interesting because it demonstrates that, while keeping the polymer and hybrid chemistry and composition constant, we can still control and alter the printability of the hybrids by varying the polymer architecture and more specifically the position of the TMSPMA groups.
 | ||
| Fig. 9 Photographs of 3D printed inner and random poly(MMA-TMSPMA)-star/silica hybrid scaffolds of 70:30 inorganic:organic wt% composition. Random = poly(MMA-stat-TMSPMA)-star/silica; Inner = poly(MMA-b-TMSPMA)-star/silica. | ||
The scaffold architecture was investigated through SEM analysis of top surface of inner (Fig. 10a and b) and random (Fig. 10c and d) scaffolds which showed an interconnected pore channel structure after 3D printing. As expected from the more consistent extrusion of the filament, the scaffolds obtained with the poly(MMA-b-TMSPMA)-star (inner)/silica hybrid ink (Fig. 10a and b) showed a more regular structure than the ones obtained with poly(MMA-stat-TMSPMA)-star (random)/silica hybrid ink, confirming that the poly(MMA-b-TMSPMA)-star (inner)/silica hybrid is the most promising material for the 3D printing of porous structures. Struts were very repeatable, with widths of 204 ± 20 μm (the nozzle dimension). The channel size was (160 ± 64 μm), the higher deviation comes from the fact that the struts were not as regular. Scaffolds made with the random-star hybrid had similar strut (201 ± 17 μm) and channel (179 ± 61 μm) dimensions.
 | ||
| Fig. 10 SEM images of top surface (x–y) of 3D printed inner (a and b) and random (c and d) poly(MMA-TMSPMA)-star/silica hybrid scaffolds. | ||
Conclusions
We present the first enzyme cleavable polymer–silica sol–gel hybrids that degrade in response to endogenous tissue specific enzymes that are involved in natural remodelling of bone, enabling sustained degradation of 10% after two months of exposure to the enzymes. Three polymers of different architecture were synthesised, keeping the chemistry and composition constant. Specifically, two star block copolymers and one statistical star copolymer of TMSPMA and of MMA were successfully prepared by RAFT polymerisation connecting the linear arms of the stars via a collagenase sensitive peptide-based cross linker. The enzyme sensitive star copolymers were used as organic source to synthesize polymer/glass hybrids by a sol–gel route, that allowed them to be 3D printed successfully. The effect of the position of the TMPSMA group within the structure of polymers and consequently on the hybrid formation, degradation, mechanical properties and printability was demonstrated. Interestingly, the most promising polymers was the star polymer with the TMSPMA inner-star architecture that led to the production of the more flexible and tougher material compared to the other star architectures. The same polymer architecture also demonstrated the most promising for the 3-D printability of the hybrid as it had the most suitable gelation time for the extrusion process. Thus, it was demonstrated that the polymer architecture, in particular the position of the TMSPMA groups, is crucial for the hybrids properties and can be used to tailor these properties and even the fabrication process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge EPSRC (EP/M019950/1) for funding part of this project. Raw data is available from http://rdm-enquiries@imperial.ac.uk.References
- F. J. O'Brien, Biomaterials & scaffolds for tissue engineering, Mater. Today, 2011, 14(3), 88–95 CrossRef.
- N. Patel, S. M. Best, W. Bonfield, I. R. Gibson, K. A. Hing and E. Damien, et al., A comparative study on the in vivo behavior of hydroxyapatite and silicon substituted hydroxyapatite granules, J. Mater. Sci. Mater. Med., 2002, 13(12), 1199–1206 CrossRef CAS.
- J. R. Jones, D. S. Brauer, L. Hupa and D. C. Greenspan, Bioglass and bioactive glasses and their impact on healthcare, Int. J. Appl. Glass Sci., 2016, 7(4), 423–434 CrossRef CAS.
- K. A. Hing, P. A. Revell, N. Smith and T. Buckland, Effect of silicon level on rate, quality and progression of bone healing within silicate-substituted porous hydroxyapatite scaffolds, Biomaterials, 2006, 27(29), 5014–5026 CrossRef CAS.
- A. Nommeots-Nomm, S. Labbaf, A. Devlin, N. Todd, H. Geng and A. K. Solanki, et al., Highly degradable porous melt-derived bioactive glass foam scaffolds for bone regeneration, Acta Biomater., 2017, 57, 449–461 CrossRef CAS.
- X. Shi, A. Nommeots-Nomm, N. M. Todd, A. Devlin-Mullin, H. Geng and P. D. Lee, et al., Bioactive glass scaffold architectures regulate patterning of bone regeneration in vivo, Appl. Mater. Today, 2020, 20, 100770 CrossRef.
- P. Habibovic, H. P. Yuan, C. M. van der Valk, G. Meijer, C. A. van Blitterswijk and K. de Groot, 3D microenvironment as essential element for osteoinduction by biomaterials, Biomaterials, 2005, 26(17), 3565–3575 CrossRef CAS.
- G. Poologasundarampillai, P. D. Lee, C. Lam, A. M. Kourkouta and J. R. Jones, Compressive Strength of Bioactive Sol–Gel Glass Foam Scaffolds, Int. J. Appl. Glass Sci., 2016, 7(2), 229–237 CrossRef CAS.
- Q. Fu, E. Saiz and A. P. Tomsia, Direct ink writing of highly porous and strong glass scaffolds for load-bearing bone defects repair and regeneration, Acta Biomater., 2011, 7(10), 3547–3554 CrossRef CAS.
- A. Nommeots-Nomm, P. D. Lee and J. R. Jones, Direct ink writing of highly bioactive glasses, J. Eur. Ceram. Soc., 2018, 38(3), 837–844 CrossRef CAS.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering, Biomaterials, 2006, 27(18), 3413–3431 CrossRef CAS.
- S. H. Rhee, Y. K. Lee and B. S. Lim, Evaluation of a novel poly(epsilon-caprolactone)-organosiloxane hybrid material for the potential application as a bioactive and degradable bone substitute, Biomacromolecules, 2004, 5(4), 1575–1579 CrossRef CAS.
- J. R. Jones, Review of bioactive glass: From Hench to hybrids, Acta Biomater., 2013, 9(1), 4457–4486 CrossRef CAS.
- M. S. Saveleva, K. Eftekhari, A. Abalymov, T. E. L. Douglas, D. Volodkin and B. V. Parakhonskiy, et al., Hierarchy of Hybrid Materials - The Place of Inorganics-in-Organics in it, Their Composition and Applications, Front. Chem., 2019, 7 Search PubMed.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Applications of advanced hybrid organic–inorganic nanomaterials: from laboratory to market, Chem. Soc. Rev., 2011, 40(2), 696–753 RSC.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, Applications of hybrid organic–inorganic nanocomposites, J. Mater. Chem., 2005, 15(35–36), 3559–3592 RSC.
- S. Lin, C. Ionescu, K. J. Pike, M. E. Smith and J. R. Jones, Nanostructure evolution and calcium distribution in sol-gel derived bioactive glass, J. Mater. Chem., 2009, 19(9), 1276–1282 RSC.
- G. Poologasundarampillai, C. Ionescu, O. Tsigkou, M. Murugesan, R. G. Hill and M. M. Stevens, et al., Synthesis of bioactive class II poly(gamma-glutamic acid)/silica hybrids for bone regeneration, J. Mater. Chem., 2010, 20(40), 8952–8961 RSC.
- O. Mahony, O. Tsigkou, C. Ionescu, C. Minelli, L. Ling and R. Hanly, et al., Silica-Gelatin Hybrids with Tailorable Degradation and Mechanical Properties for Tissue Regeneration, Adv. Funct. Mater., 2010, 20(22), 3835–3845 CrossRef CAS.
- O. Mahony, S. Yue, C. Turdean-Ionescu, J. V. Hanna, M. E. Smith and P. D. Lee, et al., Silica–gelatin hybrids for tissue regeneration: inter-relationships between the process variables, J. Sol-Gel Sci. Technol., 2014, 69(2), 288–298 CrossRef CAS.
- G. Poologasundarampillai, B. Yu, O. Tsigkou, E. M. Valliant, S. Yue and P. D. Lee, et al., Bioactive silica–poly(g-glutamic acid) hybrids for bone regeneration: effect of covalent coupling on dissolution and mechanical properties and fabrication of porous scaffolds, Soft Matter, 2012, 8(17), 4822–4832 RSC.
- G. Poologasundarampillai, B. Yu, O. Tsigkou, D. Wang, F. Romer and V. Bhakhri, et al., Poly(gamma-glutamic acid)/silica hybrids with calcium incorporated in the silica network by use of a calcium alkoxide precursor, Chem. – Eur. J., 2014, 20(26), 8149–8160 CrossRef CAS.
- E. M. Valliant, F. Romer, D. Wang, D. S. McPhail, M. E. Smith and J. V. Hanna, et al., Bioactivity in silica/poly(gamma-glutamic acid) sol–gel hybrids through calcium chelation, Acta Biomater., 2013, 9(8), 7662–7671 CrossRef CAS.
- L. Gabrielli, L. Connell, L. Russo, J. Jimenez-Barbero, F. Nicotra and L. Cipolla, et al., Exploring GPTMS reactivity against simple nucleophiles: chemistry beyond hybrid materials fabrication, RSC Adv., 2014, 4(4), 1841–1848 RSC.
- L. Gabrielli, L. Russo, A. Poveda, J. R. Jones, F. Nicotra and J. Jimenez-Barbero, et al., Epoxide Opening versus Silica Condensation during Sol–Gel Hybrid Biomaterial Synthesis, Chem. – Eur. J., 2013, 19(24), 7856–7864 CrossRef CAS.
- L. S. Connell, L. Gabrielli, O. Mahony, L. Russo, L. Cipolla and J. R. Jones, Functionalizing natural polymers with alkoxysilane coupling agents: reacting 3-glycidoxypropyl trimethoxysilane with poly(gamma-glutamic acid) and gelatin, Polym. Chem., 2017, 8(6), 1095–1103 RSC.
- S. H. Rhee, J. Y. Choi and H. M. Kim, Preparation of a bioactive and degradable poly(epsilon-caprolactone)/silica hybrid through a sol-gel method, Biomaterials, 2002, 23(24), 4915–4921 CrossRef CAS.
- D. Tian, S. Blacher, J. P. Pirard and R. Jerome, Biodegradable and biocompatible inorganic-organic hybrid materials. 3. A valuable route to the control of the silica porosity, Langmuir, 1998, 14(7), 1905–1910 CrossRef CAS.
- F. Tallia, L. Russo, S. Li, A. L. H. Orrin, X. Shi and S. Chen, et al., Bouncing and 3D printable hybrids with self-healing properties, Mater. Horiz., 2018, 5, 849 RSC.
- J. J. Chung, Y. Fujita, S. W. Li, M. M. Stevens, T. Kasuga and T. K. Georgiou, et al., Biodegradable inorganic-organic hybrids of methacrylate star polymers for bone regeneration, Acta Biomater., 2017, 54, 411–418 CrossRef CAS.
- J. J. Chung, J. R. Jones and T. K. Georgiou, Toward Hybrid Materials: Group Transfer Polymerization of 3-(Trimethoxysilyl)propyl Methacrylate, Macromol. Rapid Commun., 2015, 36(20), 1806–1809 CrossRef CAS.
- J. J. Chung, S. W. Li, M. M. Stevens, T. K. Georgiou and J. R. Jones, Tailoring Mechanical Properties of Sol–Gel Hybrids for Bone Regeneration through Polymer Structure, Chem. Mater., 2016, 28(17), 6127–6135 CrossRef CAS.
- P. Gurnani and S. Perrier, Controlled radical polymerization in dispersed systems for biological applications, Prog. Polym. Sci., 2020, 102 Search PubMed.
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization-A User Guide, Macromolecules, 2017, 50(19), 7433–7447 CrossRef CAS.
- I. Kurtulus, G. Yilmaz, M. Ucuncu, M. Emrullahoglu, C. R. Becer and V. Bulmus, A new proton sponge polymer synthesized by RAFT polymerization for intracellular delivery of biotherapeutics, Polym. Chem., 2014, 5(5), 1593–1604 RSC.
- A. Kuroki, A. K. Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock and M. Unnikrishnan, et al., Targeting intracellular, multi-drug resistant Staphylococcus aureus with guanidinium polymers by elucidating the structure-activity relationship, Biomaterials, 2019, 217 Search PubMed.
- H. Po, H. Liang, J. Xu, M. Xue and C. J. Jackson, Matrix metalloproteinases in bone development and pathology: current knowledge and potential clinical utility, Metalloproteinases Med., 2016, 3, 93–102 CrossRef.
- M. D. Bond and H. E. Vanwart, Characterization of the individual collagenases from clostridium–histolyticum, Biochemistry, 1984, 23(13), 3085–3091 CrossRef CAS.
- E. Themistou and C. S. Patrickios, Synthesis and characterization of polymer networks and star polymers containing a novel, hydrolyzable acetal-based dimethacrylate cross-linker, Macromolecules, 2006, 39(1), 73–80 CrossRef CAS.
- D. Kafouris, E. Themistou and C. S. Patrickios, Synthesis and characterization of star polymers and cross-linked star polymer model networks with cores based on an asymmetric, hydrolyzable dimethacrylate cross-linker, Chem. Mater., 2006, 18(1), 85–93 CrossRef CAS.
- M. Elladiou and C. S. Patrickios, A dimethacrylate cross-linker cleavable under thermolysis or alkaline hydrolysis conditions: synthesis, polymerization, and degradation, Chem. Commun., 2016, 52(15), 3135–3138 RSC.
- T. J. Gibson, P. Smyth, M. Semsarilar, A. P. McCann, W. J. McDaid and M. C. Johnston, et al., Star polymers with acid-labile diacetal-based cores synthesized by aqueous RAFT polymerization for intracellular DNA delivery, Polym. Chem., 2020, 11(2), 344–357 RSC.
- S. Li, F. Tallia, A. A. Mohammed, M. M. Stevens and J. R. Jones, Scaffold channel size influences stem cell differentiation pathway in 3-D printed silica hybrid scaffolds for cartilage regeneration, Biomater. Sci., 2020, 8, 4458–4466 RSC.
- D. M. Wang, F. Romer, L. Connell, C. Walter, E. Saiz and S. Yue, et al., Highly flexible silica/chitosan hybrid scaffolds with oriented pores for tissue regeneration, J. Mater. Chem. B, 2015, 3(38), 7560–7576 RSC.
- C. Gao, M. N. Rahaman, Q. Gao, A. Teramoto and K. Abe, Robotic deposition and in vitro characterization of 3D gelatinbioactive glass hybrid scaffolds for biomedical applications, J. Biomed. Mater. Res., Part A, 2013, 101(7), 2027–2037 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1. Fig. S1. 1H NMR characterisation of poly(MMA-b-TMSPMA), poly(TMSPMA-b-MMA) and poly(MMA-stat-TMSPMA) linear arms. 2. Fig. S2. 1H NMR characterisation of dimethacryloyl peptide (MaCh-pep) crosslink. 3. Scheme S1. Chemical structures of: (a) the starting di-amino ended peptide ({GLY}{PRO{LEU}{GLY}{PRO{LYS}}}); and (b) MaCh-peptide. See DOI: 10.1039/d0ma00674b |
| This journal is © The Royal Society of Chemistry 2020 |