
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assemblies of a 1,8-naphthalimide conjugate and its aggregation-induced emission property†
Souvik
Misra
 a,
Pijush
Singh
b,
Ankita
Das
c,
Paula
Brandão
d,
Pathik
Sahoo
e,
Nayim
Sepay
f,
Gourab
Bhattacharjee
g,
Pallab
Datta
c,
Ajit K.
Mahapatra
a,
Biswarup
Satpati
g and
Jayanta
Nanda‡
*a
a,
Pijush
Singh
b,
Ankita
Das
c,
Paula
Brandão
d,
Pathik
Sahoo
e,
Nayim
Sepay
f,
Gourab
Bhattacharjee
g,
Pallab
Datta
c,
Ajit K.
Mahapatra
a,
Biswarup
Satpati
g and
Jayanta
Nanda‡
*a
aDepartment of Chemistry, Indian Institute of Engineering Science and Technology, Shibpur, P.O. – Botanic Garden, Howrah-711103, West Bengal, India. E-mail: drjayantananda16@gmail.com
bDepartment of Biochemistry and Biophysics, University of Kalyani, Kalyani, Nadia, West Bengal, India
cCentre for Healthcare Science and Technology, Indian Institution of Engineering Science and Technology, Shibpur, P.O. – Botanic Garden, Howrah, India
dDepartamento de Química/CICECO, Universidade de Aveiro, 3810-193 Aveiro, Portugal
eInternational Center for Materials and Nanoarchitectronics (MANA) and Research Center for Advanced Measurement and Characterization (RCAMC), National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Japan
fDepartment of Chemistry, Jadavpur University, Jadavpur, Kolkata-700032, India
gSaha Institute of Nuclear Physics, 1/AF Bidhannagar, Kolkata, West Bengal 700064, India
First published on 11th November 2020
Abstract
The aggregation-induced emission (AIE) property has attracted considerable research interest since its discovery. Several small molecule-based AIE fluorogens (AIEgens) have shown unique properties. In this work, we are reporting for the first time a 1,8-naphthalimide (NMI) conjugate of a dipeptide, which is found to exhibit J-type aggregation in the solid and solution state, and also in the gel state. Unsubstituted naphthalimide units usually exhibit aggregation-caused quenching due to face-to-face stacking compared to ring-substituted (V-shaped) derivatives. Here, the amide hydrogen bonding interaction present in the molecule guides the packing of unsubstituted naphthalimide units in the crystal structure and it is consistent with J-type aggregation. The self-assembly and gelation properties of the NMI derivative were studied in several pure and water-miscible mixed solvents. In the mixed solvents, gelation is caused by the anti-solvent effect. Aggregation-induced emission properties were observed upon addition of water in the water-miscible solvent solutions of the gelator. Finally, the AIE property of the NMI-peptide conjugate was used extensively for live-cell imaging.
Introduction
In the last two decades, the aggregation-induced emission (AIE)1–7 properties of different fluorophore-based systems have been explored extensively owing to their wide-range of applications in different research areas including sensing, imaging, and theranostics. Most of the common and conventional organic aromatic fluorophores with planar structure show strong intermolecular H-type π–π stacking interactions and relax via a non-radiative pathway, leading to aggregation-caused quenching (ACQ). Using such organic fluorophores in the fabrication of organic light-emitting diodes (OLEDs), scientists faced adverse problems, as these materials are aggregated in the thin films state, leading to the quenching of the fluorescence emission. For the first time in 2001, Tang et al. discovered an interesting photophysical phenomenon which was opposite to the effect of ACQ, known as aggregation-induced emission (AIE).8 Following this discovery, Park et al. (in 2002) reported an aggregation-induced emission enhancement (AIEE) phenomenon,9 caused by the synergetic effect of J-type aggregation and planarization. Recently, 1,8-naphthalimide (NMI), a naphthalene core with N-imide functionality, is considered as a promising fluorophore for various AIE-based applications10–20 as the NMI moiety possesses extraordinary thermal and chemical stability with a high fluorescence quantum yield.21 In a recent review, Gopikrishna et al. summarized that ring-substituted NMI compounds, which adopt mainly a V-shape, are highly fluorescent in the aggregated state.21 On the contrary, planar unsubstituted NMI compounds, generally highly fluorescent in solution, undergo strong face to face stacking in the aggregated state, which leads to quenching of the fluorescence due to the ACQ effect.To date, very few report on aggregation-induced emission of unsubstituted NMI-based molecule is known in the literature specifically in the solid state.13 Liu and co-workers reported a 1,8-naphthalimide-based molecule which showed intense blue emission in the solid-state. The molecules are arranged in a J-type manner due to the presence of weak non-covalent interactions.13 In this manuscript, we are presenting for the first time an unsubstituted NMI-conjugate of a dipeptide (NMI-GW-OMe) which shows J-type aggregation22,23 and remarkable fluorescence properties in the solid state as well as in the self-assembled state.
Results and discussion
The chemical structure of NMI-GW-OMe is given in Scheme 1 and a detailed synthetic procedure and its characterization are reported in the ESI.† A light yellow coloured needle-shaped crystal was developed in CHCl3–methanol medium and the solid-state structure of NMI-GW-OMe was solved by the single crystal X-ray diffraction (SXRD) technique at 150 K. It crystallizes in the orthorhombic space group P212121 with unit cell dimensions of a = 4.8611(3) Å, b = 15.2401(11) Å, c = 28.170(2) Å and V = 2086.9(3) Å3 (ESI,† Table S1). In the solid-state, an intermolecular amide hydrogen bonding (N–H···O, see Table S2, ESI†) interaction is noticed. This strong hydrogen bonding interaction guides aromatic–aromatic interactions24 of unsubstituted NMI units (Fig. 1a). The π–π stacking interaction distances (centroid to centroid) between naphthalimide and naphthalimide units and benzopyrrole and benzopyrrole units of the Trp group are 4.86 Å in both cases. The slippage angle between the naphthalimide units is 46.37° (ESI,† Fig. S6), which is less than the magic angle, 54.7°. Such a type of molecular packing is consistent with J-type aggregates.22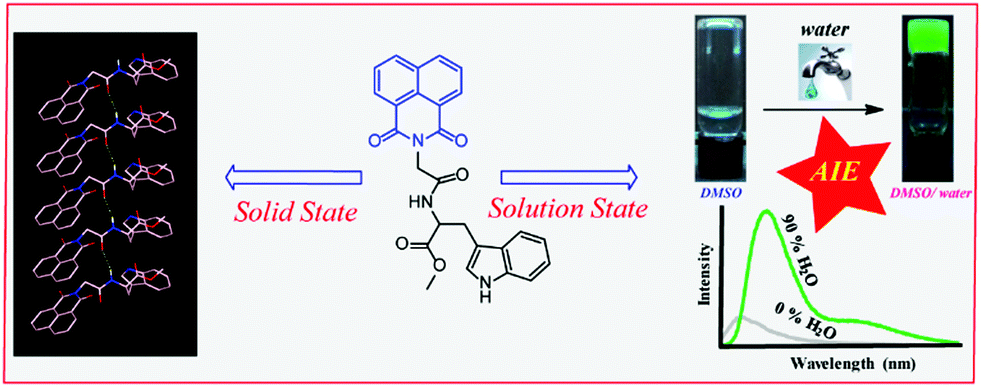 | ||
| Scheme 1 Chemical structure of the studied naphthalimide derivative (NMI-GW-OMe) and its supramolecular assemblies in the solid and solution state. | ||
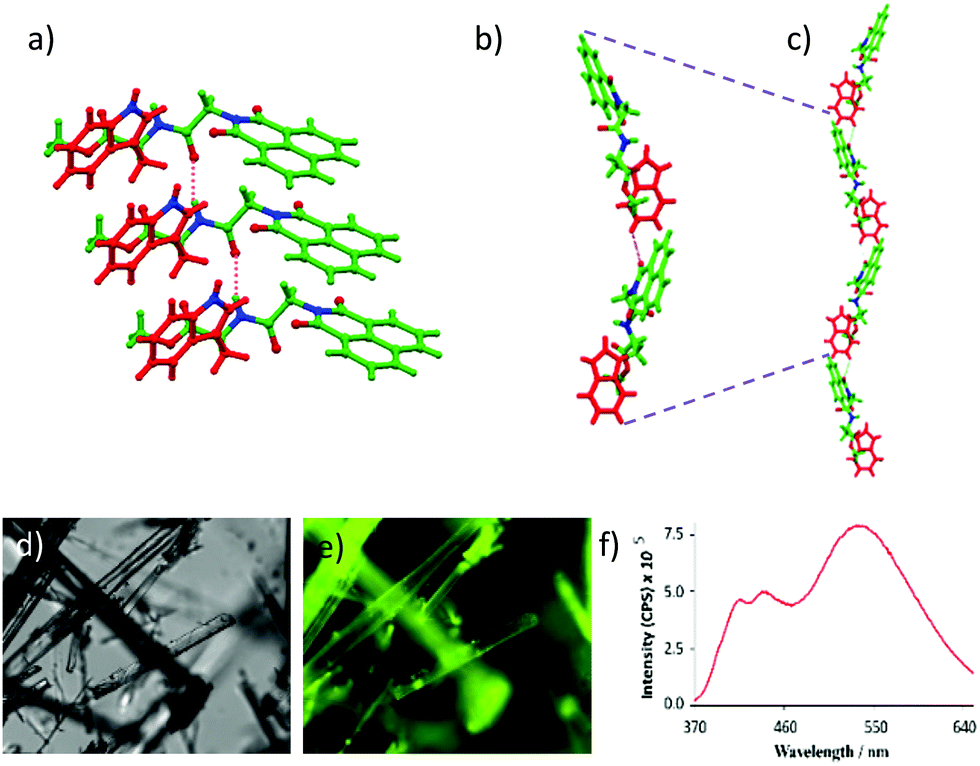 | ||
| Fig. 1 (a) Amide hydrogen bonding mediated J-type aggregation of NMI-GW-OMe in the solid-state. (b) Head to tail interactions of two NMI derivatives through C–H⋯O hydrogen bonding interactions (oxygen of the naphthalimide unit and tryptophan ring hydrogen). (c) Formation of a helical structure in the solid-state. (d) Optical and (e) fluorescence microscopic images of NMI-OMe crystals and (f) solid-state fluorescence spectra of NMI-GW-OMe at 30 °C. | ||
NMI-GW-OMe molecules are connected in a head to tail manner via weak intermolecular (C–H⋯O hydrogen bonding) interactions between the aromatic naphthalimide unit and the Trp moiety of another molecule. The distance between the oxygen of naphthalimide and the tryptophan ring hydrogen is 2.612 Å (Fig. 1b). On careful observation, it introduces a bend in the higher-order assembly, which leads to the formation of a supramolecular helical structure in the solid-state (Fig. 1c).
It is interesting to study the fluorescent property of a naphthalimide derivative which is arranged in the J-type fashion in the solid-state. The compound emitted (λex = 343 nm) characteristic monomeric peaks at 409 and 440 nm along with a more intense broad excimer peak at 539 nm.12 The highly fluorescent nature of the crystals was also visualized using a fluorescence microscope (Fig. 1e). Temperature-dependent solid-state fluorescence spectroscopy was performed to explore the thermochromic nature of the crystal. The intensity of the excimer peak at 539 nm was quenched with an increase in temperature from 30 to 100 °C. At 100 °C, the excimer peak (at 539 nm) was blue-shifted and its intensity reduced25 1.6 times compared to that at 30 °C (ESI,† Fig. S7). However, controlled-cooling leads to almost full retention of the fluorescence intensity and peak position.
Planar aromatic NMI group containing molecules are also known to form supramolecular gels26–31 due to strong aromatic–aromatic interactions among the naphthalimide units. We studied the self-assembly and gelation behaviour of NMI-GW-OMe in several common organic solvents (Table 1). Initially, the heating–cooling technique was used to trigger gelation. Samples were dissolved in organic solvents and left for cooling at room temperature. In most of the cases, the mixture remained as a solution upon cooling. However, in a few solvents (aromatic solvents and long-chain aliphatic alcohols) viscous precipitations were observed. Later, we noticed that sonication27,30,32–35 is an interesting stimulus to induce gelation in this system.
| Solvents | Physical state | Solvents | Physical state |
|---|---|---|---|
| Chloroform | S | N,N-Dimethyl formamide | S |
| Methanol | I | Dimethyl sulphoxide | S |
| n-Butanol | G (18 mM) | Tetrahydrofuran | S |
| n-Hexanol | G (16 mM) | 1,4-Dioxane | S |
| n-Octanol | G (13 mM) | Toluene | G (15 mM) |
| Allyl alcohol | S | Xylene | G (10 mM) |
| Benzyl alcohol | S | Chlorobenzene | G (13 mM) |
| Acetonitrile | S | Cyclopentanone | S |
Here, gel formation was noticed in these aromatic solvents and long-chain alcohols after sonication (ESI,† Fig. S8). The details of gelation of NMI-GW-OMe are given in Table 1.
Recently, several emerging properties26,36–43 of planar fluorophores have been observed in mixed solvents. One of them is supramolecular gelation.44–61 It is clear from the gelation table that the gelator is insoluble in water and soluble in DMSO. We serendipitously observed that the quick addition of water into DMSO solution led to the formation of a self-supported gel.40 This is an example of anti-solvent induced gelation.62 We then systematically studied the gelation properties of NMI-GW-OMe by gradually increasing (10%) the water fraction in each vial (left to right) containing the DMSO solution of the gelator [gelator conc. = 5 mM] (Fig. 2a and b). It was observed that with an increase in the water content (>50%), the solution became hazy and viscous. A self-supported supramolecular gel material was observed upon addition of 70% water in the solution. Moreover, on addition of higher fraction of water, self-supported gels were obtained. The gel formation was supported by a rheological experiment. In a frequency sweep experiment, the storage modulus (G′) value is higher than the loss modulus (G′′) value. This is indicative of supramolecular gel formation.32,63 The storage modulus value is in the order of 103 Pa. The gel formed after the addition of 90% water was also investigated by a similar experiment. The storage modulus values in both cases are comparable (Fig. 3).
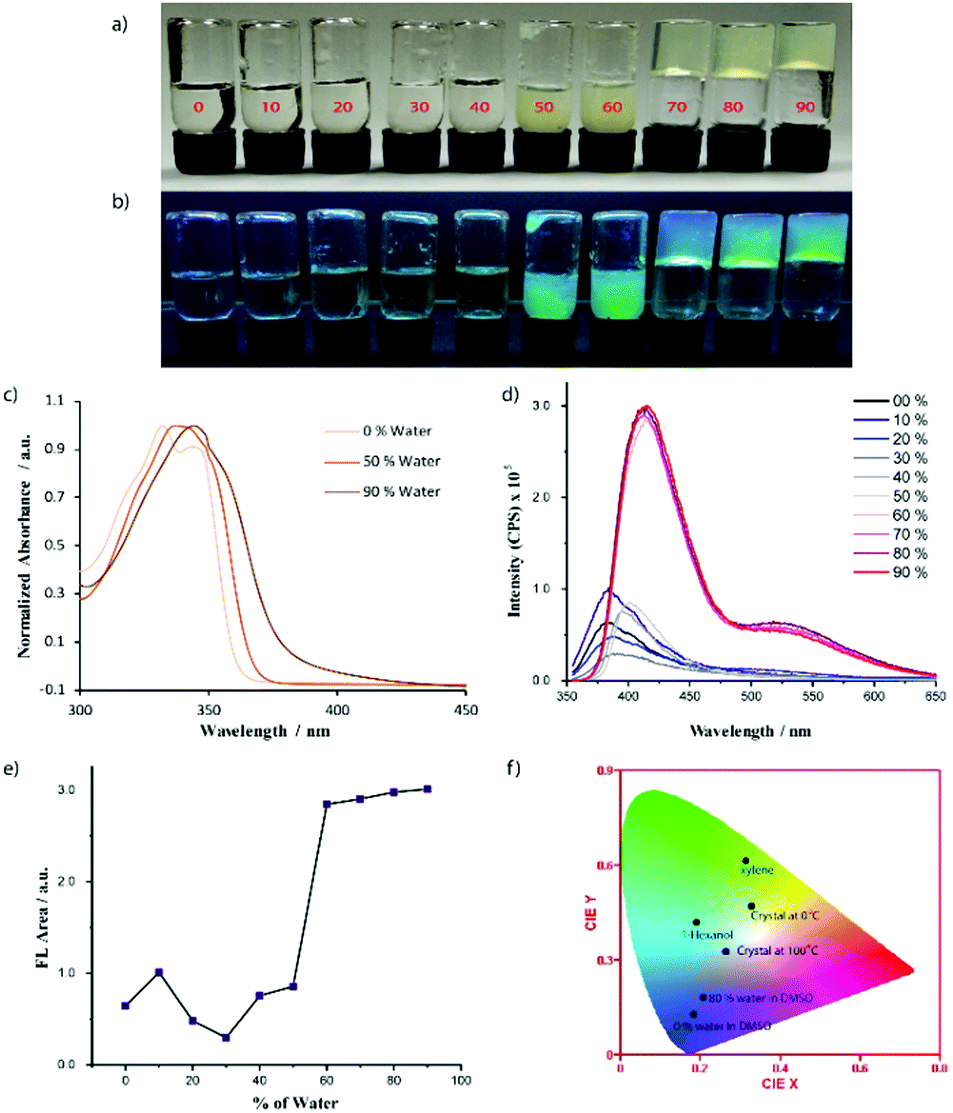 | ||
| Fig. 2 Solvent induced gelation properties of NMI-GW-OMe (5 mM) in a DMSO–water mixed solvent. From left to right the water content is increased. The % of water is written on the glass vial. Image of glass vials under (a) normal daylight and (b) UV-light. (c) Normalized absorbance spectra of NMI-GW-OMe at 40 μM. (d) Solvent-dependent fluorescence spectra of NMI-GW-OMe (3 mM). The % of water in DMSO is shown in the figure legends. Here, the AIE-property is noticed. (e) Area under the fluorescence spectra (Fig. 2d) of NMI-GW-OMe with the % of water in the DMSO solution. (f) CIE plot of the fluorescence spectra of NMI-GW-OMe in different assembled states. | ||
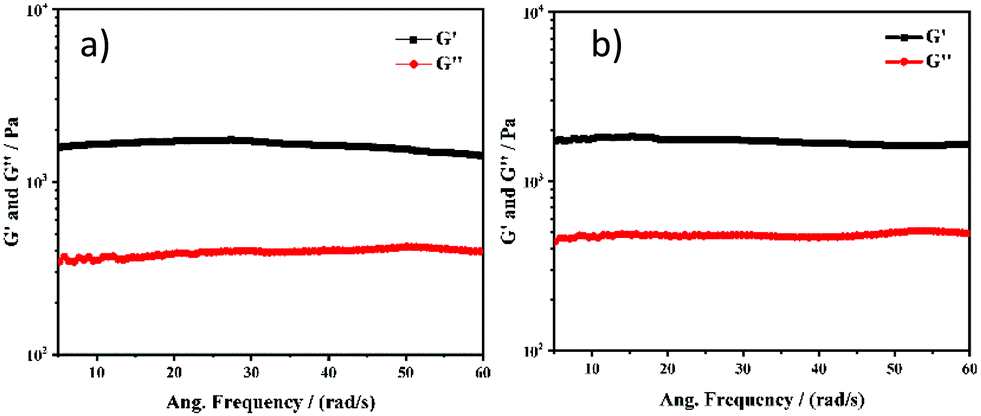 | ||
| Fig. 3 Frequency sweep experiments of NMI-GW-OMe based supramolecular gels prepared after addition of (a) 70% water and (b) 90% water in the DMSO solution of the gelator. | ||
We studied the gel formation by varying the concentration of the gelator and the % of added water in the DMSO–water solution. It was observed that the gelator concentration and water content varied inversely. Under UV light (365 nm), cyan-blue coloured fluorescence was observed in the aggregated solution and gel state (Fig. 2b). We then explored this process to check whether addition of water can also gelify the solution of the gelator in acetonitrile, DMF, and THF or not. In every case, self-supported gels were obtained (Fig. S9, ESI†). The details of the gelation study in mixed solvents is given in Table 2.
| % of water | DMSO | ACN | DMF | THF |
|---|---|---|---|---|
| 10 | S | S | S | S |
| 20 | S | S | S | S |
| 30 | S | S | S | S |
| 40 | S | S | S | S |
| 50 | V | V | S | S |
| 60 | V | G | V | V |
| 70 | G | G | G | G |
| 80 | G | G | G | G |
| 90 | G | G | G | G |
| 100 | I | I | I | I |
With an increase of the water content in the mixed solvents, the self-assembly properties of NMI-GW-OMe were enhanced and this motivated us to investigate the assembly properties using different spectroscopic techniques. NMI-GW-OMe showed an absorption maximum (λmax) at 333 nm in pure DMSO medium at 40 μM concentration. The molecules were in a non-aggregated state and this absorption maximum corresponded to the π–π* electronic transition of the naphthalimide unit. In the presence of 50% and 90% water in DMSO–water solution, the absorption maximum was shifted to 343 nm and 347 nm, respectively.
So, a red-shift of the absorption maximum in the UV-vis spectra was observed with the increase of water. In the presence of 90% water, the absorption maximum shifted almost 14 nm. This red-shift can be considered as a J-type interaction20,64–66 among the naphthalimide units. The addition of water into a DMSO solution of the gelator prompts the self-assembly process through π–π stacking interactions, which favours the red-shift of the absorption peak. To check the stability of the self-assembled structure, we performed a variable-temperature UV-vis study of the gelator (90% water in DMSO–water solution) and found that the spectral signature of aggregated NMI-GW-OMe remained almost invariant even at 50 °C, suggesting its appreciable thermal stability (Fig. S10, ESI†). We studied the circular dichroism (CD) spectra of the gelator in pure DMSO and DMSO–water mixed solvent. Though a chiral amino acid is present in the system, the CD spectrum is silent in pure DMSO. On addition of water into the DMSO solution of the gelator, new peaks were evolved (Fig. S11, ESI†). This is an example of chiral induction in the supramolecular state.28,29,67
The solvent-dependent fluorescent property of NMI-GW-OMe was studied in a mixed solvent (DMSO–water) at 3 mM concentration (Fig. 2d). In DMSO, the emission spectrum exhibited a maximum at 386 nm. In the presence of 50% water in the DMSO–water solution, the maximum of the emission spectrum was shifted to 402 nm. However, a drastic change in the emission spectrum was noticed with a further increase of the water content in the solution. In the presence of 60% water, the intensity of the emission spectrum was significantly enhanced. The maximum of the spectrum was further shifted to 416 nm along with a broad excimer peak at 527 nm. Similar observations were noticed with a further increase in the water content. Here, AIE phenomena were noticed in the presence of a higher content of water. This is an example of a unique molecule that forms J-type aggregates in the solid and solution states. The fluorescent properties of NMI-GW-OMe gels in different aliphatic alcohols and aromatic solvents were studied (Fig. S12, ESI†). In aliphatic solvents, the maximum of the emission peak centred around 410–425 nm, while in electron-rich aromatic solvents a significant red-shift of the emission spectrum was observed.65 Based on the different fluorescent properties in the solid and gel (in organic and aqueous solvent) states, a CIE plot of the NMI derivative is shown in Fig. 2f.
To gain more insight into the AIE properties in the mixed solvent, time-correlated single-photon-counting (TCSPC) experiments (Fig. 4) were performed. In DMSO, the emission was monitored at 350 nm (λex = 343 nm). A very short-lived decay (2.3 ps) was noted. A similar experiment was performed in 90% water in DMSO and a substantially longer average lifetime of 463 ps was seen, which is consistent with literature precedent for J-aggregation.24,68
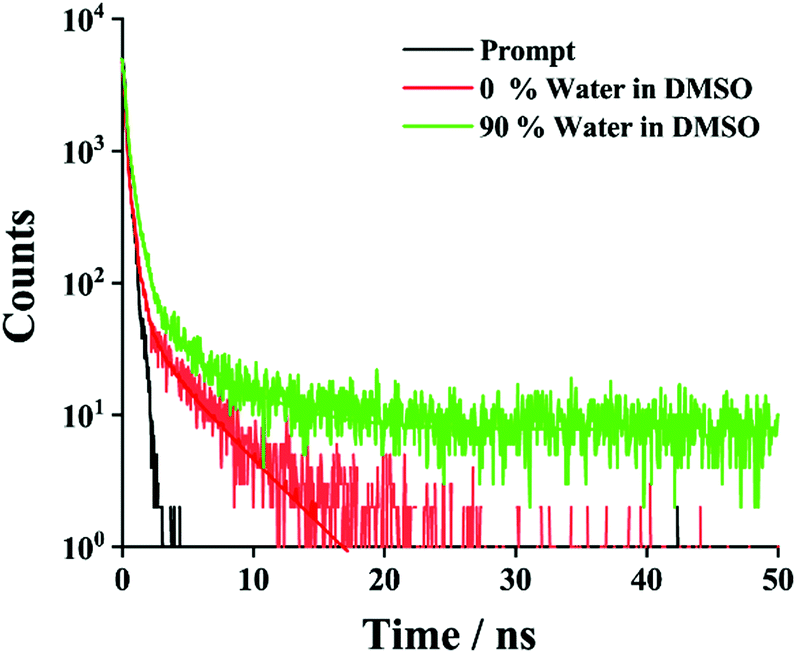 | ||
| Fig. 4 TCSPC decay profile of NMI-GW-OMe in DMSO and DMSO–water solvent. All samples were excited at 343 nm, and the decay was recorded at 350 nm. In the mixed solvent a longer average lifetime value was noticed. | ||
The structural features of NMI-GW-OMe have been investigated with the help of DFT calculations.20 The energy minimized structures of the compound at the B3LYP and 6-311g(d,p) levels of theory show a structural resemblance with its X-ray crystallographically determined structure. However, the optimization of the dimeric structure of NMI-GW-OMe reveals that non-covalent interactions between two molecules are playing the key roles for J-type association. The amide hydrogen and C–H of NMI attached glycine are involved in conventional and carbon hydrogen bonding with the amide carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). The π-stacking between two NMI rings is disturbed somewhat due to this kind of hydrogen bonding and they stayed in a transverse fashion to each other. It is the main driving force of their J-type association. The π-stacking interaction between two indole moieties also favours this association.
O). The π-stacking between two NMI rings is disturbed somewhat due to this kind of hydrogen bonding and they stayed in a transverse fashion to each other. It is the main driving force of their J-type association. The π-stacking interaction between two indole moieties also favours this association.
To explore the electronic properties of NMI-GW-OMe, TD-DFT calculations have been utilized. The gas phase calculation showed that the HOMO–LUMO band gap of dimeric NMI-GW-OMe has a considerable value. The HOMO of the dimer is concentrated over the electron rich indole moiety whereas the LUMO is contributed by the NMI part.
The nanoscale morphologies of the gels were studied in a mixed solvent (90% water in a DSMO–water solution) and xylene using transmission electron microscopy (TEM). In both cases, a nanofibrillar morphology was noticed. It is clear from Fig. 5 that the nanofibers formed in the mixed solvent are shorter in length compared to the nanofibers formed in xylene. The average diameters of the gel nanofibers in the DMSO–water and xylene solvents are 15.6 ± 3.1 and 19.0 ± 3.2 nm, respectively. Xerogel materials (prepared from mixed solvents) were investigated using fluorescence microscopy. A greenish-coloured fibrillar morphology was observed through the fluorescence microscopy due to the AIE property (Fig. S13, ESI†).
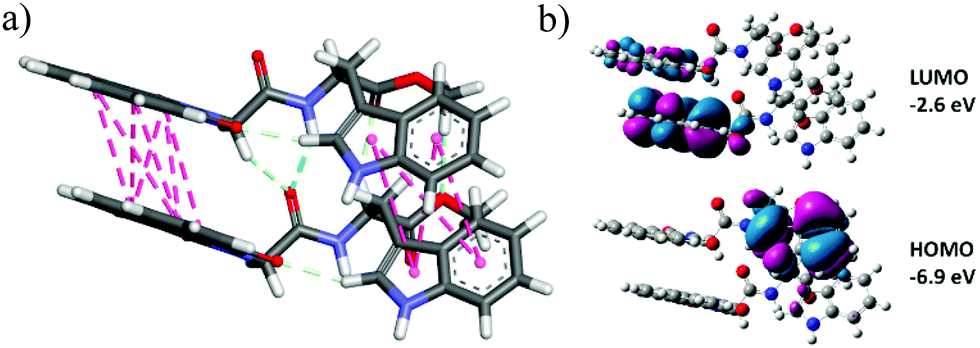 | ||
| Fig. 5 (a) DFT [B3LYP/6-311g(d,p)]-optimised dimeric structure of NMI-GW-OMe. (b) Frontier molecular orbitals of the NMI derivative. | ||
To envisage the supramolecular structure in the gel network,69 we employed powder X-ray diffraction (PXRD) on the gel obtained from DMSO–H2O medium (Fig. 7). Gel state PXRD study often exhibits poor crystallinity, but reveals the authentic packing pattern in the gel state.70 Here, the poor crystallinity widens the PXRD pattern in the gel state. While comparing it with the simulated XRD pattern of the NMI-GW-OMe crystal, the major peak positions remain intact. It depicts the presence of a quite similar packing pattern in the gel state (Fig. 6).
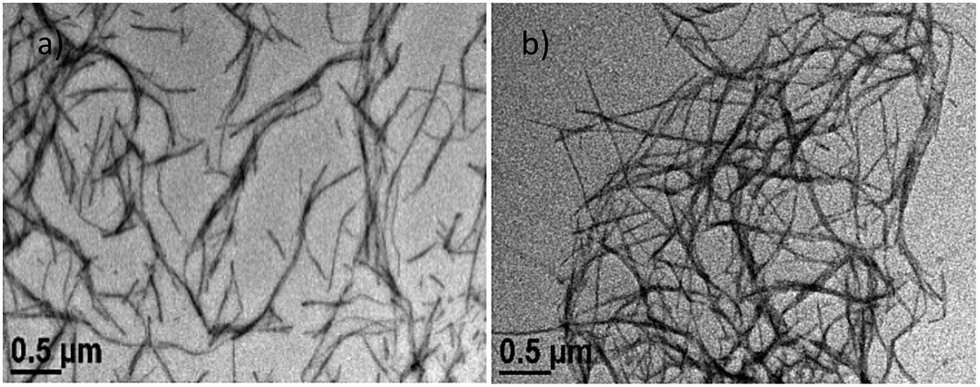 | ||
| Fig. 6 Nanofibrillar morphology of the gel in (a) DMSO–water and (b) xylene solvent. | ||
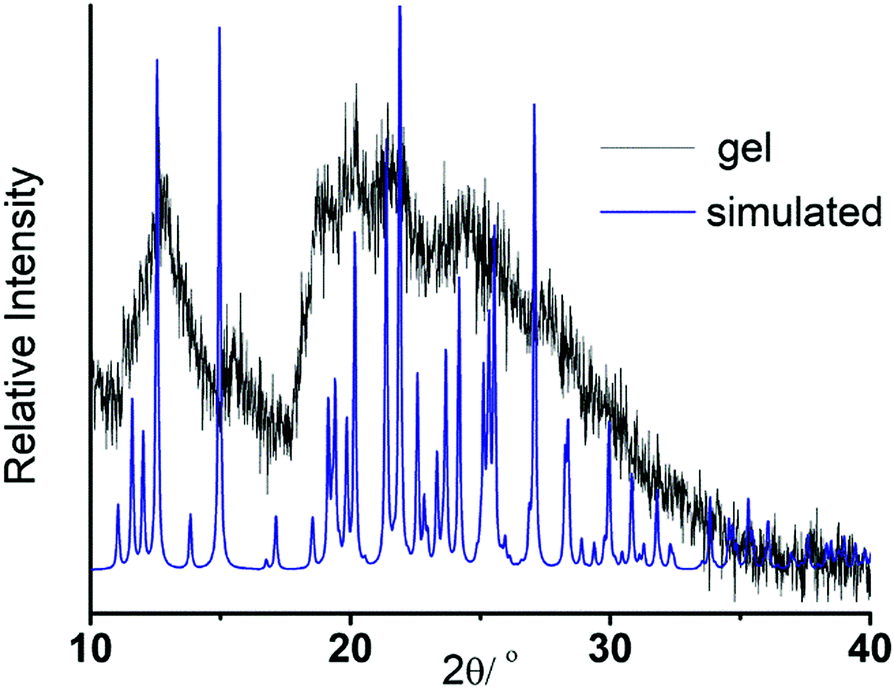 | ||
| Fig. 7 Comparison of the PXRD pattern of the gel in DMSO–water solvent with the simulated XRD pattern of the NMI-GW-OMe crystal. | ||
Inspired by the AIE property, the gelator was used in a cell imaging study. Before utilizing it for cells, the cytotoxicity of the gelator was evaluated using MTT assay at 37 °C by incubating with HADF cells for 24 h. The results depicted that the cellular viability at 65, 130 and 260 μM remains ∼98, 84 and 75%, respectively, indicating less cytotoxicity (Fig. 8a). The cellular viability was found to decrease to 60% after incubating the cells with 325 μM NMI-GW-OMe. The cells are almost viable in the presence of 65 μM gelator, indicating its biocompatibility for living cells. In vitro assessment of HADF cells was done by incubating the gelator for 24 h at 37 °C. It is evident from Fig. S14 (ESI†) that the HADF cells were not showing any background fluorescence signal. On incubating with 65 μM NMI derivative, the HADF cells showed prominent green fluorescence, Fig. 8c. This result implied that this naphthalimide derivative could be used as an efficient candidate for live cell imaging.
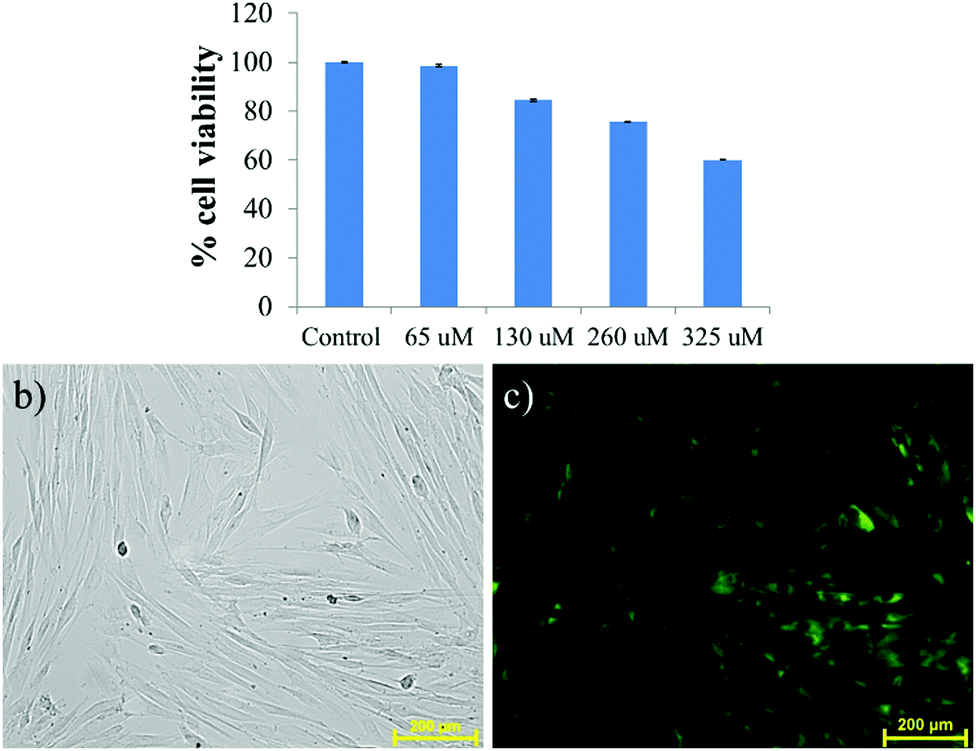 | ||
| Fig. 8 (a) Viability of HADF cells after 24 h of treatment with different concentrations of NMI-GW-OMe (65 to 325 μM), determined by MTT assay. (b) Bright field and (c) fluorescence images of HADF cells were captured (20×) after incubation with 65 μM NMI derivative. | ||
Conclusions
In summary, we designed and synthesized an unsubstituted naphthalimide based dipeptide to demonstrate its photophysical properties in the solid and self-assembled state. We noticed that the structurally simple fluorogen exhibits J-type aggregation in the solid, solution, and gel state. We discussed the different structural aspects of this naphthalimide derivative in the solid-state. Here, an amide hydrogen bonding interaction guides the arrangement of unsubstituted naphthalimide groups in the J-type fashion. In the solution and gel states, J-type aggregation was confirmed by different spectroscopic techniques. The aggregation-induced enhancement of emission phenomenon in aqueous solution was used for live cell imaging. The conjugates of the 1,8-naphthalimide unit with a naturally occurring peptide can be an excellent combination as a fluorescent probe for future applications in biology. The naphthalimide units can be easily modified by chemical reactions to incorporate additional functionality for tailor-made fluorescence and self-assembly properties. In the future, these kinds of AIEgens having low cytotoxicity and longer lifetime can be used as a probe for studying the biological functions and pathological effects in the sub-cellular structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JN gratefully acknowledges DST-INSPIRE Faculty Grant (IFA16-CH246) and Department of Chemistry, IIEST-Shibpur, for financial assistance. P. Brandão acknowledges the financial support from the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 & UIDP/50011/2020, financed by national funds through the FCT/MEC and by FEDER under the PT2020 Partnership Agreement.Notes and references
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- C. Gui, E. Zhao, R. T. K. Kwok, A. C. S. Leung, J. W. Y. Lam, M. Jiang, H. Deng, Y. Cai, W. Zhang, H. Su and B. Z. Tang, Chem. Sci., 2017, 8, 1822–1830 RSC.
- N. Meher, S. Panda, S. Kumar and P. K. Iyer, Chem. Sci., 2018, 9, 3978–3985 RSC.
- C. Zhu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Appl. Bio Mater., 2018, 1, 1768–1786 CrossRef CAS.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- N. Meher and P. K. Iyer, Nanoscale, 2019, 11, 13233–13242 RSC.
- Z. Chen, D. Wu, X. Han, Y. Nie, J. Yin, G.-A. Yu and S. H. Liu, RSC Adv., 2014, 4, 63985–63988 RSC.
- L. Zhu, X. Li, Q. Zhang, X. Ma, M. Li, H. Zhang, Z. Luo, H. Ågren and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 5175–5182 CrossRef CAS.
- N. Meher and P. K. Iyer, Angew. Chem., Int. Ed., 2018, 57, 8488–8492 CrossRef CAS.
- Y. Ni, Z. Sun, Y. Wang, H. F. Nour, A. C. H. Sue, N. S. Finney, K. K. Baldridge and M. A. Olson, J. Mater. Chem. C, 2019, 7, 7399–7410 RSC.
- M. Poddar, G. Sivakumar and R. Misra, J. Mater. Chem. C, 2019, 7, 14798–14815 RSC.
- C. Balachandra and T. Govindaraju, J. Org. Chem., 2020, 85, 1525–1536 CrossRef CAS.
- S. Jena, P. Dhanalakshmi, G. Bano and P. Thilagar, J. Phys. Chem. B, 2020, 124, 5393–5406 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Chem. – Eur. J., 2014, 20, 8012–8023 CrossRef CAS.
- P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef CAS.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS.
- A. Gładysiak, T. N. Nguyen, R. Bounds, A. Zacharia, G. Itskos, J. A. Reimer and K. C. Stylianou, Chem. Sci., 2019, 10, 6140–6148 RSC.
- C. Felip-León, F. Galindo and J. F. Miravet, Nanoscale, 2018, 10, 17060–17069 RSC.
- X. Pang, X. Yu, H. Lan, X. Ge, Y. Li, X. Zhen and T. Yi, ACS Appl. Mater. Interfaces, 2015, 7, 13569–13577 CrossRef CAS.
- S.-M. Hsu, F.-Y. Wu, H. Cheng, Y.-T. Huang, Y.-R. Hsieh, D. T.-H. Tseng, M.-Y. Yeh, S.-C. Hung and H.-C. Lin, Adv. Healthcare Mater., 2016, 5, 2406–2412 CrossRef CAS.
- Q. N. Pham, N. Brosse, C. Frochot, D. Dumas, A. Hocquet and B. Jamart-Grégoire, New J. Chem., 2008, 32, 1131–1139 RSC.
- K. Liu, L. Meng, S. Mo, M. Zhang, Y. Mao, X. Cao, C. Huang and T. Yi, J. Mater. Chem. C, 2013, 1, 1753–1762 RSC.
- M. Kumar, D. Sementa, V. Narang, E. Riedo and R. V. Ulijn, Chem. – Eur. J., 2020, 26, 8372–8376 CrossRef CAS.
- B. Adhikari, J. Nanda and A. Banerjee, Chem. – Eur. J., 2011, 17, 11488–11496 CrossRef CAS.
- C. G. Pappas, P. W. J. M. Frederix, T. Mutasa, S. Fleming, Y. M. Abul-Haija, S. M. Kelly, A. Gachagan, D. Kalafatovic, J. Trevino, R. V. Ulijn and S. Bai, Chem. Commun., 2015, 51, 8465–8468 RSC.
- S. Maity, P. Das and M. Reches, Sci. Rep., 2015, 5, 16365 CrossRef CAS.
- J. M. Malicka, A. Sandeep, F. Monti, E. Bandini, M. Gazzano, C. Ranjith, V. K. Praveen, A. Ajayaghosh and N. Armaroli, Chem. – Eur. J., 2013, 19, 12991–13001 CrossRef CAS.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS.
- D. Ivnitski, M. Amit, O. Silberbush, Y. Atsmon-Raz, J. Nanda, R. Cohen-Luria, Y. Miller, G. Ashkenasy and N. Ashkenasy, Angew. Chem., Int. Ed., 2016, 55, 9988–9992 CrossRef CAS.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS.
- P. Singh, S. Misra, N. Sepay, S. Mondal, D. Ray, V. K. Aswal and J. Nanda, Soft Matter, 2020, 16, 6599–6607 RSC.
- J. S. Valera, R. Sánchez-Naya, F. J. Ramírez, J. L. Zafra, R. Gómez, J. Casado and L. Sánchez, Chem. – Eur. J., 2017, 23, 11141–11146 CrossRef CAS.
- S. Biswas, M. Kumar, A. M. Levine, I. Jimenez, R. V. Ulijn and A. B. Braunschweig, Chem. Sci., 2020, 11, 4239–4245 RSC.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- K. Hawkins, A. K. Patterson, P. A. Clarke and D. K. Smith, J. Am. Chem. Soc., 2020, 142, 4379–4389 CrossRef CAS.
- J. Ruíz-Olles and D. K. Smith, Chem. Sci., 2018, 9, 5541–5550 RSC.
- P. Slavík, D. W. Kurka and D. K. Smith, Chem. Sci., 2018, 9, 8673–8681 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS.
- G. Liu, C. Zhou, W. L. Teo, C. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 9366–9372 CrossRef CAS.
- D. E. Clarke, C. D. J. Parmenter and O. A. Scherman, Angew. Chem., Int. Ed., 2018, 57, 7709–7713 CrossRef CAS.
- C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700 CrossRef CAS.
- S. Ghosh, V. K. Praveen and A. Ajayaghosh, Annu. Rev. Mater. Res., 2016, 46, 235–262 CrossRef CAS.
- S. Cherumukkil, S. Ghosh, V. K. Praveen and A. Ajayaghosh, Chem. Sci., 2017, 8, 5644–5649 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332–335 CrossRef CAS.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- S. Saha, J. Bachl, T. Kundu, D. Díaz Díaz and R. Banerjee, Chem. Commun., 2014, 50, 3004–3006 RSC.
- C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- N. Bäumer, K. K. Kartha, J. P. Palakkal and G. Fernández, Soft Matter, 2020, 16, 6834–6840 RSC.
- P. Sahoo, D. K. Kumar, S. R. Raghavan and P. Dastidar, Chem. – Asian J., 2011, 6, 1038–1047 CrossRef CAS.
- P. Singh, S. Misra, A. Das, S. Roy, P. Datta, G. Bhattacharjee, B. Satpati and J. Nanda, ACS Appl. Bio Mater., 2019, 2, 4881–4891 CrossRef CAS.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2013, 49, 6891–6893 RSC.
- S. Basak, N. Nandi, A. Baral and A. Banerjee, Chem. Commun., 2015, 51, 780–783 RSC.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sanchez, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef CAS.
- D. A. Shejul, S. M. Wagalgave, R. W. Jadhav, M. A. Kobaisi, D. D. La, L. A. Jones, R. S. Bhosale, S. V. Bhosale and S. V. Bhosale, New J. Chem., 2020, 44, 1615–1623 RSC.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS.
- P. Sahoo and P. Dastidar, Cryst. Growth Des., 2012, 12, 5917–5924 CrossRef CAS.
- P. Sahoo, V. G. Puranik, A. K. Patra, P. U. Sastry and P. Dastidar, Soft Matter, 2011, 7, 3634–3641 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1991131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00584c |
| ‡ Present address: Department of Chemistry, Kalimpong College, Kalimpong, West Bengal 734301, India. |
| This journal is © The Royal Society of Chemistry 2020 |