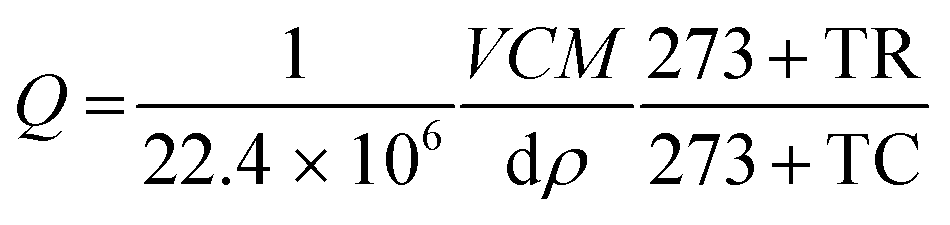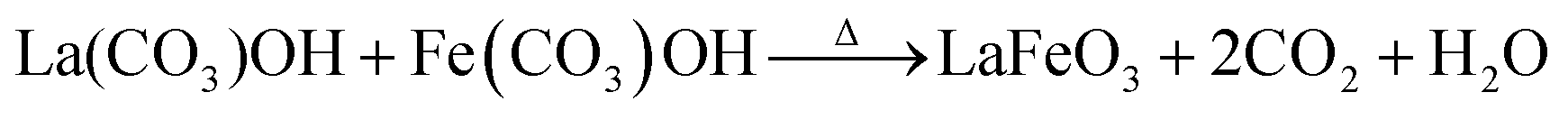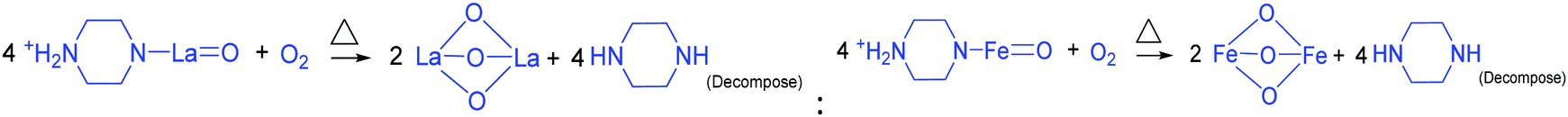DOI:
10.1039/D0MA00602E
(Paper)
Mater. Adv., 2020,
1, 2971-2982
Precisely tailored LaFeO3 dendrites using urea and piperazine hexahydrate for the highly selective and sensitive detection of trace level acetone†
Received
14th August 2020
, Accepted 15th October 2020
First published on 5th November 2020
Abstract
Trace level sensing of chemical vapours using metal oxides with fascinating morphologies has attracted much attention in many fields. Leaf-like dendrites have been investigated as a potential morphology for sensors because of their fast surface reactivity and transducer–receptor function. However, the preparation and understanding of these unique and homogenized morphological metal oxides are still far from sufficient. Herein, simple additives urea (9 mM) and piperazine hexahydrate (0–21 mM, 3 mM increments) were used to tailor homogenized lanthanum ferrite (LaFeO3) dendrites under hydrothermal conditions. The sequential reactions revealed that the urea by-products NH4+ and CO32− controlled the formation of the La2O3/Fe2O3 impurity phases and improved the formation of LaFeO3. The protonated piperazine molecules reacted with the metal hydroxides to form a bridge-like La–2O–Fe structure between two protonated piperazine molecules. This complex structure induced the growth of dendrites and led to the formation of LaFeO3 dendrites upon calcination. By utilizing these LaFeO3 dendrites for trace level detection at low temperature, an acetone sensing device was fabricated, which experimentally achieved the detection range from 10 ppm to 10 ppb with excellent stability and low noise to signal ratio at a chamber and device temperature of 100 °C. The maximum and minimum sensitivity values were 157.88 and 1.05, respectively. The sensitivity, response and recovery time for 1 ppm acetone were 37.63, 1.19 s and 95.81 s, respectively, and the device exhibited outstanding repeatability with long term stability. Furthermore, the selective detection of acetone in the presence of comparable sensitive molecules ethanol and formaldehyde was achieved by varying the chamber and device temperatures.
1. Introduction
In recent years, the development and application of advanced sensing materials for the highly sensitive and trace level (ppb) detection of chemical vapours in the environmental safety and human health care sectors have gained much research attention.1 Especially, the detection of commonly used chemical solvents in industries and laboratories is essential because of their volatility, combustibility and toxicity, which make them a hazard. On the other hand, some of these chemicals are excreted from the human body through various metabolic conditions and are considered as biomarkers. Thus, the detection of these chemicals in the form of vapour from exhaled air is highly demanded in clinical monitoring.2 Acetone is one of the primary solvents and potential breath biomarkers for diabetes. Prolonged exposure to acetone can cause skin sensitivity, eye irritation, headaches, fatigue, nausea and nervous system knock-out. Further, the detection of acetone in exhaled air greater than 1.8 ppm confirms diabetes.3,4 Diabetes mellitus is one of the common metabolic disorders occurring in different age groups due to inactive lifestyle and food practices.5,6 However, despite the availability of advanced glucometers, their invasiveness causes discomfort to patients. Thus, the detection of acetone vapour is useful for the non-invasive diagnosis of diabetes and monitoring personal safety.
However, the sensitive detection of acetone is strongly influenced by the structural, morphological, compositional and crystal facet construction of materials.7 Several reports have shown that lanthanum ferrite (LaFeO3) with interesting morphologies is a potential material for the sensitive and selective detection of chemical species such as xylene,8 methanol,9 acetone,10 formaldehyde,11 and ethanol.12 LaFeO3 with a desirable morphology was obtained using physical and chemical templates. In the physical template method, lotus leaves,8 polystyrenes spheres12 and biowaste13 have been used to produce nanoparticles, honeycomb-like periodic porous and powder form of LaFeO3, respectively. In the chemical approach metal organic frameworks,10 hexylsalicylate,14 Pluronic P123-based SBA-15,15 and citric acid16 were used to produce porous nano octahedrons, nanoparticles, mesoporous and porous LaFeO3, respectively. Further, several methods have been applied in the formation of LaFeO3 such as molecular beam epitaxy,17 sol–gel,18 pulsed laser deposition,19 microwave-assisted synthesis,20 molecular imprinting,21 and the hydrothermal method.22 However, the understanding of the formation process of LaFeO3 with a homogenized morphology is still far from sufficient because the presence of two metal cations in LaFeO3 leads to the production of individual metal oxides with independent morphologies during the preparation process. Thus, the selection of a suitable preparation method with novel chemical reactants and a detailed formation process is necessary.
Herein, we formulated a systematic approach to derive LaFeO3 dendrites using urea and piperazine hexahydrate via the hydrothermal method. The presence of two amide groups and a carbonyl group in urea helped to control the formation of La2O3/Fe2O3 impurity phases and enhance the crystal structure of LaFeO3. The other additive, piperazine hexahydrate, protonated and made the solution alkaline, which caused La3+ and Fe3+ to precipitate into their metal hydroxides. These metal hydroxides interacted with the protonated piperazine to form a bridge-like La–2O–Fe structure between two protonated piperazine molecules and then produced LaFeO3 dendrites upon calcination. The prepared LaFeO3 dendrites were used to fabricate a low-temperature operable sensor for the trace level detection of acetone. Here, the developed acetone sensor exhibited detection limits in the range of 10 ppm to 10 ppb. This sensing range is better than that of the recently reported acetone sensors using other metal oxides such as Co–In2O3 (100 ppm),23 WO3 (12.5 ppm),24 Pd–SmFe1−xMgxO3 (0.5 ppm),25 Co3O4 (200 ppm),26 NiO/NiCo2O4 (100 ppm),27 and Au–ZnO (100 ppm).28 Due to the low detection limit of the present LaFeO3 dendrite-based acetone sensor, it could be used to monitor acetone vapour even at trace level (ppb) concentrations. Besides sensitivity, the selectivity of a sensor is also important. This is because industrial waste and breath vapour contain some volatile organic compounds (VOC) together with acetone. Interestingly, the present sensor was found to be insensitive towards VOC. However, formaldehyde and ethanol were shown to have an acceptable sensing response as that of acetone. Nevertheless, by varying the chamber and device temperatures, the selective detection of acetone could be achieved.
2. Experimental section
2.1 Materials
The primary precursors including lanthanum(III) nitrate hydrate, iron(III) nitrate nonahydrate and urea were purchased from Sigma-Aldrich. Piperazine hexahydrate, Hi-LR was purchased from HiMedia Laboratories Pvt. Ltd India. The accessories for the fabrication of the sensor including gold paste (Siltech Corporation Inc. India), ceramic cylindrical tubes (OD 2 mm × ID 1 mm × L 5 mm, Eltech Ceramics, India), platinum wire, 0.1 mm diameter and nickel chromium wire, 0.143 mm diameter (Alfa Aesar) were commercially procured. The vapour sensing solvents including acetone, ACS (Merk Life Science Private Ltd, India), ethanol absolute, AR, formaldehyde (37–41%), hydrogen peroxide (30%) (SD Fine-Chem Limited, India), benzene, Hi-AR and toluene, AR (HiMedia Laboratories Pvt. Ltd India) were procured from commercial sources. All the precursors and solvents were used without further purification. The carrier gas nitrogen (UHP grade) and interference gas CO2 (1000 ppm mixed with Argon) were acquired from Bhuruka Gases Limited, India.
2.2 Preparation of LaFeO3 dendrites
Perovskite oxide-type LaFeO3 dendrites were tailored through aqueous hydrothermal conditions. Initially, a stoichiometric amount of 1 mM (324.94 mg) lanthanum(III) nitrate hydrate and 1 mM (404.00 mg) iron(III) nitrate nonahydrate were dissolved in 10 mL of double-distilled water (DDW) under constant stirring for 30 min. The reactants, 9 mM (540.54 mg) urea and an increasing concentration of piperazine hexahydrate ranging from 0 to 21 mM at 3 mM increments, were dissolved in 10 mL of DDW separately under constant stirring for 30 min. Then, the urea solution followed by the different piperazine hexahydrate solutions were dripped into the metal nitrate solution and stirred continuously for 30 min. The obtained mixture solution was transferred to a Teflon-lined stainless steel autoclave (40 mL) and heated at 180 °C for 24 h in a hot air oven followed by cooling. The product was washed, and pH neutralization was performed via centrifugation using DDW. The final product was dried at 110 °C for 1 h followed by calcination at 900 °C for 3 h. This synthetic procedure was also carried out for the other piperazine hexahydrate concentrations. The detailed reaction conditions and their outcomes are tabulated in Table S1 (ESI†).
2.3 Fabrication of trace level sensing device for the detection of acetone
A ceramic cylindrical tube with the dimensions of OD 2 mm × ID 1 mm × L 5 mm was used and an aliquot of gold paste was painted over both ends of the tube, which was made a parallel electrode. The heating element, nickel chromium wire, 0.143 mm diameter, was made into a cylindrical coil and inserted into the ceramic tube. Then platinum wire, 0.1 mm diameter, was used to form a contact with both gold electrodes. The entire setup was mounted on top of a hard plastic material with the support of six nickel-plated bronze pins for electrical contact. By applying power, the heating coil was heated up to 200 °C, which removed the nickel–chromium wire protective plating and binder in the gold paste. Finally, 10 mg of LaFeO3 dendrites was dispersed in DDW and made into a slurry. The slurry was coated over the ceramic tube and allowed to dry naturally at room temperature (30 °C). No further heat treatment was required for device stability. The entire vapour sensing response was investigated at 5 V bias and the device working temperature was set at 100 °C, which was obtained from 1.65 W.
2.4 Characterization techniques
The crystalline phases of the prepared materials were examined using a PANalytical X’pert Pro X-ray diffractometer (XRD) with a monochromatized wavelength of λ = 1.5406 Å Cu Kα radiation. The derived morphology and tailoring processes were identified using a FEI Quanta 200 scanning electron microscope (SEM) with an operating voltage of 20 kV and working distance of 10 mm. The morphology, structural lattices and diffraction patterns of the final optimized LaFeO3 dendrites were studied via transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and selected area electron diffraction (SAED) on a JEOL JEM 2100 high-resolution transmission electron microscope at an acceleration voltage of 200 kV. The compositional homogeneity was confirmed through energy dispersive spectroscopy (EDS) and EDX element mapping analysis using a Bruker Quantax 200. The mass spectrum of the as-prepared sample was recorded on a Waters Micromass Q-TOF mass spectrometer. X-Ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha Surface Analysis instrument. The thermal stability was investigated using a PerkinElmer Simultaneous Thermal Analysis 6000 at a heating rate of 10 °C min−1 under a 20 mL min−1 nitrogen atmosphere. N2 ad/desorption isotherms were recorded using a Belsorp Mini II surface area analyzer under −196 °C. The vibrational properties of both the untreated and calcinated samples were measured using a Bruker Tensor 27 in KBr mode. All the gas sensing measurements were performed using a Keysight B2912A Precision Source/Measure unit with a customized sensing chamber arrangement.
2.5 Sensing measurement
The various concentrations (ppm) of the vapours such as acetone, ethanol, formaldehyde and other interference substances were obtained from the respective liquid volume using the following relation (eqn (1))29 and injected into the chamber using a Hamliton® syringe.| | 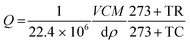 | (1) |
where Q is the corresponding chemical volume and the numerators, V, M, and C are the chamber volume, molecular weight of the chemical and concentration (ppm), respectively. The denominators d and ρ represent the volume fraction (percentage) and density of the target chemical, respectively. The temperature factors TR and TC represent the room and chamber temperatures, respectively. The number 22.4 is the molar volume of gas. The sensitivity was defined as S = Rg/Ra for the reducing vapour, whereas the oxidizing species was defined as S = Ra/Rg, where Rg and Ra are the resistance of the device in the presence of the target gas and air, respectively.
3. Result and discussion
3.1 Structural and morphological tailoring of LaFeO3 dendrites
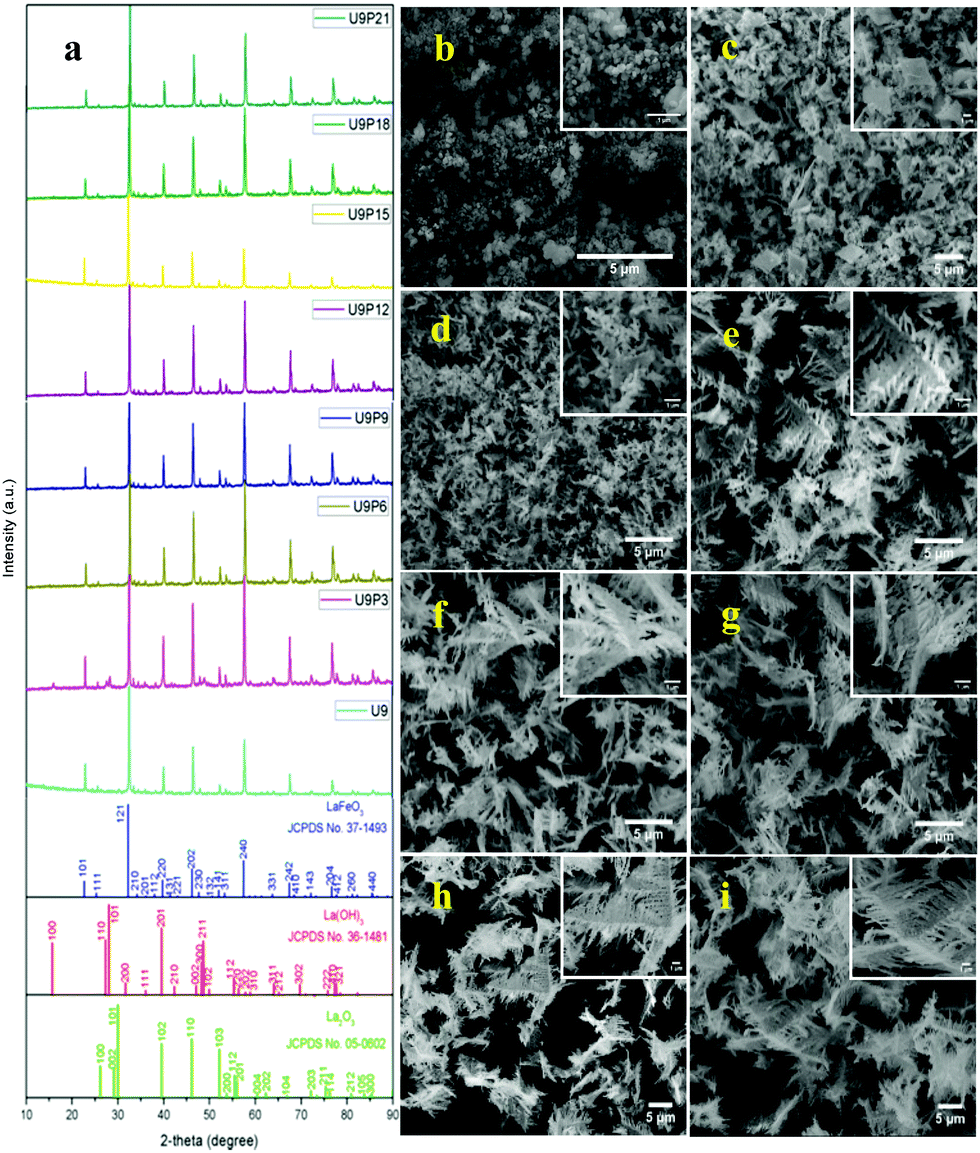
The tailoring process of the LaFeO3 dendrites started with the dissociation of lanthanum(III) nitrate hydrate and iron(III) nitrate nonahydrate into La3+, Fe3+ and NO3− ions in double distilled water, as shown in eqn (2) and (3), respectively. Then, 9 mM urea was added in the absence of piperazine hexahydrate to the metal nitrate solution, which dissociated into NH4+, CO2 and OH− under the hydrothermal conditions, as shown in eqn (4).30 The in situ generated OH− ions gradually increased the solution pH from 2.67 to 9.30. This progressive increase in alkalinity induced the nucleation of La3+ and Fe3+ into La(OH)3/LaOOH and Fe(OH)3/FeOOH precipitates, respectively, as shown in eqn (5). Under the alkaline conditions, the metal hydroxide precipitates possibly produced La2O3/Fe2O3 impurity phases together with LaFeO3 according to eqn (6) and (7) upon calcination. Interestingly, the XRD pattern of the 9 mM urea-based sample U9 (Fig. 1a) exhibited the LaFeO3 phase without any impurity phases, and it is completely consistent with the standard LaFeO3 (JCPDS No. 37-1493). However, in the diffraction pattern a small peak at 30.144° was observed, which could have originated from the La–O surface termination of the LaFeO3 structure, not from La2O3 (JCPDS No. 05-0602).31| | | La(NO3)3·xH2O + H2O → La3+ + 3NO3− + xH2O | (2) |
| | | Fe(NO3)3·9H2O + H2O → Fe3+ + 3NO3− + 10H2O | (3) |
| | | CH4N2O + 3H2O → 2NH4+ + CO2 + 2OH− | (4) |
| La3+ + 3OH− → La(OH)3/LaOOH + H2O: |
| | | Fe3+ + 3OH− → Fe(OH)3/FeOOH + H2O | (5) |
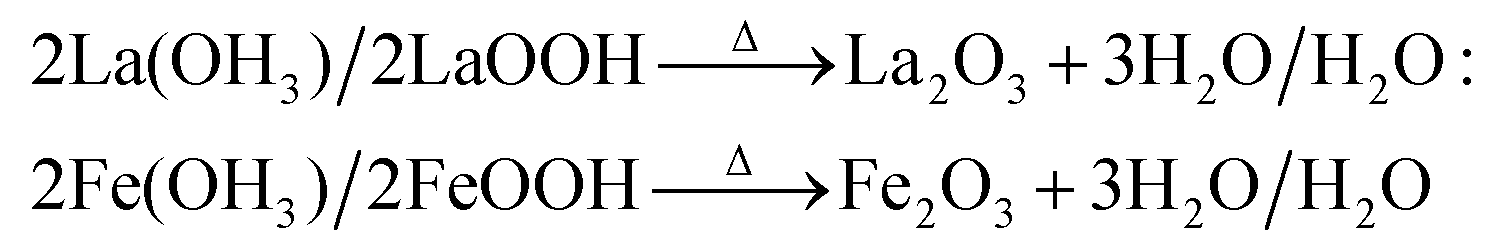
| 2La(OH)3/2LaOOH → Δ La2O3 + 3H2O/H2O: |
| | 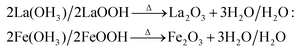 | (6) |
| | 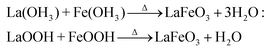 | (7) |
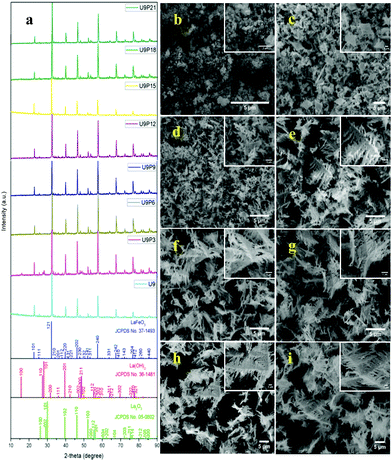 |
| | Fig. 1 (a) XRD patterns and (b–i) SEM images of fixed urea concentration (9 mM) with an increase in the concentration of piperazine hexahydrate (0, 3, 6, 9, 12, 15, 18 and 21 mM) in the LaFeO3 dendrite tailoring process. | |
It is believed that the impurity phases were controlled by the urea by-product NH4+. The mildly acidic NH4+ could react with the base OH− to produce NH3 and H2O according to eqn (8)via an acid–base reaction, and as evidence, an NH3 smell was observed in the Teflon autoclave. This acid–base reaction stabilized the formation of OH− ions, and thus the excessive formation of metal hydroxides, leading to the controlled formation of the La2O3/Fe2O3 impurity phases. The other urea by-product, CO2, in the reaction solution could react with water to produce CO32− and H+ under the hydrothermal condition, as shown in eqn (9).32 CO32− also saturated the solution and induced the crystal growth process via the interaction of unreacted La3+/Fe3+ to form La2(CO3)3/Fe2(CO3)3, as displayed in eqn (10), and both metal carbonates could interact to produce LaFeO3 with the elimination of CO2 upon calcination (eqn (11)). However, this is a less favourable reaction because under alkaline conditions most of the metal ions are in the form of their hydroxides. Thus, the remaining CO32− could replace the OH− in the metal hydroxides and form the respective metal carbonate hydroxide structure33,34 (eqn (12)). This reduced the formation of individual metal oxides and enhanced the crystallization of LaFeO3 by carbonate decomposition35 with the elimination of CO2 and H2O, as shown in eqn (13).
| | | NH4+ + OH− ⇌ NH3 + H2O | (8) |
| | | CO2 + H2O → CO32− + 2H+ | (9) |
| 3CO32− + 2La3+ → La2(CO3)3: |
| | | 3CO32− + 2Fe3+ → Fe2(CO3)3 | (10) |
| | 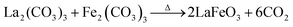 | (11) |
| La(OH)3 + CO32− → La(CO3)OH + 2OH−: |
| | | Fe(OH)3 + CO32− → Fe(CO3)OH + 2OH− | (12) |
| | 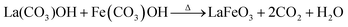 | (13) |
| |  | (14) |
| | 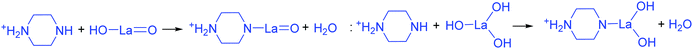 | (15) |
| | 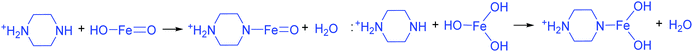 | (16) |
| |  | (17) |
| | 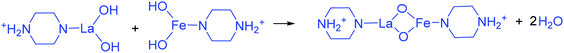 | (18) |
| |  | (19) |
| | 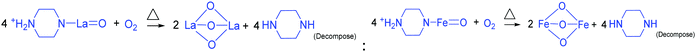 | (20) |
| | 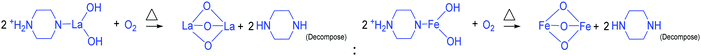 | (21) |
The SEM image of sample U9 in Fig. 1b shows irregular particles with an aggregated structure. These irregular particles are due to the two different crystallization processes of LaFeO3, i.e. the metal hydroxide and metal carbonate hydroxide process. It is known that the use of surfactants such as CTAB, HMTA and SDS can produce homogenized structures with interesting morphologies. However, it was essential to preserve the single-phase LaFeO3, and thus, herein, piperazine hexahydrate was used to improve the growth of the homogenized morphology. The organic molecule piperazine could easily interact with the nucleated metal hydroxide seeds and reduce the interfacial energy, thus preventing their aggregation. Further, the alkaline nature of piperazine hexahydrate helped to induce the LaFeO3 crystallization process.
The addition of 3 mM piperazine hexahydrate to the above 9 mM urea and metal nitrate reaction mixture led to an increase in the solution pH from 2.67 to 6.74 due to the protonation of piperazine with water and release of OH− ions (eqn (14)) even before the hydrothermal treatment. The increased pH and corresponding OH− ions could convert La3+/Fe3+ into their hydroxides, which possibly bonded with the protonated piperazine, as illustrated in eqn (15) and (16). The protonated piperazine-bonded individual metal hydroxides could interact with each other to form a bridge-like La–2O–Fe bond between two protonated piperazine molecules (eqn (17) and (18)). This protonated piperazine-bonded bridge-like La–2O–Fe compound remained stable even after the hydrothermal treatment and was converted into LaFeO3 upon calcination in the presence of oxygen according to eqn (19). The XRD pattern of the corresponding sample (U9P3) is shown in Fig. 1a, which confirms the presence of LaFeO3. Furthermore, the diffraction peaks at 15.927° (100), 27.486° (110), 28.220° (101), 48.854° (211), 55.446° (112) and 58.944° (310) correspond to the standard La(OH)3 (JCPDS No. 36-1481). However, interestingly no Fe(OH)3 was observed. It is believed that the low ionic radii Fe3+ was precipitated into iron hydroxide even before the hydrothermal treatment with low pH (6.74) and bonded with the protonated piperazine, thereby leaving no Fe(OH)3 after the hydrothermal reaction. The presence of La(OH)3 could be due to the increase in pH from 6.74 to 9.35 upon the dissociation of urea under the hydrothermal conditions, which caused the unreacted La3+ to react with OH−. However, the hygroscopic La(OH)3 did not produce La2O3 even after calcination.36,37 Further, no La2O3/Fe2O3 impurity phases were observed, which may be due to the presence of the urea by-products NH4+ and CO32−. The obtained SEM images of the parallelogram and irregular structure in Fig. 1c represent the presence of two independent phases, such as LaFeO3 and La(OH)3. The piperazine-based complex structure was converted into a parallelogram under the hydrothermal conditions. Simultaneously, the urea-based reaction involved in the formation of an irregular structure.
To derive a homogenized morphology, the piperazine hexahydrate concentration was further increased from 6 mM to 21 mM in steps of 3 mM, thereby causing an increase in the pH before/after the hydrothermal treatment of 9.49/9.56 (U9P6), 9.54/9.60 (U9P9), 9.88/9.92 (U9P12), 9.90/9.90 (U9P15), 10.01/10.03 (U9P18) and 10.10/10.12 (U9P21). In all the above cases, the pH values remained almost the same before and after the hydrothermal treatment, indicating that there was no dissociation or further protonation of piperazine even after the hydrothermal treatment. Further, the dissociation of urea during the hydrothermal treatment was not sufficient to change the pH of the existing alkaline solution. The XRD patterns of the samples are shown in Fig. 1a, which indicate the complete formation of LaFeO3, and thus confirmed the piperazine-based LaFeO3 formation mechanism (eqn (14)–(19)). Even at higher concentrations, 6 mM piperazine hexahydrate and pH 9.49 were found to be sufficient to transform all the metal cations into metal hydroxide, thereby bonding with them to produce LaFeO3 upon calcination, and thus no La(OH)3 was produced via the urea process. However, it was necessary to determine the role of 9 mM urea in the reaction. Thus, 15 mM piperazine hexahydrate alone was mixed with metal nitrates (P15) without urea, and the corresponding XRD pattern is shown in Fig. S1a (ESI†). Most of the diffraction peaks are indexed to LaFeO3. However, the diffraction peaks at 26.332° (100), 29.252° (002), 30.126° (101), 55.586° (112), 56.181° (201), 62.616° (202), 73.300° (203), and 75.486° (211) correspond to the presence of La2O3. Further, the diffraction peaks observed at 24.426° (012), 33.344° (104), 41.108° (113), 49.746° (024) and 54.275° (116) correspond to Fe2O3 (JCPDS No. 89-8103). These individual metal oxides may be from the protonated piperazine-bonded respective metal hydroxides upon calcination, as shown in eqn (20) and (21). Thus, the presence of urea and its by-products NH4+ and CO32− is essential for enhancing the heterogeneous interaction of protonated piperazine-bonded metal hydroxides and to form the bridge-like La–2O–Fe between two protonated piperazines, and thus reduced the presence of individual protonated piperazine-bonded metal hydroxides. Therefore, no La2O3 and Fe2O3 impurity phases were observed in the above piperazine hexahydrate with constant urea-based reactions.
The morphologies of the samples with piperazine hexahydrate concentrations in the range of 6 mM to 21 mM with a constant urea concentration (9 mM) in Fig. 1d–i summarize the dendritic growth tailoring process, which starts from dendrite growth initiation to fully grown dendrites upon an increase in piperazine hexahydrate concentration. This observation suggests the fact that during the crystallization process, the organic protonated piperazine molecules bonded with the nucleated seed metal hydroxides and induced dendritic growth. Therefore, it is believed that piperazine acts as a morphology-directing agent, and thus produced LaFeO3 dendrites upon calcination. Further, the SEM image in Fig. S1b (ESI†) of the sample with 15 mM piperazine hexahydrate alone (P15) shows the matured dendritic structure. This matured dendritic growth was due to the crystallization of other impurity phases over the dendrites, which covered the dendritic sub-branches.
3.2 Conformational studies for the formation mechanism and growth process of fully grown LaFeO3 dendrites
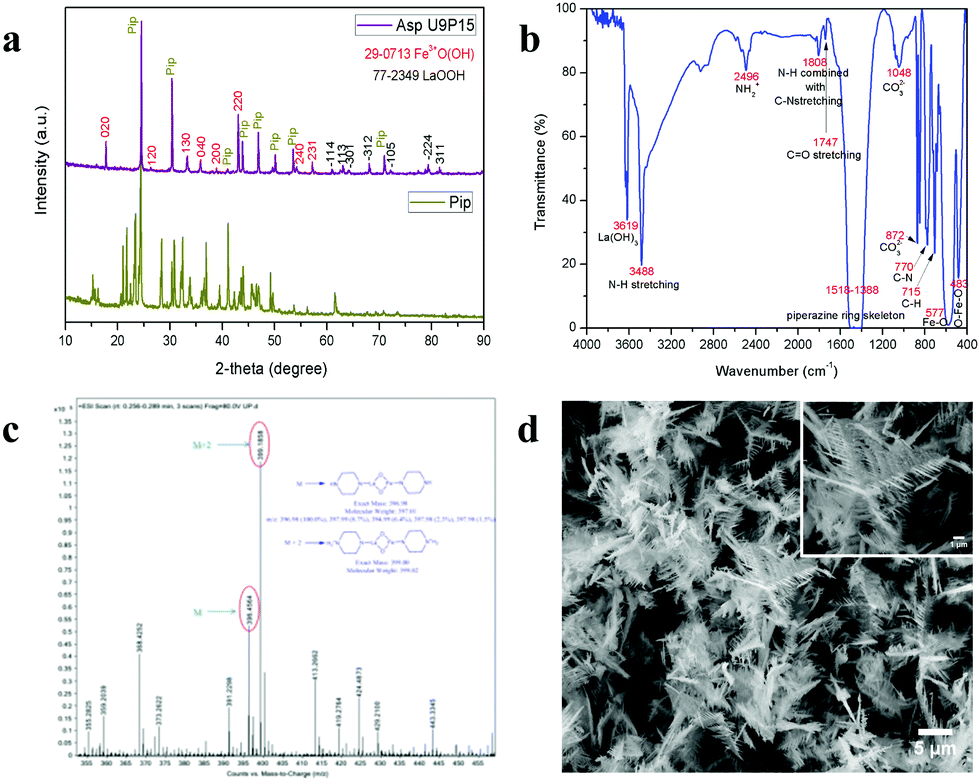
To validate the above discussed reaction mechanisms and to confirm the formation of LaFeO3 dendrites, the fully grown dendritic sample prepared using 9 mM urea and 15 mM piperazine hexahydrate (U9P15) was subjected to various characterizations. The XRD pattern of the sample not subjected to calcination is shown in Fig. 2a. The presence of metal hydroxides is confirmed by the different sets of diffraction peaks. The diffraction peaks at 17.851° (020), 26.332° (120), 33.361° (130), 36.072° (040), 38.922° (200), 43.171° (220), 54.275° (240) and 57.231° (231) correspond to Fe3+O(OH) (JCPDS No. 29-0713) and the other diffraction peaks at 61.007° (−114), 63.071° (113), 64.225° (−301), 68.177° (−312), 72.128° (−105), 79.298° (−224) and 81.728° (311) correspond to LaOOH (JCPDS No. 77-2349). However, these diffraction peaks showed some inconsistent shifts compared to their respective metal hydroxides standards and their prominent peaks were also not observed. Further, the diffraction peaks at, 24.583°, 30.441°, 40.968°, 43.888°, 46.966°, 50.183°, 53.558°, and 70.957° have remained un-indexed, which are correlated with the piperazine (Pip) diffraction peaks. This finding confirmed that the un-calcinated sample is not an individual metal hydroxide, but metal hydroxides bonded with piperazine molecule in a periodic arrangement. The FTIR spectrum corresponding to the un-calcinated sample in Fig. 2b shows the presence of metal hydroxides and piperazine in the sample through various modes. The sharp mode located at 3619 cm−1 indicates the presence of bulk hydroxyl group in the form of La(OH)3.36,37 The modes located at 3488, 1808, 770 and 715 cm−1 are due to the N–H stretching, N–H combined with C–N stretching, C–N and C–H vibrations in piperazine, respectively. Further, the broad mode in the range of 1518 to 1388 cm−1 confirms the presence of the piperazine ring skeleton.38–40 The mode at 2496 cm−1 shows the protonated piperazine in one of the NH sites to NH2+.41 The mode at 1747 cm−1 is the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, which originated from the carbonates, and the modes at 1048 and 872 cm−1 confirm the presence of carbonate-like species.13,37 The characteristic modes at 577 and 483 cm−1 confirm the presence of the Fe–O asymmetric stretching and O–Fe–O bending, respectively.13 The Q-time of flight (TOF) mass spectrum of the un-calcinated sample in Fig. 2c shows the maximum intensity of m/z at 399.1858, which is equivalent to the C8H202+FeLaN4O2 composition with the calculated mass value of 399.00. This again confirms the formation of two protonated piperazine molecules bonded via bridge-like La–2O–Fe according to eqn (17) and (18). The morphology of the un-calcinated sample in Fig. 2d confirms the presence of homogenized dendrites. Fig. 3a shows the thermogravimetry and differential scanning calorimetry profiles of the un-calcinated sample. The first weight loss and endothermic peak in the range of 40 °C to 425 °C is assigned to the removal of surface absorbed water. The next weight loss and continuous exothermic peak from 425 °C to 490 °C is related to the decomposition of piperazine and the plateau region up to 640 °C is attributed to the crystallization process. The final weight loss from 640 °C to 750 °C corresponds to the decomposition of carbonate. Thus, the calcination temperature of 900 °C is suitable for the formation of LaFeO3 dendrites.
O stretching vibration, which originated from the carbonates, and the modes at 1048 and 872 cm−1 confirm the presence of carbonate-like species.13,37 The characteristic modes at 577 and 483 cm−1 confirm the presence of the Fe–O asymmetric stretching and O–Fe–O bending, respectively.13 The Q-time of flight (TOF) mass spectrum of the un-calcinated sample in Fig. 2c shows the maximum intensity of m/z at 399.1858, which is equivalent to the C8H202+FeLaN4O2 composition with the calculated mass value of 399.00. This again confirms the formation of two protonated piperazine molecules bonded via bridge-like La–2O–Fe according to eqn (17) and (18). The morphology of the un-calcinated sample in Fig. 2d confirms the presence of homogenized dendrites. Fig. 3a shows the thermogravimetry and differential scanning calorimetry profiles of the un-calcinated sample. The first weight loss and endothermic peak in the range of 40 °C to 425 °C is assigned to the removal of surface absorbed water. The next weight loss and continuous exothermic peak from 425 °C to 490 °C is related to the decomposition of piperazine and the plateau region up to 640 °C is attributed to the crystallization process. The final weight loss from 640 °C to 750 °C corresponds to the decomposition of carbonate. Thus, the calcination temperature of 900 °C is suitable for the formation of LaFeO3 dendrites.
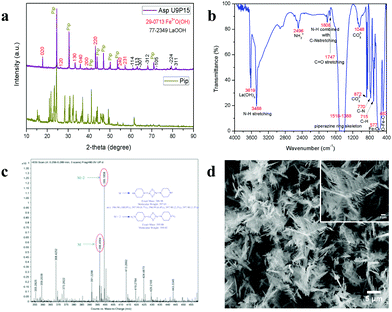 |
| | Fig. 2 (a) XRD pattern of piperazine hexahydrate (Pip) with un-calcinated LaFeO3 dendrites (Asp U9P15). (b) FTIR spectrum, (c) Q-TOF mass spectrum and (d) SEM image of un-calcinated LaFeO3 dendrites (Asp U9P15). | |
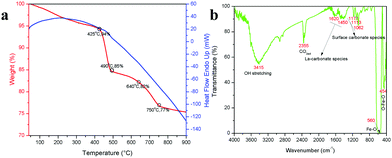 |
| | Fig. 3 (a) TG/DSC graph of un-calcinated LaFeO3 dendrites (Asp U9P15) and (b) FTIR spectrum of calcinated LaFeO3 dendrites (U9P15). | |
The XRD pattern of the calcinated U9P15 sample matches the standard data for LaFeO3, as displayed in Fig. 1a. All the peaks are completely correlated, confirming the formation of the LaFeO3 orthorhombic crystal structure with the Pnma space group. No other impurity phases and piperazine diffraction peaks can be observed. The calculated lattice parameters are a = 5.530 Å, b = 7.928 Å and c = 5.562 Å with the measured volume and grain size of 243.84 Å3 and 52.36 nm, respectively. The FTIR spectrum in Fig. 3b shows the characteristic mode at 454 and 560 cm−1 for the O–Fe–O bending and Fe–O asymmetric stretching in the FeO6 structure, respectively. The modes at 1062, 1113 and 1171 cm−1 are attributed to the surface carbonate species in the atmosphere. Further, the modes at 1450 and 1620 cm−1 are due to the atmospheric adsorbed La–carbonate species, which confirm the presence of La in the LaFeO3 dendrites. The modes at 2355 and 3415 cm−1 correspond to the surfaces adsorbed CO2(ad) and OH(ad) stretching, respectively.13 Here, no vibrational modes corresponding to the presence of piperazine molecules and bulk metal hydroxides were observed, which confirmed the complete removal of piperazine and the formation of LaFeO3 upon calcination. The morphology of the calcinated U9P15 sample in Fig. 4a shows few tens of micrometer-sized LaFeO3 dendrites consisting of a dark centre, trunks, branches and sub-branches. The lengths of the branches decreased towards the tip of the trunk. This confirmed that the nucleation occurred from the centre to the trunks, branches and sub-branches. Fig. 4b shows the magnified TEM image of a dendrite with different sized and directed branches, which confirms that both sides of the branches grew independently. Fig. 4c clearly shows that the small irregular grains were attached to produce dendrites without altering their grain boundaries. This unique formation process occurs through the attachment of organic piperazine molecules. Further, the single crystalline SAED pattern with zone axis [−12−1] and HRTEM image with the crystal plane (121) in Fig. 4d and e also confirm the formation of single crystalline LaFeO3. The EDS spectrum in Fig. 4f shows the presence of only La, Fe and O without impurities, and the corresponding atomic percentages of 17.21, 19.88 and 62.92 is consistent with the composition of LaFeO3, respectively. The EDX element mapping (shown in the inset of Fig. 4(f)) images in Fig. 4g–i show the uniform distribution of La, Fe and O in the LaFeO3 dendrites.
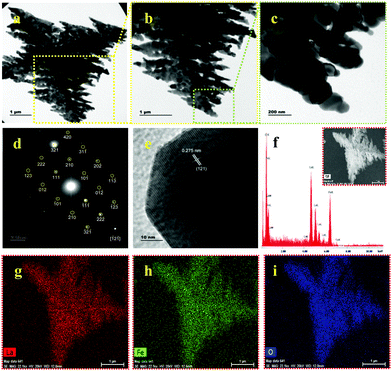 |
| | Fig. 4 (a–c) TEM images with increasing magnifications, (d) SAED pattern, (e) HRTEM with (121) plane, (f) EDS spectrum and (g–i) EDX element mapping of LaFeO3 dendrites (U9P15). Inset (f): FESEM image of LaFeO3 dendrites. | |
The XPS spectrum in Fig. 5a shows the survey scan of the surface state of LaFeO3, which contains La 3d, Fe 2p, O 1s and reference C1s peaks. The deconvoluted La 3d spectrum in Fig. 5b consists of two sets of peaks, where the lower binding energy set at 833.9 eV and 838.0 eV is attributed to La 3d5/2 and the satellite peak and the higher binding energy set at 850.8 eV and 854.8 eV is La 3d3/2 and the satellite peak, respectively. This information about the core level of La indicates that the La ions exhibit the La3+ valence state in the LaFeO3 dendrites. Fig. 5c shows the Fe 2p spectrum with two main peaks at 710.2 eV and 724.1 eV for Fe 2p3/2 and Fe 2p1/2, and the corresponding satellite peaks are observed at 718.6 eV and 732.6 eV, respectively. Thus, the peak fittings confirmed that the Fe ions are in the form of mixed valence states of Fe3+ and Fe4+ in the LaFeO3 dendrites. The presence of Fe4+ is due to the weakly pre-adsorbed oxygen species (O− and O2−) on Fe3+ site. The O 1s spectrum with two peaks at 529.3 eV and 531.3 eV in Fig. 5d confirms the presence of lattice oxygen and weakly bound surface oxygen in the LaFeO3 dendrites, respectively. The presence of surface oxygen and the interesting LaFeO3 dendritic morphology can enhance the sensing property, but information on the surface area is essential for any gas sensing material. Therefore, the N2 adsorption–desorption isotherm and Brunauer–Emmett–Teller (BET) plot were measured, as shown in Fig. 6a (inset: BET plot). The measured specific surface area value is 4.40 m2 g−1. Fig. 6b shows the Barrett–Joyner–Halenda (BJH) plot and the measured pore volume is 1.22 nm, where this low value is due to the high crystalline orientation.
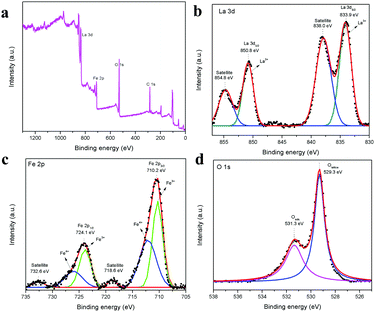 |
| | Fig. 5 XPS spectra of LaFeO3 dendrites (U9P15), (a) survey scan, (b) La 3d, (c) Fe 2p, and (d) O 1s. | |
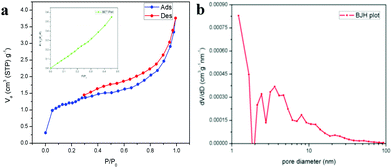 |
| | Fig. 6 (a) N2 adsorption–desorption isotherms (inset: BET plot) and (b) BJH plot for LaFeO3 dendrites (U9P15). | |
3.3 Acetone sensing properties
The planar micrometer-sized LaFeO3 dendrites can interact well with acetone molecules. Thus, a sensing device for the trace level detection of acetone was fabricated using the LaFeO3 dendrites, as described in the Experimental section. Here, N2 was used as a carrier gas throughout the sensing measurement because of its insensitive behaviour with the LaFeO3 dendrites. The whole sensing arrangement is shown in Fig. S2 (ESI†). The device and chamber temperatures were kept at 100 °C and the acetone sensing experiment was conducted by varying the acetone concentration from 10 ppm to 0.01 ppm (10 ppb). These concentrations were obtained using a stock solution of 5% acetone in water because in industrial waste and human breath acetone is always mixed with high water concentrations. Further, the stock solution was diluted to low level concentrations for easy sample handling. Fig. 7a–c show the measured sensing response (S = Rg/Ra) curves obtained by monitoring the changes in the device base resistance value upon exposure to different concentration of acetone. The dynamic sensing response curve in Fig. 7a shows the sensitivity in the range of 10 ppm to 1 ppm at 1 ppm intervals in decreasing order and the corresponding values are 157.88, 139.09, 127.02, 115.36, 111.17, 102.84, 91.58, 87.84, 76.87 and 37.63, respectively. This response curve indicates the fact that a decrease in acetone concentration led to a decrease in the sensor response value because of the reduced interaction of acetone over the sensing surface. Fig. 7b shows the dynamic sensing response in the range of 2 ppm to 0.1 ppm at 0.2 ppm intervals in decreasing order, and the sensitivity values are 76.87, 69.09, 63.91, 50.45, 42.48, 37.63, 30.25, 25.51, 16.89, 10.49 and 2.90, respectively. This observation clearly shows that even 0.2 ppm variations in the acetone concentration have a great impact on the sensing response, and thus the device can easily monitor the environmental acetone in the workplace. Further, this sensing range covers the lower diabetes acetone limit of 1.8 ppm with a sensitivity value of 69.09, which is a very high value, and thus it can detect the breath-exhaled acetone from diabetic patients. Further, this sensor showed good sensitivity even at 0.1 ppm, therefore the acetone concentration was reduced to below 0.1 ppm to determine the lowest detection limit of this sensing device. The detection of trace acetone levels is also important for effective monitoring. Fig. 7c shows the trace level sensing response in the range of 0.1 ppm to 0.01 ppm acetone with a 0.01 ppm decrement and the resulting sensitivity values are 2.90, 2.36, 2.18, 1.73, 1.54, 1.42, 1.24, 1.20, 1.15 and 1.05, respectively. This result demonstrates that the current device can detect acetone molecules at the minimum concentration of 10 ppb, which is the lowest recorded limit for the detection of acetone at a low operating temperature (100 °C) compared to that in recent reports. The overall measured sensing response values are plotted in Fig. 7d to determine the device linearity. It can be seen that the response values in the range of 0.1 ppm to 2 ppm show the highest linear regression value (R2 = 0.99167), which suggests that this range of acetone molecules is optimum to interact well with the reactive oxygen species on the sensing surface. An increase in the concentration from 2 ppm to 10 ppm reduced the R2 value (0.96909) because of the lack of reactive oxygen species over the sensing surface. On the other hand, the low concentrations in the range of 0.1 ppm to 0.01 ppm in the inset Fig. 7d show the less linearity (R2 = 0.87916) due to the minimal acetone molecule interaction with the sensing material. Subsequently, the advantage of the dendritic morphology in the sensitivity of the sensor was also investigated by comparing its performance with an irregular LaFeO3 particle-based sensing device. The irregular LaFeO3 particle-based sensor was tested several times to determine the cross sensitivity. Fig. S3 (ESI†) shows the 1 ppm acetone sensing response using the LaFeO3 irregular particles at the device and chamber temperatures of 100 °C. The device exhibited the sensitivity value of S = 24.10, which is less than that of S = 37.63 for the LaFeO3 dendrites. This is because of less charge carrier transport from one particle to another. However, in the case of the LaFeO3 dendrites, the planar structure and the self-assembled grains (see Fig. 4c) improved acetone interaction and the carrier transport properties, respectively. Therefore it showed high sensitivity towards the target acetone molecules. Further, the irregular LaFeO3 particles showed a slow response and recovery time (Rs = 14.84 s and Rc = 104.62 s) compared to the LaFeO3 dendrites (Rs = 1.19 s and Rc = 95.81 s, see Fig. S10a (ESI†)), respectively, which is consistent with their slow interaction with the acetone molecules.
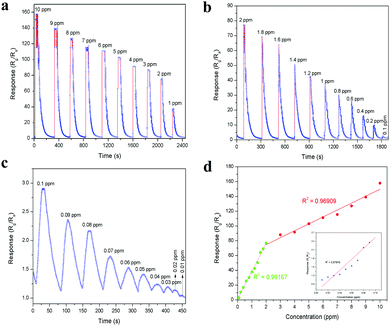 |
| | Fig. 7 Dynamic acetone sensing response in the range of (a) 10–1 ppm, (b) 2–0.1 ppm and (c) 0.1–0.01 ppm at 100 °C device and chamber temperature. (d) Concentration-dependent acetone sensing response with linear regression fits in the range of 0.1–2 ppm and 2–10 ppm (inset: 0.01–0.1 ppm). | |
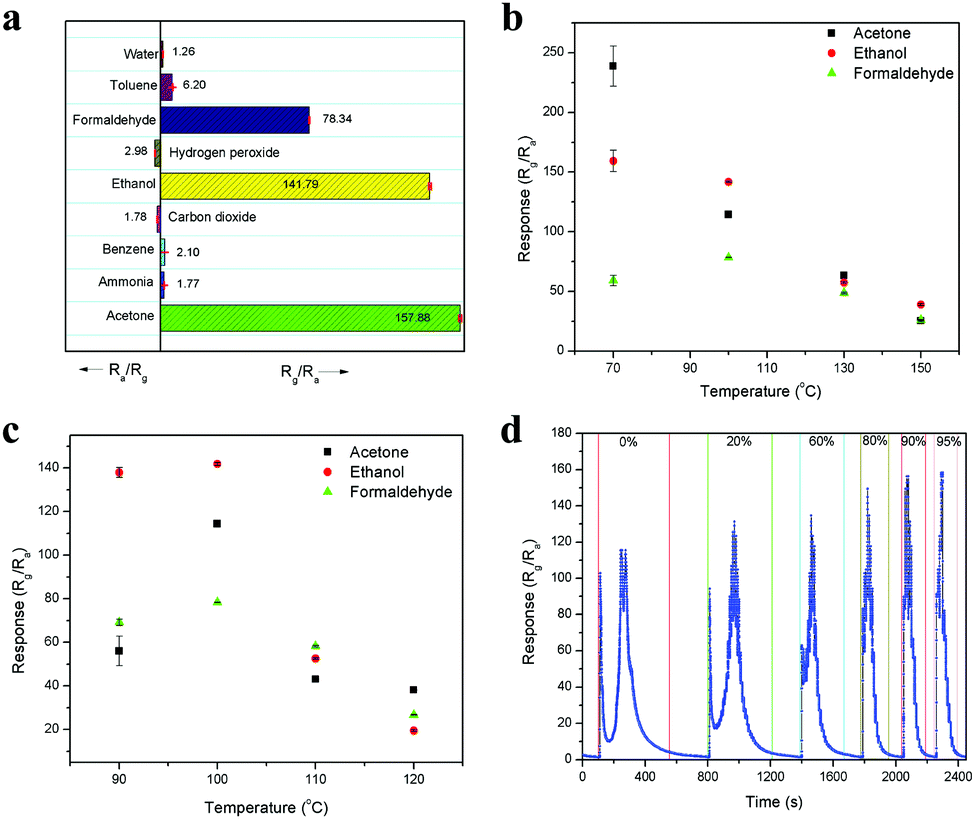
Selectivity is also a major concern in any sensing device, and thus the selectivity of this sensor was tested with 10 ppm of various analyte molecules, including acetone, ammonia, benzene, ethanol, formaldehyde, toluene, water (1000 ppm), carbon dioxide (1000 ppm) and hydrogen peroxide. The sensitivity values for these analyte molecules are 157.88, 1.77, 2.10, 141.79, 78.34, 6.20, 1.26, 1.78 and 2.98, respectively, and they are compared in Fig. 8a. Among them, acetone showed the highest sensitivity because it produced more electrons on the sensor surface upon interaction, which interacted with the majority of charge carriers, leading to an enhancement in the sensing response. In addition, the presence of weakly pre-adsorbed oxygen species (O− and O2−) on the Fe site in the LaFeO3 surface can interact well with the acetone molecules and produce high sensitivity.42 Thus, based on the sensitivity, the selective detection can be identified. Further, it is noted that the oxidizing species such as hydrogen peroxide and carbon dioxide showed the opposite sensing response to the other molecules because these molecules accept the surface electrons and leave holes on the sensing surface, causing an increase in conductivity, which confirms the p-type semiconducting property of the LaFeO3 dendrites. It was noted that the analyte molecules ethanol and formaldehyde showed significant sensitivity values compared with acetone. However, based on their vapourization temperature, the selective acetone detection was improved by varying the chamber and device temperatures. The operating temperature is highly important for any sensing device to achieve high sensitivity.43,44Fig. 8b shows the sensitivities of 10 ppm pure acetone, ethanol and formaldehyde at different chamber temperatures (70–150 °C) with a constant device temperature (100 °C). The chamber temperature of 70 °C showed the highest acetone sensitivity due to the low boiling point of acetone (56.06 °C), which vapourized acetone more for it to interact with the sensing surface. In contrast, ethanol (78.24 °C) and formaldehyde (100 °C) have a higher boiling point than the chamber temperature, and thus they could not show similar sensitivity. When the chamber temperature was increased to 100 °C, a decrease in acetone and ethanol sensitivity was observed, whereas an improved sensitivity value was observed for formaldehyde, which can be due to their equal boiling points with the chamber temperature. A further increase in the temperature to 130 °C and 150 °C led to a decrease in the sensitivity for all three vapours because of the fast vapourization, which caused the analytes to be immediately flushed out from the chamber by the carrier gas. The fast vapourization was reflected by the decrease in the response/recovery times upon an increase in chamber temperature. It was observed that there was a decrease in the response/recovery time for 10 ppm acetone upon increasing the chamber temperature from 70 °C to 150 °C, with the values of 2.77 s/627.04 s, 1.83 s/374.80 s, 1.16 s/312.83 s and 0.83 s/262.16 s, as obtained from Fig. S4a–d (ESI†), respectively. Similarly, Fig. S5a–d (ESI†) show that the response/recovery times for 10 ppm ethanol were 1.49 s/376.78 s, 8.07 s/317.07 s, 7.47 s/270.53 s and 5.70 s/194.22 s upon increasing the chamber temperature from 70 °C to 150 °C, respectively. Here, the unsuitable response time at 70 °C is due to the partial vapourization of ethanol. In the case of 10 ppm formaldehyde, the sensing response/recovery times are 34.02 s/256.59 s, 7.98 s/200.17 s, 7.00 s/108 s and 4.64 s/83.65 s, for the above temperature range and the sensing curves are shown in Fig. S6a–d (ESI†), respectively.
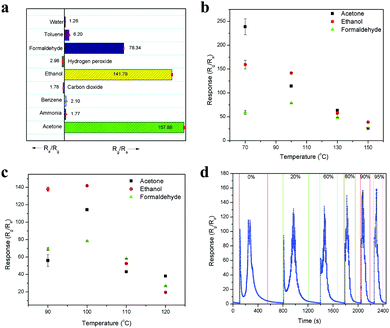 |
| | Fig. 8 (a) Sensing response of 10 ppm of various chemical species. (b) Chamber and (c) device temperature-dependent sensing response to 10 ppm of pure acetone, ethanol and formaldehyde. (d) Dynamic sensing response for 10 ppm concentration of pure acetone and acetone in different percentages of water. | |
In another set of experiments, the chamber temperature was kept at 100 °C but the device temperature was varied from 90 °C to 120 °C at increments of 10 °C, and the responses are shown in Fig. 8c. At the device temperature of 90 °C, ethanol showed high sensitivity due to the sufficient chemical activation of the sensing material for ethanol molecules. However, an increase in temperature to 100 °C enhanced the sensitivity of all three molecules because of suitable surface condition was maintained for the all the analyte molecules to interact effectively on the surface of the device. A further increase in temperature to 110 °C and 120 °C led to an unfavorable surface condition for the analyte molecules to interact, resulting in low sensitivity. This device temperature-dependent sensing behavior can be explained according to the adsorption and desorption kinetics on the sensing surface. When the device temperature is low, the chemical activation for the adsorption of oxygen species (O− and O2−) on the sensing surface is low, which results in lower conductivity. Then, an increase in temperature increases the oxygen species on the sensing surface, which leads to an increase in the sensor conductivity because the oxygen species chemically interact with electrons from the material, leaving holes on its surface. These holes are responsible for the conductivity of the p-type semiconducting LaFeO3 dendrites. Further, the oxygen species are responsible for the analyte molecule adsorption and desorption process on the sensing material surface. The adsorption and desorption kinetics can be explained based on the response and recovery times. The sensing response of 10 ppm acetone obtained at different device temperatures of 90 °C, 100 °C, 110 °C and 120 °C are shown in Fig. S7a–d (ESI†), respectively. The response curves confirmed that an increase in device temperature led to a reduction in the base resistance, with the values of 3.1 × 106, 1.3 × 106, 3.5 × 105 and 2.3 × 105 Ω respectively. The response/recovery times for acetone were 2.35 s/2925.37 s, 1.83 s/374.80 s, 1.74 s/182.43 s and 1.63 s/174.65 s at 90 °C, 100 °C, 110 °C and 120 °C, respectively. The increase in device temperature led to an enhancement in the acetone adsorption/desorption process, which resulted in a decrease in the response/recovery time. In the case of the ethanol, the response/recovery times at the above temperatures are 5.05 s/3194.23 s, 8.07 s/317.07 s, 8.20 s/226.19 s and 8.95 s/179.35 s, as obtained from Fig. S8a–d (ESI†), respectively. Here, there was an increase in the response time and decrease in the recovery time, which can be due to the slow interaction and fast desorption of ethanol from the sensor surface. This contradictory result of slow interaction upon an increase in device temperature confirms that the adsorption kinetics depends on the analyte molecules, and thus high sensitivity was observed for ethanol at 90 °C. The formaldehyde sensing response curves are shown in Fig. S9a–d (ESI†) and the response/recovery times are 10.67 s/1071.98 s, 7.98 s/200.17 s, 7.90 s/173.36 s and 7.13 s/111.07 s for 90 °C, 100 °C, 110 °C and 120 °C, respectively. Therefore, a similar response was observed for formaldehyde to that for acetone upon an increase in device temperature. It was noted that the device temperature of 90 °C is not enough to desorb all three analytes molecule, and thus their recovery times are very high. By comparing the above temperature-dependent sensitivities, it was found that 90 °C chamber and 100 °C device temperatures can deliver the highest acetone sensitivity among the molecules for the selective detection of acetone. However, the recovery time was greater compared to that other temperatures. Nevertheless, the chamber and device temperature of 100 °C showed relatively good results in all aspects, and thus considered as optimized temperature.
The effect of water interference in acetone was also tested because in the workplace and clinical samples, acetone in mostly in diluted form with high water concentrations. Fig. 8d shows the dynamic sensing response of 10 ppm concentration of pure acetone and acetone diluted with different percentages of water including 20%, 60%, 80%, 90% and 95%. The resulting sensitivity values are 116.12, 126.10, 133.60, 145.05, 156.98 and 157.88, respectively. The increase in sensitivity upon an increase in water percentage is due to the formation of more oxygen species on the surface, which helps more acetone molecules interact on the surface. This is because the water vapour predominantly interacts with the surface lattice oxygen of LaFeO3, leading to the formation of oxygen vacancies, which deliver electrons to the surface. These electrons are the active sites for the oxygen molecules and interact with them to produce surface adsorbed oxygen species. This process was confirmed when water alone reacted with the LaFeO3 sensing device to produce an increase in resistance due to the donation of an electron to the p-type semiconducting material (Fig. 8a). Further, the sensitivity value was very low with even 1000 ppm water, which confirms that the electrons are immediately trapped by oxygen molecules to produce oxygen species, which causes an increase in the amount of holes and recovery back to the base resistance. Thus, this device can deliver high acetone sensitivity under high water vapour, which is very favourable for the detection of industrial waste and breath exhaled acetone with a high water concentration. This sensing behaviour is correlated with recently reported results.24,45 Further, it was observed that the sensitivity values for 90% and 95% water were very close, which confirms that saturation level was reached. Thus, 95% water with acetone is considered as an optimized amount because under this condition, any increase in sensitivity is only from acetone. Furthermore, there were variations in the sensing curve from a split peak to a single peak for pure acetone with an increase in the water percentage. These changes in the peak profile are due to the interaction of water with the acetone by-product CO2 and production of carbonic acid, which reduces the oxidizing species CO2, and thus no decrease in sensitivity was observed in the peak profile upon an increase in water percentage. This interesting behaviour can be used to quantify the acetone percentage in industrial waste and breath-exhaled acetone. The other advantage with an increase in the water percentage is that the water can physically interact more on the sensing surface, which causes acetone to desorb faster. Therefore, the overall sensing time, which includes the response, saturation and recovery times, was reduced from pure acetone to acetone with an increase in water percentage, with the values of 452.90 s, 408.72 s, 280.93 s, 177.27 s, 152.54 s and 147.83 s, respectively.
3.4 Sensing mechanism
Under atmospheric conditions, the p-type semiconducting LaFeO3 dendrites chemically interact with oxygen molecules to produce chemisorbed oxygen species (O− and O2−) on their surface. The chemisorbed oxygen species accept electrons in the sensing surface and generate holes, as shown in eqn (22). These holes create a hole accumulation layer on the surface of the LaFeO3 dendrites. This process improved the conductivity of the sensing device. A detailed schematic illustration is shown in Fig. 9a.| | | O2(g) + 2e− → 2O(ads)− + 2h+ | (22) |
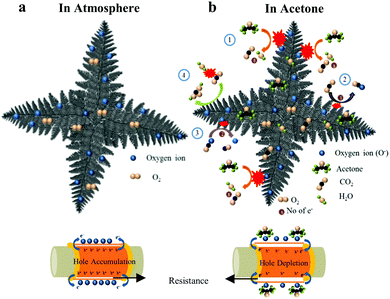 |
| | Fig. 9 Schematic sensing mechanism of LaFeO3 dendritic surface (U9P15) in (a) atmosphere and (b) acetone environment. | |
In an acetone environment, the interaction of acetone molecules over the sensing surface can be explained by four reaction mechanisms, as shown in Fig. 9b. Initially, the acetone interacts with the surface oxygen species and delivers CO2 and electrons (eqn (23)). The electrons recombine with the holes, leading to a decrease in the majority charge carrier (holes) on the surface of the LaFeO3 dendrites (eqn (24)). Thus, there is a sudden increase in resistance in the sensing device. Simultaneously, the by-product CO2(g) accepts the electron from the sensing material and turns into a CO2(ads)− ion (eqn (25)). Then, the CO2(ads)− ion dissociates into CO and O2− upon interaction with the surface oxygen species and an electron from the material (eqn (26)). The above two processes generate holes, which induce an immediate decrease in resistance. However, this does not last long, and thus an increase in resistance is observed because of the increase in product electrons from acetone compared to the CO2 reactant electrons. Finally, the acetone is flushed out by the N2 carrier gas and the base resistance is recovered. Therefore, a pure acetone molecule produces a split sensing peak. However, in the presence of water, acetone shows a single peak (Fig. 8d). This is because the water vapour controls the availability of CO2 by converting it into carbonic acid, and thus no split sensing peak can be observed (eqn (27)).
| | | CH3COCH3(g) + 8O(ads)− → 3CO2(g) + 3H2O + 8e− | (23) |
| | | Recombination process: e− + h+ → null | (24) |
| | | CO2(g) + e− → CO2(ads)− | (25) |
| | | CO2(ads)− + O(ads)− + 2e− → CO(g) + 2O(ads)2− | (26) |
3.5 Device stability
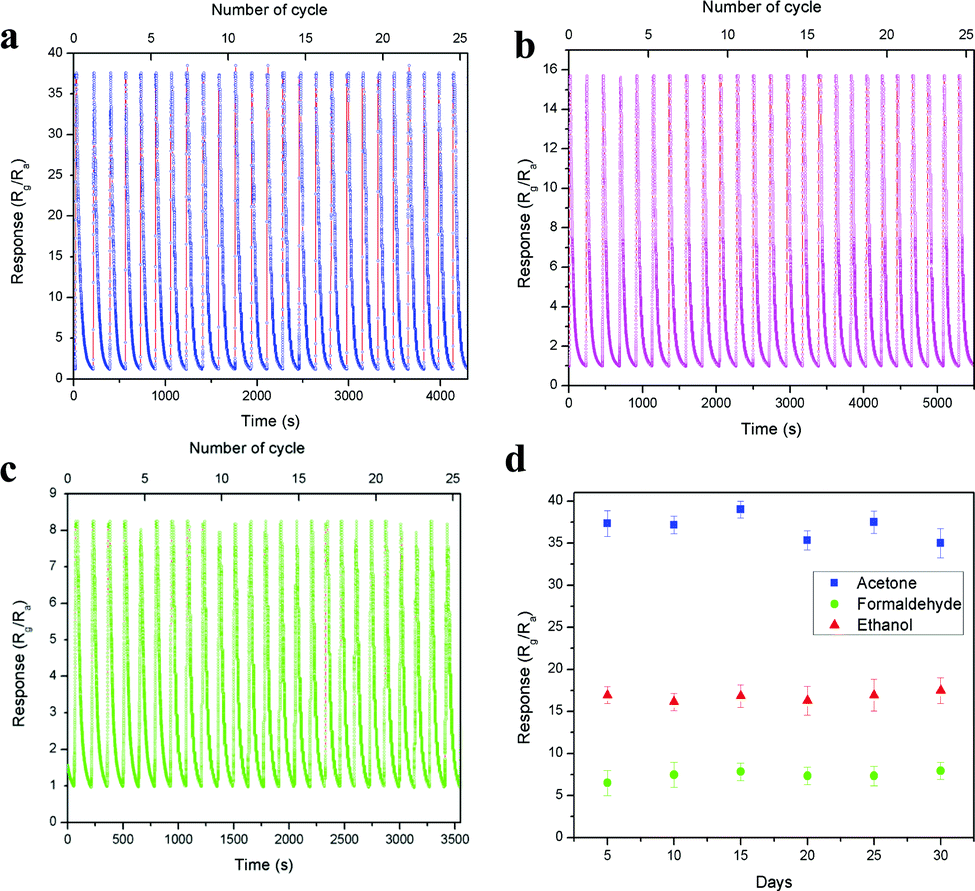
The device short term stability was evaluated through the cyclic sensing responses of all three vapours for 25 cycles over 1 ppm concentration, as shown in Fig. 10a–c. The sensitivity values of acetone, ethanol and formaldehyde are 37.63, 15.74 and 8.29 and the corresponding response/recovery times are 1.19 s/95.81 s, 8.35 s/150.81 s and 12.62 s/68.23 s (Fig. S10a–c, ESI†), respectively. All three vapours showed good stability over 25 cycles in a single measurement. Here, acetone showed more than 2-fold and 4-fold sensitivity compared with ethanol and formaldehyde, respectively. Further, the long-term stability of the device was also tested over 1 ppm acetone, ethanol and formaldehyde, as shown in Fig. 10d. The result showed that the device performed well and was not affected by any surface oxidation process. The stability of the base resistance and the sensing response for 1 ppm acetone on the 1st and 30th day remained the same, and the graph is shown in Fig. S11 (ESI†). The above results exhibit that the LaFeO3 dendrites displayed good sensitivity, fast response time, low operating temperature and real-time detection of trace-level acetone. These results are compared with various recently reported acetone sensors in Table S2 (ESI†).
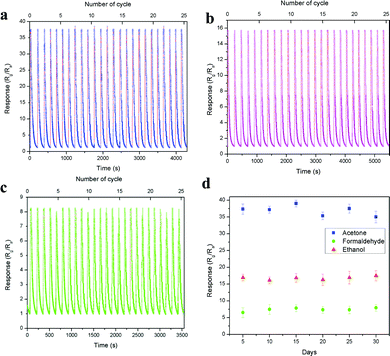 |
| | Fig. 10 (a–c) Short and (d) long term device stability over 1 ppm of acetone, ethanol and formaldehyde, respectively. | |
4. Conclusions
LaFeO3 dendrites were tailored through the hydrothermal approach in the presence of urea and piperazine hexahydrate. The detailed reaction mechanisms confirmed that the urea and piperazine hexahydrate helped to control the La2O3/Fe2O3 impurity phases and induced the growth of the LaFeO3 dendrites, respectively. By utilizing the LaFeO3 dendrites, a sensing device was fabricated for the trace level detection of acetone at low temperature, and the optimized device and chamber temperatures were 100 °C. The present sensor exhibited the highest sensitivity value of S = 157.88 at 10 ppm acetone. Additionally, the LaFeO3 dendrite-based device exhibited excellent sensitivity, fast response/recovery time, reproducibility and stability over an acetone concentration of 1 ppm. Further, trace level (10 ppb) acetone detection was also achieved. The present work discussed the selective detection of acetone through the variation in the chamber and device temperatures. This device possesses high sensitivity and fast recovery time under high water interference, which is beneficial for the detection of acetone in industry and exhaled breath with high water concentrations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors K. Palani Thiruppathi and Devaraj Nataraj would like to acknowledge the RUSA Phase – II Program, Government of India for financial support. The author K. Palani Thiruppathi extends to acknowledge Defense Research Development Organization, Government of India under DRDO – Bharathiar University Centre for Life sciences (DRDO – BU CLS) for using their resources. The author Devaraj Nataraj acknowledges the RUSA Phase I Program (Component – 8, Research & Innovation) and (UGC-CPEPA) Centre for Advanced Studies in Physics for the development of Solar Energy Materials and Devices (2-8/2016(NS/PE)) for providing financial support to develop the characterization facilities. K. Palani Thiruppathi thanks to Dr. K. Kadirvelu for providing SEM and XRD facilities and also thanks Dr. G. Radha, Mrs. M. Srividhya and Miss. S. Umamaheswari for their contribution towards scientific discussion and manuscript preparation.
References
- S. Giannoukos, B. Brkic, S. Taylor, A. Marshall and G. F. Verbeck, Chem. Rev., 2016, 116, 8146 CrossRef CAS.
- G. Konvalina and H. Haick, Acc. Chem. Res., 2014, 47, 66 CrossRef CAS.
- N. Makisimovich, V. Vorotyntsev, N. Nikitina, O. Kaskevich, P. Karabun and F. Martynenko, Sens. Actuators, B, 1996, 36, 419 CrossRef CAS.
- W. Cao and Y. Duan, Clin. Chem., 2006, 52, 800 CrossRef CAS.
- M. D. Campbell, T. Sathish, P. Z. Zimmet, K. R. Thankappan, B. Oldenburg, D. R. Owens, J. E. Shaw and R. J. Tapp, Nat. Rev. Endocrinol., 2020, 16, 395 CrossRef CAS.
- Y. Zheng, S. H. Ley and F. B. Hu, Nat. Rev. Endocrinol., 2018, 14, 88 CrossRef.
- M. Wang, Z. Shen, Y. Chen, Y. Zhang and H. Ji, CrystEngComm, 2017, 19, 6711 RSC.
- M. Chen, Y. Zhang, J. Zhang, K. Li, T. Lv, K. Shen, Z. Zhu and Q. Liu, J. Mater. Chem. C, 2018, 6, 6138 RSC.
- J. Qin, Z. Cui, X. Yang, S. Zhu, Z. Li and Y. Liang, Sens. Actuators, B, 2015, 209, 706 CrossRef CAS.
- N. Zhang, S. Ruan, Y. Yin, F. Li, S. Wen and Y. Chen, ACS Appl. Nano Mater., 2018, 1, 4671 CrossRef CAS.
- Y. Zhang, J. Zhang, J. Zhao, Z. Zhu and Q. Liu, Phys. Chem. Chem. Phys., 2017, 19, 6973 RSC.
- Z. Dai, C. S. Lee, B. Y. Kim, C. H. Kwak, J. W. Yoon, H. M. Jeong and J. H. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 16217 CrossRef CAS.
- F. Deganello, M. L. Tummino, C. Calabrese, M. L. Testa, P. Avetta, D. Fabbri, A. B. Prevot, E. Montoneri and G. Magnacca, New J. Chem., 2015, 39, 877 RSC.
- K. Manseki, A. Amelia and T. Sugiura, CrystEngComm, 2018, 20, 6382 RSC.
- R. Zhang, P. Li, N. Liu, W. Yue and B. Chen, J. Mater. Chem. A, 2014, 2, 17329 RSC.
- P. Xiao, J. Zhu, D. Zhao, Z. Zhao, F. Zaera and Y. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 15517 CrossRef CAS.
- M. D. Scafetta and S. J. May, Phys. Chem. Chem. Phys., 2017, 19, 10371 RSC.
- X. Ren, H. Yang, S. Gen, J. Zou, T. Yang, X. Zhang, Z. H. Cheng and S. Sun, Nanoscale, 2016, 8, 752 RSC.
- K. J. May, D. P. Fenning, T. Ming, W. T. Hong, D. Lee, K. A. Stoerzinger, M. D. Biegalski, A. M. Kolpak and Y. S. Horn, J. Phys. Chem. Lett., 2015, 6, 977 CrossRef CAS.
- L. M. Misch, A. Birkel, C. A. Figg, B. P. Fors, C. J. Hawker, G. D. Stucky and R. Seshadri, Dalton Trans., 2014, 43, 2079 RSC.
- Y. Zhang, Q. Liu, J. Zhang, Q. Zhu and Z. Zhu, J. Mater. Chem. C, 2014, 2, 10067 RSC.
- S. Thirumalairajan, K. Girija, V. Ganesh, D. Mangalaraj, C. Viswanathan and N. Ponpandian, Cryst. Growth Des., 2013, 13, 291 CrossRef CAS.
- X. Zhang, D. Song, Q. Liu, R. Chen, J. Liu, H. Zhang, J. Yu, P. Liu and J. Wang, CrystEngComm, 2019, 21, 1876 RSC.
- M. Imran, S. S. A. A. H. Rashid, Y. Sabri, N. Motta, T. Tesfamichael, P. Sonar and M. Shafiei, J. Mater. Chem. C, 2019, 7, 2961 RSC.
- H. Zhang, H. Qin, P. Zhang and J. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 15558 CrossRef CAS.
- R. Zhang, T. Zhou, L. Wang and T. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 9765 CrossRef CAS.
- T. Zhou, X. Liu, R. Zhang, Y. Wang and T. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 37242 CrossRef CAS.
- H. Fu, X. Wang, P. Wang, Z. Wang, H. Ren and C. Wang, Dalton Trans., 2018, 47, 9014 RSC.
- P. Shankar and J. B. B. Rayappan, ACS Appl. Mater. Interfaces, 2016, 8, 24924 CrossRef CAS.
- N. K. Chaudhari, H. C. Kim, D. Son and J. S. Yu, CrystEngComm, 2009, 11, 2264 RSC.
- C. Hou, W. Feng, L. Yuan, K. Huang and S. Feng, CrystEngComm, 2014, 16, 2874 RSC.
- L. Wang, Y. Sun, S. Zeng, C. Cui, H. Li, S. Xu and H. Wang, CrystEngComm, 2016, 18, 8072 RSC.
- L. Hui, D. Jia, H. Yu, Y. Xue and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11(3), 2618 CrossRef CAS.
- G. Wang, H. Wang, J. Bai, Z. Ren and J. Bai, Chem. Eng. J., 2013, 214, 386 CrossRef CAS.
- W. Zheng, R. Liu, D. Peng and G. Meng, Mater. Lett., 2000, 43, 19 CrossRef CAS.
- P. Fleming, R. A. Farrell, J. D. Holmes and M. A. Morris, J. Am. Ceram. Soc., 2010, 93, 1187 CrossRef CAS.
- B. P. Gangwar, V. Palakollu, A. Singh, S. Kanvah and S. Sharma, RSC Adv., 2014, 4, 55407 RSC.
- W. M. Al-Asbahy, M. Usman, F. Arjmand, M. Shamsi and S. Tabassum, Inorg. Chim. Acta, 2016, 445, 167 CrossRef CAS.
- X. Cao, P. Deng, S. Hu, L. Ren, X. Li, P. Xiao and Y. Liu, Nanomaterials, 2018, 8, 336 CrossRef.
- P. Saranya, S. Swarnalatha and G. Sekaran, RSC Adv., 2014, 4, 34144 RSC.
- D. B. Hassan, W. Rekik, H. Naıli and T. Mhiri, Chem. Pap., 2014, 68, 210 CAS.
- Y. Chen, H. Qin, X. Wang, L. Li and J. Hu, Sens. Actuators, B, 2016, 235, 56 CrossRef CAS.
- W. Zhou, Y. P. Wu, J. Zhao, W. W. Dong, X. Q. Qiao, D. F. Hou, X. Bu and D. S. Li, Inorg. Chem., 2017, 56, 14111 CrossRef CAS.
- X. Q. Qiao, Z. W. Zhang, D. F. Hou, D. S. Li, Y. Liu, Y. Q. Lan, J. Zhang, P. Feng and X. Bu, ACS Sustainable Chem. Eng., 2018, 6, 12375 CrossRef CAS.
- Y. J. Jeong, W. T. Koo, J. S. Jang, D. H. Kim, M. H. Kim and I. D. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 2016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 – detailed reactant concentrations and corresponding outcomes; Fig. S1 – XRD and SEM images of P15 sample. Fig. S2 – schematic sensing arrangement; Fig. S3 – irregular LaFeO3 particles based 1 ppm acetone sensing response. Fig. S4–S6 – 10 ppm of acetone, ethanol and formaldehyde sensing responses at different chamber temperatures (70–150 °C) with constant device temperature (100 °C); Fig. S7–S9 – 10 ppm of acetone, ethanol and formaldehyde sensing responses at different device temperatures (90–120 °C) with constant chamber temperature (100 °C); Fig. S10 – response/recovery time of 1 ppm (a) acetone, (b) ethanol and (c) formaldehyde at 100 °C device and chamber temperatures; Fig. S11 – comparison of 1st and 30th day acetone sensing response. Table S2 – comparison of recently reported acetone sensors with the present LaFeO3 dendrites based device. See DOI: 10.1039/d0ma00602e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Devaraj
Nataraj
ab and
Devaraj
Nataraj