
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of Fe3O4 integrated polymer/phosphate glass composite scaffolds for bone tissue engineering
Raji
Govindan
ab,
Sekar
Karthi†
c,
Govindan Suresh
Kumar
 d and
Easwaradas Kreedapathy
Girija
*b
d and
Easwaradas Kreedapathy
Girija
*b
aKey Laboratory of Theoretical Chemistry of Environment, Ministry of Education, School of Chemistry, South China Normal University, Guangzhou 510006, P. R. China
bDepartment of Physics, Periyar University, Salem, 636 011, India
cCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng 475001, P. R. China. E-mail: girijaeaswaradas@gmail.com
dDepartment of Physics, K.S. Rangasamy College of Arts and Science (Autonomous), Tiruchengode 637215, India
First published on 13th October 2020
Abstract
Magnetic nanoparticle (MNP) integrated biomimetic scaffolds are receiving a lot of attention for the repair of bone defects and in bone tissue engineering applications. In the present work, Fe3O4 MNP integrated polymer/phosphate glass (CG/PG/MNP) composite scaffolds developed using a freeze drying technique are reported. MNP integrated CG/PG composite scaffolds were highly porous in nature with pores of size ranging between 20 and 150 μm and the pores were typically interconnected. Integration of Fe3O4 with CG/PG significantly influenced the swelling and degradation behavior constructively. The CG/PG/MNP composite scaffold exhibited a soft ferromagnetic nature and the compressive modulus increased significantly with increasing MNP content. In addition, the MNP containing CG/PG composite scaffolds demonstrated good bioactivity and cytocompatibility. Based on the results, MNP integrated CG/PG composite scaffolds developed in the present study may be potential scaffolds for bone tissue engineering applications.
1. Introduction
The development of biomimetic scaffolds with the ability to regenerate bone and adequate mechanical properties is a promising area of research in bone tissue engineering. In particular, three dimensional porous scaffolds are the primary choice for bone regeneration since they offer vital support for cell migration and proliferation and stimulate different cellular activities.1,2 Besides, for ideal bone regeneration, scaffolds should resemble natural bone in structure, morphology, function and chemical constituents. Thus, biopolymer/calcium phosphate composite scaffolds have received much attention as bone tissue engineering scaffolds.3,4Chitosan, a biopolymer, has been used widely in tissue engineering as a scaffold owing to its excellent biodegradability, biocompatibility and ability to stimulate wound healing.5 However, the hydrophobic nature of chitosan leads to poor cell attachment, which can be overcome by blending it with another biopolymer, such as gelatin, possessing a highly hydrophilic nature. Gelatin, being a biocompatible and biodegradable biomaterial, is widely used as a scaffold for bone tissue engineering applications.6,7 Evidently, the blending of gelatin with chitosan has enhanced biological activity including cell adhesion, cell migration and also hydrophilicity.8 Nevertheless, natural polymer scaffolds suffer in load bearing applications due to the lack of adequate mechanical strength. In order to obtain a potential scaffold with high porosity, suitable mechanical strength and superior cellular activity, integration of an inorganic bioceramic material into the polymer matrix is one of the effective strategies for tissue engineering. Bioceramics such as hydroxyapatite (HAp),9–13 β-tricalcium phosphate (β-TCP),14–16 biphasic calcium phosphate (BCP),17 dihydrogen calcium phosphate anhydrous (DCP),18 LAPONITE®19 and bioglass (silica-based glass)20–22 integrated with gelatin–chitosan composites have been explored for bone regeneration applications.
Bioactive glasses are non-crystalline materials that have been proven to have superior in vivo osteoconductivity compared to HAp, enhanced biodegradability compared to β-TCP and excellent bioactivity with good biocompatibility.23 Phosphate-based glass (PG: the basic components are P2O5, CaO and Na2O) materials have been extensively studied as a graft material in tissue engineering due to their tunable dissolution rate by changing the glass network and excellent cellular response.24–26 So far, only a few studies on PG/polymer composites have been reported for tissue engineering and drug delivery applications. For instance, Barbeck et al.27 reported a 3D printed bi-layered PLA/G5 bioglass (G5: P2O5–CaO–Na2O–TiO2) composite scaffold for osteochondral regeneration. Vancomycin loaded PG/poly(ε-caprolactone) (PCL) composites have been developed using solvent extraction and thermal pressing methods for tissue regeneration.28 These composites have demonstrated better biocompatibility, favorable cell proliferation and sustained release of vancomycin drug. However, polymer/bioceramic scaffolds developed for both soft and hard tissue regeneration cannot effectively control cell differentiation and angiogenesis processes.29,30 Hence, it is necessary to incorporate stimulators for cell attachment, proliferation and bone formation.
The idea of magnetic guidance in biomedical applications extends from hyperthermia for the destruction of cancer cells to thermo responsive drug release and tissue engineering (magnetic cell seeding, magneto-mechanical stimulation of mechanosensitive ion channels and cell seeded constructs, controlled cell proliferation and differentiation, etc.). Recently, magnetite (Fe3O4) has attracted much attention in the fields of tissue engineering, thermal-induced drug delivery, magnetic resonance imaging and magnetic hyperthermia due to its good biocompatibility, nontoxicity, ease of functionalization and high magnetization at room temperature.31,32 In cancer treatment, MNPs act as a hyperthermia agent capable of generating heat energy at the targeted sites for the destruction of cells. Cancer cells are destroyed in the temperature range from 41 to 43 °C, while healthy cells can withstand this temperature range.33 Furthermore, studies on incorporating MNPs into composite scaffolds such as gelatin/bioglass and on Fe3O4 nanoparticles incorporated in mesoporous bioactive glass/PCL demonstrated prolonged anticancer drug delivery, hyperthermia properties, and stimulated cell proliferation and differentiation in addition to excellent apatite forming ability and enhanced compressive strength.34–36 Recently, HAp/Fe3O4 incorporated chitosan/collagen composite scaffold exhibited enhanced mechanical strength with compressive modulus of 2.515 MPa compared to the chitosan/collagen/HAp composite scaffold.37 Furthermore, the developed composite scaffold possessed superior cytocompatibility for cell adhesion, osteogenic differentiation and proliferation in both in vitro and in vivo models. These results clearly indicated that the addition of MNPs not only assists in hyperthermia and drug release but also accelerates cell proliferation and differentiation with enhanced mechanical strength of the composite scaffolds. Nevertheless, there is no report on MNP integrated natural polymer/PG composite scaffolds for both hyperthermia and tissue engineering applications.
The aim of this work is to fabricate Fe3O4 integrated chitosan-gelatin/phosphate glass (CG/PG/MNP) composite scaffolds via a freeze drying method, and to investigate the effects of MNP addition on the physicochemical properties, heating ability and cellular properties of the developed scaffolds.
2. Materials and methods
2.1. Preparation of phosphate glass (PG) and Fe3O4 nanoparticles (MNP)
PG of composition 45P2O5-29CaO-21Na2O-5SrO (mol%) was prepared using the traditional melt quenching method.38 The average particle size of the obtained PG was 40 μm. Fe3O4 nanoparticles were prepared using the co-precipitation method.392.2. Fabrication of CG/PG/MNP composite scaffold
2% (w/v) chitosan was dissolved in 1% aqueous acetic acid and 0.5 g of gelatin was added and stirred overnight at room temperature. Then, 1 wt% of PG and Fe3O4 (0.5, 1 and 1.5 wt%) with respect to the total polymer quantity were added to the above mixture and continuously stirred for 24 h. The homogeneous complex was transferred into 96 well culture plates and pre-frozen at −5 °C overnight followed by lyophilizing at −50 °C for 12 h. Then, the scaffolds were neutralized in NaOH for 2 h and further washed with deionized water. For convenience, these MNP containing CG/PG scaffolds are labeled as CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP for 0, 0.5, 1 and 1.5% w/w MNP concentrations, respectively.2.3. Characterization of the prepared samples and composite scaffold
Powder X-ray diffraction (PXRD) patterns of the samples were recorded using a Rigaku MiniFlex II powder X-ray diffractometer operating at a voltage of 40 kV with 10° ≤ 2θ ≤ 70° and Cu Kα monochromatic radiation (1.5406 Å). The functional group analyses of the synthesized nanoparticles and developed scaffolds were performed using a Bruker Tensor 27 spectrometer with the potassium bromide (KBr) pellet technique in the operating range of 400–4000 cm−1. The morphology of Fe3O4 was assessed using TEM (JEOL-JEM2100F). The morphology and pore size of the scaffolds were observed using FESEM (FEI – QUANTA-FEG 250). Scaffolds were sectioned using a razor blade into thin sections and coated with gold for 2 min before imaging. The mean pore size of the scaffolds was determined by measuring the size of 30 pores.The magnetic properties of the Fe3O4 nanoparticles and scaffolds were determined using a vibrating sample magnetometer (VSM, Oxford, UK). Easy Heat 8310 (Ambrell, Gloucester-shire, UK) was used to study the heating performance of the powdered MNP incorporated CG/PG scaffold (CG/PG/10MNP). An alternating magnetic field was generated under a field strength of ∼500 Oe and frequency 300 kHz. For the measurements, 2, 5 and 10 mg of powdered samples were dispersed in 1 mL of PEG (10%) solution in a plastic tube and were kept at the midpoint of the coil and the temperature change was noted every 30 s interval up to 10 min.
2.4. Porosity of CG/PG/MNP scaffolds
The porosity of the scaffolds was measured using the liquid displacement method. The scaffolds were immersed in ethanol for 5 min in a cylinder containing a known volume of ethanol (V1). The final volume after impregnation of the scaffolds was noted as V2. Then, the scaffolds were removed from the ethanol and the remaining volume was noted as V3. The porosity of the scaffold was calculated from the following equation:| P = (V1 − V3)/(V1 − V2) |
2.5. Swelling behaviour of CG/PG/MNP scaffolds
The swelling study was performed using phosphate buffered saline (PBS) at 37 °C. The initial weight of the scaffolds was noted (Wo). The scaffold was immersed in PBS for 1 h at 37 °C. Then, the surface adsorbed water was removed and the wet weight was noted (Ww). The swelling ratio was calculated using the following formula: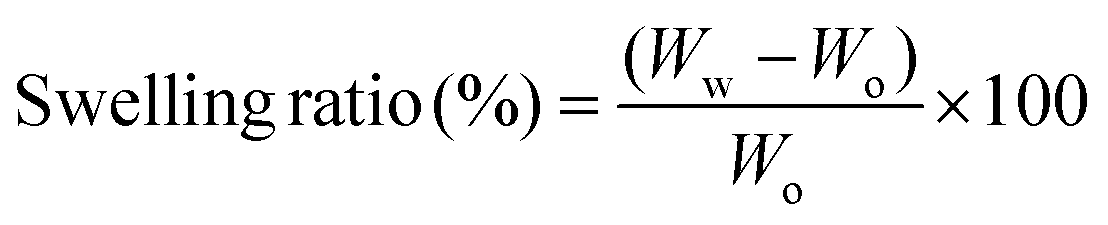
2.6. In vitro enzymatic degradation studies of CG/PG/MNP scaffolds
The degradation of the scaffolds was performed in PBS medium comprising lysozyme (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1) at 37 °C for 7 days. After 7 days, scaffolds were withdrawn from the medium and freeze dried at −50 °C for 12 h. The enzymatic degradation of the scaffolds was determined using the following formula:
000 U mL−1) at 37 °C for 7 days. After 7 days, scaffolds were withdrawn from the medium and freeze dried at −50 °C for 12 h. The enzymatic degradation of the scaffolds was determined using the following formula:where Wo and Wt are the initial and final weights of the scaffold, respectively.
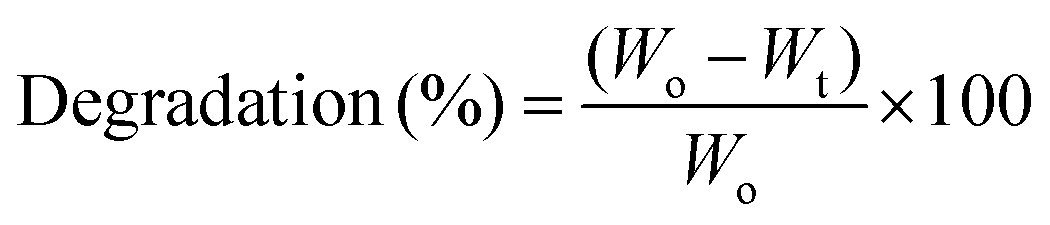
2.7. Compressive strength of CG/PG/MNP scaffolds
The mechanical strength of the scaffolds was determined using a 3366, Instron® UTM (Instron® Co. Ltd Norwood, MA, USA). The compressive strength was calculated from the compressive load (F, Newton) and surface area of the scaffold (A, square millimeters) using the following equation:| S = F/A |
2.8. In vitro bioactivity of CG/PG/MNP scaffolds
The apatite forming ability over the scaffolds was studied by incubating in simulated body fluid (SBF) at 37 °C. The SBF was prepared according to the procedure of Tas.40 Then, the scaffolds were incubated in 25 mL of SBF in plastic containers that was renewed every three days for a period of 28 days. Then, the composite scaffolds were washed thoroughly with distilled water to remove unwanted salts. After freeze drying the scaffolds, the mineralization of apatite over the scaffold surface was investigated using FESEM (FEI – QUANTA-FEG 250).2.9. Cytocompatibility of CG/PG/MNP scaffolds
The cytocompatibility test was done for sliced scaffolds with human osteoblast-like MG-63 grown in eagles minimum essential medium comprising 10% fetal bovine serum (FBS) and 100 U mL−1 penicillin/streptomycin under standard culture conditions with 100% relative humidity. The monolayer cells were isolated with trypsin–EDTA to make single cell suspensions and viable cells were totaled using a hemocytometer and diluted with medium containing 5% FBS to provide a final density of 1 × 105 cells mL−1. 100 μL per well of cell suspension was added into 96-well plates with a density of 10000 cells/well and incubated under standard culture conditions. After 24 h of incubation, scaffolds (sterilized with 70% ethanol) with 1 mm thickness were added to the culture medium. The medium without the composite scaffold was considered as the positive control. Subsequently, the plates were incubated for 48 h at 37 °C in 5% CO2, 95% air and 100% relative humidity. The assay was performed in triplicate. After 48 h of incubation, 15 μL of 3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide (MTT) (5 mg mL−1) in PBS was added to each well and incubated at 37 °C for 4 h. Then, the medium was removed from each well and the formazan crystals were dissolved in 100 μL of dimethyl sulfoxide (DMSO) and then the absorbance was measured at 570 nm using a microplate reader.
3. Results and discussion
The crystalline nature, functional groups, elemental composition, microstructure, particle size distribution and magnetic properties of the Fe3O4 nanoparticles were studied and reported in our previous work.41,42Fig. 1 shows a schematic representation of the fabrication of the CG/PG and MNP integrated CG/PG composite scaffolds.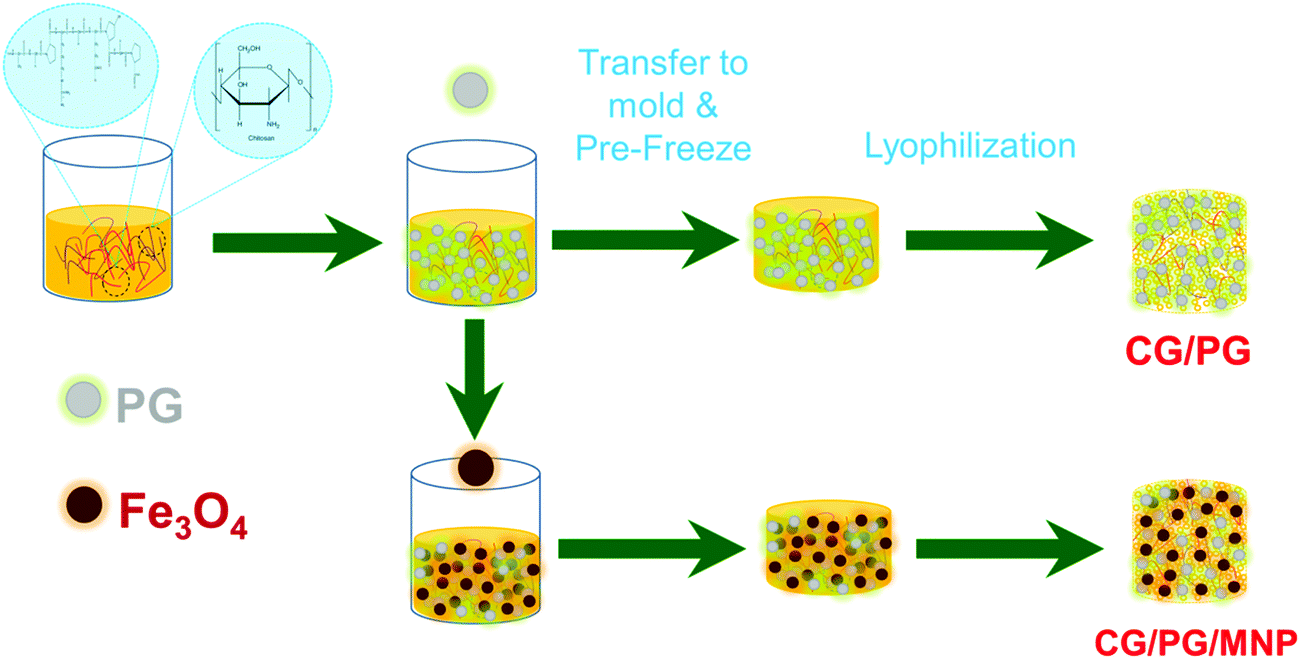 | ||
| Fig. 1 Schematic illustration of the fabrication procedure of the CG/PG and CG/PG/MNP composite scaffolds. | ||
3.1. XRD
Fig. 2a shows the XRD patterns of the CG/PG and CG/PG/MNP composite scaffolds. The CG/PG composite scaffold displays a broad peak between 15 and 30°, which reflects the amorphous nature of PG and the polymer blend. After the introduction of MNPs into the CG/PG composite scaffold, characteristic diffraction peaks of Fe3O4 were observed; meanwhile, the intensity of the broad peak was suppressed. Interestingly, the intensity of the diffraction peaks of the CG/PG/MNP composite scaffolds increased with increasing MNP content.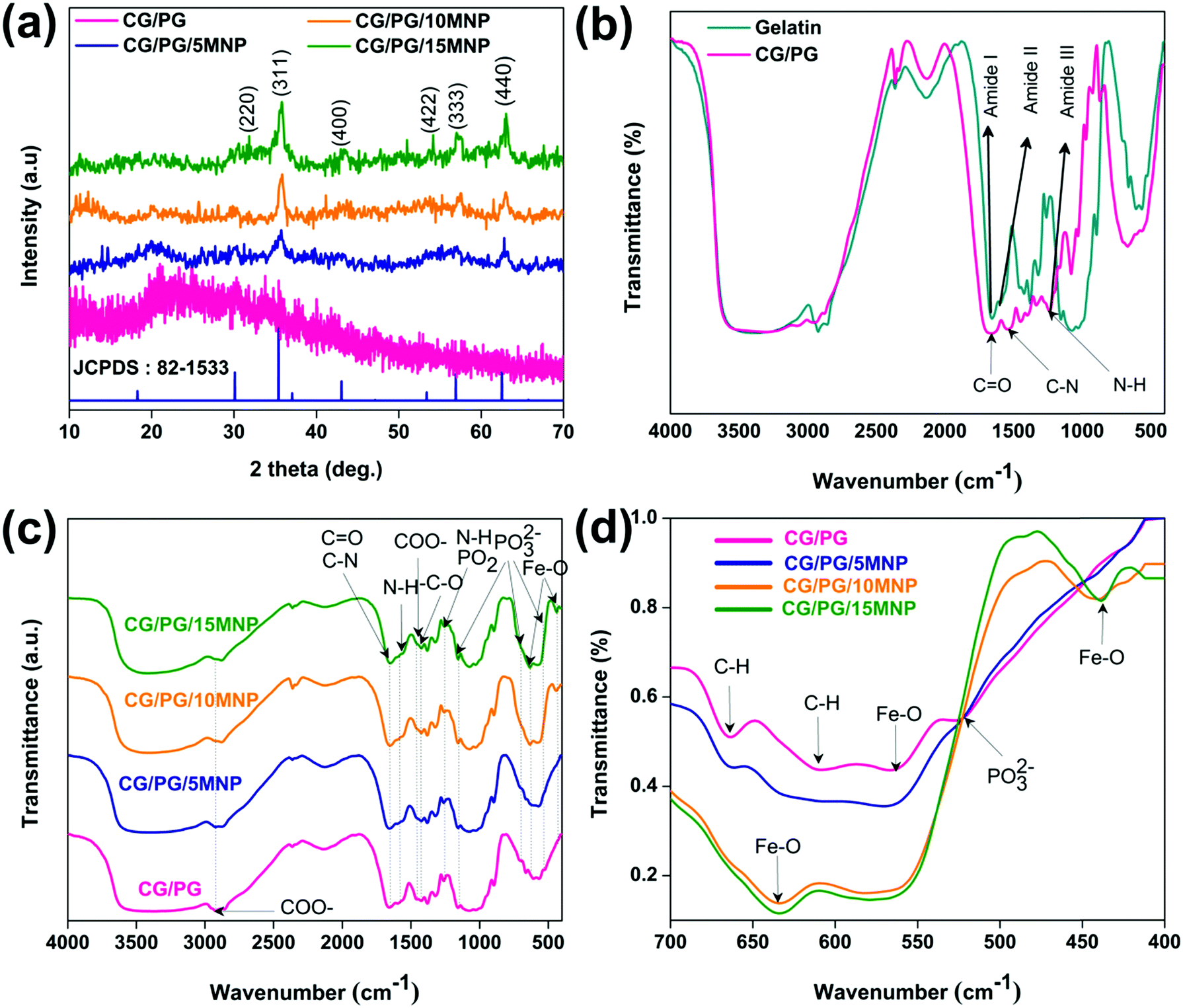 | ||
| Fig. 2 (a) XRD pattern. (b) FTIR spectra of gelatin and CG/PG composite scaffold and (c and d) FTIR spectra of developed composite scaffolds. | ||
3.2. FTIR spectroscopy
Fig. 2b illustrates the FTIR spectra of gelatin and the CG/PG composite scaffold. The bands that appeared at 1664, 1537 and 1238 cm−1 in the spectrum of gelatin correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, N–H deformation, and C–N stretching and N–H deformation of the amide I, II and III vibrations of gelatin, respectively.43 The peaks at 1449, and 2930 and 3076 cm−1 are attributed to COO− and C–H stretching vibrations, respectively. On blending chitosan with gelatin, the carbonyl characteristic band at 1665 cm−1 red shifted and the gelatin peak broadened with decreased intensity.43,44 Moreover, the amide-I and amide-II peaks also shifted as well as reducing in intensity compared to that of the gelatin, which may be due to the modification of the interior structure of gelatin caused by the chitosan.45 The intensity of the amide III bands of the composite scaffold decreased after blending with chitosan due to the interaction between the amino group of gelatin and carbonyl group of chitosan through electrostatic interactions.46
O stretching, N–H deformation, and C–N stretching and N–H deformation of the amide I, II and III vibrations of gelatin, respectively.43 The peaks at 1449, and 2930 and 3076 cm−1 are attributed to COO− and C–H stretching vibrations, respectively. On blending chitosan with gelatin, the carbonyl characteristic band at 1665 cm−1 red shifted and the gelatin peak broadened with decreased intensity.43,44 Moreover, the amide-I and amide-II peaks also shifted as well as reducing in intensity compared to that of the gelatin, which may be due to the modification of the interior structure of gelatin caused by the chitosan.45 The intensity of the amide III bands of the composite scaffold decreased after blending with chitosan due to the interaction between the amino group of gelatin and carbonyl group of chitosan through electrostatic interactions.46
The FT-IR spectra of CG/PG and CG/PG/MNP composite scaffolds are presented in Fig. 2c. The bands that appeared at 1665 and 1593 cm−1 of the CG/PG composite scaffold are ascribed to the CO and C–N stretching and N–H deformation of gelatin and the CO stretching and N–H bending vibrations of chitosan for amide I and II, respectively. The bands that appeared at 1262 cm−1 and 1423 cm−1 of the CG scaffold are attributed to the N–H bending and C–O stretching vibrations of gelatin and chitosan, respectively.47,48 The peaks at 1460 and 2927 cm−1 are ascribed to the COO− vibration of gelatin. The vibration bands at 524, 707, and 1156 and the one at 1262 cm−1 present in the spectrum of the GC/PG composite scaffolds are related to the symmetric stretching vibrations of PO32− and asymmetric vibrations of PO2, respectively.49
The appearance of amide I and amide II peaks at 1664 and 1537 cm−1 confirms that the gelatin in the composite scaffolds adopts predominantly the α-helical configuration. The band appearing at 1325cm−1 represents covalent bond formation between the COO− group in the polymers and Ca2+ in PG in the particulate reinforced composite scaffold.50 The CG/PG and MNP composite scaffold spectra showed additional peaks at 438 and 631 cm−1 that correspond to the characteristic Fe–O stretching vibration peaks of magnetite.48
The interaction between MNPs and the CG/PG composite scaffold can be explained as follows (Fig. 2d). The band at 607 cm−1 disappeared with the addition of MNPs and also the intensity of the peak at 664 cm−1 decreased with the introduction of MNPs, and it subsequently disappeared with a further increase in MNP content. Moreover, the PO32− stretching band of PG at 524 cm−1 disappeared and the intensity of the peaks at 638 and 571 cm−1 increased with the addition of MNPs. A broad characteristic band that appeared between 3200 cm−1 and 4000 cm−1 is due to the OH− stretching of CG/PG and CG/PG/MNP composite scaffolds. The changes in these peaks suggest the formation of a CG/PG/MNP network in the composite scaffold due to strong hydrogen bonding interaction between the polymers and Fe3O4 and PG.48 Consequently, the polymer network not only serves as a matrix for the PG and MNPs but also offers an anchoring site for inorganic particles to form a complex structure.51
3.3. Morphology of the composite scaffold
Fig. 3a displays the morphology of CG/PG and CG/PG/MNP composite scaffolds. As shown in Fig. 3a, all four composite scaffolds are highly porous in nature with pores of size ranging between 20 and 150 μm and the pores are typically interconnected but are highly inhomogeneous in nature. Furthermore, the CG/PG scaffold exhibits less surface roughness than the other three scaffolds. The CG/PG composite scaffold exhibits reduced pore size, which may be due to the increased viscosity of the solution with the addition of chitosan. The surface becomes rough with the introduction of Fe3O4 and the roughness further increased with increasing Fe3O4 content in the CG/PG composite scaffold.52 It is obvious from previous studies that the scaffolds with a pore size in the range of 20–150 μm support chondrocyte differentiation.53 Moreover, a minimum pore size in the range of 20–250 μm, interconnectivity and roughness are essential for tissue ingrowth, nutrient supply and better cell proliferation.54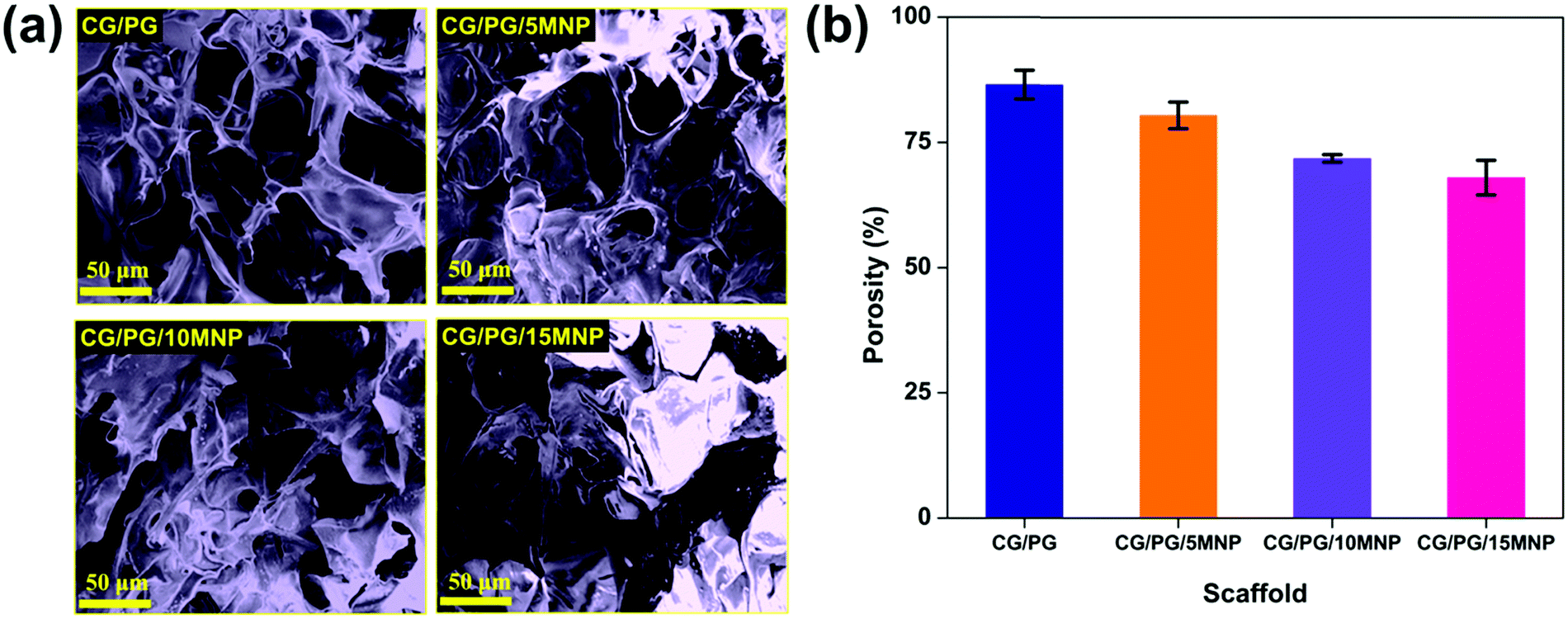 | ||
| Fig. 3 (a) SEM images and (b) porosity of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds. | ||
3.4. Porosity of the composite scaffold
Fig. 3b shows the percentage porosity of the prepared composite scaffolds and it was observed that the porosity of the CG/PG scaffold decreased with an increase in MNP content. The porosity of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds is 86.5 ± 2.87, 80.4 ± 2.67, 71.8 ± 0.76 and 68.0 ± 3.46%, respectively. The decreased porosity may be due to the gradual increase in the MNP concentration in the polymer matrix. The decrease in porosity may be attributed to the strong interaction between chitosan and inorganic constituents.55 The porosity generated in the composite scaffolds resembles the porosity of cancellous bone (50–90%), which facilitates cell ingrowth, proliferation and nutrition supply.56,573.5. Swelling of the composite scaffold
Swelling increases the pore size and porosity of the scaffolds, which are essential for cell infusion and attachment. The swelling behavior of CG/PG and CG/PG/MNP composite scaffolds is shown in Fig. 4a. Swelling of all the composite scaffolds increased initially and attained almost saturation after 3 days. The CG/PG composite scaffold shows a higher swelling than the CG/PG/MNP composite scaffolds. The swelling behavior of the scaffolds mainly depends on the hydrophilic nature of the constituents. Owing to the hydrophilic nature of gelatin, the CG/PG composite scaffold exhibited superior swelling behaviour. After the introduction of MNPs, the swelling ratio of the CG/PG composite scaffold decreased and the ratio was found to further decrease with increasing MNP concentration, which may be attributed to the strong interactions between the cationic sites of the inorganic phase and the carboxylate groups of the polymer, which reduced the hydrophilic nature of the scaffold, leading to decreased water sorption.58 The integration of PG and MNPs with polymers also caused less swelling and contributed to greater structural integrity of the scaffold, which favors cell proliferation.59 Conversely, the favorable swelling properties of the MNP integrated CG/PG composite scaffolds support the diffusion of nutrients and metabolites into newly developed tissues.60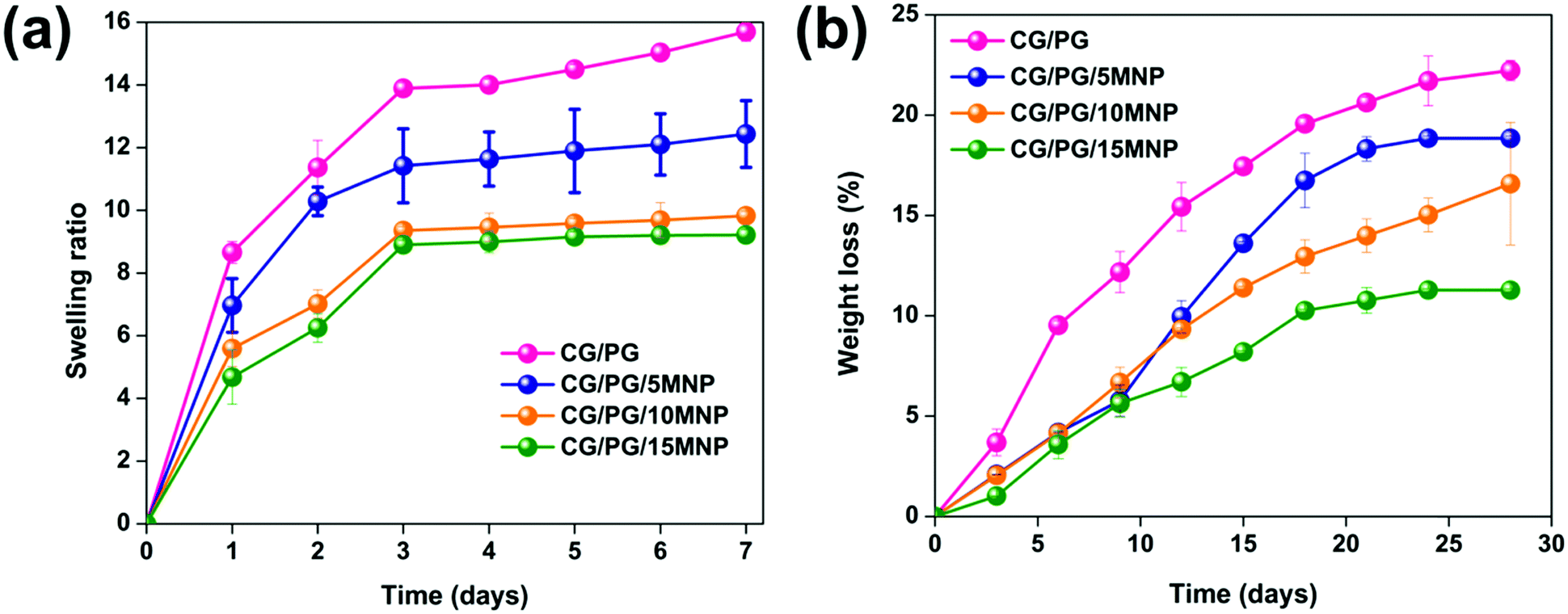 | ||
| Fig. 4 (a) Swelling behavior and (b) weight loss of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds. | ||
3.6. Enzymatic degradation of the composite scaffold
The degradation rate of an ideal scaffold must be analogous to the rate of new tissue regeneration. Fig. 4b displays the in vitro enzymatic degradation of CG/PG and CG/PG/MNP composite scaffolds. The percentage degradation values of the CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds are 22.22 ± 0.5, 18.84 ± 0.13, 16.59 ± 3.0 and 11.28 ± 0.08, respectively. The degradation rate of all the scaffolds gradually increased with increasing soaking time. The CG/PG composite scaffold exhibited higher degradation rate than MNP integrated CG/PG composite scaffolds; this may be ascribed to the rapid hydrolysis of the macromolecular chains in gelatin. The rate of degradation decreased with the addition of MNPs and further decreased with increasing MNP addition, which was due to the reduced hydrophilic nature of the polymer in PBS.58,61 The obtained result is in good agreement with the previous work by Aliramaji et al.62 In this work, the degradation study was performed only for 28 days. After 28 days, the 10% MNP integrated CG/PG composite scaffold showed 16.59 ± 3.0% degradation, with the slow degradation rate suggesting that the developed composite scaffold may be suitable for new tissue regeneration. However, the degradation rate of composite scaffolds is anticipated to be faster in vivo compared to in vitro because of the fluid flow of human tissue and faster bone tissue formation.63 Hence, further in vivo investigations are required to understand the degradation rate of the composite scaffolds.3.7. Mechanical properties of the composite scaffolds
The compressive modulus of the CG/PG and CG/PG/MNP scaffolds was calculated at 40% deformation and the stress–strain curves are shown in Fig. 5a. The values of compression modulus obtained for CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds are 3.1, 7.2, 9.7 and 13.3 MPa, respectively (Fig. 5b).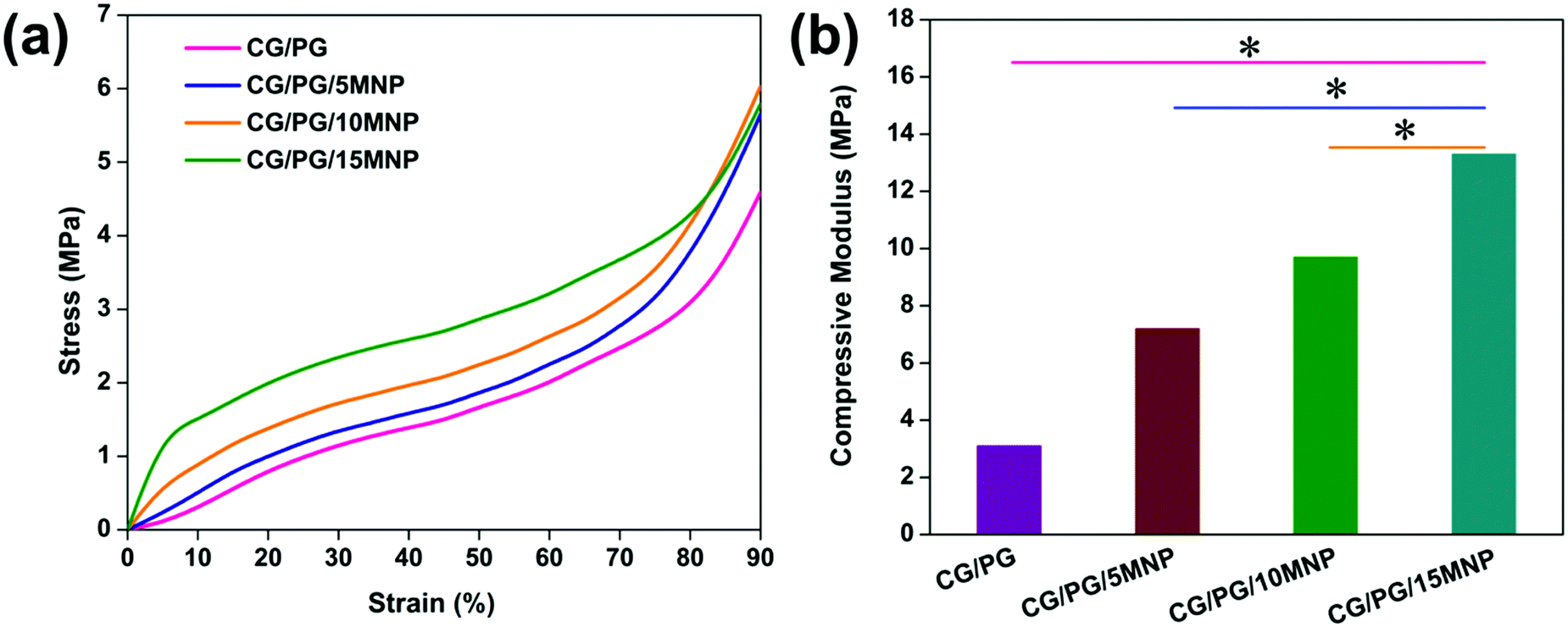 | ||
| Fig. 5 (a) Stress–strain curve and (b) compressive modulus of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds. | ||
The stress–strain curve reveals an enhancement in the mechanical properties of the CG/PG composite scaffold with the addition of MNPs and also that the CG/PG and CG/PG/MNP composite scaffolds did not fracture up to 90% strain. The compression modulus of gelatin–chitosan can be ascribed to both electrostatic and hydrogen bonding that leads to a denser matrix.64,65 The reason for the enhanced mechanical strength of the CG scaffold is the anti-plasticization effect of gelatin added to chitosan.64 The compressive modulus increases with increasing MNP content and reaches a value four times greater than that of the CG/PG composite scaffold. The presence of higher content of inorganic phase in the gelatin–chitosan matrix tends to absorb the applied stress and disperse it without cracking the composite structure. The strong interaction between gelatin–chitosan and the ceramics decreased the brittleness and increased the toughness of the CG/PG/MNP scaffolds.66 The compressive modulus of the developed composite scaffolds is in the range of 3.1–13.3 MPa, which is an acceptable range when compared to the compressive modulus of human articular cartilage (1.16 ± 0.20–13.0 ± 4.2 MPa).67,68
3.8. Magnetic properties of the composite scaffold
The curves of applied field vs. magnetization of the CG/PG and CG/PG/MNP scaffolds are shown in Fig. 6a. The CG/PG/MNP scaffolds exhibited soft ferromagnetic behavior whereas CG/PG was found to be diamagnetic. The magnetization curve indicates magnetization as a function of applied magnetic field for the MNP integrated composite scaffolds. The saturation magnetization of the CG/PG/MNP scaffolds increased with increasing MNP content, and the values of saturation magnetization of CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP scaffolds are 0.180, 0.432 and 0.496 emu g−1, respectively. The coercivity (Hc) values for CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds are 25.46, 7.576 and 11.15 Oe, respectively. The reduced saturation magnetization when compared to pristine MNPs is due to the interaction between magnetic MNPs and the non-magnetic CG/PG, which leads to a decreased magnetic moment of the composite scaffolds.69,70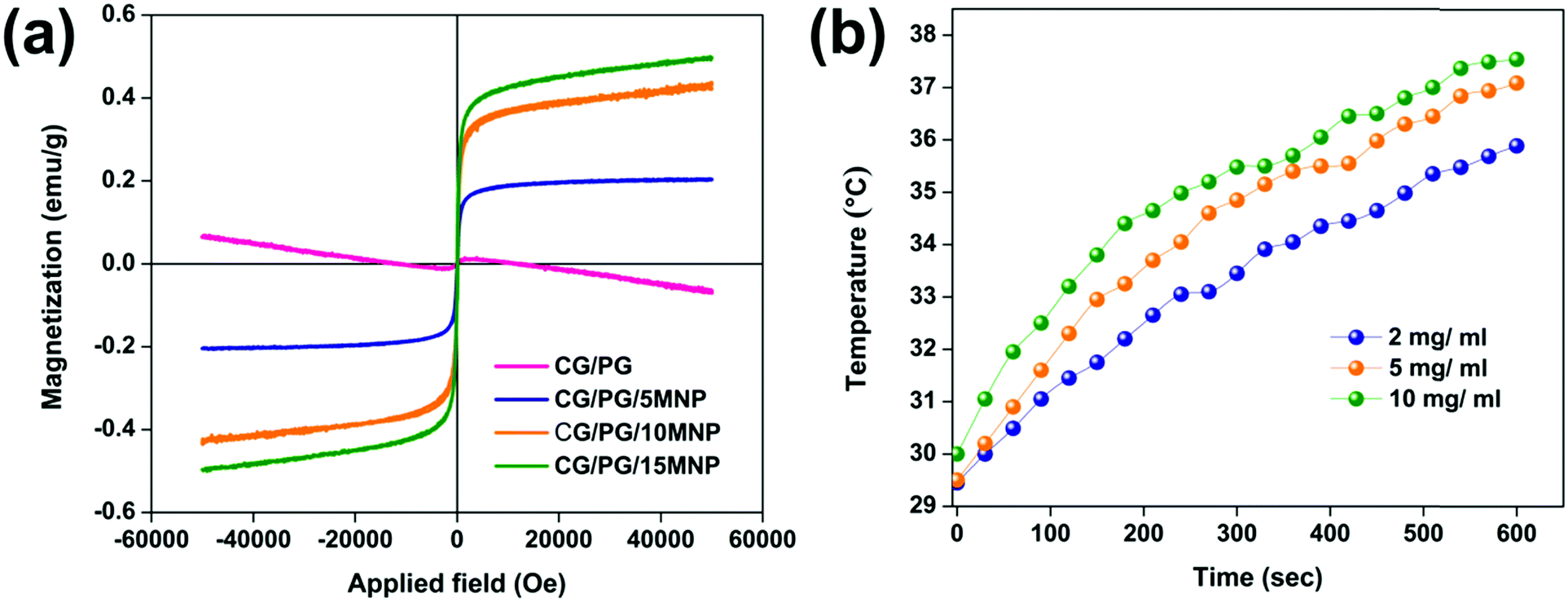 | ||
| Fig. 6 (a) Hysteresis loop of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds and (b) the magnetic heating ability of the GC/PG/10MNP composite scaffold evaluated under an alternating magnetic field with different concentrations. | ||
3.9. Induction heating ability
Fig. 6b shows the temperature changes in a 10% PEG solution containing powdered CG/PG/10MNP scaffolds measured in an alternating magnetic field (frequency at 300 kHz) for three different (2, 5 and 10 mg mL−1) concentrations. In the case of the 2 mg mL−1 concentration, the solution temperature raised to 36 °C at 500 Oe. The temperatures of the solutions with 5 and 10 mg mL−1 concentrations were 37.1 °C and 37.6 °C, respectively. Unfortunately, all the three samples in the solution did not attain the hyperthermia temperature of 43 °C within 10 min, which may be due to the fact that the magnetic phase of the sample is extremely suppressed by the presence of nonmagnetic ceramic and polymer phases. Nanoparticle based hyperthermia is strongly dependent on the Brownian and Néel relaxation processes.71 In addition, the poor dispersible nature of the polymer containing magnetic nanoparticles intensely affects the total heating ability.72 In this work, MNPs are dispersed in the polymer matrix and the polymer may act as a coating agent over the magnetic nanoparticles, which changes the viscosity of the medium and leads to reduced dispersibility of the magnetic nanoparticles.73 The hyperthermia temperature can be probably achieved by varying the MNP concentration in the polymer matrix, time, etc., and further optimization is needed to address these issues that are critical for real time applications.3.10. Bioactivity
Fig. 7a shows the FESEM image of the CG/PG and CG/PG/MNP scaffolds before and after soaking in SBF for 21 days. As shown in Fig. 7a, the rich apatite deposits observed after soaking in SBF indicate the in vitro bioactivity of CG/PG and CG/PG/MNP scaffolds. There is no observable difference in apatite deposit formation among the pristine and MNP containing scaffolds, indicating that the MNPs in the scaffolds did not affect the bioactivity of the scaffolds. The formation of rich apatite over the composite scaffolds can be attributed to the interaction between NH2 and OH− groups present in the polymers and the phosphate groups of the ceramics with Ca2+ ions of SBF.74 MNPs are reported to effectively stimulate osteoinductivity, which results in improved bone formation and bonding.75,76 Since apatite nanocrystals are the major inorganic component of human bones, the bioactive nature of the scaffold is believed to be favorable for better cell attachment, cell proliferation and superior tissue growth in vivo.77,78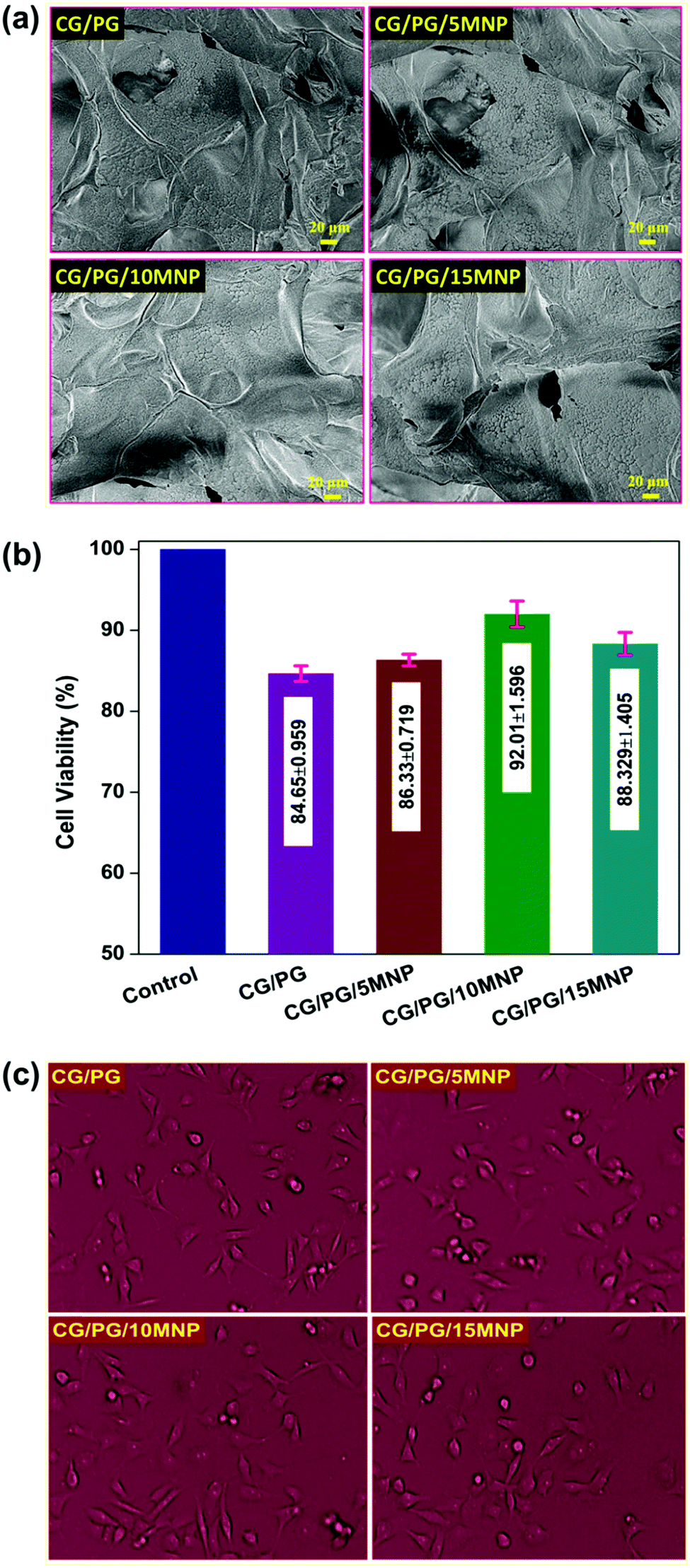 | ||
| Fig. 7 (a) FESEM images of CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds after soaking in SBF for 21 days. (b) Cell viability and (c) optical microscope images of MG-63 cells on CG/PG, CG/PG/5MNP, CG/PG/10MNP and CG/PG/15MNP composite scaffolds. | ||
3.11. Cytocompatibility
Fig. 7b shows the result of MG-63 cell viability study on the CG/PG and CG/PG/MNP composite scaffolds as evaluated by MTT assay. After 24 h of incubation, the number of viable cells on the developed scaffolds with respect to the control seems to be less, whereas the number of viable cells is higher on the MNP containing scaffolds than on the pristine one. Nevertheless, the cell viability of MNP integrated scaffolds increased up to 10% MNPs and decreased for 15% MNPs. Interestingly, CG/PG and MNP integrated GC/PG composite scaffolds did not show adverse effects, suggesting that all the samples exhibited good biocompatibility towards MG-63 cells. According to ISO 10993 – 5:2009, all the four scaffolds showed cell viability not lower than 70%, demonstrating that all the scaffolds are compatible with MG-63 cells. After 24 h of incubation, quasi spherical shaped MG-63 cells became flattened on CG/PG and CG/PG/MNP composite scaffolds with decreased cell thickness (Fig. 7c).79,80 MNP integrated polymer/ceramic composite scaffolds exhibited a better cell–material interaction because MNPs stimulate cell proliferation, cell differentiation and bone formation.81 Yang et al.82 developed a poly-L-lactic scaffold containing oleic acid functionalized Fe3O4 nanoparticles that showed enhanced cell adhesion, proliferation and differentiation. Hence, MNP integrated GC/PG composite scaffolds are believed to be suitable candidates for bone tissue regeneration.4. Conclusions
Highly porous CG/PG and MNP integrated CG/PG composite scaffolds were reported for bone tissue engineering and hyperthermia treatment. Extensive physicochemical, morphological, and magnetic characterization as well as in vitro degradation and swelling studies was performed. The compressive modulus of the MNP integrated CG/PG composite scaffolds in the range of 3.1–13.3 MPa was achieved. Unfortunately, hyperthermia temperature could not be achieved even for the scaffold containing a high MNP content (CG/PG/10MNP), due to the presence of the non-magnetic polymer and ceramic matrix. The preliminary in vitro experiments indicated the apatite forming ability of the scaffolds and the biocompatible nature for MG-63 cells. Overall, the above results suggest that the Fe3O4 NP integrated polymer/PG composite scaffold has great potential as a bone tissue engineering material. Nevertheless, further optimization is needed to achieve hyperthermia temperature. Hence, the CG/PG/MNP scaffolds developed in this study have much potential for bone regeneration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Dr S. Karthi for his active involvement with this work, data analysis and in the initial stages of manuscript preparation. The author E. K. Girija expresses sincere thanks to Department of Science and Technology (DST), New Delhi, India (Project Ref. No. DST/TSG/WM/2015/576/G) for financial support.References
- Y. Wang, C. Yang, X. Chen and N. Zhao, Macromol. Mater. Eng., 2006, 291, 254–262 CrossRef CAS.
- I. Bružauskaite, D. Bironaite, E. Bagdonas and E. Bernotiene, Cytotechnology, 2016, 68, 355–369 CrossRef.
- S. Mann, Nature, 1993, 365, 499–505 CrossRef CAS.
- C. V. M. Rodrigues, P. Serricella, A. B. R. Linhares, R. M. Guerdes, R. Borojevic, M. A. Rossi, M. E. L. Duarte and M. Farina, Biomaterials, 2003, 24, 4987–4997 CrossRef CAS.
- M. S. R. Rajoka, L. Zhao, H. M. Mehwish, Y. Wu and S. Mahmood, Appl. Microbiol. Biotechnol., 2019, 103, 1557–1571 CrossRef.
- M. Tadsen, R. P. Friedrich, S. Riedel, C. Alexiou and S. G. Mayr, ACS Appl. Mater. Interfaces, 2019, 11, 7450–7458 CrossRef CAS.
- M. C. Echave, L. Saenz del Burgo, J. L. Pedraz and G. Orive, Curr. Pharm. Des., 2017, 23, 3567–3584 CrossRef CAS.
- A. Oryan, S. Alidadi, A. Bigham-Sadegh and A. Moshiri, J. Mater. Sci.: Mater. Med., 2016, 27, 155 CrossRef.
- M. Peter, N. Ganesh, N. Selvamurugan, S. V. Nair, T. Furuike, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2010, 80, 687–694 CrossRef CAS.
- P. Chen, L. Liu, J. Pan, J. Mei, C. Li and Y. Zheng, Mater. Sci. Eng., C, 2019, 97, 325–335 CrossRef CAS.
- Y. Dan, O. Liu, Y. Liu, Y.-Y. Zhang, S. Li, X.-B. Feng, Z.-W. Shao, C. Yang, S.-H. Yang and J.-B. Hong, Nanoscale Res. Lett., 2016, 11, 487 CrossRef.
- K. Maji and S. Dasgupta, Trans. Indian Ceram. Soc., 2014, 73, 110–114 CrossRef CAS.
- K. Maji, S. Dasgupta, B. Kundu and A. Bissoyi, J. Biomater. Sci., Polym. Ed., 2015, 26, 1190–1209 CrossRef CAS.
- I. R. Serra, R. Fradique, M. C. S. Vallejo, T. R. Correia, S. P. Miguel and I. J. Correia, Mater. Sci. Eng., C, 2015, 55, 592–604 CrossRef CAS.
- Y. Zhou, L. Xu, X. Zhang, Y. Zhao, S. Wei and M. Zhai, Mater. Sci. Eng., C, 2012, 32, 994–1000 CrossRef CAS.
- Y. Yin, F. Ye, J. Cui, F. Zhang, X. Li and K. Yao, J. Biomed. Mater. Res., Part A, 2003, 67A, 844–855 CrossRef CAS.
- L. Nie, Q. Wu, H. Long, K. Hu, P. Li, C. Wang, M. Sun, J. Dong, X. Wei, J. Suo, D. Hua, S. Liu, H. Yuan and S. Yang, J. Biomater. Sci., Polym. Ed., 2019, 30, 1636–1657 CrossRef CAS.
- Y. P. Singh, S. Dasgupta and R. Bhaskar, J. Biomater. Sci., Polym. Ed., 2019, 30, 1756–1778 CrossRef CAS.
- L. Tao, L. Zhonglong, X. Ming, Y. Zezheng, L. Zhiyuan, Z. Xiaojun and W. Jinwu, RSC Adv., 2017, 7, 54100–54110 RSC.
- E. Sharifi, M. Azami, A.-M. Kajbafzadeh, F. Moztarzadeh, R. Faridi-Majidi, A. Shamousi, R. Karimi and J. Ai, Mater. Sci. Eng., C, 2016, 59, 533–541 CrossRef CAS.
- M. Peter, N. S. Binulal, S. V. Nair, N. Selvamurugan, H. Tamura and R. Jayakumar, Chem. Eng. J., 2010, 158, 353–361 CrossRef CAS.
- K. Maji, S. Dasgupta, K. Pramanik and A. Bissoyi, Int. J. Biomater., 2016, 2016, 9825659 Search PubMed.
- A. L. Boskey, J. Dent. Res., 1997, 76, 1433–1436 CrossRef CAS.
- E. A. Abou-Neel, D. M. Pickup, S. P. Valappil, R. J. Newport and J. C. Knowles, J. Mater. Chem., 2009, 19, 690–701 RSC.
- M. Navarro, M.-P. Ginebra and J. Planell, J. Biomed. Mater. Res., Part A, 2003, 67A, 1009–1015 CrossRef CAS.
- E. A. Abou Neel, T. Mizoguchi, M. Ito, M. Bitar, V. Salih and J. C. Knowles, Biomaterials, 2007, 28, 2967–2977 CrossRef CAS.
- M. Barbeck, T. Serra, P. Booms, S. Stojanovic, S. Najman, E. Engel, R. Sader, C. J. Kirkpatrick, M. Navarro and S. Ghanaati, Bioact. Mater., 2017, 2, 208–223 CrossRef.
- H.-W. Kim, E.-J. Lee, I.-K. Jun, H.-E. Kim and J. C. Knowles, J. Biomed. Mater. Res., Part B, 2005, 75B, 34–41 CrossRef CAS.
- A. R. Amini, C. T. Laurencin and S. P. Nukavarapu, CRC Crit. Rev. Bioeng., 2012, 40, 363–408 Search PubMed.
- A. Marshall, M. S. Salerno, M. Thomas, T. Davies, C. Berry, K. Dyer, J. Bracegirdle, T. Watson, M. Dziadek, R. Kambadur, R. Bower and M. Sharma, Exp. Cell Res., 2008, 314, 1013–1029 CrossRef CAS.
- J. M. Kanczler and R. O. C. Oreffo, Eur. Cells Mater., 2008, 15, 100–114 CrossRef CAS.
- K. Lee, E. A. Silva and D. J. Mooney, J. R. Soc., Interface, 2011, 8, 153–170 CrossRef CAS.
- X. Yao, X. Niu, K. Ma, P. Huang, J. Grothe, S. Kaskel and Y. Zhu, Small, 2017, 13, 1602225 CrossRef.
- S. B. Lang, Nature, 1996, 212, 704–705 CrossRef.
- A. C. Jayalekshmi, S. P. Victor and C. P. Sharma, Colloids Surf., B, 2013, 101, 196–204 CrossRef CAS.
- J. Zhang, S. Zhao, M. Zhu, Y. Zhu, Y. Zhang, Z. Liu and C. Zhang, J. Mater. Chem. B, 2014, 2, 7583 RSC.
- Y. Zhao, T. Fan, J. Chen, J. Su, X. Zhi, P. Pan, L. Zou and Q. Zhang, Colloids Surf., B, 2019, 174, 70–79 CrossRef CAS.
- R. Govindan and E. K. Girija, J. Mater. Chem. B, 2014, 2, 5468–5477 RSC.
- N. V. Jadhav, A. I. Prsad, A. Kumar, R. Misra, S. Dhara, K. R. Babu, C. I. Prajapat, N. I. Misra, R. S. Ningthoujam, B. N. Pandey and R. K. Vatsa, Colloids Surf., B, 2013, 108, 158–168 CrossRef CAS.
- A. C. Tas, Biomaterials, 2000, 21, 1429–1438 CrossRef CAS.
- S. Karthi, G. A. Kumar, D. K. Sardar, G. C. Dannangoda, K. S. Martirosyan and E. K. Girija, Mater. Chem. Phys., 2017, 193, 356–363 CrossRef CAS.
- S. Karthi, R. Govindan, A. K. Gangadharan, G. C. Dannangoda, K. S. Martirosyan, D. K. Sardar, S. Chidangil and E. K. Girija, J. Am. Ceram. Soc., 2019, 102, 2558–2568 CAS.
- N. Cai, C. Li, C. Han, X. Luo, L. Shen, Y. Xue and F. Yu, Appl. Surf. Sci., 2016, 369, 492–500 CrossRef CAS.
- L. Cui, J. Jia, Y. Guo, Y. Liu and P. Zhu, Carbohydr. Polym., 2014, 99, 31–38 CrossRef CAS.
- J. H. Muyonga, C. G. B. Cole and K. G. Duodu, Food Hydrocolloids, 2004, 18, 581–592 CrossRef CAS.
- M. Pereda, A. G. Ponce, N. E. Marcovich, R. A. Ruseckaite and J. F. Martucci, Food Hydrocolloids, 2011, 25, 1372–1381 CrossRef CAS.
- R. Golubevas, Z. Stankeviciute, A. Zarkov, R. Golubevas, L. Hansson, R. Raudonis, A. Kareiva and E. Garskaite, Mater. Adv., 2020, 1, 1675–1684 RSC.
- M. Yadav, K. Y. Rhee, S. J. Park and D. Hui, Composites, Part B, 2014, 66, 89–96 CrossRef CAS.
- J. O. Byun, B. H. Kim, K. S. Hong, H. J. Jung, S. W. Lee and A. A. Izyneev, J. Non-Cryst. Solids, 1995, 190, 288–295 CrossRef CAS.
- A. Boskey and N. P. Camacho, Biomaterials, 2007, 28, 2465–2478 CrossRef CAS.
- Y. J. Yin, F. Zhao, X. F. Song, K. D. Yao, W. W. Lu and J. C. Leong, J. Appl. Polym. Sci., 2000, 77, 2929–2938 CrossRef CAS.
- L. Zhao and J. Chang, J. Mater. Sci.: Mater. Med., 2004, 15, 625–629 CrossRef CAS.
- H. Stenhamre, U. Nannmark, A. Lindahl, P. Gatenholm and M. Brittberg, J. Tissue Eng. Regener. Med., 2011, 5, 578–588 CrossRef CAS.
- J. Zeltinger, J. K. Sherwood, D. A. Graham, V. Müeller and L. G. Griffith, Tissue Eng., 2001, 7, 557–572 CrossRef CAS.
- S. K. Lan Levengood, S. J. Polak, M. B. Wheeler, A. J. Maki, S. G. Clark, R. D. Jamison and A. J. Wagoner Johnson, Biomaterials, 2010, 31, 3552–3563 CrossRef CAS.
- L.-C. Gerhardt and A. R. Boccaccini, Materials, 2010, 3, 3867–3910 CrossRef CAS.
- L. Polo-Corrales, M. Latorre-Esteves and J. E. Ramirez-Vick, J. Nanosci. Nanotechnol., 2014, 14, 15–56 CrossRef CAS.
- P. Gentile, M. Mattioli-Belmonte, V. Chiono, C. Ferretti, F. Baino, C. Tonda-Turo, C. Vitale-Brovarone, I. Pashkuleva, R. L. Reis and G. Ciardelli, J. Biomed. Mater. Res., Part A, 2012, 100A, 2654–2667 CrossRef CAS.
- S. Shahriarpanah, J. Nourmohammadi and G. Amoabediny, Int. J. Biol. Macromol., 2016, 93, 1069–1078 CrossRef CAS.
- C. Sharma, A. K. Dinda, P. D. Potdar, C. F. Chou and N. C. Mishra, Mater. Sci. Eng., C, 2016, 64, 416 CrossRef CAS.
- A. Tampieri, M. Iafisco, M. Sandri, S. Panseri, C. Cunha, S. Sprio, E. Savini, M. Uhlarz and T. Herrmannsdörfer, ACS Appl. Mater. Interfaces, 2014, 6, 15697–15707 CrossRef CAS.
- S. Aliramaji, A. Zamanian and M. Mozafari, Mater. Sci. Eng., C, 2017, 70, 736–744 CrossRef CAS.
- H. Jin, Y. Zhuo, Y. Sun, H. Fu and Z. Han, Adv. Mech. Eng., 2019, 11, 1–10 Search PubMed.
- N. Benbettaïeb, M. Kurek, S. Bornaz and F. Debeaufort, J. Sci. Food Agric., 2014, 94, 2409–2419 CrossRef.
- M. Cheng, J. Deng, F. Yang, Y. Gong, N. Zhao and X. Zhang, Biomaterials, 2003, 24, 2871–2880 CrossRef CAS.
- K. Maji, S. Dasgupta, K. Pramanik and A. Bissoyi, Mater. Sci. Eng., C, 2018, 86, 83–94 CrossRef CAS.
- S. S. Chen, Y. H. Falcovitz, R. Schneiderman, A. Maroudas and R. L. Sah, Osteoarthrotic Cartilage, 2001, 9, 561–569 CrossRef CAS.
- R. A. Magnussen, F. Guilak and T. P. Vail, J. Orthop. Res., 2005, 23, 576–583 CrossRef.
- J. Cheng, X. Ni, H. Zheng, B. Li, X. Zhang and D. Zhan, Mater. Res. Bull., 2006, 41, 1424–1429 CrossRef CAS.
- R. V. Ramanujan and Y. Y. Yeow, Mater. Sci. Eng., C, 2005, 25, 39–41 CrossRef.
- G. Vallejo-Fernandez, O. Whear, A. G. Roca, S. Hussain, J. Timmis, V. Patel and K. O’Grady, J. Phys. D: Appl. Phys., 2013, 46, 312001 CrossRef.
- K. Y. Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS.
- X. L. Liu, H. M. Fan, J. B. Yi, Y. Yang, E. S. G. Choo, J. M. Xue, D. D. Fan and J. Ding, J. Mater. Chem., 2012, 22, 8235–8244 RSC.
- U. Anjaneyulu, V. K. Swaroop and U. Vijayalakshmi, RSC Adv., 2016, 6, 10997–11007 RSC.
- D. M. Huang, J. K. Hsiao, Y. C. Chen, L. Y. Chien, M. Yao, Y. K. Chen, B. S. Ko, S. C. Hsu, L. A. Tai, H. Y. Cheng, S. W. Wang, C. S. Yang and Y. C. Chen, Biomaterials, 2009, 30, 3645–3651 CrossRef CAS.
- K. Dashnyam, R. A. Perez, R. K. Singh, E. J. Lee and K. W. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 15697–15707 CrossRef.
- C. Shuai, W. Yang, C. He, S. Peng, C. Gao, Y. Yang, F. Qi and P. Feng, Mater. Des., 2020, 185, 108275–108286 CrossRef CAS.
- E. Tanasa, C. Zaharia, A. Hudita, I.-C. Radu, M. Costache and B. Galateanu, Mater. Sci. Eng., C, 2020, 110, 110714 CrossRef CAS.
- G. Orsini, B. Assenza, A. Scarano, M. Piattelli and A. Piattelli, Int. J. Oral Maxillofac. Implants, 2000, 15, 779–784 CAS.
- M. Li, J. Liu, X. Cui, G. Sun, J. Hu, S. Xu, F. Yang, L. Zhang, X. Wang and P. Tang, Regener. Biomater., 2019, 6, 373–381 CrossRef CAS.
- J.-W. Lu, F. Yang, Q.-F. Ke, X.-T. Xie and Y.-P. Guo, J. Nanomed. Nanotechnol., 2018, 14, 811–822 CrossRef CAS.
- W. Yang, Y. Zhong, P. Feng, C. Gao, S. Peng, Z. Zhao and C. Shuai, Polym. Test., 2019, 76, 33–42 CrossRef CAS.
Footnote |
| † Deceased. |
| This journal is © The Royal Society of Chemistry 2020 |