
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compact polyelectrolyte hydrogels of gelatin and chondroitin sulfate as ion's mobile media in sustainable all-solid state electrochemical devices†
Jimena S.
González
 ab,
Arsen
Burlaka
b,
José
Paz
b,
Horacio J.
Salavagione
b,
Javier
Carretero-González
*b and
Rebeca
Hernández
*b
ab,
Arsen
Burlaka
b,
José
Paz
b,
Horacio J.
Salavagione
b,
Javier
Carretero-González
*b and
Rebeca
Hernández
*b
aInstitute of Materials Science and Technology (INTEMA), University of Mar del Plata and National Research Council (CONICET), Colón 10890, 7600 Mar del Plata, Argentina. E-mail: rhernandez@ictp.csic.es; jcarretero@ictp.csic.es
bInstitute of Polymer Science and Technology (ICTP-CSIC), Juan de la Cierva, 3 Madrid, Spain
First published on 17th September 2020
Abstract
The creation of flexible and high strength hydrogel materials from natural polymers as low cost and safe solid electrolytes is an area of intense research nowadays. We present a novel approach for the preparation of gelatin and chondroitin sulfate hydrogel complexes by using a simple centrifugation process. The innovative dual-bio-gel-network is able to swell and shrink upon changes on the pH and NaCl concentration. The solid bio-gels sandwiched between two macroporous carbon electrode materials are assembled in symmetric cells and their electrochemical properties are evaluated by cyclic voltammetry, galvanostatic, and impedance spectroscopy measurements. The cells exhibit areal capacitance values up to 2.74 mF cm−2 (3.1 F g−1) and a low resistance value of 12 Ohm cm2 for graphene electrode materials. These properties are the consequence of the successful infiltration of the solid gel inside the porous structure of the carbon electrode that boosts the charge transfer at the biopolymer/carbon electrode interphase. The results obtained may provide additional inspiration in the emerging field of bioelectronics, where biocompatible and powered systems are of the utmost importance.
Introduction
The development of new materials as compact electrolytes in all-solid-state electrochemical cells is currently an area of intense research.1,2 Solid electrolytes might solve specific problems such as leakage and solvent evaporation that exist in devices using liquid electrolytes, increase the device compactness and minimize the explosion risks related to solvent flammability. Among the various types of solid-state electrolytes, gel polymer electrolytes (GPEs) present the advantages of both solid and liquid electrolytes and their relatively high ionic conductivity (10−4 to 10−3 S cm−1 under ambient conditions).3 In particular, great efforts are devoted to the preparation of sustainable biohydrogels, composed of hydrophilic networks of biopolymers that are renewable, biocompatible, biodegradable, and of low cost. The hybridization of biohydrogels with conducting polymers such as poly(3,4-ethylenedioxythiophene) (PEDOT) or polyaniline (PANI) has resulted in innovative materials for high-safety solid-state rechargeable metal batteries4 and supercapacitors (SCs)5 as well as for emerging applications in bioelectronics like biocompatible and biodegradable printable and lightweight batteries, high-sensitivity photosensors, soft actuators and flexible wearable devices.6–8Here, we report on the preparation and characterization of an innovative hydrogel formed by polyelectrolyte complexation (PEC) of oppositely charged biopolymers, gelatin (gel) and chondroitin sulfate (ChS), and its demonstration as a safe and sustainable electrolyte material in an all-solid-state electrochemical double-layer capacitor (EDLC). Both biopolymers are able to assemble through electrostatic interactions between the polycation (gelatin) and the polyanion (ChS), leading to the formation of polyelectrolyte complexes (PECs).9 Chemical crosslinking is often employed in order to increase the mechanical properties of conducting biohydrogels.10 However, in our approach, a centrifugation step was applied to the gel–ChS PECs in order to obtain gel-like materials based on compact PECs, thus avoiding the employment of chemical crosslinkers. In this process, salt water acts as a plasticizer and allows the compact polyelectrolyte complex to reorganize and fuse into a material with solid-like mechanical properties that are characteristic of a hydrogel and with the ability to swell in aqueous solutions.11
Due to the presence of sulfate groups, ChS-based materials have been recently tested for single ion conducting hydrogels in proton batteries.12 Besides, the ChS–gelatin hydrogels exhibit a single anion and cation covalently bonded to the polymer backbone, and then both counterions can be in movement. This dual conductivity is an important aspect of the applicability of the novel compounds as electrolyte materials in EDLCs. In EDLCs, both ions are mobile, so the charges of opposite sign created at the surface of each porous carbon electrode during the charge and discharge of the symmetric cell are counterbalanced.13 For that, the electrolyte's ions rapidly migrate to the surface of the electrodes stabilizing the ionic double layer by electrostatic interactions and then reversibly storing electrical energy in the form of chemical energy.14
Here, we have determined the effect of bio-solid-gels on the electrochemical properties of symmetric electrochemical cells comprising porous carbons (double layer mechanism) as electrode materials. The porous carbons that we have used are porous graphene and fibrous carbon mats (carbon gas diffusion layer). Both materials ensured a fast conductivity and diffusion of the ions through the gel electrolyte and also at the electrolyte/electrode interphase, while keeping the dual-polymer-gel-network structure practically intact. The strategy reported herein allows for the assembly of the three main components in the device: the negative and positive carbon electrodes and a polymer biohydrogel prepared through a mild potentially scalable process. This approach does not require in situ formation nor an additional crosslinking step of the hydrogel for integration with the electrodes and formation of the electrochemical all-solid state cell, which opens new avenues for the development of sustainable materials with potential for application in biotechnological devices.
Experimental section
Materials
Chondroitin sulfate A, sodium salt (ChS, 90% pure, Alfa Aesar), and gelatin derived from porcine skin (gel, Type A, Sigma-Aldrich) were used as received. For the preparation of graphene oxide, powdered graphite (Aldrich, grain size < 45 μm, purity ≥99.99%) was employed. For the preparation of swelling media, sodium chloride (NaCl), acetic acid and phosphate buffer saline (PBS) (pH = 7.4) in the form of powder and later dissolved in 1 L of MilliQ water were all purchased from Aldrich and used as received.Zeta (ζ) potential and precipitation studies
In order to optimize the experimental conditions for the formation of a polyelectrolyte complex between gelatin and chondroitin sulfate, a thorough study of the ζ potential of aqueous solutions of gelatin and chondroitin sulfate as a function of pH, NaCl and polymer concentration was carried out.For the preparation of polymer aqueous solutions, ChS and gelatin powders were dissolved separately in ultrapure water (Milli-Q) at different polymer concentrations: 1, 5, 10 and 15 mg mL−1. Gelatin was prepared by the addition of the powder to Milli-Q solution immersed in a thermal bath of silicone at 50 °C. ChS was prepared directly by the addition of the powder to MilliQ water at room temperature. The pH of the final solutions was adjusted through addition of 1 M aqueous solutions of acetic acid and NaCl concentrations were fixed at 0, 0.15 and 0.5 M. As a result, a total number of 144 samples were obtained, each of them with a unique combination of the 4 determinant factors: type of polymer, concentration of polymer, concentration of NaCl and pH.
The ζ potential was measured for each of the solutions at 25 °C using a Nano-ZS (Malvern Panalytical-UK). Precipitation experiments were performed by mixing aliquots of the solutions prepared under selected experimental conditions.
Preparation of ChS–gelatin hydrogels
The preparation of ChS–gelatin hydrogels involved two steps. First, aliquots (10 mL) of aqueous solutions of gelatin and ChS were mixed by means of a Gilson Miniplus 3 Peristaltic Pump, employing PVC 0.5 mm tubes and a flow rate of 0.8 mL min−1. The precipitate formed was stored at 4 °C for 24 h. Then, the precipitate was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min in a Sigma 2-16p Centrifuge. The hydrogels obtained were immersed in milliQ water for 24 h and the swelling ratio, SR (%), was determined as
000 rpm for 30 min in a Sigma 2-16p Centrifuge. The hydrogels obtained were immersed in milliQ water for 24 h and the swelling ratio, SR (%), was determined as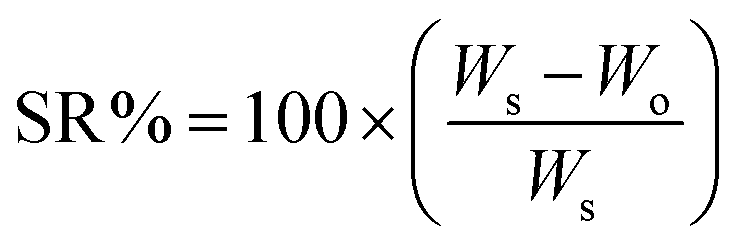 | (1) |
Swelling experiments
The gravimetric method was employed to measure the swelling ratios of the hydrogels immersed in aqueous media at different pH at a fixed 0.15 M NaCl concentration and at different NaCl concentrations at a fixed pH of 4. PBS was also employed as the swelling media. The SR% was determined after 24 h of immersion in the media according to eqn (1).Mechanical properties
Mechanical properties were measured by a dynamic compression test using the universal materials testing machine Instron 3366. Measurements were performed at room temperature on gels hydrated for 24 h in aqueous solutions at different NaCl concentrations and in PBS. Cylindrical samples of 8 mm of diameter and an average height of 3.5 mm were employed. The cell load was 100 N and the samples were compressed at 2 mm min−1. The elastic compressive modulus was determined from the slope of the stress–strain curve at 10% of strain.Oscillatory frequency sweeps between 10 and 0.1 Hz at T = 20 °C were carried out on a AR1000 rheometer (TA Instruments, USA) using parallel steel plates of 20 mm. A solvent trap was employed to prevent solvent evaporation during the course of the experiments. The experiments were performed at a constant torque of 10 mN inside the linear viscoelastic region (LVR) of the gels, determined with the aid of stress sweeps carried out at 1 Hz and 20 °C. The results were analyzed using the Rheology Advantage Data Analysis software (TA Instruments, USA).
Porous carbon electrode materials
Two different types of porous carbon electrode materials were used for the assessment of the ChS–gel hydrogels as an electrolyte material in an EDLC cell: (i) carbon gas diffusion layer (CGDL) and (ii) macroporous graphene. Both materials were infiltrated with the hydrogels, and tested as positive and negative electrodes in a symmetric supercapacitor cell. The CGDL is a non-woven fabric supplied by SIGRACET® (type 39AA). Electrodes were easily obtained from these carbon sheets by cutting several circular pieces with a diameter of 12 mm and an average weight of 15 mg. For the preparation of porous graphene electrodes, graphite oxide, synthesized by the Hummers’ method, was employed.15 A brief explanation of the method of preparation is provided in the ESI.† The obtained powdered graphite oxide presents the typical Raman features of oxidized graphitic species (Fig. S1, ESI†) and it is easily dispersed in water to give stable graphene oxide dispersions.16 A concentrated aqueous dispersion of graphite oxide (30 mg mL−1) was bath sonicated to get a homogeneous dispersion. TEM analysis of an appropriate dilution of this dispersion suggests the presence of thin GO flakes (principally monolayers and bilayers) with lateral dimension in the order of microns (Fig. S1 in the ESI†). In order to build the electrodes, 60 μL of the graphene oxide dispersion was deposited on top of 12 mm circular pieces of stainless-steel mesh employing a micropipette and immediately frozen with liquid nitrogen and lyophilized. After lyophilization, the electrodes were taken to the oven where they received heat treatment at 500 °C for 2 hours in order to reduce the graphene oxide into conducting reduced graphene oxide. The final mass of the reduced graphene oxide electrodes obtained after the whole process was ∼1 mg.Assembly of the electrochemical cell
ChS–gelatin hydrogels were cut into cylinders of 8 mm of diameter and an average height of 3.5 mm. To achieve optimum contact with the surface of the porous carbon electrode, the hydrogel was infiltrated within the electrodes using a vacuum pump. The morphological characterization of the electrodes and gel assembly was carried out through Scanning Electron Microscopy (SEM) using the Philips XL30 equipment. For visualization of the morphology of the electrochemical cell, lyophilized samples were frozen in liquid nitrogen and snapped immediately to obtain cross-sectioned materials and sputtered with gold using a Thermo VG scientific Polaron SC7640 Sputter Coater.Electrochemical measurements
The performance of the electrochemical cells was measured in a two-electrode system. The porous electrodes were directly used as positive and negative electrodes for the electrochemical tests. For that, porous carbons, CGDL and graphene, were assembled in a symmetric Swagelok-type cell, of 12.7 mm in diameter, and placed in direct contact with titanium plungers. Electrochemical cyclic voltammetry (CV) measurements at different scan rates from 100 to 500 mV s−1 as well as measurements in galvanostatic mode at different current densities ranging from 0.1 to 1 A g−1 were performed under ambient conditions using a multichannel potentiostat/galvanostat (Biologic VMP3, France). The specific capacitance was evaluated per active mass of a single electrode. The areal capacitance was calculated considering the surface of the plunger. Data were analysed using the software EC LAB commercialized by Bio-Logic Science Instruments. Electrochemical impedance spectroscopy (EIS) measurements were carried out by applying a low sinusoidal amplitude alternating voltage of 10 mV to the cell at frequencies from 1 MHz to 10 mHz using the above mentioned multichannel potentiostat/galvanostat. Measurements were performed at the open circuit voltage (OCV). The specific capacitance (Cgrav) was calculated from the CVs at each scan rate using the following equation: Cgrav = 2I/(dV/dt m), where I is the current, m is the mass of the active material and dV/dt is the scan rate.Results and discussion
Preparation of ChS–gel hydrogels
The study of interactions between gelatin, a protein with amphoteric characteristics, and polysaccharides of either anionic or cationic character (e.g. gum arabic, pectins, carboxymethylcellulose, chitosan, etc.) is currently the focus of intensive research work.17 Gelatin and chondroitin sulfate are expected to establish electrostatic interactions between the positive charges of amino-acids in the chemical backbone of gelatin (lysine, hydroxylysine, histidine and arginine) and the negative charges present in the disaccharide unit of ChS, the carboxylic and the sulfonic group (Fig. 1). Such interactions result in the formation of polyelectrolyte complexes and depend on parameters such as pH, ionic strength, molar ratio, total concentration and temperature.18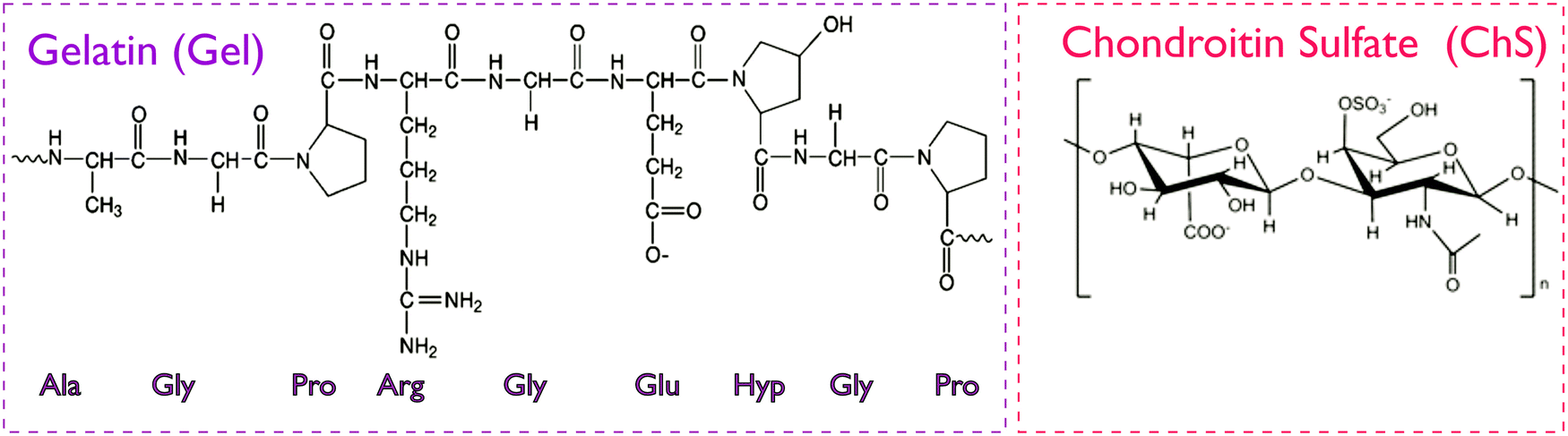 | ||
| Fig. 1 Chemical structure of gelatin and chondroitin sulfate. The gelatin structure is based on repeating tri-peptide sequences of glycine-aa1-aa2, where amino acids aa1 and aa2 are mainly proline and hydroxyproline.19 In ChS, the disaccharide unit presents carboxylic and sulfonic groups prone to protonation.12 | ||
In the present study, a thorough analysis of the zeta (ζ) potential of precursor aqueous solutions of gelatin and ChS was carried out as a function of pH and polymer and NaCl concentration in order to select optimal experimental variables to conduct gelatin/ChS complexation. A range of pH values from pH = 3.0 to pH = 5.5 were set based on the isoelectric point reported for gelatin A (pH 6–7), implying that in the pH range under study it has positive character.17 On the other hand, ChS presents carboxylic (pKa 3.5–4.6) and sulfonic groups (pKa 2.6) that are deprotonated and attain negative charge at pH > pKa.20,21 Therefore, the pH range under study is close to the pKa of the weakly acidic carboxylic groups, which implies that these groups are slightly negatively charged whereas the strongly acidic sulfonic groups are fully deprotonated. The concentration of gelatin and ChS was varied in the range 1–15 mg mL−1 and NaCl concentration was varied from 0 to 0.5 M.
The variation of the ζ potential with the experimental parameters is represented in Fig. 2. Gelatin precursor solutions showed positive ζ potential values under all conditions tested, correlating with positively charged amide groups in the polypeptidic chain. ζ potential values approximated to 0 as the pH increased, which determines the gelatin isoelectric point (Fig. S2 in the ESI†). On the other hand, ChS showed negative ζ potential values, corresponding to charged carboxylic and sulfate groups.
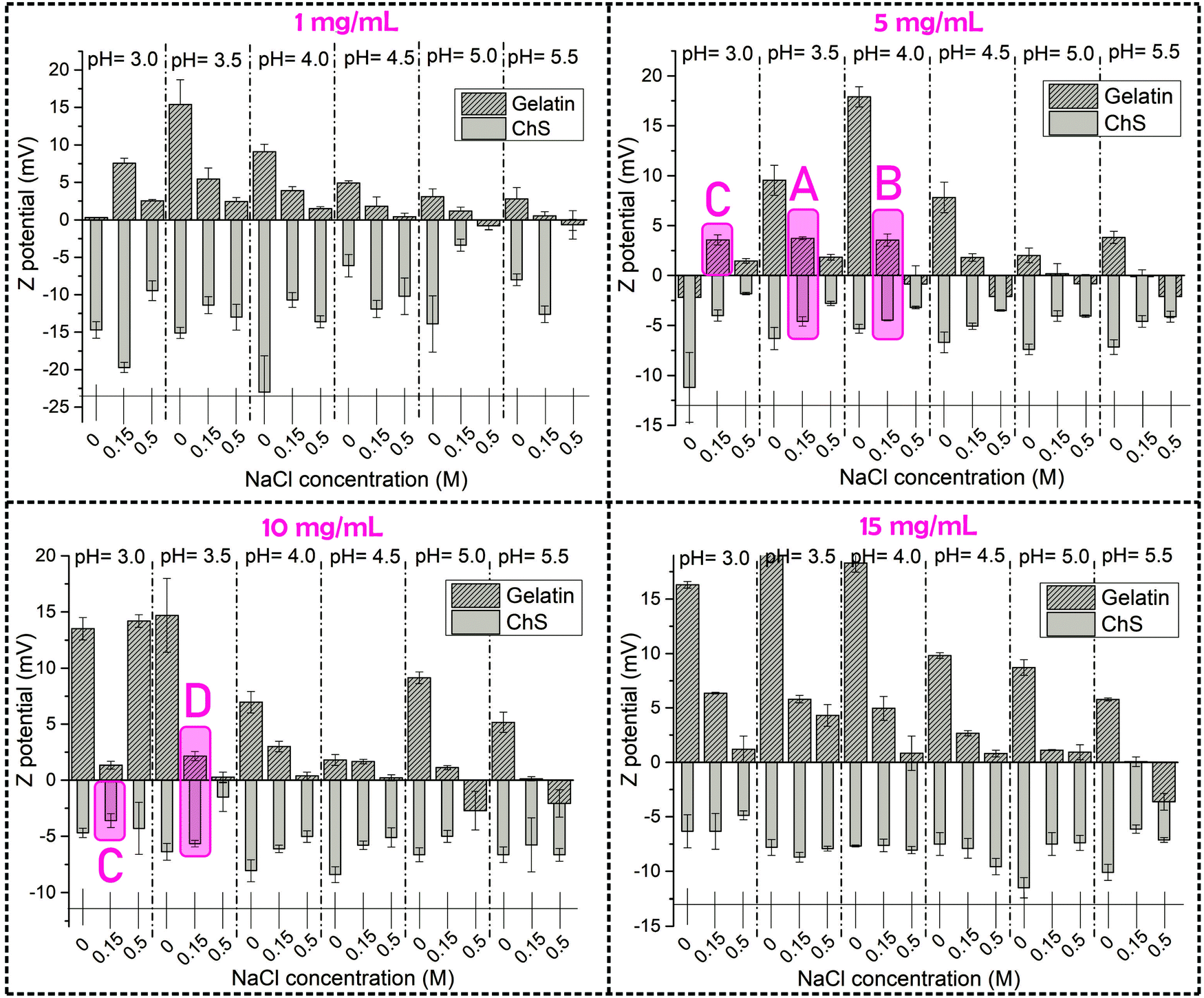 | ||
| Fig. 2 Graphical representation of the ζ potential of aqueous solutions of gelatin and chondroitin sulfate at different polymer concentrations: 1, 5, 10 and 15 mg mL−1. For easy visualization, the experimental parameters selected for the formation of PECs have been marked in purple. | ||
The concentration of NaCl in the media clearly changes the ζ potential of the aqueous solutions of ChS and gelatin, this effect being more evident for the latter. For ChS an increasing ionic strength of salt solutions resulted in the screening of the electrostatic repulsion between negatively charged groups, thus resulting in a decrease in the ζ potential with the increase of NaCl concentration. For the highest polymer concentrations (15 mg mL−1), the ζ potential remained unaffected by the NaCl concentration, which suggests that concentrations of NaCl higher than 0.5 M would be needed to produce appreciable differences in the screening of negatively charged groups.22 For gelatin at all the studied concentrations the trend is similar; that is, an increase in NaCl concentration resulted in a decrease of ζ potential which can be attributed to the fact that an increasing ionic strength of salt solutions results in screened attraction between ionized carboxyl and amine groups.23
The experimental conditions selected for the formation of ChS–gelatin PECs are highlighted in purple in Fig. 2 and summarized in Table 1. Aqueous solutions of gelatin and ChS with the closest zeta potential in absolute value and opposite sign were chosen for a selected NaCl concentration of 0.15 M. A gelatin concentration of 1 mg mL−1 yielded a very low amount of precipitate whereas a gelatin concentration of 15 mg mL−1 formed physical gels after being stored at 4 °C for 24 h. Hence, gelatin concentrations of 5 and 10 mg mL−1 were selected as optimum concentrations and mixed with equal concentrations of chondroitin sulfate for the formation of ChS–gelatin PECs (samples A, B and D in Table 1). Sample C was prepared through mixing of starting aqueous solutions of gelatin and ChS at 5 and 10 mg mL−1 respectively, and having ζ potentials within the experimental error and of opposite sign.
| Sample designation | [gel] (mg mL−1) | pH | ζ Potential (mV) | [ChS] (mg mL−1) | pH | ζ Potential (mV) |
|---|---|---|---|---|---|---|
| A | 5 | 3.5 | +3.7 ± 0.1 | 5 | 3.5 | −4.6 ± 0.5 |
| B | 5 | 4.0 | +3.6 ± 0.6 | 5 | 4.0 | −4.5 ± 0.1 |
| C | 5 | 3.0 | +3.6 ± 0.5 | 10 | 3.0 | −3.6 ± 0.6 |
| D | 10 | 4.0 | +3.0 ± 0.5 | 10 | 4.0 | −6.1 ± 0.3 |
The assessment of the formation of a ChS–gelatin PEC was done through precipitation tests carried out by mixing of aqueous solutions of gelatin and ChS at the previously chosen pH and salt concentration values. The results of the whole series of gelation experiments are provided in the ESI† (Fig. S3 in the ESI†). The formation of a precipitate could be ascertained as a result of the formation of a ChS–gelatin PEC for all the experimental conditions selected in Table 1.
Hydrogels were prepared after centrifugation of ChS–gelatin PECs following the experimental method shown in Scheme 1. This is a procedure analogous to that reported for the compaction of PECs constituted of alginate and chitosan which allows for the preparation of highly hydrated hydrogels in the presence of salt.24,25
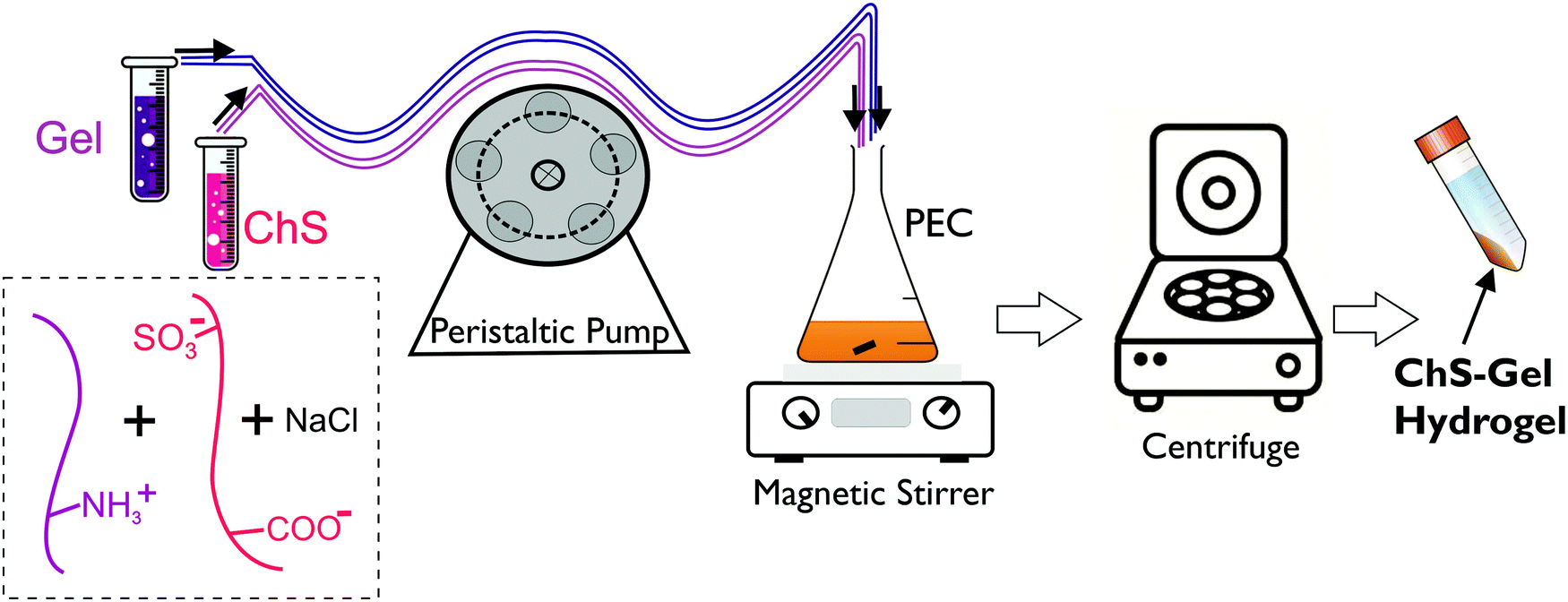 | ||
| Scheme 1 Representation of the experimental procedure followed to prepare ChS–gel hydrogels from gelatin and chondroitin sulfate PECs. | ||
After centrifugation (compaction stage), and removing the supernatant, the precipitates from all the samples presented a hydrogel-like aspect as can be observed in Fig. 3. Except for sample C, the SR (%) of all hydrogels increased after being immersed in water for 24 h. The negative SR (%) value obtained for sample C implied partial dissolution of the sample in water. This might be attributed to the fact that the concentration of the pristine ChS aqueous solution (10 mg mL−1) is double with respect to that of the pristine gelatin solution (5 mg mL−1). Therefore, the excess of negative charges of sulfate groups (ionized 100%) and carboxylic groups (degree of ionization ∼60% at pH = 4 employed for the preparation of the gel)26 might not be involved in hydrogel formation.
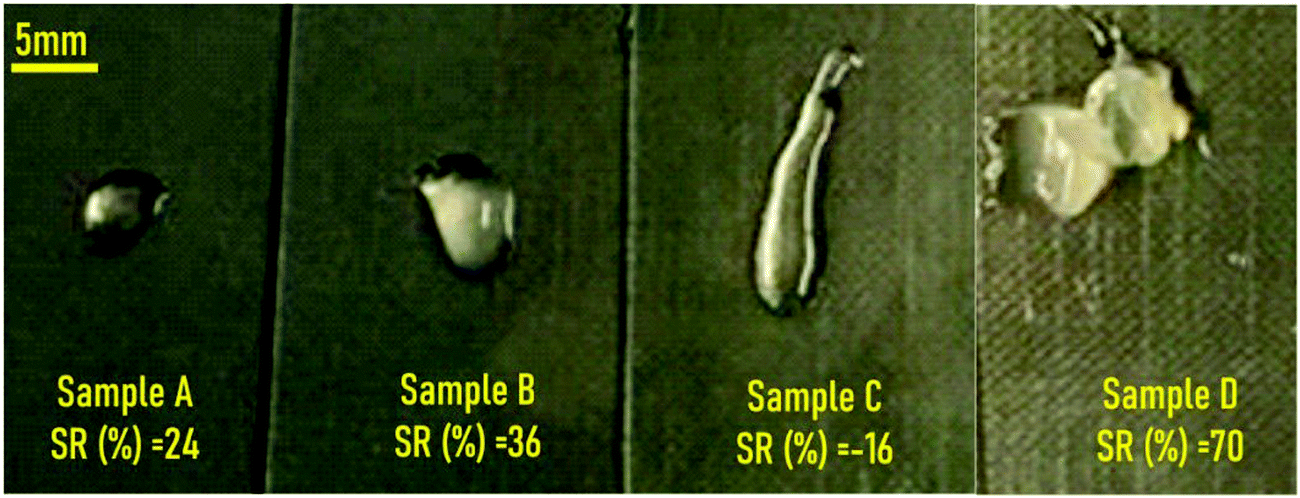 | ||
| Fig. 3 Visual appearance of the materials obtained after centrifugation of ChS–gelatin precipitates with indication of the swelling ratios obtained after immersion in water for 24 h. | ||
As it is well known, gelatin hydrogels can be formed by physical crosslinking in water above a certain concentration (around 20 mg mL−1) and below 30–35 °C. During the process, gelatin molecules aggregate and undergo a conformational change from a random coil to a triple helix.27 In order to determine whether the event of gelation was possible just by physical gelation of gelatin or the addition of ChS was a determinant factor for the formation of a ChS–gelatin hydrogel, a control experiment was conducted. It consisted of preparing samples A–D without ChS, this is, by mixing using a peristaltic pump an aqueous solution of gelatin and MilliQ water at fixed pH and NaCl concentration followed by cooling the precipitate at 5 °C overnight. No gel structure was observed, highlighting the crucial role of ChS in the formation of ChS–gelatin hydrogels.
ATR-FTIR spectroscopy allows determining the presence of interactions in ChS–gel hydrogels by comparison with the ATR-FTIR spectra of individual components (Fig. S4 in the ESI†). The analysis of these ATR-FTIR spectra is complicated due to the overlapping of many of the absorption bands corresponding to vibrational modes of chemical bonds in gelatin and ChS in the regions of interest. Nevertheless, the spectra corresponding to samples A, B and D show a clear shift of the characteristic band of gelatin amide II to higher wavenumbers (from 1529 cm−1 to 1547 cm−1). This shift indicates the presence of interactions (hydrogen bonding and electrostatic interactions) involving the gelatin amide groups.28 Similar results have been obtained for complexes of gelatin an ι-Carrageenan.29
Based on the highest SR (%) value achieved for sample D, gels obtained from aqueous solutions of ChS and gelatin of 10 mg mL−1 and pH = 4 were selected for all subsequent studies and samples were designated as ChS–gel hydrogels.
Swelling and mechanical properties as a function of pH and NaCl concentration
Firstly, the swelling properties of ChS–gel hydrogels in aqueous solutions were measured as a function of pH at a fixed NaCl concentration (0.15 M) and in PBS (pH = 7.4 and NaCl = 0.15 M) and the results are shown in Fig. 4A. At pH = 3, the negative swelling ratio observed can be attributed to the ionic crosslinks between the positively charged groups of gelatin and the negatively charged sulfonic groups of chondroitin sulfate. At pH > 3, the ChS–gel hydrogels slightly swelled as a result of the excess of negatively charged groups of ChS, as carboxylic groups become ionized. In fact, there appears to be a small increase of the SR (%) values for pH in the range of 4–5. This might be attributed to the isoelectric point of gelatin, where the electrostatic attraction between opposite charges makes the hydrogel collapse and thus, the swelling observed is only due to the small excess of negative charges in ChS (see Fig. S2 in the ESI†). At pH > 6, both gelatin and ChS are negatively charged leading to an increase in the SR (%). Fig. 4B represents the changes in the swelling ratio (SR (%)) and the Young's modulus (E), calculated from compressive stress–strain curves, with the NaCl concentration for ChS–gel hydrogels immersed in water at a fixed pH (pH = 4) and in PBS (pH = 7.4 and NaCl = 0.15 M). As can be observed, the SR (%) increased with the concentration of NaCl for values >0.01 M, which could be explained by the anti-polyelectrolyte effect.30 Salt ions break electrostatic crosslinks and become arranged around the polyelectrolyte charged groups. Weakening the structure brings more water into the bulk and the swelling increases, whereas at low salt concentrations the external osmotic pressure dehydrates residual pores and thus the swelling decreases.31 ChS–gel hydrogels were also immersed in PBS, which contains 0.15 M NaCl concentration; however, the fact that pH = 7.4 in PBS explains the higher SR (%) found for gels swollen in PBS with respect to gels swollen in aqueous solutions at pH = 4 and 0.15 M NaCl. To explain the negative swelling ratio encountered at 0.01 M NaCl concentration and in the absence of salt in the doping solution, it is necessary to consider that salt ions used in the gel preparation may not remain associated with the charged polyelectrolyte groups32 and, hence, they would diffuse outside of the hydrogel to compensate the internal osmotic pressure, thus promoting the formation of ionic bonds between gelatin and ChS.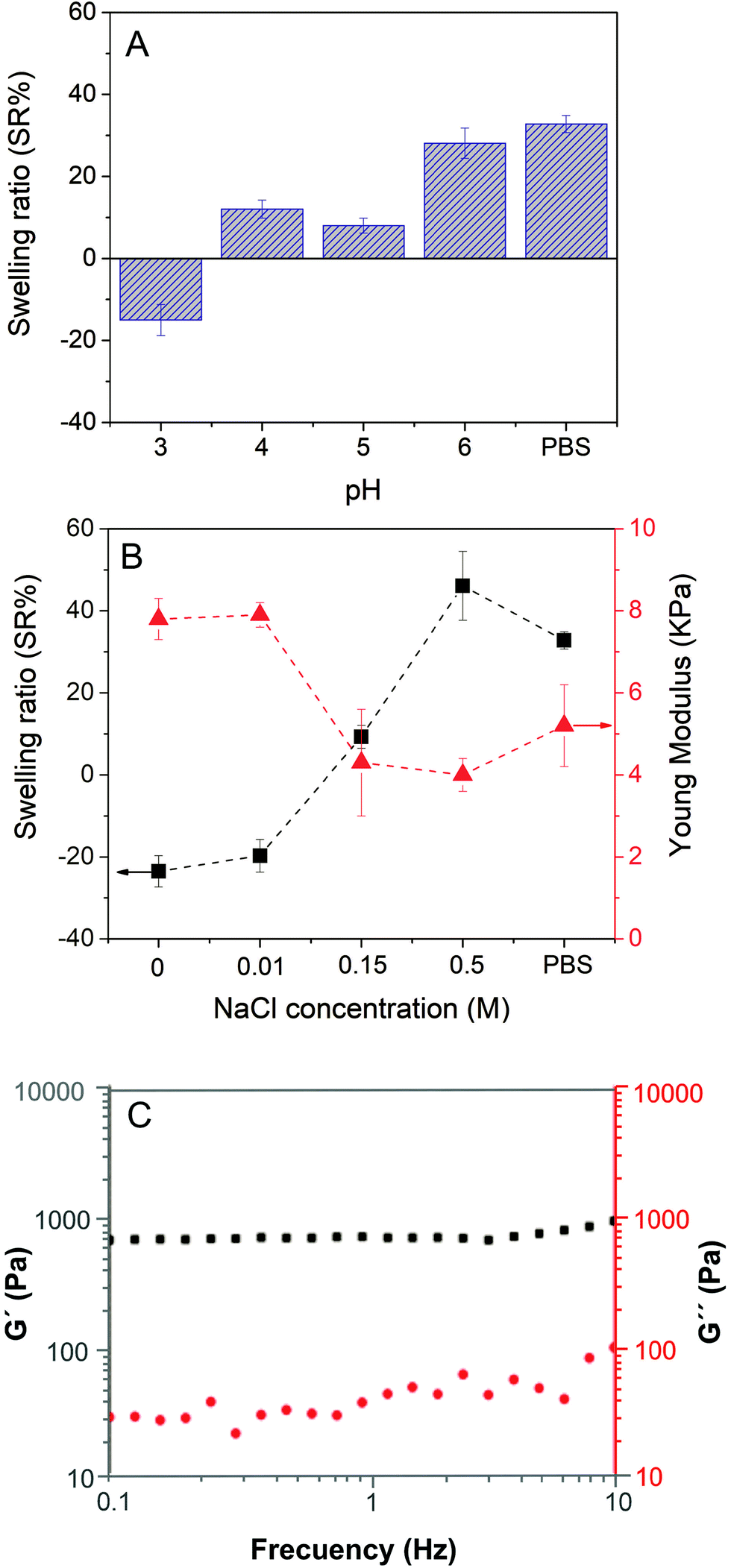 | ||
| Fig. 4 (A) Swelling ratio of ChS–gel hydrogels as a function of pH (NaCl = 0.15 M). (B) Swelling ratio and Young's modulus of ChS–gel hydrogels as a function of NaCl concentration (pH = 4) and (C) oscillatory frequency sweeps corresponding to ChS–gelatin hydrogels swollen in PBS. | ||
The effect of salt doping on the structure of ChS–gel hydrogels was ultimately reflected in their compression moduli. Hydrogels swelled in swelling media at 0 and 0.01 M NaCl concentrations, which showed negative values for SR (%), presented the highest compression moduli (7–8 kPa). At NaCl concentrations higher than 0.01 M, the uptake of water leads to a decrease of the compression moduli. The Young's moduli for native soft tissues and organs are within the range from 0.1 kPa to 1 MPa.33 Specifically, the moduli obtained for ChS–gel hydrogels can be compared to those obtained for skin measured at 5–10% strain in tension (5 kPa) and are reported to be similar to that measured in compression.34
Fig. 4C shows the representative results corresponding to the elastic moduli (G′) and the loss moduli (G′′) as a function of frequency for ChS–gelatin hydrogels swollen in PBS. As can be observed, G′ is higher than G′′ over the entire frequency range and both moduli are essentially frequency independent, a typical behaviour of hydrogels.35 The value of G′ obtained for ChS–gelatin hydrogels (G′ = 639 ± 90 Pa) is similar to G′ values reported for 5% w/v gelatin gels (G′ = 800 ± 100 Pa) and much higher than those reported for 1% w/v gelatin gels (140 ± 25).36 The fact that the ChS–gelatin hydrogels are prepared from aqueous solutions of gelatin and ChS at 10 mg mL−1 (1% w/v) indicates that the process of compaction of ChS–gel PECs into hydrogels results in a significant increase of the elastic properties of hydrogels with respect to pure gelatin gels.
Evaluation of ChS–gelatin hydrogels as the electrolyte material in all-solid state electrochemical symmetric cells
Solid-state electrochemical devices containing a hydrogel electrolyte usually suffer from a large interface contact resistance between the electrode and the hydrogel electrolyte due to poor contact between them because of the solid-like nature of the hydrogels.5 The lack of an effective electrolyte/electrode contact has a negative impact on the system as it produces an increase in the electrode/electrolyte resistance as well as a deterioration of the mechanical properties.Therefore, one of the most crucial parameters in this context is the design and creation of an effective electrode/electrolyte interface. In order to address this issue, polymer gelation is usually carried out through different approaches. One strategy involves in situ polymerization of aniline monomers inside an insulating polymer gel matrix such as polyvinyl alcohol (PVA) acting as a host-network for the electrode material. In this approach, to create an all-in-one supercapacitor the two polyaniline (PANI)–PVA electrodes are sandwiched around an electrolyte layer of PVA gel.37 Another approach is based on the physical gelation of k-carrageenan hydrogels doped with PANI to induce electrochemical activity assembled within poly(3,4-ethylenedioxythiophene) (PEDOT) thin films acting as electrodes.38For the materials under study here, in order to promote an appropriate contact between the ChS–gel hydrogel electrolyte and the electrodes, the gels were infiltrated within the porous structure of the macroporous graphene (Fig. 5A and B) and CGDL (Fig. S5 in the ESI†). Electrodes constructed by a three-dimensional (3D) porous graphene architecture have demonstrated excellent performances in charge-storage applications due to their large specific surface area, high electrical conductivity, and excellent mechanical properties.39,40 The gel infiltration protocol is quite simple; generally, a disc of the hydrated biohydrogel was assembled between the two porous electrodes. As can be seen in Fig. 5C the infiltration is achieved by the occupation of practically all the porous graphene structure by the gel. An intermediate hydrated biohydrogel's layer of about 200 μm thick was evidenced by SEM (Fig. 5D) in which the difference between the porous electrode structure and the non-porous structure corresponding to the hydrated biohydrogel can be observed. The results corresponding to the analysis of the pore size and the pore size distribution of the biohydrogel after lyophilization are provided in the ESI† (Fig. S6). Then, the layered system was placed inside a Swagelok type-cell with a symmetric configuration (Fig. S7 in the ESI†), for full electrochemical characterization.
 | ||
| Fig. 5 SEM characterization of the macroporous graphene electrode material. (A and B) SEM images corresponding to the graphene before gel infiltration. (C) Surface of the graphene after infiltration with the biohydrogel electrolyte. (D) Cross-section of the graphene/biohydrogel/graphene electrochemical cell. The dash-dot lines in the figure are guide for the eye. Inset (A): Graphene electrodes supported on stainless steel mesh. Length of the scale bar: 11 mm. | ||
Fig. 6A shows the cyclic voltammetry response of the porous graphene infiltrated with a series of ChS–gel hydrogels and recorded in a supercapacitor symmetric cell at 100 mV s−1 and room temperature. As can be evidenced all graphene/biohydrogel/graphene electrochemical cells show CVs with a rectangular shape, typical of capacitive electrochemical storage. The characteristic charge and discharge current densities had similar values to those observed in rechargeable solid-state supercapacitors for soft and smart contact lenses with continuous operation.41 The CVs corresponding to CGDL/biohydrogel/CGDL cells also show a capacitive behaviour but under a smaller potential window with respect to graphene-biogel cells (Fig. S8 in the ESI†). Besides, the gravimetric capacitance values for CGDL/biohydrogel/CGDL cells were two orders of magnitude lower than that for the graphene counterparts most probably due to the small electrode surface area available for the electrolyte phase as a consequence of the less effective contact between the biohydrogel and the CGDL. These results confirm the positive initial assessment of the ChS–gel hydrogel as a potential solid electrolyte material for EDLCs. Fig. 6B and C show the evolution of the specific capacitance as a function of the scan rate ranging between 100 mV s−1 and 500 mV s−1. All graphene/biohydrogel/graphene cells presented a capacitance retention value close to 70%, except the sample containing the lowest NaCl salt concentration that showed a value slightly lower than 60%, as indicated in Fig. 6B. The presence of a higher amount of salt might increase both capacitance and rate capability values because of the potential higher ionic conductivity that could be achieved in the polymer gel electrolytes. A maximum value of areal capacitance of 2.74 mF cm−2 (3.1 F g−1) was achieved for the cell containing the biohydrogel swollen in 0.5 M NaCl and cycled at 100 mV s−1. A slightly smaller value of 2.3 mF cm−2 (2.6 F g−1) was found for the solid–electrolyte biogels swollen with a PBS solution for the same scan rate. This value is of the same order of magnitude as that obtained for ultrathin epidermal capacitors based on gold nanowires/PANI composites, recently published.29Fig. 6C shows a comparison between the capacitance retention detected in the two porous carbon electrode systems infiltrated with the solid biohydrogel swollen in PBS. As it is clearly observed, the capacity values varied similarly with the scan rate in both cells, but it is one order of magnitude higher for the graphene-biohydrogel cell than for the CGDL-biohydrogel one.
 | ||
| Fig. 6 (A) CVs of graphene/biohydrogel/graphene supercapacitors measured at 100 mV s−1 in a symmetric configuration. (B) Rate capability measurements as a function of the scan rate for the series of graphene-biohydrogel cells. (C) Comparison between graphene and CGDL symmetric cells based on the polyelectrolyte gel containing PBS. (D) Nyquist plots for graphene and CGDL biohydrogel-supercapacitors at the open circuit voltage. | ||
Fig. 6D shows the Nyquist graph of the electrochemical cells obtained from PEIS data. Contrary to what should be expected for a solid gel electrolyte material,42 the impedance spectrum of the graphene/biohydrogel/graphene cell does not appear as a depressed semicircle. Instead, slightly inclined straight lines intercepting the real axis on the high-frequency side like in liquid electrolytes characterize the spectrum. This situation is maintained even after the galvanostatic cycling of the supercapacitor, except at high frequencies where the capacitive behavior deviated from its initial slope. The latter fact was most probably due to a small reduction in the diffusion of the ions into the polymeric network since the effective ion size has not changed. The situation was slightly different for the CGDL/biohydrogel/CGDL supercapacitor cell where the impedance spectrum already shows a semicircle indicating a higher value of the electrochemical series resistance (ESR) component where the bulk conductivity of the hydrogel is overlapped. This might be due to the thicker layer of the gel electrolyte already existing inside the pores/voids of the carbon electrode mat. Impedance analysis of the hydrogel supercapacitors allowed the calculation of their ESR values. The electrolyte resistance was calculated by interception of the branch to the real axis. The biohydrogel supercapacitor with macroporous graphene electrodes exhibited similar ESR values, ∼12 Ohm cm2, before and after being cycled. The CGDL/biohydrogel/CGDL supercapacitor cell exhibited a higher ESR value of 55 Ohm cm2, i.e., almost five times higher resistance than their graphene analogues. The results obtained clearly indicate that the combination of macroporous graphene electrodes with the innovative composite solid biohydrogels developed here is fully compatible with use in bioelectronics applications as a scalable and rechargeable source of energy.43
Conclusions
Using a simple and scalable centrifugation approach, hydrogels obtained from gelatin–chondroitin sulfate polyelectrolyte complexes have been successfully developed through the optimization of experimental variables such as pH and polymer and NaCl concentration. According to the results obtained from zeta potential experiments and precipitation studies, electrostatic interactions between gelatin and ChS in water were favoured for starting solutions of the polymers at equal pH and concentration. The formation of hydrogels was assessed from centrifugation of gel–ChS PECs obtained through mixing of aqueous solutions of gelatin and ChS at 10 mg mL−1 and a pH of 4. The biohydrogels obtained present compression moduli in the range between 4kPa and 8kPa as a function of pH and NaCl concentration. Such values have been reported for soft biological tissues such as skin. In addition, the increase of the elastic moduli of ChS–gel hydrogels with respect to pure gelatin gels confirms, on the one hand, the role of electrostatic interactions between the biopolymers and, on the other hand, the effectiveness of the compaction process in the formation of the hydrogel. An application to safe and sustainable all-solid-state electrochemical systems has been detailed, as a demonstration of the ideal areal capacitance and rate capability properties that are the consequence of a very high effective interaction between the biopolymer hydrogel electrolyte and the porous graphene electrode material at the interphase. The cells exhibit areal capacitance values up to 2.74 mF cm−2 (3.1 F g−1) and a low resistance value of 12 Ohm cm2 for graphene electrode materials. The capacity values are one order of magnitude higher than those observed for cells using the porous carbon gas diffusion layer as the electrode material. These facts confirm the importance of the very high effective interaction between the biopolymer hydrogel electrolyte and the porous graphene electrode material at the interphase. Because of the benign nature of the materials and systems developed here they might be used in the next generation of biodevices designed to function alongside the human body.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank David Gómez for assistance regarding scanning electron microscopy (SEM). Rebeca Hernández is a member of the SusPlast platform from the Spanish National Research Council (CSIC). This study was supported by CONICET (National Scientific and Technical Research Council), ANPCyT (National Agency of Scientific and Technology Promotion, PICT 2016-1905), UNMdP (National University of Mar del Plata) and Spanish Ministry of Economy, Industry and Competitiveness (MINECO) (grants: MAT2017-83014-C2-2-P and MAT2017-88232-P). Javier Carretero-González acknowledges the Ramon y Cajal Fellow (RYC-2015-17722) from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO).References
- N. Boaretto, L. Meabe, M. Martinez-Ibañez, M. Armand and H. Zhang, J. Electrochem. Soc., 2020, 167, 070524 CrossRef CAS.
- Y. Wang and W.-H. Zhong, ChemElectroChem, 2015, 2, 22–36 CrossRef CAS.
- D. P. Dubal, N. R. Chodankar, D.-H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- Q. Han, X. Chi, S. Zhang, Y. Liu, B. Zhou, J. Yang and Y. Liu, J. Mater. Chem. A, 2018, 6, 23046–23054 RSC.
- J. Zhao, Y. Chen, Y. Yao, Z.-R. Tong, P.-W. Li, Z.-M. Yang and S.-H. Jin, J. Power Sources, 2018, 378, 603–609 CrossRef CAS.
- L. Wang, D. Chen, K. Jiang and G. Shen, Chem. Soc. Rev., 2017, 46, 6764–6815 RSC.
- M. J. Donahue, A. Sanchez-Sanchez, S. Inal, J. Qu, R. M. Owens, D. Mecerreyes, G. G. Malliaras and D. C. Martin, Mater. Sci. Eng., R, 2020, 140, 100546 CrossRef.
- S.-S. Kim, J.-H. Jeon, H.-I. Kim, C. D. Kee and I.-K. Oh, Adv. Funct. Mater., 2015, 25, 3560–3570 CrossRef CAS.
- M. Criado, J. M. Rey, C. Mijangos and R. Hernández, RSC Adv., 2016, 6, 105821–105826 RSC.
- I. del Agua, S. Marina, C. Pitsalidis, D. Mantione, M. Ferro, D. Iandolo, A. Sanchez-Sanchez, G. G. Malliaras, R. M. Owens and D. Mecerreyes, ACS Omega, 2018, 3, 7424–7431 CrossRef CAS.
- C. H. Porcel and J. B. Schlenoff, Biomacromolecules, 2009, 10, 2968–2975 CrossRef CAS.
- F. M. Santos, P. C. Barbosa, R. F. P. Pereira, M. M. Silva, H. M. R. Gonçalves, S. C. Nunes, F. L. Figueiredo, A. J. M. Valente and V. de Zea Bermudez, Eur. Polym. J., 2020, 124, 109453 CrossRef.
- A. M. Navarro-Suárez, J. Carretero-González, V. Roddatis, E. Goikolea, J. Ségalini, E. Redondo, T. Rojo and R. Mysyk, RSC Adv., 2014, 4, 48336–48343 RSC.
- B. E. Conway, Electrochemical Supercapacitors. Scientific Fundamentals and Technological Applications, Springer, US, 1999 Search PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. Castelaín, G. Martínez, P. Merino, J. Á. Martín-Gago, J. L. Segura, G. Ellis and H. J. Salavagione, Chem. – Eur. J., 2012, 18, 4965–4973 CrossRef.
- N. Devi, C. Deka, T. Maji and D. Kakati, Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, 2016, 3557–3569, DOI:10.1081/E-EBPP-120049954.
- N. Devi, M. Sarmah, B. Khatun and T. K. Maji, Adv. Colloid Interface Sci., 2017, 239, 136–145 CrossRef CAS.
- I. J. Haug and K. I. Draget, in Handbook of Hydrocolloids, ed. G. O. Phillips and P. A. Williams, Woodhead Publishing, 2nd edn, 2009, pp. 142–163 DOI:10.1533/9781845695873.142.
- B. Chakrabarti, J. W. Park and E. S. Stevens, Crit. Rev. Biochem., 1980, 8, 225–313 CrossRef CAS.
- A. Denuziere, D. Ferrier and A. Domard, Carbohydr. Polym., 1996, 29, 317–323 CrossRef CAS.
- R. Barbucci, M. Casolaro, N. Danzo, V. Barone, P. Ferruti and A. Angeloni, Macromolecules, 1983, 16, 456–462 CrossRef CAS.
- D. Pinto Ramos, S. Sarjinsky, M. Alizadehgiashi, J. Möbus and E. Kumacheva, ACS Omega, 2019, 4, 8795–8803 CrossRef CAS.
- T. Phoeung, M. V. Spanedda, E. Roger, B. Heurtault, S. Fournel, A. Reisch, A. Mutschler, F. Perrin-Schmitt, J. Hemmerlé, D. Collin, M. Rawiso, F. Boulmedais, P. Schaaf, P. Lavalle and B. Frisch, Chem. Mater., 2017, 29, 10418–10425 CrossRef CAS.
- M. B. Oliveira, H. X. S. Bastos and J. F. Mano, Biomacromolecules, 2018, 19, 2742–2749 CrossRef CAS.
- W.-B. Chen, L.-F. Wang, J.-S. Chen and S.-Y. Fan, J. Biomed. Mater. Res., Part A, 2005, 75A, 128–137 CrossRef CAS.
- F. Bode, M. A. da Silva, A. F. Drake, S. B. Ross-Murphy and C. A. Dreiss, Biomacromolecules, 2011, 12, 3741–3752 CrossRef CAS.
- R. S. Derkach, A. Y. Kuchina, S. D. Kolotova and G. N. Voron’ko, Polymers, 2020, 12(2), 266 CrossRef.
- L. G. Gómez-Mascaraque, B. Llavata-Cabrero, M. Martínez-Sanz, M. J. Fabra and A. López-Rubio, J. Colloid Interface Sci., 2018, 517, 113–123 CrossRef.
- J. Fu, H. M. Fares and J. B. Schlenoff, Macromolecules, 2017, 50, 1066–1074 CrossRef CAS.
- X. Feng and R. Pelton, Macromolecules, 2007, 40, 1624–1630 CrossRef CAS.
- J. B. Schlenoff, M. Yang, Z. A. Digby and Q. Wang, Macromolecules, 2019, 52, 9149–9159 CrossRef CAS.
- J. Liu, H. Zheng, P. S. P. Poh, H.-G. Machens and A. F. Schilling, Int. J. Mol. Sci., 2015, 16, 15997–16016 CrossRef CAS.
- Y. Wang, K. L. Marshall, Y. Baba, G. J. Gerling and E. A. Lumpkin, PLoS One, 2013, 8, e67439 CrossRef CAS.
- R. Hernández and C. Mijangos, Rheology, 2013, 383–407 Search PubMed.
- I. Samba, R. Hernandez, N. Rescignano, C. Mijangos and J. M. Kenny, Nanocomposites, 2015, 1, 46–50 CrossRef CAS.
- K. Wang, X. Zhang, C. Li, X. Sun, Q. Meng, Y. Ma and Z. Wei, Adv. Mater., 2015, 27, 7451–7457 CrossRef CAS.
- M. M. Pérez-Madrigal, F. Estrany, E. Armelin, D. D. Díaz and C. Alemán, J. Mater. Chem. A, 2016, 4, 1792–1805 RSC.
- X. Xu, W. Shi, P. Li, S. Ye, C. Ye, H. Ye, T. Lu, A. Zheng, J. Zhu, L. Xu, M. Zhong and X. Cao, Chem. Mater., 2017, 29, 6058–6065 CrossRef CAS.
- A. M. García, V. L. Budarin, Y. Zhou, M. De bruyn, A. J. Hunt, L. Lari, V. K. Lazarov, H. J. Salavagione, E. Morales, G. J. Ellis, J. H. Clark and P. S. Shuttleworth, J. Mater. Chem. A, 2018, 6, 1119–1127 RSC.
- J. Park, D. B. Ahn, J. Kim, E. Cha, B.-S. Bae, S.-Y. Lee and J.-U. Park, Sci. Adv., 2019, 5, eaay0764 CrossRef CAS.
- Impedance Spectroscopy Theory, Experiment, and Applications, ed. E. Barsoukov and J. R. Macdonald, John Wiley & Sons, Inc., 2005 Search PubMed.
- L. Yin, X. Huang, H. Xu, Y. Zhang, J. Lam, J. Cheng and J. A. Rogers, Adv. Mater., 2014, 26, 3879–3884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00514b |
| This journal is © The Royal Society of Chemistry 2020 |