
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diketopyrrolopyrrole linked porphyrin dimers for visible-near-infrared photoresponsive nonfullerene organic solar cells†
Venkatesh
Piradi
a,
Xiaopeng
Xu
b,
Qiang
Peng
*b and
Xunjin
Zhu
 *a
*a
aInstitute of Molecular Functional Materials, Department of Chemistry, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong, P. R. China. E-mail: xjzhu@hkbu.edu.hk
bDepartment of Chemistry, Sichuan University, Chengdu, Sichuan 610000, P. R. China. E-mail: qiangpeng@scu.edu.cn
First published on 15th September 2020
Abstract
Two new A2–π–D–A1–D–π–A2 structural porphyrin small molecules denoted as TDPP-2P and ETDPP-2P are constructed from dimeric porphyrin centers, in which the two porphyrin moieties are linked by either the electron deficient 3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (TDPP) for the former, or diethynyl substituted TDPP for the latter, further symmetrically π-extended with electron withdrawing rhodanine units through phenylenethynylene π-bridges. For both, 2-butyloctyl and 1-octylundecyl branched alkyl chains are attached to TDPP and porphyrin moieties, respectively, to improve their solubility and self-assembly properties. Compared to TDPP-2P, ETDPP-2P shows a broader absorption in the near-infrared (NIR) range over 1000 nm due to the enhanced coplanarity of the dimeric porphyrin center. Using IT-M as the acceptor, the blend film ETDPP-2P:IT-M accomplishes a panchromatic photoresponse from 300–900 nm, and as a result, the device exhibits a prominent PCE of 5.69%, whereas, the film TDPP-2P:IT-M shows a lower PCE of 4.12% due to the lack of NIR photoresponses. These primary results open up a promising avenue for constructing porphyrin-based all small molecule organic solar cells with a panchromatic photoresponse.
Introduction
Solution-processed organic solar cells (OSCs) have eventually been considered as one of the most promising next-generation photovoltaics due to their lightweight, optimistic flexibility, and low production cost.1–5 Over the past few decades, the power conversion efficiencies (PCEs) of OSCs have exceeded 14% for binary layers consisting of polymer-based donors and fullerene derivative acceptors.6–9 On the other hand, non-fullerene small-molecule (NFSM) acceptors have also attracted early interest to overcome the disadvantages of fullerene derivatives such as purification techniques, high-cost synthetic procedures, and unfavourable morphology.10,11 Impressively, the PCEs of NF-OSCs have enhanced rapidly over 15%, owing to their complementary absorption, low energy loss, adequate and balanced electron mobility, and flexible molecular structures.12–17Among the various candidates, A–D–A structural SM acceptors built from fused indacenodithiophene (IDT)18 rings and their analogues, such as ITIC,17 IEIC,19 IT-4F,20 IT-M,21 and IDIC22 (see abbreviations in the ESI†), have shown the most promising performance in NF-OSCs because of their intensive near-infrared (NIR) absorption, easily processable capability, well-defined crystallinity, and ideal bandgaps.8,23,24 However, SM donors sporadically succeeded with several advantages such as high crystallinity, rapid aggregation pathways, controllable energy levels, etc., and as a result, assisting the significant enhancement of PCEs in recent years.25–38 However, the combination of a SM donor and a SM acceptor has been a bottleneck hindering the performance so far due to their high crystallinity and unfavourable phase separation and morphologies.25,39 Recently, Wei et al. reported an A–π–D–π–A structural narrow bandgap SM donor based on a larger coplanar core DTBDT and achieved the highest PCE of 14.34% at that time for binary all SM OSCs by optimizing their hierarchical morphologies.35 This significant success has led to an enhanced performance of all SM donors and acceptors which are comparable to that of NF-OSCs based on polymer donors. However, there are still very limited SM donors developed with the combination of NF acceptors for all SM-OSCs and those are with a fully responsive spectrum in the visible-NIR region.
On the other side, porphyrins have been extensively explored as a SM donor or acceptor in OPVs with impressive performances.40,41 Over the past several years, we and other groups have steadily worked for the development of porphyrin SM electron donors in OSCs due to their excellent super π-conjugation properties, tremendous light-harvesting capabilities, exceptional charge transport mobilities, and easy processability with fullerene acceptors, as a result, achieving high PCEs up to 10%.42–55 One major disadvantage of porphyrin derivatives for OSCs is that they usually show a deep valley between the intense Soret band (400–500 nm) and Q-bands (650–800 nm).45,56 It is still challenging to design a porphyrin molecule with a full spectrum from 300 nm to 900 nm. Recently, ternary OSCs were proposed to be used as the third component as an absorption complementary donor or acceptor to achieve a full absorption spectrum.57–59 For example, we have recently constructed a ternary OSC using a narrow bandgap porphyrin (DPP-2TTP) and an absorption complementary wide-bandgap SMs (DR3TBDTTF) as donors, and PCBM as the acceptor. Impressively, the photon-current conversion of the device crossed over UV-visible and NIR regions, and as a result, a PCE of 11% was recorded.45 At the same time, Jang et al.,60 Jen et al.,29 and Wan et al.61 have succeeded without using a third component to construct a binary layer system of porphyrin donors and NF acceptors for all SM-OSCs. However, only a very few binary layer systems featuring porphyrin donors and NFSM acceptors have been reported so far for OSCs. It is very challenging to construct porphyrins with NIR light absorbing nature and compensate the valley between the Soret and Q-bands by a NFSM acceptor. Thus, we are motivated to develop NIR porphyrin-based donors and investigate their compatibility with NFSM acceptors targeting for high-performance all SM-OSCs in the absence of a third component.
In this study, we designed and synthesized two new porphyrin-based SMs as ETDPP-2P and TDPP-2P with A2–π–D–A1–D–π–A2 architecture, in which vertically meso-alkyl-substituted porphyrin (D) dimers are linked by thiophene-flanked diketopyrrolopyrrole (A1 = TDPP) and ethynylene-thiophene-flanked diketopyrrolopyrrole (A1 = ETDPP), respectively, and further horizontally π-extended via a π-bridge of ethynylene–phenylene and end-capped with electron-deficient 3-ethylrhodanine (A2). As compared with TDPP-2P, ETDPP-2P shows a longer π-conjugated planar structure with an exceptionally red-shifted absorption over 1000 nm. To achieve panchromatic light-harvesting, SM acceptor IT-M with a perfect complementary absorption to ETDPP-2P was selected for all SM binary OSCs, resulting in a PCE of 5.69%. And a lower PCE of 4.12% was recorded for the binary OSC based on the TDPP-2P:IT-M active layer due to the less light harvesting in the NIR region.
Results and discussion
The synthesis of the intermediates 3 and 6 is described in the ESI† in Scheme S1. Briefly, ETDPP-2P and TDPP-2P (Scheme 1) can be prepared through Sonogashira/Stille coupling and Knoevenagel condensation reactions. Both porphyrin SMs are well soluble in most organic solvents for solution processing OSCs. Moreover, all the new compounds were characterized by 1H NMR, 13C NMR, and MALDI-TOF techniques (ESI†).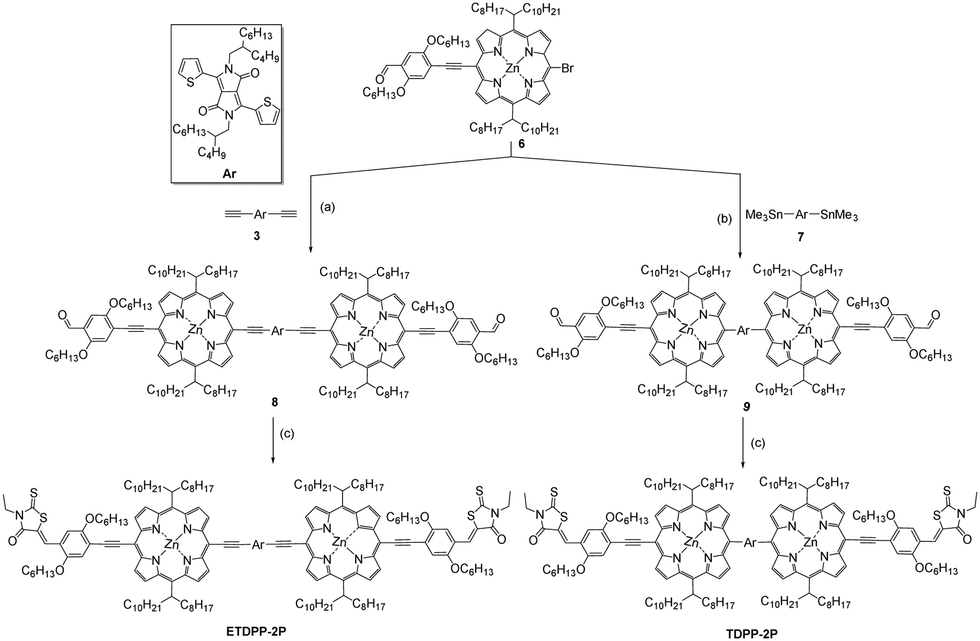 | ||
| Scheme 1 Synthetic routes and structures of ETDPP-2P and TDPP-2P. (a) CuI, Pd(PPh3)4, tetrahydrofuran, triethylamine, (b) Pd(PPh3)4, toluene, reflux, 48 h (c) 3-ethylrhodanine, dry CHCl3, piperidine, reflux, overnight. | ||
Photophysical properties
Fig. 1a shows the absorption spectra of ETDPP-2P and TDPP-2P recorded in chloroform solution and the data are summarized in Table 1. Both the compounds majorly display two broad and intense bands. The bands at ca. 390–560 nm for TDPP-2P and at ca. 390–570 nm for ETDPP-2P correspond to the Soret band. Similarly, the peaks at ca. 590–725 nm for TDPP-2P and at ca. 620–900 nm for ETDPP-2P are attributed to the Q-band absorption. Compared with TDPP-2P, ETDPP-2P with ethynylene conjugated DPP exhibits very broad and redshifted (175 nm) Q-band absorption over the NIR region because the elongated π-conjugation enhances the strong intramolecular charge transfer (ICT) interactions within the porphyrin molecules. However, the thin films of both compounds exhibit much red-shifted absorption spectra than those in solution as shown in Fig. 1b. In contrast, ETDPP-2P shows a significant broad peak of Q-band over 1000 nm with a shoulder peak at 760 nm. This could be ascribed to the extended π-conjugation of ETDPP-2P possessing stronger intermolecular π–π stacking within the donor molecules.62,63 On the other hand, the absorption spectra of the IT-M thin-film disclose an optical response from 470 to 830 nm with a shoulder peak at ca. 640 nm. Fig. 1c depicts the absorption spectra of blend films of ETDPP-2P or TDPP-2P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IT-M (1:1, w/w) spin-coated from chloroform solution and processed under solvent-vapor annealing (SVA) and thermal annealing (TA) treatments. However, both the blend films exhibit panchromatic absorption, and the film ETDPP-2P:IT-M shows a wider range of NIR absorption over 900 nm than the film TDPP-2P:IT-M (750 nm). Furthermore, the optical band gaps (Eoptg) were estimated to be 1.40 eV for ETDPP-2P and 1.72 eV for TDPP-2P from the onset wavelength (λonset) in solution. The broad and panchromatic response of the ETDPP-2P:IT-M film is favourable for efficient charge carrier generation and thus a higher current density (Jsc) is achieved in the NF based active device (vide infra).
:IT-M (1:1, w/w) spin-coated from chloroform solution and processed under solvent-vapor annealing (SVA) and thermal annealing (TA) treatments. However, both the blend films exhibit panchromatic absorption, and the film ETDPP-2P:IT-M shows a wider range of NIR absorption over 900 nm than the film TDPP-2P:IT-M (750 nm). Furthermore, the optical band gaps (Eoptg) were estimated to be 1.40 eV for ETDPP-2P and 1.72 eV for TDPP-2P from the onset wavelength (λonset) in solution. The broad and panchromatic response of the ETDPP-2P:IT-M film is favourable for efficient charge carrier generation and thus a higher current density (Jsc) is achieved in the NF based active device (vide infra).
 | ||
| Fig. 1 Vis-NIR absorption spectra of ETDPP-2P and TDPP-2P in chloroform solution (a), thin films including IT-M (b), (c) blend films of ETDPP-2P:IT-M and TDPP-2P:IT-M (inset: molecular diagram of IT-M) and (d) energy level diagram of TDPP-2P, ETDPP-2P, and IT-M materials, respectively. | ||
| Comp. | λ max (nm, CHCl3) (ε/105 M−1 cm−1) | λ max (nm) (film) | E HOMO (eV) | E LUMO (eV) | E g (eV) |
|---|---|---|---|---|---|
| ETDPP-2P | 474 (1.63), 774 (1.91) | 510, 764, 844 | −5.12 | −3.72 | 1.40 |
| TDPP-2P | 455 (2.10), 671 (1.47) | 462, 506, 688 | −5.09 | −3.37 | 1.72 |
Electrochemical properties
The redox potentials of ETDPP-2P and TDPP-2P were calculated by performing the cyclic voltammetry (CV) measurements in CHCl3 solution (Fig. S1, ESI†) and the corresponding data are noted in Table 1. The onset oxidation potentials were used to calculate the highest occupied molecular orbitals (HOMOs), and the lowest unoccupied molecular orbitals (LUMOs) were then determined according to ELUMO = EHOMO + Eoptg eV. The HOMO/LUMO energy levels of ETDPP-2P and TDPP-2P were approximated to be −5.12/−3.72 eV and −5.09/−3.37 eV, respectively. Obviously, the ethynylene linkage between porphyrin and TDPP led to a more planar A2–π–D–A1–D–π–A2 structure of ETDPP-2P with the elongated π-conjugation, as well as the deeper HOMO and LUMO levels. Fig. 1d presents the energy level diagrams of TDPP-2P, ETDPP-2P, and IT-M molecules, respectively. The compound ETDPP-2P shows perfect alignment energy levels with the IT-M acceptor to transfer hole and electrons when compared to the TDPP-2P compound due to the low lying HOMO and LUMO levels.Photovoltaic properties
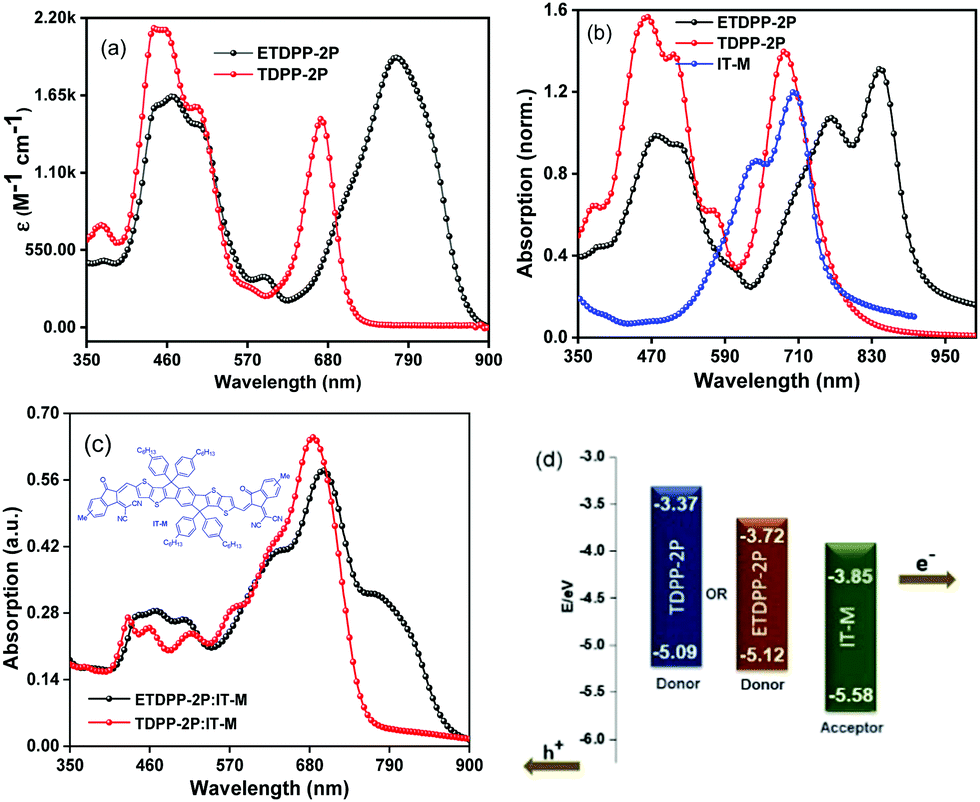
The photovoltaic performance of ETDPP-2P and TDPP-2P compounds as donors with IT-M acceptors was evaluated for the conventional device structure of ITO/PEDOT:PSS/donor:acceptor/ZrAcac/Al under simulated AM 1.5 G illumination at 100 mW cm−2. The optimal blend films were processed from chloroform (CF) as the solvent and 1,8-diiodooctane (DIO) as an additive (99.5:0.5 v/v) in a weight ratio of donor:acceptor = 1:0.6 (w/w). Furthermore, the films underwent TA treatment at 150 °C for about 15 min. Fig. 2a shows the current density–voltage (J–V) curves of the devices and the key parameters are summarized in Table 2. The optimal device ETDPP-2P:IT-M exhibits an open circuit voltage (Voc) of 0.84 V, along with a short circuit current (Jsc) of 11.05 mA cm−2, and a fill factor (FF) of 61.3%, thus resulting in a promised maximum PCE of 5.69%. On the other hand, the device TDPP-2P:IT-M shows a comparatively lower performance with a Voc of 0.79 V, a Jsc of 8.96 mA cm−2, and a FF of 58.2%, corresponding to a PCE of 4.12%. The overall better photovoltaic performance of the device ETDPP-2P:IT-M could be attributed to efficient charge dissociation, charge transfer, and well-ordered molecular packing morphology of the active layer (vide infra).
 | ||
| Fig. 2 (a) Current density–voltage (J–V) curves and (b) the EQE curves of the devices ETDPP-2P/TDPP-2P:IT-M based devices under optimized conditions. | ||
Fig. 2b shows the external quantum efficiency (EQE) curves of the optimized devices for photocurrent generation. Notably, the device ETDPP-2P:IT-M reaches a broad photo-response range from 300–910 nm with a higher maximum EQE (EQEmax) value of 45% at ca. 700 nm. However, the device TDPP-2P:IT-M shows a comparatively lower photo-response from 300–780 nm with an EQEmax value of 45% at ca. 620 nm. The calculated current density values (JscEQE) are found to be 10.71 mA cm−2 and 8.68 mA cm−2 for ETDPP-2P:IT-M and TDPP-2P:IT-M, respectively. These results are consistent with those collected from J–V measurements with a little deviation, as shown in Table 2a. The higher photo-response and EQE value of the ETDPP-2P:IT-M device indicates the sufficient exciton separation, charge transport, and collection (vide infra).
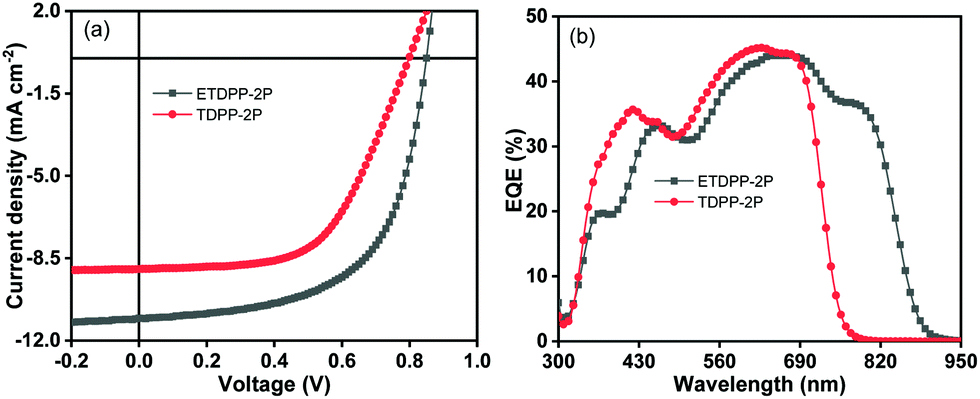
Fig. 3a shows the photocurrent density (Jph) (Jph = JL − JD) for ETDPP-2P and TDPP-2P devices, where JL and JD are the current density of the devices under illumination and dark, respectively, as a function of effective voltage Veff (Veff = V0 − V), where V0 is the voltage at Jph = 0. The Jsat is the saturation current density which is equal to Jph when Veff ≥ 2.0 V.64,65 Thus, the charge collection probabilities P(E,T) can be characterized by using the ratio Jph/Jsat. Under the optimized conditions and maximal power output, the calculated ratios are found to be 0.92% for ETDPP-2P:IT-M and 0.83% for TDPP-2P:IT-M. The high P(E,T) value of ETDPP-2P:IT-M shows that the device has more efficient exciton dissociation and charge collection efficiency compared to the TDPP-2P:IT-M device.
 | ||
| Fig. 3 Plots of photocurrent density (Jph) versus effective voltage (Veff) curve (a), current density (Jsc) versus light intensity (Plight) (b) of the ETDPP-2P and TDPP-2P devices. | ||
Furthermore, the Jsc and Voc measurements were performed under different light intensities (Plight) to identify the charge recombination mechanisms of these devices. Fig. 3b shows the relationship of Jsc and (Plight) which can be described as Jsc ∝ (Plight)γ,66 where γ is the bimolecular recombination exponential factor. The recorded γ values were found to be 0.91 for ETDPP-2P:IT-M and 0.88 for TDPP-2P:IT-M. When the γ value is close to 1, indicating that bimolecular recombination in the ETDPP-2P:IT-M device is negligible.67 On the other hand, to analyse the trap-assisted recombination of these devices, as shown in Fig. S2 (ESI†) the plot of Voc against the light intensity (Plight) is constructed in log scales, which can be expressed as Voc ∝ (SkT/q)ln(Plight), using the slope equation where k is Boltzmann's constant, T is the temperature, and q is the elementary charge. Generally, the S parameter value is in the range of 1 to 2. When S >1, it indicates the existence of trap-assisted recombination and when S = 1, it indicates that it is free of trap-assisted recombination. The recorded S values are found to be 1.36 (kT/q) for ETDPP-2P:IT-M and 1.51 (kT/q) for TDPP-2P:IT-M. Both the devices show trap-assisted recombination, however, the device ETDPP-2P:IT-M has less extent of traps than the device of TDPP-2P:IT-M, leading to an improved FF of 61.3% and a Voc of 0.84 V.
In addition, the charge mobilities of the devices were measured by using the space charge limited current (SCLC) method. The optimal device structure of glass/ITO/PEDOT:PSS/donor:acceptor/MoO3/Au was used for the hole only device and glass/Al/donor:acceptor/Al was used for the electron only device. The hole and electron mobilities (μh and μe) were calculated from the slopes of J1/2–V curves as shown in Fig. 4, respectively, and the data are summarized in Table 2. The μh mobilities of the blend films were fitted to be 7.16 × 10−5 cm2 V−1 s−1 for ETDPP-2P:IT-M and 3.64 × 10−5 cm2 V−1 s−1 for TDPP-2P:IT-M. And the μe mobilities were recorded to be 5.83 × 10−5 cm2 V−1 s−1 for ETDPP-2P:IT-M and 1.47 × 10−5 cm2 V−1 s−1 for TDPP-2P:IT-M. The balanced μe/μh mobility ratio was calculated to be 0.81 for ETDPP-2P:IT-M and 0.40 for TDPP-2P:IT-M. The higher and more balanced mobilities of the ETDPP-2P:IT-M blend film contribute to its high Jsc and FF values.
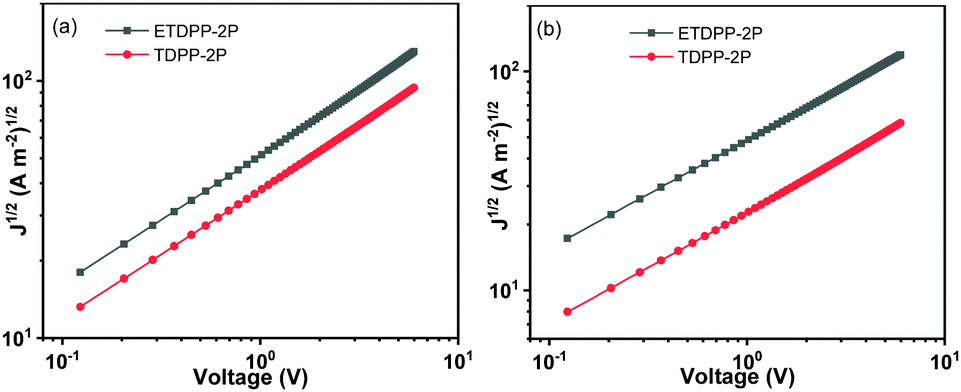 | ||
| Fig. 4 Dark current–voltage characteristics of (a) hole only device on Glass/ITO/PEDOT: PSS/active layer/MoO3/Au and (b) electron only device on glass/Al/active layer/Al under optimized conditions. | ||
The morphology of the active layers was measured by using trapping-mode atomic force microscopy (AFM). As illustrated in Fig. 5a and b, the height images of ETDPP-2P:IT-M and TDPP-2P:IT-M exhibit a smooth surface morphology with a root-mean-square (RMS) roughness of 2.01 nm and 2.46 nm, respectively. The lower RMS of the ETDPP-2P:IT-M film would be beneficial to construct bi-continuous interpenetrating networks when compared to that of the TDPP-2P:IT-M counterpart. This is essential to promote the exciton dissociation and charge transport, as discussed previously, that enables relatively high Jsc and FF values. On the other hand, the phase images in Fig. 5c and d show a more well-ordered morphology of the donor and acceptor interlayers in the blend ETDPP-2P:IT-M film, which facilitates the efficient charge dissociation and transfer in the device.
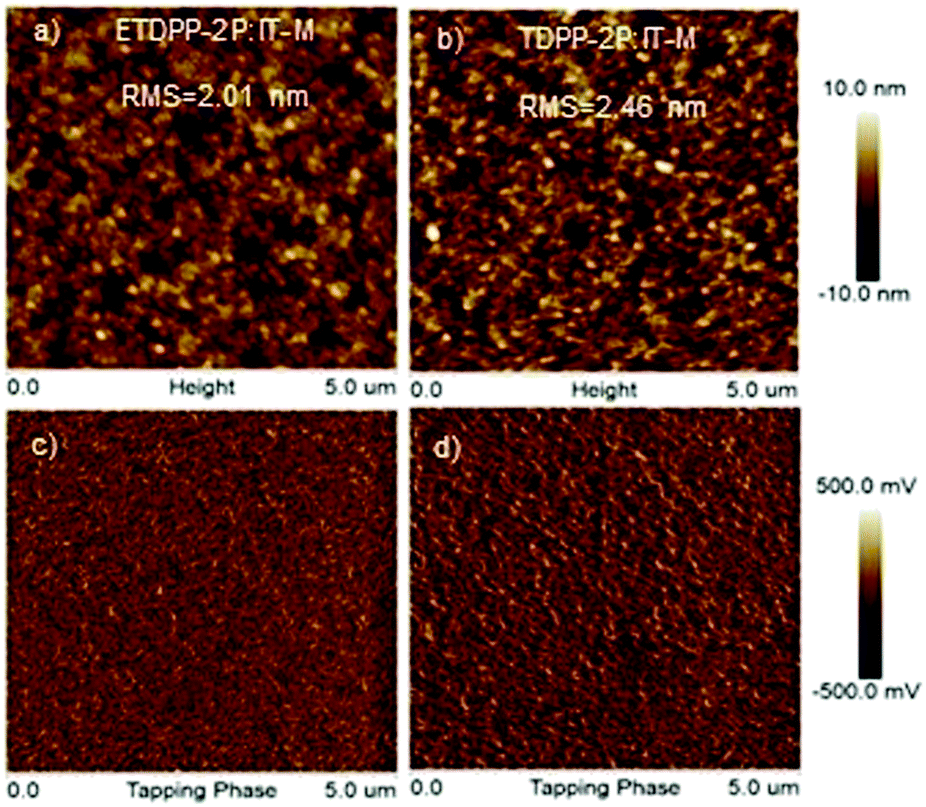 | ||
| Fig. 5 AFM height images (a and b) and phase images (c and d) of the binary blend films ETDPP-2P:IT-M and TDPP-2P:IT-M from left to right. The scan size is 5 μm × 5 μm for all images. | ||
Conclusions
In summary, we have designed and synthesized two new porphyrin dimers TDPP-2P and ETDPP-2P constructed from the dimeric porphyrin core linked by TDPP and diethynyl substituted TDPP, respectively. Due to the longer π-conjugation and more planar structure, the ETDPP-2P solid film exhibits a stronger and much red-shifted absorption spectrum ranging from 300 to 1000 nm, whereas TDPP-2P shows a narrower absorption from 300 to 750 nm due to its less π-conjugation and planarity. And the deep absorption valley between the Soret band and Q-band of the ETDPP-2P film could be perfectly compensated by the SM acceptor IT-M. Consequently, the optimal device based on the active layer of ETDPP-2P:IT-M possesses a panchromatic photoresponse in the range of 300–900 nm and a PCE of 5.69% with a Voc of 0.84 V, a Jsc of 11.05 mA cm−2 and an FF of 61.3%. In contrast, the device based on the active layer of TDPP-2P:IT-M shows a lower PCE of 4.12% with a Voc of 0.79 V, a Jsc of 8.96 mA cm−2, and an FF of 58.2%. To date, the molecular behaviours and photovoltaic properties of a porphyrin donor with a NFSM acceptor are still underexplored; thus, our study can offer a crucial guidance for the development of porphyrin-based all small molecule organic solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hong Kong Baptist University (FRG2-16-17-024, FRG2-17-18-068) and the Inter-Institutional Collaborative Research Scheme (RC-ICRS/15-16/02E, RC-ICRS/1617/02C-CHE).References
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding and R. Xia, Science, 2018, 361, 1094–1098 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- H. Yin, J. Yan, J. K. W. Ho, D. Liu, P. Bi, C. H. Y. Ho, X. Hao, J. Hou, G. Li and S. K. So, Nano Energy, 2019, 64, 103950 CrossRef CAS.
- B. Kan, H. Feng, H. Yao, M. Chang, X. Wan, C. Li, J. Hou and Y. Chen, Sci. China: Chem., 2018, 61, 1307–1313 CrossRef CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef.
- B. C. Thompson and J. M. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef.
- G. Zhang, G. Yang, H. Yan, J. H. Kim, H. Ade, W. Wu, X. Xu, Y. Duan and Q. Peng, Adv. Mater., 2017, 29, 1606054 CrossRef.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng and P. A. Johnson, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119 CrossRef CAS.
- Y. Li, L. Zhong, B. Gautam, H.-J. Bin, J.-D. Lin, F.-P. Wu, Z. Zhang, Z.-Q. Jiang, Z.-G. Zhang and K. Gundogdu, Energy Environ. Sci., 2017, 10, 1610–1620 RSC.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- H. Fu, Z. Wang and Y. Sun, Angew. Chem., Int. Ed., 2019, 58(14), 4442–4453 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- Y. Li, M. Gu, Z. Pan, B. Zhang, X. Yang, J. Gu and Y. Chen, J. Mater. Chem. A, 2017, 5, 10798–10814 RSC.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 RSC.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS.
- Y. Li, N. Zheng, L. Yu, S. Wen, C. Gao, M. Sun and R. Yang, Adv. Mater., 2019, 31, 1807832 CrossRef.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS.
- S. Dai and X. Zhan, Adv. Energy Mater., 2018, 8, 1800002 CrossRef.
- X. Chen, Y. Sun, Z. Wang, H. Gao, Z. Lin, X. Ke, T. He, S. Yin, Y. Chen, Q. Zhang and H. Qiu, Dyes Pigm., 2018, 158, 445–450 CrossRef CAS.
- T. Duan, H. Tang, R.-Z. Liang, J. Lv, Z. Kan, R. Singh, M. Kumar, Z. Xiao, S. Lu and F. Laquai, J. Mater. Chem. A, 2019, 7, 2541–2546 RSC.
- W. Lee, J. Choi and J. W. Jung, Dyes Pigm., 2019, 161, 283–287 CrossRef CAS.
- M. Privado, P. de la Cruz, S. Biswas, R. Singhal, G. D. Sharma and F. Langa, J. Mater. Chem. A, 2018, 6, 11714–11724 RSC.
- K. Gao, S. B. Jo, X. Shi, L. Nian, M. Zhang, Y. Kan, F. Lin, B. Kan, B. Xu, Q. Rong, L. Shui, F. Liu, X. Peng, G. Zhou, Y. Cao and A. K.-Y. Jen, Adv. Mater., 2019, 31, 1807842 CrossRef.
- J. Qin, C. An, J. Zhang, K. Ma, Y. Yang, T. Zhang, S. Li, K. Xian, Y. Cui, Y. Tang, W. Ma, H. Yao, S. Zhang, B. Xu, C. He and J. Hou, Sci. China Mater., 2020, 1–9 Search PubMed.
- Q. Wu, D. Deng, R. Zhou, J. Zhang, W. Zou, L. Liu, S. Wu, K. Lu and Z. Wei, ACS Appl. Mater. Interfaces, 2020, 12(22), 25100–25107 CrossRef CAS.
- H. Chen, C. Yan, J. Huang, P. W. Fong, J. Lv, D. Hu, R. Singh, M. Kumar, Z. Xiao, Z. Kan, S. Lu and G. Li, Adv. Energy Mater., 2020, 2001076 Search PubMed.
- Y. Wang, Y. Wang, L. Zhu, H. Liu, J. Fang, X. Guo, F. Liu, Z. Tang, M. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 1309–1317 RSC.
- C. Liu, N. Qiu, Y. Sun, X. Ke, H. Zhang, C. Li, X. Wan and Y. Chen, Front. Chem., 2020, 8, 329 CrossRef CAS.
- R. Zhou, Z. Jiang, C. Yang, J. Yu, J. Feng, M. A. Adil, D. Deng, W. Zou, J. Zhang, K. Lu, W. Ma, F. Gao and Z. Wei, Nat. Commun., 2019, 10, 1–9 CrossRef CAS.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- J. Gao, R. Peng, C. Liu, L. Cao, D. Zhang, F. Billy, L. Hong, E. Zhou and Z. Ge, J. Mater. Chem. A, 2020, 8, 7405–7411 RSC.
- T. Xu, Y. Chang, C. Yan, Q. Yang, Z. Kan, R. Singh, M. Kumar, G. Li, S. Lu and T. Duan, Sustainable Energy Fuels, 2020, 4, 1789–1800 RSC.
- H. Li, Q. Wu, R. Zhou, Y. Shi, C. Yang, Y. Zhang, J. Zhang, W. Zou, D. Deng, K. Lu and Z. Wei, Adv. Energy Mater., 2019, 9, 1803175 CrossRef.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 CrossRef.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A, 2018, 6, 16769–16797 RSC.
- V. Cuesta, R. Singhal, P. de la Cruz, G. D. Sharma and F. Langa, ACS Appl. Mater. Interfaces, 2019, 11(7), 7216–7225 CrossRef CAS.
- S. Chen, L. Xiao, X. Zhu, X. Peng, W.-K. Wong and W.-Y. Wong, Chem. Commun., 2015, 51, 14439–14442 RSC.
- S. Chen, L. Yan, L. Xiao, K. Gao, W. Tang, C. Wang, C. Zhu, X. Wang, F. Liu and X. Peng, J. Mater. Chem. A, 2017, 5, 25460–25468 RSC.
- V. Piradi, X. Xu, Z. Wang, J. Ali, Q. Peng, F. Liu and X. Zhu, ACS Appl. Mater. Interfaces, 2019, 11(6), 6283–6291 CrossRef CAS.
- H. Wang, L. Xiao, L. Yan, S. Chen, X. Zhu, X. Peng, X. Wang, W.-K. Wong and W.-Y. Wong, Chem. Sci., 2016, 7, 4301–4307 RSC.
- V. Cuesta, M. Vartanian, P. de la Cruz, R. Singhal, G. D. Sharma and F. Langa, J. Mater. Chem. A, 2017, 5, 1057–1065 RSC.
- C. V. Kumar, L. Cabau, E. N. Koukaras, A. Sharma, G. D. Sharma and E. Palomares, J. Mater. Chem. A, 2015, 3, 16287–16301 RSC.
- M. Vartanian, P. de la Cruz, S. Biswas, G. D. Sharma and F. Langa, Nanoscale, 2018, 10, 12100–12108 RSC.
- T. Liang, L. Xiao, K. Gao, W. Xu, X. Peng and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 7131–7138 CrossRef CAS.
- M. Vartanian, R. Singhal, P. de la Cruz, G. D. Sharma and F. Langa, Chem. Commun., 2018, 54, 14144–14147 RSC.
- X. Zhou, W. Tang, P. Bi, L. Yan, X. Wang, W.-K. Wong, X. Hao, B. S. Ong and X. Zhu, J. Mater. Chem. A, 2018, 6, 14675–14680 RSC.
- X. Zhou, W. Tang, P. Bi, Z. Liu, W. Lu, X. Wang, X. Hao, W.-K. Wong and X. Zhu, J. Mater. Chem. C, 2019, 7, 380–386 RSC.
- S. Chen, W. Tang, H. Yin, Z. Wang, K. Zheng, L. Xie, X. Wang, S. K. So, F. Liu and X. Zhu, Org. Electron., 2019, 73, 146–151 CrossRef CAS.
- T. Lai, L. Xiao, K. Deng, T. Liang, X. Chen, X. Peng and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 10, 668–675 CrossRef.
- H. Yin, J. K. W. Ho, V. Piradi, S. Chen, X. Zhu and S. K. So, Small Methods, 2020, 2000136 CrossRef.
- M. Ren, G. Zhang, Z. Chen, J. Xiao, X. Jiao, Y. Zou,, H.-L. Yip and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12(11), 13077–13086 CrossRef.
- M. Vartanian, P. de la Cruz, S. Biswas, G. D. Sharma and F. Langa, Nanoscale, 2018, 10, 12100–12108 RSC.
- L. Nian, K. Gao, F. Liu, Y. Kan, X. Jiang, L. Liu, Z. Xie, X. Peng, T. P. Russell and Y. Ma, Adv. Mater., 2016, 28, 8184–8190 CrossRef CAS.
- W. T. Hadmojo, D. Yim, S. Sinaga, W. Lee, D. Y. Ryu, W.-D. Jang, I. H. Jung and S.-Y. Jang, ACS Sustainable Chem. Eng., 2018, 6(4), 5306–5313 CrossRef CAS.
- H.-H. Gao, Y. Sun, S. Li, X. Ke, Y. Cai, X. Wan, H. Zhang, C. Li and Y. Chen, Dyes Pigm., 2020, 176, 108250 CrossRef CAS.
- M. Turbiez, P. Frere, M. Allain, C. Videlot, J. Ackermann and J. Roncali, Chem. – Eur. J., 2005, 11, 3742–3752 CrossRef CAS.
- V. Piradi, G. Zhang, T. Li, M. Zhang, Q. Peng, X. Zhan and X. Zhu, ACS Appl. Mater. Interfaces, 2020, 12(37), 41506–41514 CrossRef.
- A. K. K. Kyaw, D. H. Wang, D. Wynands, J. Zhang, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Nano Lett., 2013, 13, 3796–3801 CrossRef CAS.
- V. Mihailetchi, L. Koster, J. Hummelen and P. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS.
- L. Feng, J. Yuan, Z. Zhang, H. Peng, Z.-G. Zhang, S. Xu, Y. Liu, Y. Li and Y. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 31985–31992 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and characterization methods, device fabrication, morphology studies, synthesis, cyclic voltammograms, 1H and 13C NMR spectra and MALDI-TOF spectra. See DOI: 10.1039/d0ma00487a |
| This journal is © The Royal Society of Chemistry 2020 |