
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel peptide-directed liposomes for targeted combination therapy of breast tumors†
Kaiyue
Zhang‡
abc,
Xiaocui
Fang‡
 ab,
Qing
You
ab,
Yuchen
Lin
ab,
Lilusi
Ma
ab,
Shilin
Xu
d,
Yangyang
Ge
d,
Haiyan
Xu
*d,
Yanlian
Yang
*abc and
Chen
Wang
*abc
ab,
Qing
You
ab,
Yuchen
Lin
ab,
Lilusi
Ma
ab,
Shilin
Xu
d,
Yangyang
Ge
d,
Haiyan
Xu
*d,
Yanlian
Yang
*abc and
Chen
Wang
*abc
aCAS Key Laboratory of Biological Effects of Nanomaterials and Nanosafety, CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: yangyl@nanoctr.cn; wangch@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSino-Danish College, University of Chinese Academy of Sciences, Beijing 100049, China
dInstitute of Basic Medical Sciences Chinese Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing 100005, China. E-mail: xuhy@pumc.edu.cn
First published on 4th November 2020
Abstract
Delivery and accumulation of therapeutic drugs into neoplastic cells distant from tumor vessels is a major challenge for antitumor therapy. Herein, we introduced a de novo peptide, p12 (QGSRRRNTVDDWISRRRALC), to conjugate onto the surface of doxorubicin (DOX) and indocyanine green (ICG) co-encapsulated nanoscale liposomes (pLipo-DOX–ICG). The p12 peptide triggered specific binding to CXC chemokine receptor 4 (CXCR4), leading to enhanced cellular uptake and improved accumulation of pLipo-DOX–ICG in CXCR4-overexpressing cancer cells. Moreover, the introduction of ICG molecules endowed pLipo-DOX–ICG with photothermal-induced structural disruption, which may be helpful for the precise and controllable release of doxorubicin at tumor tissue sites in vivo. With these advantages, the constructed pLipo-DOX–ICG demonstrated active targeting behavior for delivery and release of chemotherapeutic drugs, thereby showing much enhanced metastasis inhibition and antitumor efficacy than either drug-containing liposomes without p12 modification or free drugs in breast cancer bearing mice models. Overall, pLipo-DOX–ICG with low toxicity is expected to be a potential therapeutic agent to improve clinical benefits of breast cancer therapies, as well as treatment of a variety of CXCR4-overexpressing malignancies.
Introduction
Effective delivery of therapeutic drugs to tumor tissues and tumor cells has been challenging for the success of tumor therapy. Targeted drug delivery systems have been pursued extensively for improving drug efficacy and reducing side effects on normal tissues.1 Among them, a variety of nanometer-sized particles have been used as drug delivery systems to overcome unsatisfactory limitations of chemotherapeutic agents, such as poor water solubility, nonspecific biodistribution, and multiple side effects.2 This strategy for effective delivery of payloads into the site of interest mainly includes passive and active targeting.Passive targeting based on enhanced permeability and retention (EPR) effect has limitations due to EPR varying not only across different tumor types but also within different subregions of a single tumor tissue.3 The specific tumor microenvironment, such as dysfunctional structures of tumor vessels, and high interstitial pressure in tumor tissues, may greatly hinder the deep penetration and accumulation of drugs. With these factors, nanoparticle drug carriers based on the EPR effect have yet to meet expectations, because most of them mainly accumulate in the perivascular space of tumor tissues.4,5
The feasibility of active tumor targeting has been examined extensively. Nanoparticle drug carriers with active targeting properties, as the result of introduction of receptor-specific ligands onto their surfaces, can be developed.6–8 These active targeting systems are capable of delivering cargos into tumor parenchyma through the specific recognition of ligands including antibodies, aptamers, and peptides with tumor associated antigens.9–14 Compared with small molecules or proteins, peptides could be optimized easily by changing the composition and type of amino acids, thereby obtaining optimum binding strengths with receptors. Besides, peptides are readily made by good manufacturing practices.15 Therefore, peptides are promising ligands for targeting tumor associated antigens.
Liposomes are well-studied vehicles for encapsulating multiple agents to achieve tumor imaging and therapy. Peptide-directed liposomes consist of a nanoscale scaffold, where one or more agents are encapsulated, a corona of polymeric material that reduces their uptake by the mononuclear phagocyte system (MPS), and one or two peptide ligands that add selectivity for tumor associated antigens.16,17 With these advantages, peptide-directed liposomes could improve the selective accumulation of entrapped cargos at tumor sites.18,19
Chemokine receptor 4 (CXCR4), one of the 7-membrane G-protein-coupled receptors, is highly expressed in multiple hematopoietic and solid tumors, such as leukemia, lung tumors, prostate tumors and breast tumors.20 The interaction between functionally active CXCR4 and its cognate ligand of CXCL12 has been recognized to induce tumor cell adhesion, invasion and proliferation by triggering intracellular downstream signaling pathways, such as PI3K-Akt and Ras-Erk.20–23 Because CXCR4 overexpression correlates with tumor metastasis and development, delivering entrapped drugs to CXCR4-overexpressing tumor cells using liposomes whose surface was modified with a CXCR4 targeting peptide is a promising strategy for tumor threapy.
In this work, we develop a peptide-directed liposomal drug delivery system by introducing a de novo CXCR4 targeting peptide on the surface of liposomes that encapsulate doxorubicin (DOX) and indocyanine green (ICG) as an example for combination therapy. Targeting the peptide of p12 would help route them preferentially into tumor sites, thereby improving the antitumor efficacy but reducing adverse effects compared with either plain liposomes without p12 modification or free drugs in tumor bearing mouse models. Moreover, the photothermal property of ICG molecules could endow liposomes with an ability of precise and controllable release of DOX at tumor sites. Generally, the constructed pLipo-DOX–ICG demonstrates active targeting behavior for delivery and release of cargos, contributing to the much enhanced metastasis inhibition and antitumor efficacy for breast tumor therapy. We believe that with appropriate modifications, this peptide-directed liposomal delivery approach can be adapted for therapy for other CXCR4-overexpressing malignancies.
Results and discussion
Preparation and characterization of liposomal formulations
As illustrated in Scheme 1, pLipo-ICG was prepared by mixing the solutions of cholesterol, DPPC lipid, DSPE-PEG, DSPE-PEG-p12, and ICG via a thin-film hydration method.24 The thiol group (–SH) of p12 (QGSRRRNTVDDWISRRRALC) is coupled to the maleimide group of DSPE-PEG (DSPE-PEG-MAL) to obtain DSPE-PEG-p12. Besides, doxorubicin-encapsulating pLipo-ICG (pLipo-DOX–ICG) was further prepared by mixing the solutions of doxorubicin and pLipo-ICG via an ammonium sulfate gradient method. To obtain uniform sizes, liposomal formulations were passed through membranes of 400 nm, 200 nm and 100 nm pore sizes, sequentially. Free ICG is an amphiphilic molecule that would aggregate and sedimentate in PBS buffer, resulting in a simultaneous decline of absorption and fluorescence intensities.25 However, liposomes could incorporate ICG into their membranes in monomeric form, as the lipophilic domain of ICG is inserted into the hydrophobic phospholipid bilayer, whereas the hydrophilic domain of ICG faces the hydrophilic aqueous environment.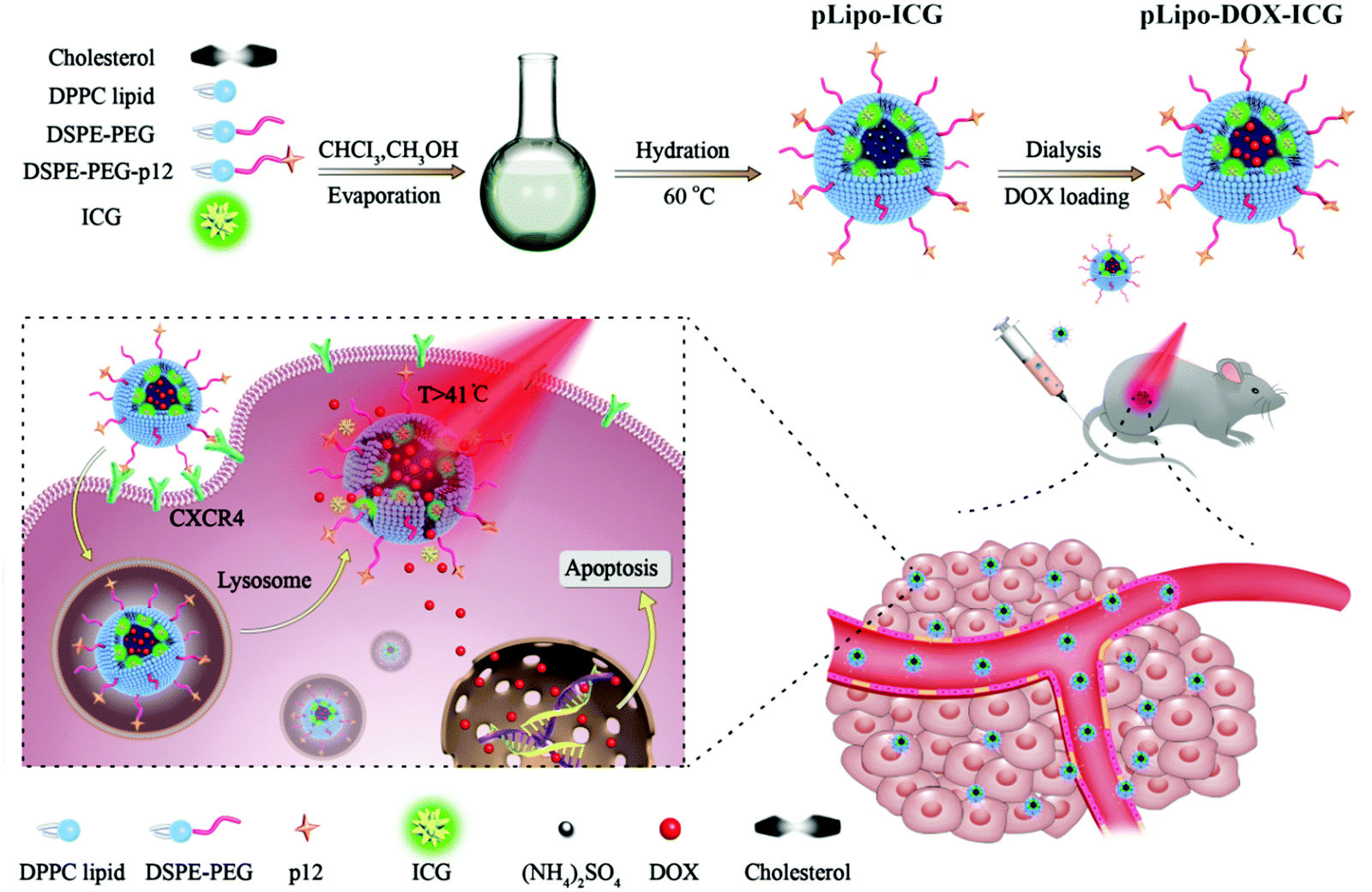 | ||
| Scheme 1 A diagram of self-assembled pLipo-DOX–ICG for specific CXCR4-overexpressing cellular uptake and effective drug delivery and release after NIR laser irradiation. | ||
The morphology, hydrodynamic diameter (nm) and zeta potential (mV) of empty liposomes (Lipo), p12-modified liposomes (pLipo), p12-modified liposomes containing ICG (pLipo-ICG), and p12-modified liposomes containing DOX/ICG (pLipo-DOX–ICG) were characterized and measured using a transmission electron microscope (TEM) and dynamic light scattering (DLS). TEM imaging and DLS results showed that these liposomal formulations were disc-shaped structures, and their sizes were within the range of 100–110 nm (Fig. 1A and B). The hydrodynamic diameter of pLipo-DOX–ICG (105.0 ± 0.6 nm) was slightly larger than either pLipo (101.7 ± 0.4 nm) or Lipo (99.9 ± 0.5 nm). Compared with control liposomes (Lipo), p12 modification significantly increased the zeta-potential of liposomes (pLipo) from −44.5 mV to 17.8 mV, which may be attributed to the positive residue of arginine (R) in p12 amino acid composition (QGSRRRNTVDDWISRRRALC).
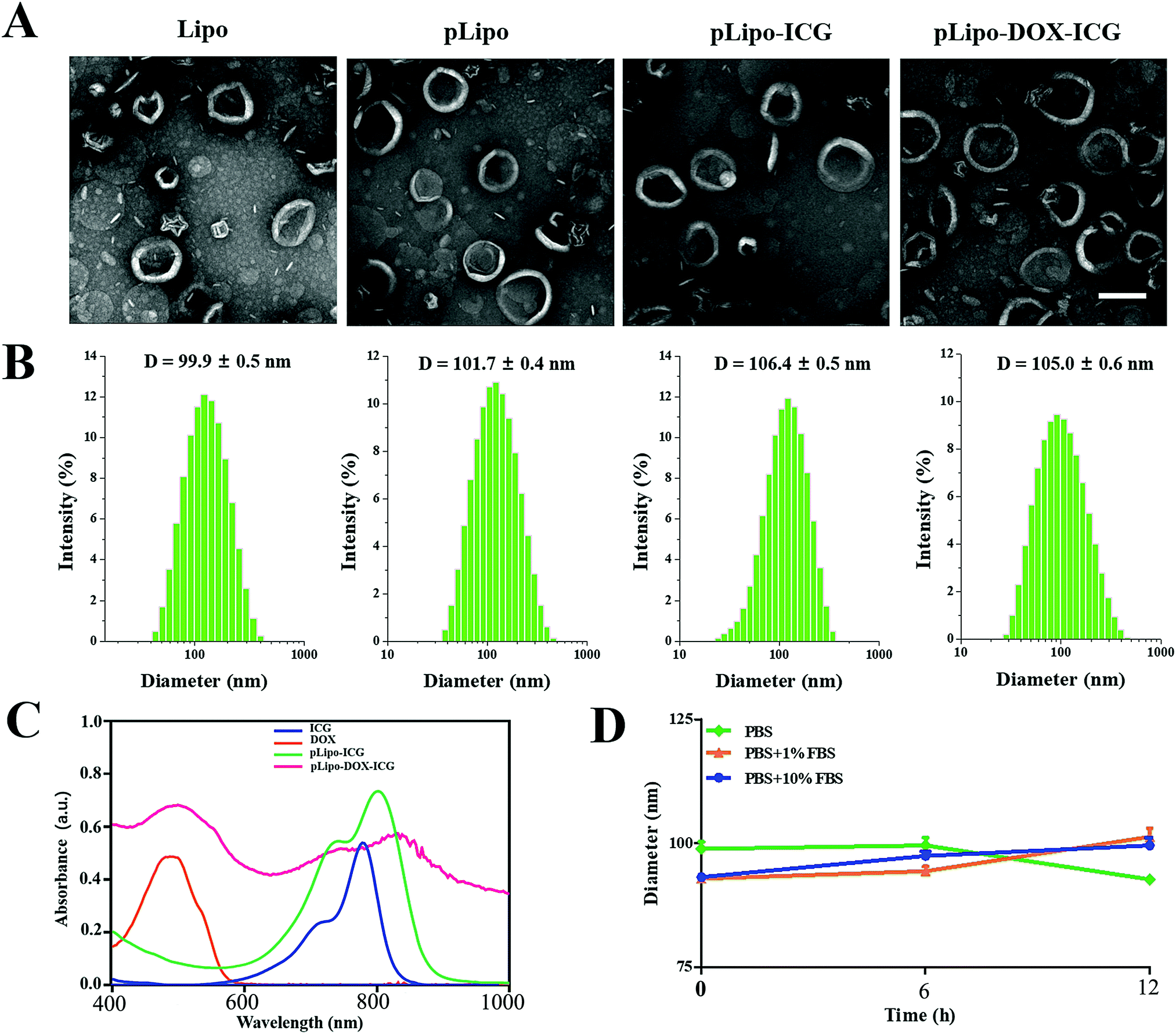 | ||
| Fig. 1 Characterization of different liposomal formulations. (A and B) The morphology and hydrodynamic diameters (nm) of samples including Lipo, pLipo, pLipo-ICG, and pLipo-DOX–ICG, respectively. (C) UV-vis-NIR absorption spectra of ICG, doxorubicin, pLipo-ICG, and pLipo-DOX–ICG, respectively. (D) Particle size changes of pLipo-DOX–ICG after storage in different solvents including PBS buffer alone, or 1% FBS- or 10% FBS-containing PBS buffer over a period of 12 h. | ||
The UV-vis-NIR absorption spectra of samples including free ICG, doxorubicin, pLipo-ICG, and pLipo-DOX–ICG were also determined (Fig. 1C). Free ICG (blue line) displayed a characteristic absorption peak at the wavelength of 780 nm, but the absorption peaks of ICG in pLipo-ICG (green line) and pLipo-DOX–ICG (red line) were red-shifted from 780 nm to 805 nm and 820 nm, respectively. Liposomal bilayers keep ICG molecules in the monomeric form, which could actively enhance their stability and photothermal efficacy. Besides, the absorption peaks of doxorubicin alone, and entrapped into liposomes (pLipo-DOX–ICG) were located at the wavelength of ∼495 nm, suggesting that doxorubicin could be intercalated into liposomes. Hence, the obtained results demonstrated that both ICG and doxorubicin were successfully encapsulated into p12-directed liposomes.
In addition, we examined the encapsulation efficiency (EE) of liposomes to encapsulate ICG and doxorubicin using fluorimetric and RP-HPLC methods. As shown in Table 1, doxorubicin encapsulation efficiency is 99% and ICG encapsulation efficiency is 68.5% in pLipo-DOX–ICG at a molar ratio of about 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50:1:5:20 (DPPC:cholesterol:DSPE-PE/DSPE-PE-p12:ICG:doxorubicin).
:50:1:5:20 (DPPC:cholesterol:DSPE-PE/DSPE-PE-p12:ICG:doxorubicin).
| Encapsulation efficiency (%) | |
|---|---|
| ICG | 68.5 |
| Doxorubicin | 99 |
Interactions occurring at the interfaces between nanoparticles and biological systems are crucial to their bio-distribution, metabolism and targeting efficiency. Upon intravenous injection, they may be coated by a protein corona, which confers a new biological identity on the nanoparticles that largely determines their biological fates.26 To check the stability of pLipo-DOX–ICG in PBS buffer alone, or 1% FBS- or 10% FBS-containing PBS buffer, the DLS method was employed to investigate their particle size changes. As shown in Fig. 1D, the diameter of freshly prepared pLipo-DOX–ICG was about 100 nm. After storage in 1% FBS- or 10% FBS-containing PBS buffer at 25 °C under dark conditions to avoid light exposure, the particle size of pLipo-DOX–ICG showed no big difference over a period of 12 h, demonstrating the high stability of these peptide-directed liposomes.
Binding affinities of p12 and pLipo to CXCR4 in vitro
Binding affinities of p12 and p12-modified empty liposomes (pLipo) to a variety of tumor cells were evaluated. Firstly, CXCR4 expression levels in different tumor cell lines including 4T1, MDA-MB-231, MDA-MB-468, and MCF-7 were analysed by a western blotting method. The results indicated that the CXCR4 protein amount expressed by 4T1 cells were almost 4 times as high as that expressed by MCF-7 cells (Fig. S1, ESI†). Hence, in this work, we selected 4T1 cells as the model of CXCR4-positive breast tumor cell line, whereas the MCF-7 cell line was treated as the model of CXCR4-negative ones.Next, we used 4T1 and MCF-7 cell lines to assess the CXCR4 targeting ability of p12. Briefly, the cells were incubated with FITC-p12 (0.5 μM, 1 μM, 2 μM, 5 μM, and 10 μM) at 37 °C in opti-MEM for 1 h, and the percent (%) of fluorescent cell samples was evaluated by flow cytometry. As shown in Fig. 2A, when concentrations of p12 were increased from 0.5 μM to 10 μM, the fluorescent percent (%) derived from 4T1 cells was much higher than that of MCF-7 cells, which illustrated that p12 had a high binding affinity to CXCR4-overexpressing tumor cells. Besides, in order to investigate the specific binding strength of p12-modified liposomes (pLipo) with CXCR4 receptors, 4T1 and MCF-7 cell lines were further incubated with different concentrations of pLipo (the molar ratio of FITC-p12:lipids is about 1:200) at 37 °C in opti-MEM for 1 h. Flow cytometry results indicated that, compared with MCF-7 cells, a correspondingly higher fluorescent percent (%) has been observed in 4T1 cells with each identical concentration of pLipo (Fig. 2B).
 | ||
| Fig. 2 Concentration-dependent binding affinities of (A) p12 and (B) pLipo to 4T1 and MCF-7 tumor cells. (C) Confocal images of 4T1 and MCF-7 tumor cells after incubation with FITC-pLipo. | ||
Binding strengths of p12 and pLipo with 4T1 tumor cells were analyzed according to the Langmuir adsorption model. This methodology is prevalently utilized for monolayer adsorption processes, and KD values of p12 and pLipo binding to 4T1 tumor cells were estimated by the following fitting equation:27
| F = Bmax × Ca/(KD + Ca) |
According to Fig. 2A and B, KD values of p12 and pLipo for CXCR4-overexpressing 4T1 tumor cells were calculated using the above-mentioned fitting equation. KD values of p12 and pLipo for 4T1 cells were about 3.57 μM and 1.47 μM, suggesting that 2–3 times the binding affinity can be observed by the introduction of liposomes for pLipo in comparison to that of free p12. Such enhancement of binding affinities of pLipo may be attributed to the additional non-specific interactions between the liposome structure and target protein, similar to the ligand-induced modulations on protein–protein binding affinities.28
To further confirm the higher binding affinity of pLipo to 4T1 cells compared with MCF-7 cells, an immunofluorescence assay was also carried out. As shown in Fig. 2C, two tumor cell lines were incubated with FITC-pLipo (p12: 1 μM, lipids: 200 μM) for 2 h at 37 °C, a strong green fluorescence signal was observed in 4T1 cells, whereas no green fluorescence signal was observed in MCF-7 cells. The result could be attributed by the higher binding strength of pLipo with CXCR4-positive cell lines than CXCR4-negative ones, which was consistent with the flow cytometry results (Fig. 2B).
Effects of p12 and pLipo on tumor cell migration
Firstly, we checked the cell viabilities of normal and tumor cells after treatment with different concentrations of pLipo for 24 h at 37 °C. As shown in Fig. S2 (ESI†), cell viabilities of both 4T1 tumor cells and HBMECs normal cells were not affected after treatment with pLipo within this concentration range (0–2.5 mg mL−1), indicating that pLipo has good biocompatibility as a drug delivery system.Numerous studies have confirmed that CXCR4 activation can mediate the migration of tumor cells and adhesion induced by CXCL12.29–31 Therefore, we next determined whether p12 and pLipo can inhibit CXCL12-induced tumor cell migration. As illustrated in Fig. 3A and B, the result from the wound healing assay showed that treatment of 4T1 and MCF-7 tumor cells with CXCL12 for 24 h led to about 70% wound closure compared to the untreated cells, indicating that CXCL12 indeed markedly accelerated CXCR4-overexpressing the migration of cells. For 4T1 cells, treatment with p12 and pLipo for 24 h significantly inhibited the CXCL12-induced cell morphological changes and wound closure. Notably, pLipo had a stronger inhibitory effect on cell migration than that of p12 alone, which may result from the higher binding strength of pLipo with 4T1 tumor cells. However, for MCF-7 cells, both p12 and pLipo treatments had little impact on their migration.
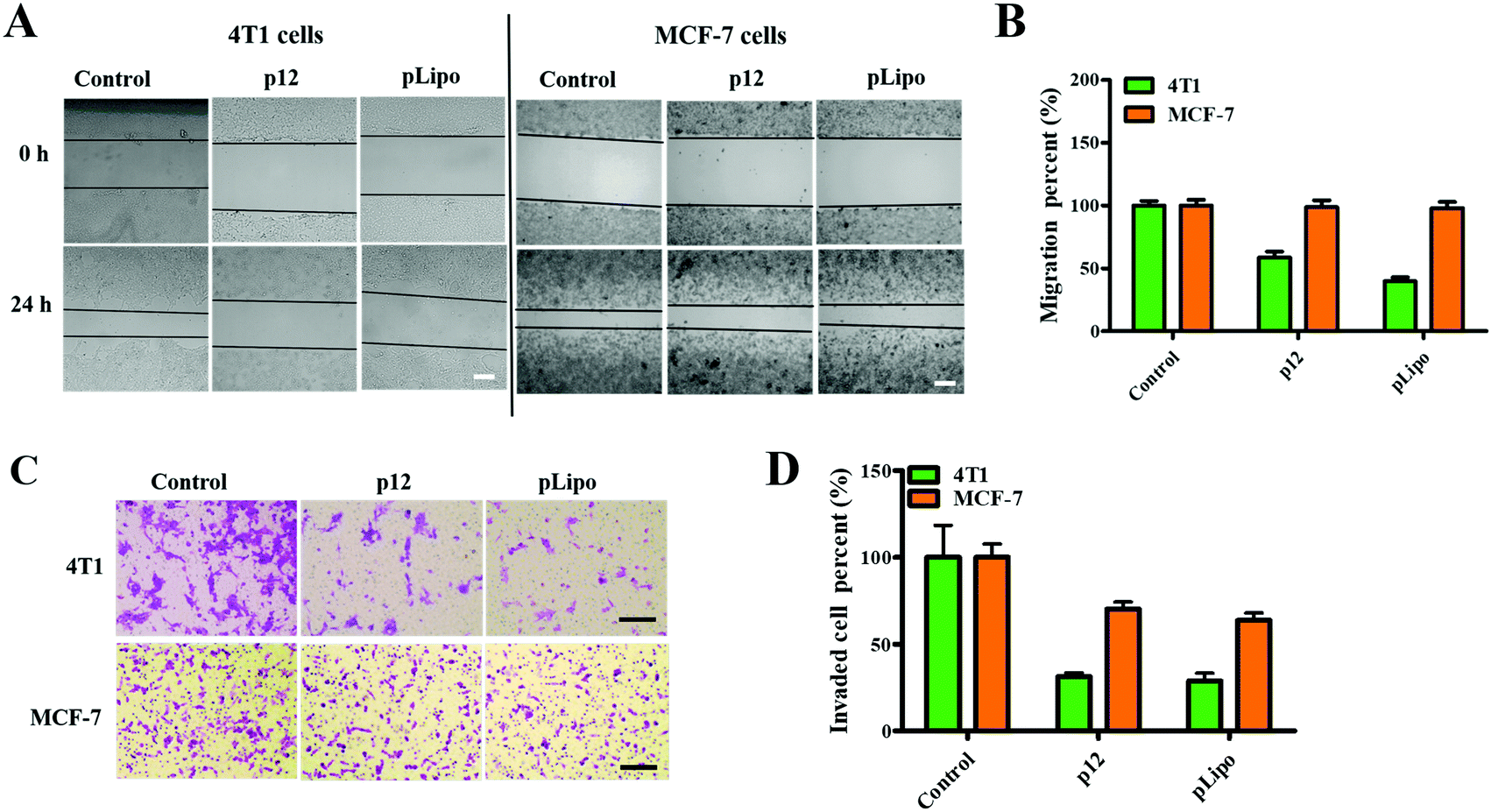 | ||
| Fig. 3 Effects of p12 and pLipo on tumor cell migration. (A) Wound healing assay of 4T1 and MCF-7 tumor cells. Photographs were taken using phase-contrast microscopy immediately after the incision (0 h) and at 24 h. Scale bar = 200 μm. (B) Quantification of the migration distance (μm) in panel (A) at the timepoint of 24 h. (C) Transwell assay of 4T1 and MCF-7 tumor cells after different treatments for 24 h. Scale bar = 100 μm. (D) The semi-quantitative analysis of migrated 4T1 and MCF-7 tumor cells after treatments with p12 and pLipo. Data are presented as the mean ± SD (n = 4). | ||
Besides, a transwell assay was also performed to examine the effect of p12 and pLipo on tumor cell migration. As shown in Fig. 3C and D, p12 and pLipo were strongly inhibited by the migration activity of 4T1 tumor cells, but showed a slightly inhibitory effect on MCF-7 cell migration, in agreement with the wound healing assay. Overall, the results from Fig. 3 demonstrated that the stronger inhibitory effect was observed in pLipo-treated 4T1 tumor cells than in p12-treated ones, suggesting that p12 conjugation with liposomes has an improved binding selectivity with CXCR4 receptors.
In vitro photothermal property of ICG
To evaluate the photothermal property of ICG and pLipo-ICG, an infrared thermal imaging camera was employed to monitor the temperature changes under 808 nm NIR laser irradiation. After laser irradiation at 1.5 W cm−2 for 3 min, temperatures of free ICG and pLipo-ICG solutions at an identical concentration of ICG (10 μg mL−1) rose sharply and maximally increased to 42 °C and 46 °C within 1 min, whereas the PBS buffer and pLipo solutions showed a slight temperature elevation (Fig. 4A and B). Notably, compared with free ICG, pLipo-ICG had a higher temperature increase in response to NIR laser irradiation, which could be attributed to the higher stability of ICG after encapsulation into liposomal bilayers. | ||
| Fig. 4 In vitro photothermal property of pLipo, ICG and pLipo-ICG. (A) Infrared thermographic images of pLipo, ICG, and pLipo-ICG under NIR laser irradiation (808 nm, 1.5 W cm−2) for 3 min. (B) Temperature increase profiles of PBS, pLipo, ICG and pLipo-ICG. (C) TEM images of pLipo-ICG before and after NIR laser irradiation (808 nm, 1.5 W cm−2) for 3 min. (D) Photothermal curve of pLipo-ICG for three repeated NIR irradiation cycles (808 nm, 1.5 W cm−2). | ||
The phase transfer temperature of DPPC-based liposomes is 41 °C, and the temperature of pLipo-ICG could move up to 46 °C after 808 nm NIR laser irradiation for 3 min at 1.5 W cm−2. TEM imaging showed that, compared with pLipo-ICG without NIR laser irradiation, photothermal-induced structural disruption can be observed in pLipo-ICG from disc-shaped structures into smaller pieces (Fig. 4C). The obtained results confirmed that the introduction of ICG molecules could endow nanocarriers with precise and controllable release of payloads at the site of interest, thereby decreasing the undesired toxicity to normal organs. Importantly, due to the fluorescence quenching of ICG molecules, the increased temperature of pLipo-ICG exhibited a slight decrease for three repeated cycles of NIR laser irradiation (Fig. 4D). However, compared with free ICG, a smaller variation in the decreased temperature was observed in pLipo-ICG (data not shown), which may be attributed to the improved stability of ICG molecules after encapsulation into liposomes.
Controlled and sustained drug release at the site of interest is very important for improving the therapeutic effect of pLipo-DOX–ICG. Therefore, the in vitro doxorubicin release behaviors were tested based on the dialysis method at 37 °C in PBS buffer (pH 7.4) under NIR laser irradiation. As shown in Fig. S3 (ESI†), compared with pLipo-DOX–ICG without NIR laser irradiation, it was observed that doxorubicin was slowly released from pLipo-DOX–ICG with time after NIR laser irradiation at 1.5 W cm−2 for 3 min (808 nm), and ∼80% of doxorubicin was released from pLipo-DOX–ICG up to 24 h. Hence, photothermal therapy (PTT) has been shown to trigger structural disruption of nanoplatform containing ICG molecules and cause the release of encapsulated doxorubicin, which may be helpful for the precise and controllable release of doxorubicin at tumor tissue sites in vivo.
Cellular uptake and cytotoxicity of pLipo-DOX–ICG in vitro
The cellular uptake of different doxorubicin formulations in the absence and presence of p12 by CXCR4-overexpressing tumor cells was determined. In brief, 4T1 tumor cells were incubated with free doxorubicin (DOX), Lipo-DOX–ICG and pLipo-DOX–ICG at 37 °C for 4 h at the identical doxorubicin concentration of 0.5 μg mL−1. As shown in Fig. 5A, compared with free doxorubicin, doxorubicin-containing liposomes entered cells more quickly, which might be due to the fact that free doxorubicin is taken up by cells through passive diffusion while the liposome is mainly taken up through endocytosis as well as membrane fusion.32 Notably, the difference in cellular uptake of doxorubicin between Lipo-DOX–ICG-treated and pLipo-DOX–ICG-treated groups may result from the high binding affinity of p12 to CXCR4-overexpressing tumor cells.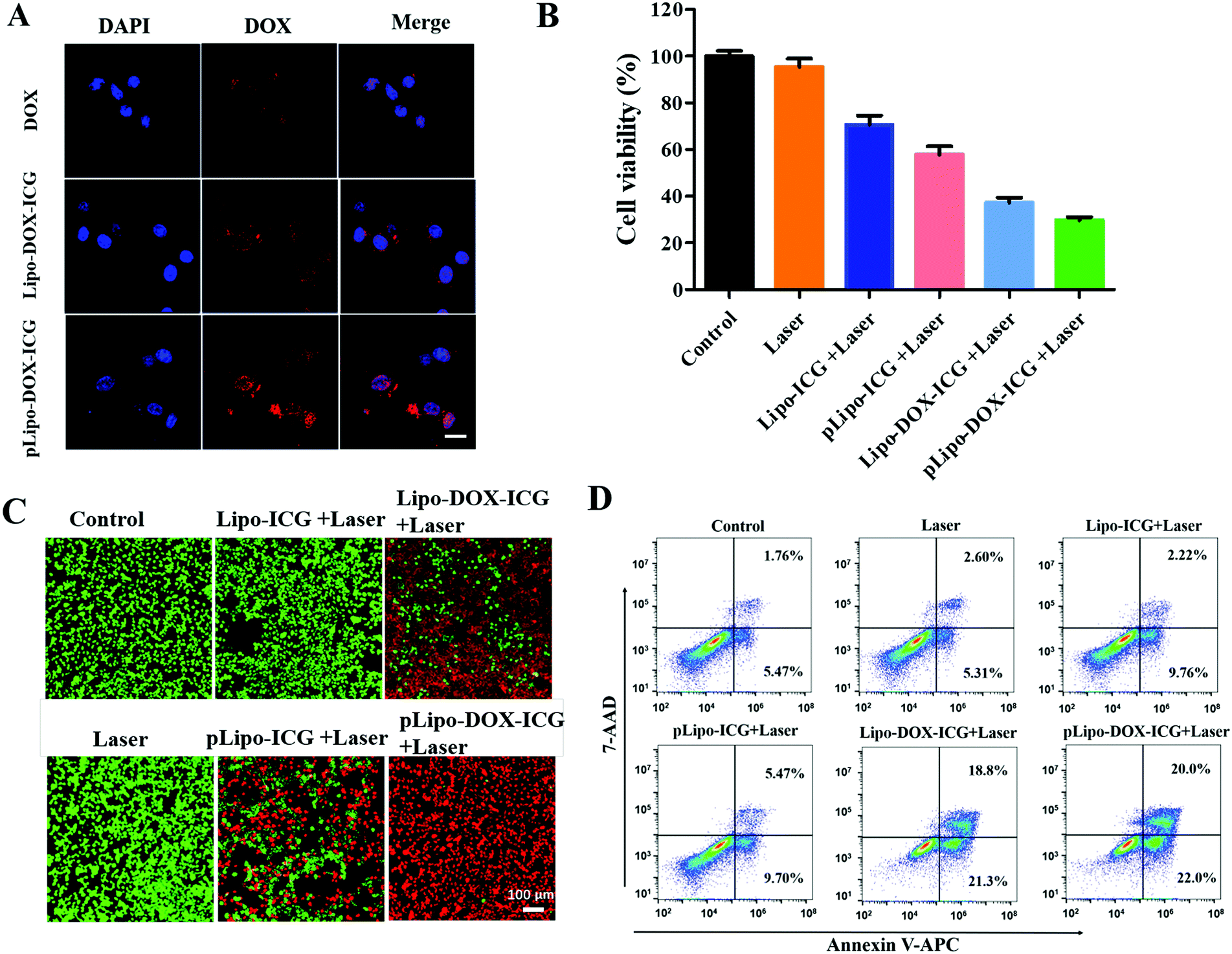 | ||
| Fig. 5 In vitro cell uptake and cytotoxicity of Lipo-ICG or Lipo-DOX–ICG in the absence and presence of p12. (A) The cell uptake of doxorubicin, Lipo-DOX–ICG and pLipo-DOX–ICG by 4T1 tumor cells. Scale bar = 20 μm. (B) Cytotoxicity of different formulations (PBS, PBS + Laser, Lipo-ICG + Laser, pLipo-ICG + Laser, Lipo-DOX–ICG + Laser and pLipo-DOX–ICG + Laser, respectively) after incubation with 4T1 cells for 24 h at 37 °C. (C) Live/dead staining images of 4T1 tumor cells after treatment with PBS, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG and pLipo-DOX–ICG under 808 nm NIR laser irradiation (1.5 W cm−2, 3 min), respectively. Cells without NIR laser irradiation were used as the control group. (D) Flow cytometry analysis of Annexin V-APC/7-AAD double-stained 4T1 cells after treatment with various formulations including PBS, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG and pLipo-DOX–ICG under 808 nm NIR laser irradiation (1.5 W cm−2, 3 min), respectively. | ||
We next determined whether the in vitro cytotoxicity of pLipo-DOX–ICG could be enhanced in comparison with other treatment groups. 4T1 cells were exposed to a series of equivalent concentrations of ICG (20 μg mL−1) or/and doxorubicin (2 μg mL−1) encapsulated liposomes with or without p12 modification for 24 h, and percentages of viable cells were quantified by the CCK-8 assay method. As illustrated in Fig. 5B, compared with the control group, the NIR laser irradiation treatment only barely affected the 4T1 cell viability, suggesting that the irradiation dose is appropriate and safe. Notably, the survival rates of 4T1 tumor cells were 70.6%, 57.9%, 37.2%, and 29.8% after treatment with Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG, and pLipo-DOX–ICG under NIR laser irradiation, respectively. This result further indicated that the co-encapsulated liposomal system with p12 modification (pLipo-DOX–ICG) exhibited a synergistic antitumor effect of doxorubicin and ICG.
Besides, cell viabilities of 4T1 tumor cells incubated with PBS buffer, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG and pLipo-DOX–ICG were also evaluated by the live/dead staining method. As shown in Fig. 5C, compared with the control group, the 4T1 cell growth was not affected after treatment with 808 nm NIR laser irradiation for 3 min at 1.5 W cm−2, suggesting that the irradiation dose is appropriate and safe. After treatment with NIR laser irradiation, p12 modification (pLipo-ICG or pLipo-DOX–ICG) significantly induced 4T1 tumor cell death compared with either Lipo-ICG or Lipo-DOX–ICG, which could be attributed to the greater cellular uptake of doxorubicin or ICG by the introduction of p12. Owing to the photothermal-induced structural disruption of pLipo-DOX–ICG after NIR laser irradiation, the entrapped doxorubicin is released in spurts, thereby exhibiting the highest cytotoxicity towards tumor cells.
The apoptosis and necrosis assay was further performed by flow cytometry with the Annexin V-APC/7-AAD kit to investigate the level and the mechanism of cell death. As shown in Fig. 5D, when 4T1 tumor cells were treated with Lipo-ICG under NIR laser irradiation, a cell survival of ∼86% was detected, suggesting slight apoptosis. The cell survival was decreased to ∼83% when the cells were treated with pLipo-ICG. After combining Lipo-ICG with doxorubicin in the absence and presence of p12, the joint treatment significantly increased the antitumor efficacy. The cell viabilities of 4T1 decreased to about 60% and 53% after treatment with Lipo-DOX–ICG and pLipo-DOX–ICG, respectively. These results demonstrated that pLipo-DOX–ICG could kill tumor cells effectively, in agreement with the in vitro cytotoxicity assay and live/dead staining assay.
Enhanced tumor accumulation of pLipo-ICG in vivo
The tumor-targeting efficacy of pLipo-DOX–ICG in vivo was further evaluated in tumor-bearing mice derived from murine 4T1 tumor cells using the ex/in vivo imaging system. Fig. 6A showed the fluorescence signal distributions at different time points (1 h, 2 h, 4 h, 12 h and 24 h) for free ICG, Lipo-ICG and pLipo-ICG delivered systemically via tail vein injections. The free ICG-treated mouse group exhibited the strongest fluorescence signals at 2 h post-injection, but the signals underwent rapid elimination after 4 h post-injection. However, compared with free ICG, the much stronger fluorescence signals can be observed in either Lipo-ICG-treated or pLipo-ICG-treated mouse groups over a period of 24 h, suggesting the long circulating effect of liposomal formulations. Lipo-ICG and pLipo-ICG were primarily located in liver organs and tumor tissues of mice after intravenous injection. As time elapsed, the fluorescence signals at tumor sites in pLipo-ICG treated mice was notably higher than that in the Lipo-ICG treated mice at 12 h and 24 h (Fig. 6A).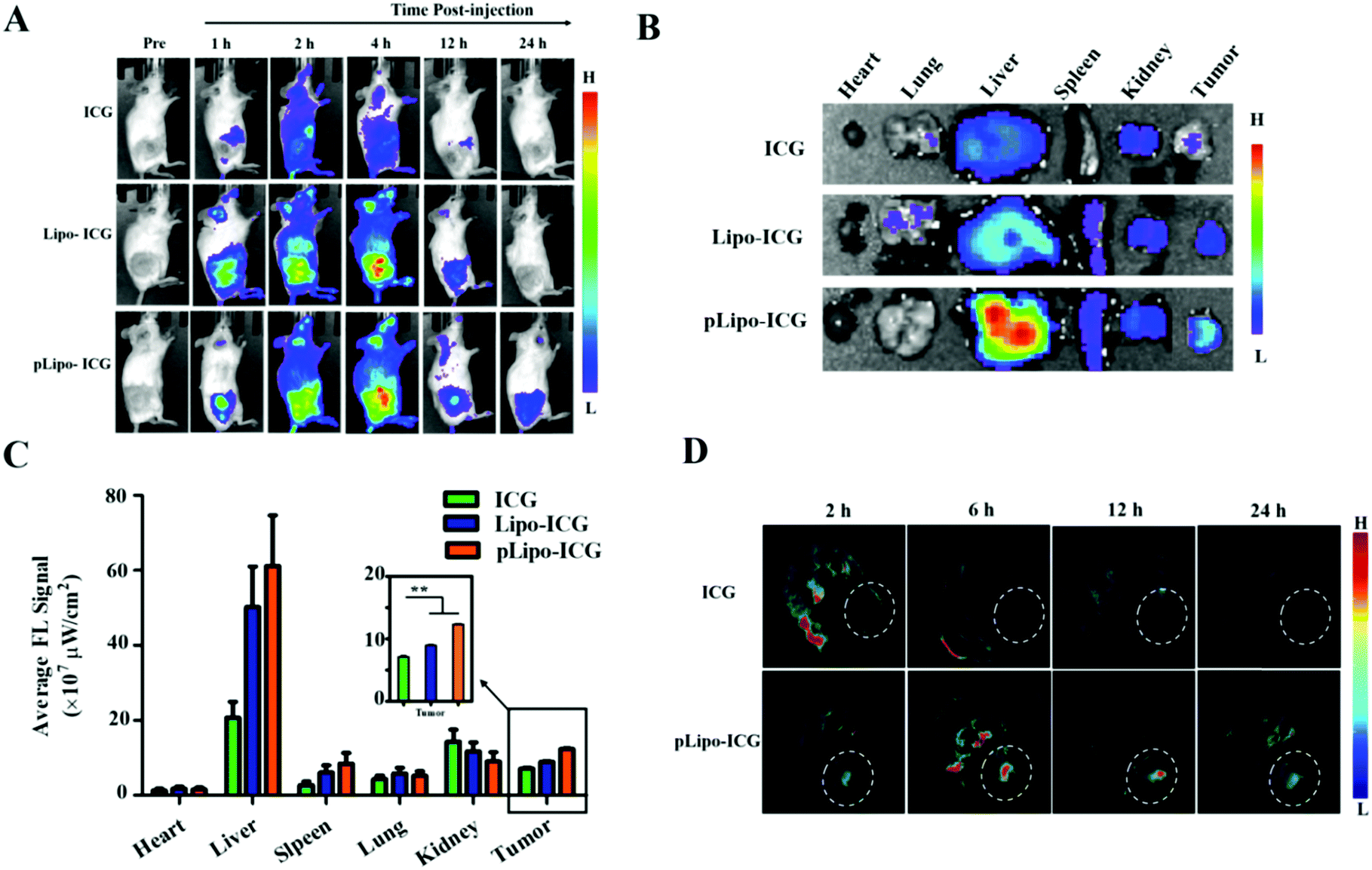 | ||
| Fig. 6 Evaluation of tumor targeting efficiency of Lipo-ICG in the absence and presence of p12. (A) In vivo fluorescence images of 4T1 tumor-bearing mice at different time points (0 h, 1 h, 2 h, 4 h, 12 h, and 24 h) after treatment with free ICG, Lipo-ICG and pLipo-ICG via intravenous injection (n = 3). (B) Ex vivo images of tumors and other tissues derived from mice after 24 h of treatment with free ICG, Lipo-ICG and pLipo-ICG, respectively. (C) Average fluorescence signals of excised tumors and major organs derived from the mice after 24 h of treatment with free ICG, Lipo-ICG and pLipo-ICG, respectively. (D) In vivo MSOT images of 4T1 tumor-bearing mice at different time points (2 h, 6 h, 12 h, and 24 h) after treatment with free ICG, Lipo-ICG, and pLipo-ICG via intravenous injection (n = 3). | ||
After 24 h, the mice were sacrificed, and tumors and major organs (heart, liver, spleen, kidneys, and lungs) were isolated. Ex vivo fluorescent images of excised tumors further confirmed the higher accumulation of pLipo-ICG at tumor sites in comparison to that of either Lipo-ICG or free ICG (Fig. 6B and C). No obvious fluorescence signal was observed in the heart and lungs, but the signal in the liver was very high, which may be attributed to the high macrophage uptake nature of livers.33
Besides, in vivo MSOT images illustrated that, compared with either free ICG or the Lipo-ICG treatment group, pLipo-ICG clearly contrasts the tumor tissue with the surrounding normal tissues, which was vigorous evidence of the high efficiency of tumor targeting of pLipo-ICG (Fig. 6D). Overall, compared with Lipo-ICG, much enhanced tumor targeting can be achieved, as the result of the introduction of p12 to the surface of liposomes (pLipo-ICG). Such effect may be attributed to the receptor clustering effect or in response to local concentration increases, but the rigorous elucidation of the affinity enhancement mechanism remains to be studied.
Improved antitumor effect of pLipo-DOX–ICG in vivo
When the tumor volume reached approximately 100 mm3, tumor bearing mice were injected intravenously with saline, free doxorubicin, Lipo-DOX, pLipo-ICG, pLipo-DOX, and pLipo-DOX–ICG with or without NIR laser irradiation at an equivalent concentration of doxorubicin (doxorubicin/body weight = 3.0 mg kg−1), respectively. Mice treated with saline comprised the control group. As exhibited in Fig. 7A–C, compared with the control group, free doxorubicin and doxorubicin-containing liposome treatment groups exhibited varying degrees of antitumor effect in vivo. In addition, pLipo-ICG with NIR laser irradiation (pLipo-ICG + Laser) also showed a remarkable antitumor effect. The average tumor volume (mm3) and weight (g) of pLipo-DOX–ICG-treated mouse group in combination with NIR laser irradiation was 436 mm3 (0.81 g), much lower than that of the saline-treated mouse group (2573 mm3, 1.96 g) at the endpoint of the experiment (on day 14). H&E staining also illustrated that, the pLipo-DOX–ICG treatment group with NIR laser irradiation showed the most excellent antitumor efficacy among all treatment groups, demonstrating doxorubicin could effectively inhibit the tumor growth when combined with photothermal therapy (Fig. 7D).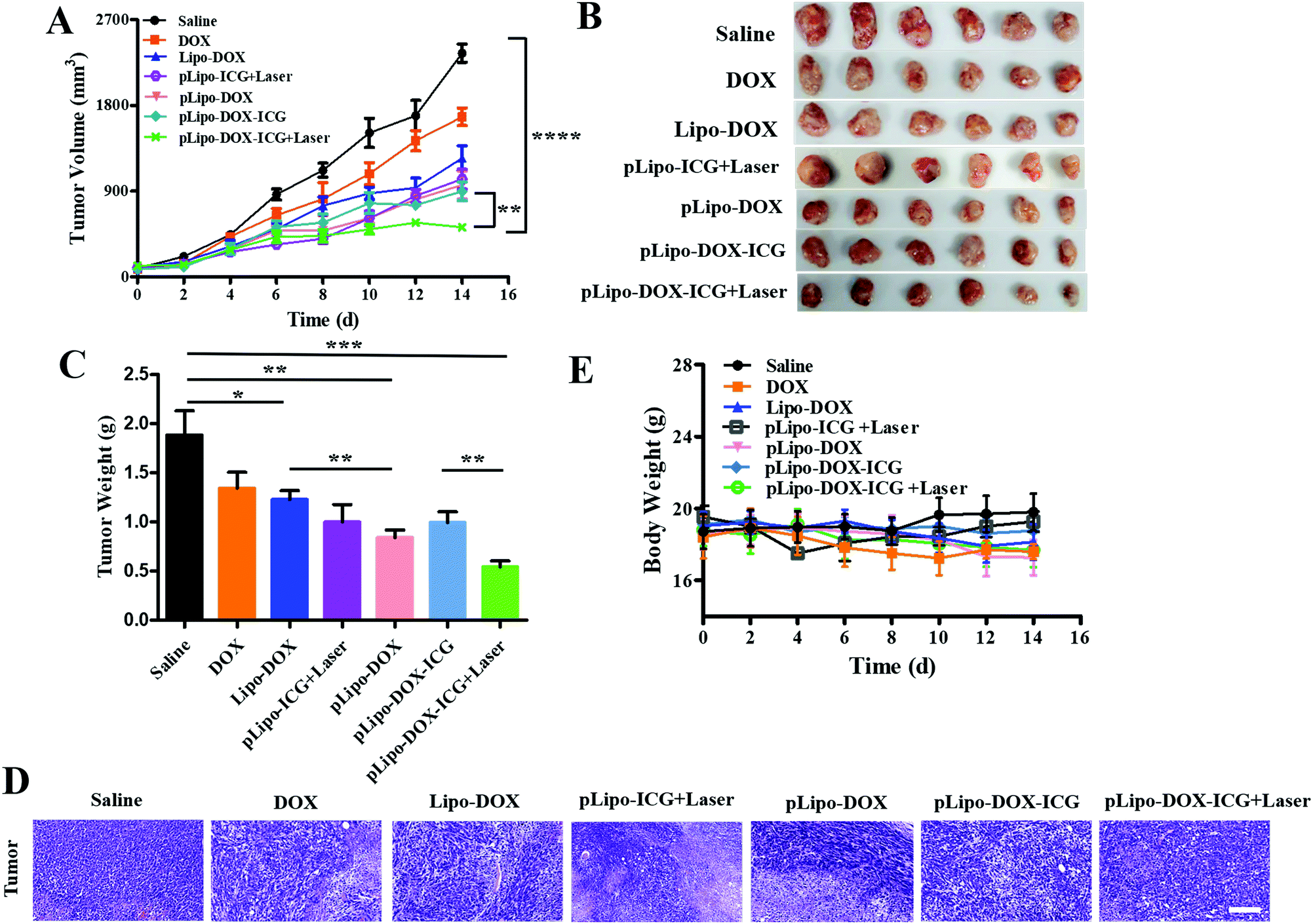 | ||
| Fig. 7 Antitumor effect of saline, free doxorubicin, Lipo-DOX, pLipo-ICG + Laser, pLipo-DOX, pLipo-DOX–ICG, and pLipo-DOX–ICG + Laser on murine breast tumor models. (A) Mean tumor volumes (mm3) of each group on days 0, 2, 4, 6, 8, 10, 12 and 14 after drug administration are shown. (B) Photographs of tumors from each treatment group excised on day 14. (C) Mean body weight (g) of excised tumors from each treatment group excised on day 14. (D) H&E staining of tumors derived from the mice with different treatments. All pictures were obtained under the same magnification (40×). Scale bar = 100 μm. (E) Mean body weight (g) of 4T1 tumor-bearing mice on days 0, 2, 4, 6, 8, 10, 12 and 14 after drug administration are shown. | ||
Free drug molecules may diffuse nonspecifically after intravenous injection, whereas drug-containing nanocarriers with diameters of <200 nm can effectively extravasate into the tumor sites through the leaky vessels via the EPR effect.34,35 Therefore, pLipo-DOX–ICG with an appropriate size of around 110 nm could successfully enter into the tumor vasculature and interstitium. The strong binding strength of p12 with CXCR4 could also contribute to the enhancement of active tumor cell targeting in vivo. Besides, pLipo-DOX–ICG can achieve precise and controllable release of doxorubicin after photothermal-induced structural disruption. The bigger size of pLipo-DOX–ICG resulted in their being retained around tumor vasculature, while the smaller size of the released doxorubicin after NIR laser irradiation made them diffused into the tumor parenchyma. Taken together, the excellent antitumor effect of pLipo-DOX–ICG in combination with NIR laser irradiation on 4T1 breast tumor progression was probably attributed to the synergistic effect of long circulation time induced by PEG chains, chemo-photothermal combination therapy of doxorubicin and ICG, and active targeting induced by p12.
Although we did not evaluate the survival in mice receiving different drug treatments, we believe the pLipo-DOX–ICG-treated mouse group could have better mouse survival than other groups. This is mainly due to two reasons. Firstly, the average tumor volume (mm3) of the pLipo-DOX–ICG-treated mouse group in combination with NIR laser irradiation was much lower than other groups. By comparing the average tumor volume of each group on day 14, the tumor volume of the “pLipo-ICG-DOX + Laser” group was about 1/6 and 1/3 as compared to the saline group and free doxorubicin group, respectively. Secondly, during the whole experiment, the withered hair, spiritual malaise, reduced activity, and reduced food intake were frequent occurrences in the saline-treated mouse group, but rarely occurred in the pLipo-DOX–ICG-treated mouse group in combination with NIR laser irradiation.
In addition, the body weight and blood biochemical indicators of each mouse group with different treatments were also analysed. There was no significant decrease in body weight over a period of 14 days in all treatment groups (Fig. 7E). As shown in Fig. S4 (ESI†), histological examination of major organs including heart, liver, spleen, lungs and kidneys were examined, and no visualized tissue damage was detected. Compared with free doxorubicin, doxorubicin-encapsulating liposomal formulations showed decreased values of alanine aminotransferase (ALT), aspartate transaminase (AST), and triacylglycerol (TG). The results indicated that this peptide-directed liposomal delivery approach could exhibit a promising therapeutic effect with relatively low systematic toxicity. Our findings are probably not unique to the treatment of breast cancer. We believe that with appropriate modifications, this peptide-directed liposomal formulation can be adapted for therapy for other CXCR4-overexpressing malignancies with doxorubicin/ICG or other drugs of interest.
Materials and methods
Materials and cell culture
Dipalmitoylphosphatidylcholine (DPPC), distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)-2000] (DSPE-PEG), and cholesterol were purchased from Jianyang Biotechnology Co., Ltd (Guangzhou, China), Avanti Polar Lipids (Alabaster, USA), and Sangon Biotech Co., Ltd (Shanghai, China), respectively. ICG and doxorubicin were provided by J&K Scientific Co., Ltd (Beijing, China) and Yuanye Biological Technology Co., Ltd (Shanghai, China), respectively. Lyophilized peptide of p12 (QGSRRRNTVDDWISRRRALC, 98% purity) and p12 coupled DSPE-PEG (DSPE-PEG-p12) with and without fluorescein isothiocyanate (FITC) labelling were chemically synthesized by Guoping Pharmaceutical Co., Ltd (Anhui, China). Dulbecco's modified Eagle's medium (DMEM), RPMI-1640 medium, streptomycin, penicillin, trypsin and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific Co., Ltd (Massachusetts, USA). The Cell counting kit-8 (CCK-8) was purchased from Fanbo Biochemicals Co., Ltd (Beijing, China). All other reagents and solvents were of analytical grade and used without any further purification unless otherwise noted.Murine and human breast tumor cell lines including 4T1 and MCF-7 were purchased from Institute of Peking Union Medical College, Basic Medical Sciences and Chinese Academy of Medical Sciences (Beijing, China). Both cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, penicillin (100 U mL−1), and streptomycin (100 U mL−1). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Synthesis of pLipo-ICG and pLipo-DOX–ICG
Firstly, DSPE-PEG, cholesterol, DSPE-PEG-p12 and DPPC were dissolved in chloroform, whereas ICG was dissolved in methanol. For Lipo-ICG preparation, certain amounts of each solution in a molar ratio of about 150:50:1:5 (DPPC:cholesterol:DSPE-PE:ICG) were mixed together in a round-bottom flask and rotary-evaporated for 1 h at 46 °C to form a lipid film. After removing organic solvents in a vacuum drying oven overnight, the dried film was hydrated with ddH2O or PBS buffer (pH 7.4) at 60 °C for 30 min. Non-encapsulated ICG was separated by dialysis of the liposome suspension through a dialysis bag with a molecular weight cut-off of 5 kDa. pLipo-ICG was prepared in an identical procedure except that DSPE-PEG was replaced with a mixture of DSPE-PEG-p12 and DSPE-PEG. Plain liposomes (Lipo) and p12-functionalized liposomes (pLipo) were also prepared as vesicle controls. The size of each liposomal formulation was further optimized by a liposome extruder through 400 nm, 200 nm, and 100 nm polycarbonate membranes, sequentially.
Next, doxorubicin-containing liposomes including Lipo-DOX, pLipo-DOX, Lipo-DOX–ICG and pLipo-DOX–ICG were obtained via an ammonium sulfate gradient method by adding doxorubicin solutions into Lipo, pLipo, Lipo-ICG or pLipo-ICG solutions in a molar ratio of about 150:50:1:5:20 (DPPC:cholesterol:DSPE-PE:ICG:doxorubicin), respectively. Then, the solutions were mixed together for 30 min at 25 °C. Non-encapsulated doxorubicin was removed by a dialysis bag with a molecular weight cut-off of 5 kDa, or by ultracentrifugation using amicon ultra centrifugal filter devices with a molecular weight cut-off of 10 kDa.
Drug encapsulation efficiency
Drug encapsulation efficiency (EE) of ICG and doxorubicin was determined by ultrafiltration. Briefly, pLipo-DOX–ICG was prepared by a film dispersion method at a molar ratio of about 150:50:1:5:20 (DPPC:cholesterol:DSPE-PE/DSPE-PE-p12:ICG:doxorubicin), respectively. Then, 500 μL of pLipo-DOX–ICG were added in amicon ultra centrifugal filter devices with a molecular weight cut-off of 10 kDa, and then centrifuged at 12000 × g for 30 min at 4 °C. The concentration of unencapsulated ICG in filtrate was quantified at the wavelength of 780 nm using the UV-vis spectrophotometer (SpectraMax i3, Molecular Devices, USA). Also, the concentration of unencapsulated doxorubicin in filtrate was quantified by reverse-phase HPLC (RP-HPLC, Agilent 1206, USA) with a C18 column (ZORBAX 300SB-C18, 5 μm, 4.6 × 150 mm, Agilent, USA). The encapsulation efficiency of ICG (or doxorubicin) was calculated using the following equation: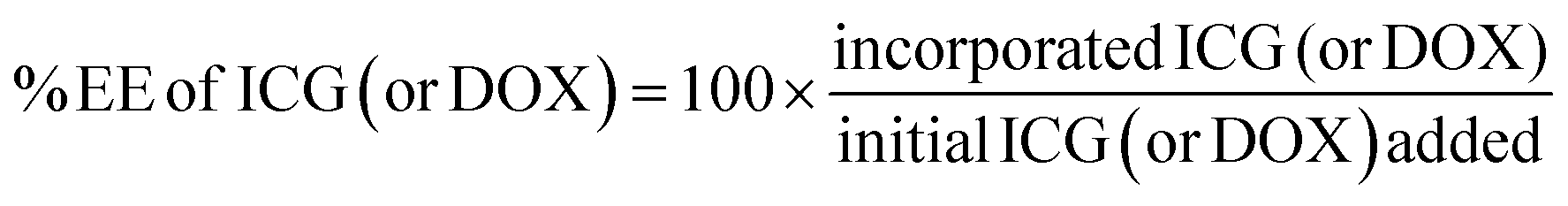
Characterization of liposomal formulations
The morphology of Lipo, pLipo, plipo-ICG, and pLipo-DOX–ICG was determined using a transmission electron microscope (TEM, HT7700, Hitachi, Japan) with 80 kV acceleration voltage. All samples were pre-stained with 1% uranyl acetate for 8 min at room temperature. Besides, particle size (nm) and zeta potential (mV) of Lipo, pLipo, plipo-ICG, and pLipo-DOX–ICG were determined using a Zeta-Sizer Nano series Nano-ZS (Malvern Instruments Ltd, Malvern, UK).Furthermore, the UV-vis-NIR spectra of ICG, doxorubicin, pLipo-ICG and pLipo-DOX–ICG were also measured using a spectrophotometer with the wavelength ranging from 400 nm to 1000 nm. The particle size changes of pLipo-DOX–ICG in PBS buffer, 1% or 10% FBS-containing PBS buffer was determined by the Zeta-Sizer Nano series Nano-ZS at different time points of 0, 6, and 12 h.
Binding affinity of p12 and pLipo to tumor cells
FITC-pLipo was prepared according to the pLipo preparation method with slight modification by the introduction of FITC-labeled DSPE-PEG-p12 instead of unlabeled DSPE-PEG-p12. The binding strengths of FITC-p12 and FITC-pLipo with MCF-7 and 4T1 cells were evaluated by concentration-effect assays. In brief, 4T1 and MCF-7 cells were incubated with FITC-p12 (0.5 μM, 1 μM, 2 μM, 5 μM, and 10 μM) and FITC-pLipo (the molar ratio of FITC-p12:lipids is about 1:200) at 37 °C in opti-MEM for 1 h. After a final wash, all cells were re-suspended in PBS buffer and subjected to an Accuri C6 flow cytometry (BD Biosciences).
Besides, binding affinities of FITC-pLipo to MCF-7 and 4T1 tumor cells were also determined by immunofluorescence. 5 × 104 MCF-7 and 4T1 cells were seeded in 35 mm confocal dishes and incubated with FITC-pLipo (FITC-p12: 1 μM, lipids: 200 μM) at 37 °C for 2 h. Then, the cells were fixed with 4% cold paraformaldehyde (PFA) for 15 min at room temperature, then incubated with DiD for 1 h at 37 °C. The nuclei were stained with 4′,6-diaminoindole (DAPI) for 15 min, and the fluorescence images of cells were obtained by using a confocal microscope (LSM 700, Carl Zeiss, Germany).
Effects of p12 and pLipo on tumor cell migration
In vitro photothermal property of ICG
The photothermal property of ICG in the absence and presence of liposomes was observed using a semiconductor power-tunable laser (Laserwave, China). Briefly, samples of PBS, pLipo (p12: 2 μM, lipids: 400 μM), ICG (10 μM), pLipo-ICG (p12: 2 μM, lipids: 400 μM, ICG: 10 μM) were irradiated at a wavelength of 808 nm (1.5 W cm−2) for 3 min, and the photothermal images and real time temperature changes at different time points were recorded by using a DT-980 infrared thermal camera (Huashengchang Co., Ltd, China). The morphology of pLipo-ICG before and after NIR laser irradiation (808 nm, 1.5 W cm−2) for 3 min was observed by TEM. Besides, the photothermal stability of pLipo-ICG was performed with cyclical laser irradiation (808 nm, 1.5 W cm−2) for 60 s, and then naturally cooled without irradiation for 300 s. This cycle was then repeated three times. The temperature changes during the whole process were recorded by using a DT-980 infrared thermal camera.Cytotoxicity studies in vitro
In order to determine the cell viabilities after treatment with different formulations, 5 × 104 per well of 4T1 tumor cells were pre-seeded into a 96-well plate. After adhesion, 4T1 cells were further incubated with PBS, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG, and pLipo-DOX–ICG under NIR laser irradiation (808 nm, 1.5 W cm−2, 3 min), respectively. The cells with PBS treatment only were used as the control. After 24 h of incubation, 4T1 cell viabilities were determined by the CCK-8 assay.
The calcein-AM/PI double stain kit provides a two-color fluorescence cell viability assay that is based on the simultaneous determination of live and dead cells with two probes. Live cells are distinguished by the presence of ubiquitous intracellular esterase activity, determined by the enzymatic conversion of the virtually nonfluorescent cell-permeant calcein-AM to the intensely fluorescent calcein. The polyanionic dye calcein is well retained within live cells, producing an intense uniform green fluorescence in live cells (ex/em ∼495 nm/∼515 nm). Propidium iodide (PI) is impermeant to live cells, but stains dead cells with red fluorescence, binding tightly to the nucleic acids in the cell (ex/em ∼535 nm/∼617 nm). Briefly, 4T1 cells were seeded at 5 × 105 cells per well in a 24-well plate for 24 h. Then, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG, and pLipo-DOX–ICG were suspended in fresh medium and added to each well. After 12 h of incubation at 37 °C, the cells were irradiated by an 808 nm NIR laser at 1.5 W cm−2 for 3 min, followed by incubation at 37 °C for another 12 h. Finally, 4T1 cells were stained with calcein-AM and PI probes, and the live and dead cells were observed using a confocal microscope (LSM 700, Carl Zeiss, Germany). The untreated cells in the medium with and without NIR laser irradiation were used as controls.
To determine the apoptosis and necrosis cells, 1 × 106 of 4T1 tumor cells were pre-seeded into a 12-well plate for adhesion. After 24 h of incubation, the cells were incubated with different formulations including PBS, Lipo-ICG, pLipo-ICG, Lipo-DOX–ICG, and pLipo-DOX–ICG (ICG: 10 μg mL−1) for 2 h. Then, the cells were irradiated with an NIR laser irradiation (1.5 W cm−2) at a wavelength of 808 nm for 3 min, and further incubated at 37 °C for another 24 h. The PBS group without laser irradiation was used as the control group. Finally, the cells were collected, stained with Annexin V-APC and 7-AAD probes according to the instruction manual, and then detected by flow cytometry.
In vivo drug accumulation at tumor sites
The breast tumor model was established by inoculating 2 × 106 4T1 cells into the left fourth mammary fat pad of each mouse. When the tumor size reached about 60 mm3, mice were intravenously injected with 100 μL of ICG (200 μg mL−1), Lipo-ICG and pLipo-ICG. For near-infrared imaging, each mouse was imaged after 1 h, 2 h, 4 h, 12 h and 24 h using an in vivo imaging system (IVIS Spectrum, PerkinElmer Instruments Co., Ltd, USA). After 24 h, the mice were sacrificed, and tumors and major organs (heart, liver, spleen, kidneys, and lungs) were removed for further ex vivo fluorescence imaging. Besides, for photoacoustic (PA) imaging, 4T1 tumor bearing mice were anesthetized with 2% isoflurane-oxygen mixture, and PA signals were monitored after 2 h, 6 h, 12 h, and 24 h using an InVison 128 MOST system (iThera Medical, Germany).Anticancer effect in vivo
A 4T1 breast tumor model was established as previously described. When the tumor volume reached approximately 100 mm3, the mice were randomly divided into 7 groups (6 mice per group) and treated as follows: control (saline), doxorubicin (3 mg kg−1), Lipo-DOX, pLipo-ICG + Laser, pLipo-DOX, pLipo-DOX–ICG, and pLipo-DOX–ICG + Laser. All drugs were administrated by tail vein injection every three days, and the tumor bearing mice were irradiated with NIR laser irradiation (1.5 W cm−2) at a wavelength of 808 nm for 5 min at 6 h post injection of pLipo-ICG or pLipo-DOX–ICG for each time point. For the tumor growth study, the tumor volume (mm3) and body weight (g) of the mice were measured every 2 days. The tumor volume of each mouse was measured by vernier calipers and calculated using the following equation:| Tumor colume (mm3) = 0.5 × L × W × W |
Ethics statement
All animal experiments reported herein were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Peking University and approved by the Animal Ethics Committee of National Center for Nanoscience and Technology, CAS.Statistical analysis
All experiments were carried out at least 3 times with 3 independent samples. The results were presented as the mean ± SD unless noted otherwise. The statistical significance between two independent groups was analyzed using Student's t-test. All statistical analyses were conducted with statistical software (SPSS 12.0), and P < 0.05 was considered as statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ***P < 0.0001).Conclusions
In this work, we have endeavored to co-deliver chemo-photothermal agents via the targeting peptide-directed liposomal delivery system to prevent breast tumor progression. The obtained results demonstrated that, compared with Lipo-DOX–ICG, p12 modification showed active targeting behavior for delivery and accumulation of payloads at the tumor site. Besides, the introduction of ICG endowed pLipo-DOX–ICG with the precise and controllable release of doxorubicin, thereby exhibiting much enhanced metastasis inhibition and antitumor efficacy in tumor bearing mice models than other treatment groups. Together with low systemic toxicity of pLipo-DOX–ICG, the constructed receptor-binding liposomal formulations may provide a strategy for efficient drug delivery, and show considerable promise as a platform for novel tumor therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0205001 and 2017YFA0205504), the National Natural Science Foundation of China (No. 51861135103, 21721002, 21673055, 21790394 and 81870133), the Key Research Program of Frontier Sciences, Chinese Academy of Science (QYZDJ-SSW-SLH048), and CAMS Innovation Fund for Medical Science (CIFMS 2016-I2M-3-004).Notes and references
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS.
- Y. N. Zhong, J. Zhang, R. Cheng, C. Deng, F. H. Meng, F. Xie and Z. Y. Zhong, J. Controlled Release, 2015, 205, 144–154 CrossRef CAS.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS.
- C. H. Heldin, K. Rubin, K. Pietras and A. Ostman, Nat. Rev. Cancer, 2004, 4, 806–813 CrossRef CAS.
- R. K. Jain, Annu. Rev. Biomed. Eng., 1999, 1, 241–263 CrossRef CAS.
- C. Zylberberg, K. Gaskill, S. Pasley and S. Matosevic, Gene Ther., 2017, 24, 441–452 CrossRef CAS.
- J. Lu, E. Jeon, B. S. Lee, H. Onyuksel and Z. J. Wang, J. Controlled Release, 2006, 110, 505–513 CrossRef CAS.
- Y. Sun, W. Ma, Y. Yang, M. He, A. Li, L. Bai, B. Yu and Z. Yu, Asian J. Pharm. Sci., 2019, 14, 581–594 CrossRef.
- L. Agemy, K. N. Sugahara, V. R. Kotamraju, K. Gujraty, O. M. Girard, Y. Kono, R. F. Mattrey, J. H. Park, M. J. Sailor, A. I. Jimenez, C. Cativiela, D. Zanuy, F. J. Sayago, C. Aleman, R. Nussinov and E. Ruoslahti, Blood, 2010, 116, 2847–2856 CrossRef CAS.
- W. Eck, A. I. Nicholson, H. Zentgraf, W. Semmler and S. Bartling, Nano Lett., 2010, 10, 2318–2322 CrossRef CAS.
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 CrossRef CAS.
- S. K. E. Messerschmidt, A. Musyanovych, M. Altvater, P. Scheurich, K. Pfizenmaier, K. Landfester and R. E. Kontermann, J. Controlled Release, 2009, 137, 69–77 CrossRef CAS.
- Z. Zhang, J. Guan, Z. Jiang, Y. Yang, J. Liu, W. Hua, Y. Mao, C. Li, W. Lu, J. Qian and C. Zhan, Nat. Commun., 2019, 10, 3561–3571 CrossRef.
- Q. Chen, Y. Yang, X. Lin, W. Ma, G. Chen, W. Li, X. Wang, Y. Yang, X. Wang, G. Liao, X. Liu, Q. Chen, H. Li, L. Lu, P. Zhao and Z. Yu, J. Colloid Interface Sci., 2017, 509, 515–521 Search PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS.
- K. Cho, X. Wang, S. Nie, Z. G. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS.
- L. Zou, W. Ding, Y. Zhang, S. Cheng, F. Li, R. Ruan, P. Wei and B. Qiu, Biomaterials, 2018, 182, 1–12 CrossRef CAS.
- T. Wei, J. Liu, H. Ma, Q. Cheng, Y. Huang, J. Zhao, S. Huo, X. Xue, Z. Liang and X. Liang, Nano Lett., 2013, 13, 2528–2534 CrossRef CAS.
- U. M. Domanska, R. C. Kruizinga, W. B. Nagengast, H. Timmer-Bosscha, G. Huls, E. G. de Vries and A. M. Walenkamp, Eur. J. Cancer, 2013, 49, 219–230 CrossRef CAS.
- J. A. Burger and A. Peled, Leukemia, 2009, 23, 43–52 CrossRef CAS.
- X. Sun, G. Cheng, M. Hao, J. Zheng, X. Zhou, J. Zhang, R. S. Taichman, K. J. Pienta and J. Wang, Cancer Metastasis Rev., 2010, 29, 709–722 CrossRef CAS.
- A. C. Spoo, M. Lübbert, W. G. Wierda and J. A. Burger, Blood, 2007, 109, 786–791 CrossRef CAS.
- C. Zhang, N. Tang, X. Liu, W. Liang, W. Xu and V. P. Torchilin, J. Controlled Release, 2006, 112, 229–239 CrossRef CAS.
- X. Xue, T. Fang, L. Yin, J. Jiang, Y. He, Y. Dai and D. Wang, Drug Delivery, 2018, 25, 1826–1839 CrossRef CAS.
- C. Corbo, R. Molinaro, F. Taraballi, N. E. Toledano Furman, M. B. Sherman, A. Parodi, F. Salvatore and E. Tasciotti, Int. J. Nanomed., 2016, 11, 3049–3063 CrossRef CAS.
- P. C. Simons, S. M. Young, M. B. Carter, A. Waller, D. Zhai, J. C. Reed, B. S. Edwards and L. A. Sklar, Nat. Protoc., 2011, 6, 943–952 CrossRef CAS.
- R. Briesewitz, G. T. Ray, T. J. Wandless and G. R. Crabtree, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1953–1958 CrossRef CAS.
- J. Juarez, L. Bendall and K. Bradstock, Curr. Pharm. Des., 2004, 10, 1245–1259 CrossRef CAS.
- C. C. Bleul, R. C. Fuhlbrigge, J. M. Casasnovas, A. Aiuti and T. A. Springer, J. Exp. Med., 1996, 184, 1101–1109 CrossRef CAS.
- A. Müller, B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verástegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32–45 CrossRef CAS.
- Y. Xiang, L. Liang, X. Wang, J. Wang, X. Zhang and Q. Zhang, J. Controlled Release, 2011, 152, 402–410 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00536c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |