
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D carbon-coated stannous sulfide-molybdenum disulfide anodes for advanced lithium-ion batteries†
Yuanyi
Luo‡
,
Chunxia
Chen‡
,
Weicheng
Sun
,
Ludi
Shi
 ,
Yemao
Lin
,
Guangtao
Cong
*,
Jiali
Yu
,
Caizhen
Zhu
* and
Jian
Xu
,
Yemao
Lin
,
Guangtao
Cong
*,
Jiali
Yu
,
Caizhen
Zhu
* and
Jian
Xu
Institute of Low-Dimensional Materials Genome Initiative, College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, P. R. China. E-mail: gtcong@szu.edu.cn; czzhu@szu.edu.cn
First published on 20th August 2020
Abstract
Metal sulfides have been extensively studied as alternative anodes for lithium-ion batteries (LIBs) due to their high specific capacity, which comes from their combined conversion and alloying/de-alloying reactions with lithium, forming Li2S and corresponding lithium alloys, respectively. However, the large volume fluctuation during cycling, low electronic/ionic conductivities of both metal sulfides and Li2S, and the electrochemical inactivity of Li2S lead to poor reversibility and stability. Here, we report a facile way to synthesize a three-dimensional carbon-coated stannous sulfide-molybdenum disulfide composite (3D SnS–MoS2@C) which is proposed to alleviate the aforementioned problems. 3D SnS–MoS2@C exhibits a highly porous structure, which enhances the contact between the electrode and the electrolyte, promotes the transport of both electrons and Li+, and buffers the volume change during cycling. MoS2 contributes to the specific capacity and catalyzes the oxidation of Li2S. The 3D SnS–MoS2@C anode demonstrates a capacity of 887.1 mA h g−1 for the first cycle at 200 mA g−1 and achieves a capacity retention of 97.1% over 200 cycles. In addition, the ternary composite exhibits a superior rate performance and cycling lifespan, achieving a gravimetric capacity of 651.2 mA h g−1 at 1000 mA g−1 after 500 cycles. The proposed facile and effective strategy could be universally applied to other metal sulfide-based electrodes.
Introduction
LIBs have dominated the area of electrochemical energy storage for the past two decades, owing to their high energy/power density, long lifespan, and low-maintenance.1–3 Although many battery systems have been indicated to be capable of delivering extremely high theoretical energy density (e.g., lithium–oxygen and lithium–sulfur), they suffer from severe parasitic reactions, leading to poor rate capability, low energy efficiency, and a short cycle life. Therefore, intensive research efforts are still devoted to the optimization of LIBs. As one of the most successful anode materials, graphitized carbon has been widely used in commercialized LIBs for its safety and reversibility; however, its application is reaching a bottleneck imposed by low specific capacity. Moreover, metallic lithium dendrites are easily formed and deposited on the graphite surface upon overcharging, which can lead to short-circuits.Alternative anodes with improved specific capacity and safety have been intensively explored.4–14 Owing to their superior redox reversibility and high specific capacity, metal sulfides (e.g., FeS, CoS, NiS, and ZnS) have been widely exploited as potential anodes for LIBs.15–24 Among them, SnS exhibits one of the highest theoretical Li+ storage capacity (1137 mA h g−1). However, there remain four key challenges to be addressed in the application of SnS anodes: (1) SnS undergoes huge volume fluctuations during cycling, leading to exfoliation and repeated formation and destruction of the solid electrolyte interphase film (SEI); (2) the inherent low electronic conductivity of SnS inevitably deteriorates the rate performance of the electrode; (3) SnS particles tend to agglomerate due to the high surface energy and the formation of large SnS particles elongates the transportation path for both electrons and Li+, which inevitably mitigates the rate performance;25–27 and (4) as one of the lithiation products of SnS, Li2S exhibits slow oxidation kinetics during de-lithiation.28–30 Many strategies have been proposed to address these challenges, including tuning the morphology and the structure,27 expanding the interlayer distance,31 and impregnating SnS into a conductive carbonaceous matrix.32 However, the poor conductivity of the SnS layer coated on the carbon support greatly mitigates its high rate performance.33,34 In addition, the bulk SnS nanosheets grown on the carbon backbone are still very thick, which acts as the main barrier to the fast transfer of both electrons and Li+.
To overcome the aforementioned issues of SnS anodes, we propose a facile method of intergrowth of a 3D SnS–MoS2@C nanostructure. The designed porous composite material demonstrates excellent electrochemical performance, benefitting from the following three advantages: (1) the unique 3D interconnected porous structure exhibits an enhanced specific surface area which will promote the transport of both electrons and Li+. The porous structure of SnS provides ample space to accommodate the volume fluctuation during cycling; (2) both SnS and MoS2 are covered by a porous carbon layer, which will mitigate the agglomeration of both the SnS and MoS2 particles; (3) the addition of MoS2 contributes to the specific capacity and the existence of the Mo intermediate not only significantly improves the oxidation kinetics of Li2S by reducing the activation potential of Li2S,35–37 but also acts as a host to the polysulfide intermediates, which inhibits the notorious polysulfide shuttling and improves the cycling stability.28–30,38–42 The proposed 3D SnS–MoS2@C porous composite demonstrates improved reversibility, capability, and cycle life over its bulky SnS@C and 3D SnS@C counterparts, representing a facile and effective solution to boost the performance of the SnS anodes and it could potentially be applied to the design of other metal sulfide-based electrodes.
Materials and methods
Preparation of SnS-based composites
Material characterization
Surface area and pore size distribution were investigated using a BELSORP-max instrument at 77 K based on the Brunauer–Emmett–Teller (BET) models. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was performed on an OPTIMA2100DV spectrometer. Field emission scanning electron microscopy (FESEM) was performed on a JSM7800F spectrometer and transmission electron microscopy (TEM) was conducted on a JEM2100 spectrometer. Elemental distribution and composition were analyzed with energy-dispersive X-ray spectroscopy (EDS). X-ray powder diffraction (XRD) measurements were conducted using MiniFlex600 (Cu-Kα) at 40 kV and 15 mA. Raman spectroscopy (Raman) was performed at a light wavelength of 532 nm and a test range of 100–8000 cm−1. X-ray photoelectron spectroscopy (XPS) data were collected on a K-Alpha+ electronic spectrometer (Thermo Scientific).Electrochemical measurement
Lithium metal half-cells were assembled using the standard 2032 lithium-ion button cells in an argon-filled glove box. The synthesized composites (70 wt%) were mixed with acetylene black (AB, 20 wt%) and sodium alginate (SA, 10 wt%), forming a uniform slurry, which was evenly coated onto Cu foil. To study the influence of the active material content on the performance of the 3D SnS–MoS2@C electrodes, electrodes with higher active material content (90 wt% 3D SnS–MoS2@C, 5 wt% AB, and 5 wt% SA) were prepared and tested. The prepared electrodes were then dried and cut out for further use. The loading of the active material on each electrode was controlled to be ∼1.5 mg cm−2. Lithium foil, 1.0 M LiPF6 in DMC/EC (v/v = 1/1), and polypropylene (PP) were used as the anode, the electrolyte, and the separator, respectively. Charge/discharge cycling tests were conducted on a LAND CT2001A battery tester from 0.01 to 3.0 V. Cyclic voltammetry (CV) was conducted on a CHI660E workstation within the same voltage range.Results and discussion
Structure and morphology of 3D SnS–MoS2@C
The synthesis of the porous 3D SnS–MoS2@C composite involved a facile and mild strategy (Fig. 1). Sodium chloride (NaCl) was employed as the template to generate the porous structure, and stannous chloride dihydrate (SnCl2·2H2O), ammonium tetrathiomolybdate ((NH4)2MoS4), thiourea, and citric acid were used as the sources of tin, molybdenum, sulfur, and carbon, respectively. A solution mixture of the precursors was first freeze-dried and then treated at a high temperature. 3D SnS–MoS2@C was generated after removing the NaCl template of the carbonation product with deionized water.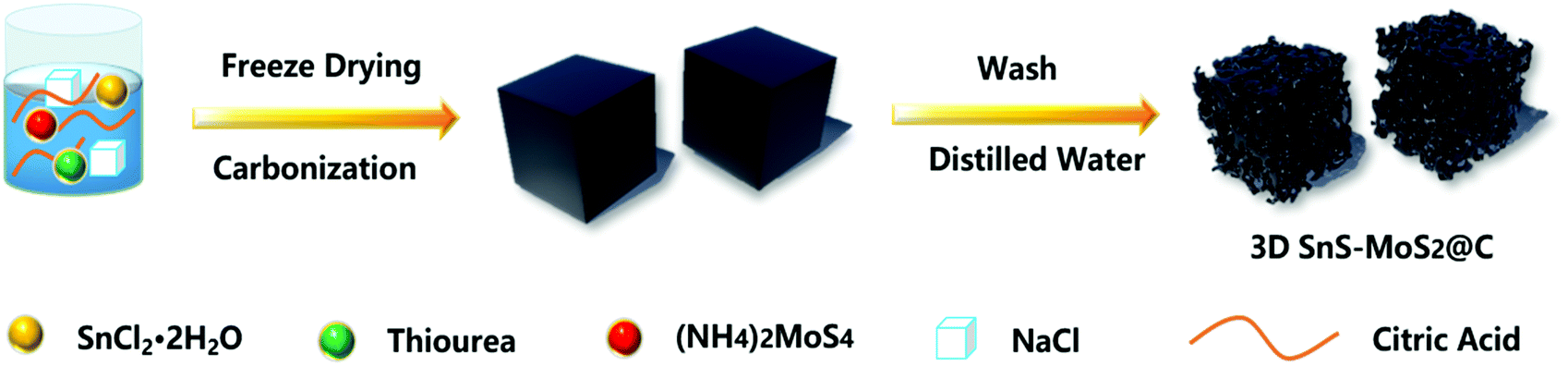 | ||
| Fig. 1 Synthetic procedure for 3D SnS–MoS2@C. | ||
First, we examined the morphology of 3D SnS–MoS2@C and SnS@C with FESEM (Fig. 2a, b and d). 3D SnS–MoS2@C has a large number of porous structures, while SnS@C has no obvious pore structure. The overall morphology of the 3D SnS–MoS2@C composite resembles that of white fungus (Fig. 2c). There are a large number of continuous three-dimensional carbon networks with a pore size distribution between 100 nm and 1 μm. The morphology of 3D SnS shares the same structure with 3D SnS–MoS2@C (Fig. S1, ESI†), which suggests the universality of the proposed strategy to prepare a highly porous structure. The specific surface areas of 3D SnS–MoS2@C, 3D SnS@C and SnS@C were found to be 196.4, 155.5, and 11.0 m2 g−1, respectively (Fig. S2, ESI†), which agrees well with the FESEM results. The enlarged surface area of the 3D SnS–MoS2@C and 3D SnS@C would enhance the contact between the electrode/electrolyte, leading to a reduced transportation path for both electrons and Li+. EDS was conducted to further study the elemental composition of 3D SnS–MoS2@C, which reveals the existence and even the distribution of C (red), N (green), Mo (purple), S (orange), and Sn (blue) over the composite (Fig. 2e–j). The contents of MoS2 and SnS in 3D SnS–MoS2@C were determined to be around 18.8 wt% and 36.5 wt%, respectively, on the basis of the ICP-AES analysis, which gives a SnS to MoS2 mole ratio of 2.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, in agreement with the mole ratio of their corresponding precursors and also agrees well with the EDS results (Fig. S3, ESI†).
:1, in agreement with the mole ratio of their corresponding precursors and also agrees well with the EDS results (Fig. S3, ESI†).
 | ||
| Fig. 2 (a) and (b) FESEM images of the as-prepared 3D SnS–MoS2@C. (c) Digital image of white fungus. (d) FESEM images of SnS@C and (e)–(j) EDS mapping (C, N, Mo, S, and Sn) images of the 3D SnS–MoS2. | ||
The crystal structures of the 3D SnS–MoS2@C, 3D SnS@C, and SnS@C were studied using XRD (Fig. 3a). All the three samples demonstrated the high crystallinity of SnS and the diffraction peaks at 22.01°, 26.01°, 27.47°, 31.53°, 31.97°, 39.05°, 45.50°, 48.51°, and 53.15° were associated with the (110), (120), (021), (111), (040), (131), (002), (211) and (122) crystal faces of SnS (PDF #39-0354), respectively.27,44 Compared to the XRD spectra of SnS@C and 3D SnS@C, an additional diffraction peak at around 14.2° was observed for 3D SnS–MoS2@C, which could be assigned to the (002) facet of MoS2.34,45 The Raman spectra of the three SnS-based composites reveal well-defined D and G bands which locate at 1357 and 1586 cm−1, respectively (Fig. 3b). The D band is assigned to the vibration of the defective carbon atoms, while the G band could be ascribed to the vibration of the sp2-hybridized carbon atoms.46 The intensity ratio (ID/IG) could be calculated to estimate the defects and the disorder degree of the carbon material. The ID/IG values of the SnS@C, 3D SnS@C, and 3D SnS–MoS2@C were found to be 0.98, 0.98, and 0.96, respectively, which indicates the abundancy of carbon defects in all the three samples. In addition, the additional peaks of 3D SnS–MoS2@C near 406 and 382 cm−1 are associated with the A1g and E2g1 vibrations of MoS2, respectively,46 which agrees well with the XRD results.
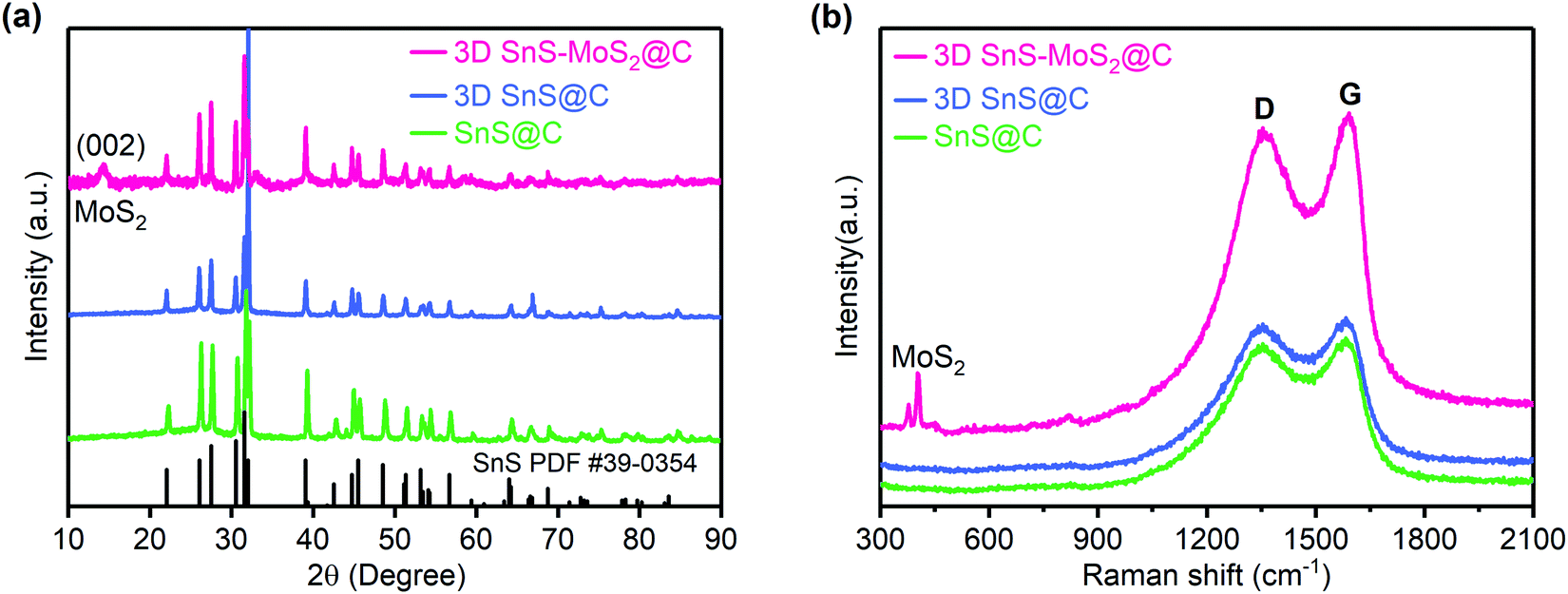 | ||
| Fig. 3 XRD (a) and Raman (b) spectra of SnS@C, 3D SnS@C, and 3D SnS–MoS2@C. The powder diffraction file (PDF) standards for SnS is included for reference. | ||
TEM was employed to further examine the structure of the as-prepared 3D SnS–MoS2@C. The ternary composite shows a continuous porous structure with SnS and MoS2 particles embedded in the carbon skeleton (Fig. 4a), which could effectively mitigate the agglomeration of both the SnS and MoS2 particles. In addition, the as-prepared 3D SnS–MoS2@C composite exhibits well-defined diffraction fringes with lattice pitches of 0.28, 0.24, and 0.68 nm, which are associated with the (111) facet of SnS and the (103) and (002) facets of MoS2, respectively (Fig. 4b).47
 | ||
| Fig. 4 TEM image (a) and high-resolution TEM (b) images of 3D SnS–MoS2@C. | ||
The XPS survey spectrum of 3D SnS–MoS2@C suggests that the as-prepared composite contains primarily Sn, Mo, N, C, S, and O elements (Fig. 5a), As shown in Fig. 5b, Sn 3d3/2 and a strong peak of Sn 3d5/2 with binding energies of 487.2 and 495.8 eV appear, which could be assigned to SnS.27 The characteristic peaks of Mo4+ in MoS2 could also be observed at 229.5 and 232.8 eV (Fig. 5c). In addition, there is a weak peak of Mo6+ 3d5/2 at 236.0 eV, indicating that trace MoO3 may exist in the material.48 The N 1s spectrum in Fig. 5d suggests that the N atom mainly exists in the form of pyridine and pyrrole type N in the composite.27 There are two main reasons for the presence of N in the material: (1) during high-temperature treatments, thiourea may react with SnCl2·H2O to form ammonia and (2) the protective gas used for high-temperature carbonization is nitrogen. Therefore, under high-temperature conditions, both ammonia and nitrogen could promote the nitrogen-doping level of the composite. The doping level of nitrogen was estimated based on the EDS analysis. A nitrogen weight ratio of 3.96 wt% was determined for the 3D SnS–MoS2@C composite (Fig. S3, ESI†). Extensive theoretical and experimental studies have showed that nitrogen doping of the carbon materials would enhance their electronic conductivity by tuning the Fermi level and increasing the fraction of the localized electronic states.49–51 In addition, the doped N atoms, especially pyridinic-N, exhibit a strong affinity to sulfide species, which would reduce the shuttling of the polysulfide intermediates.52,53
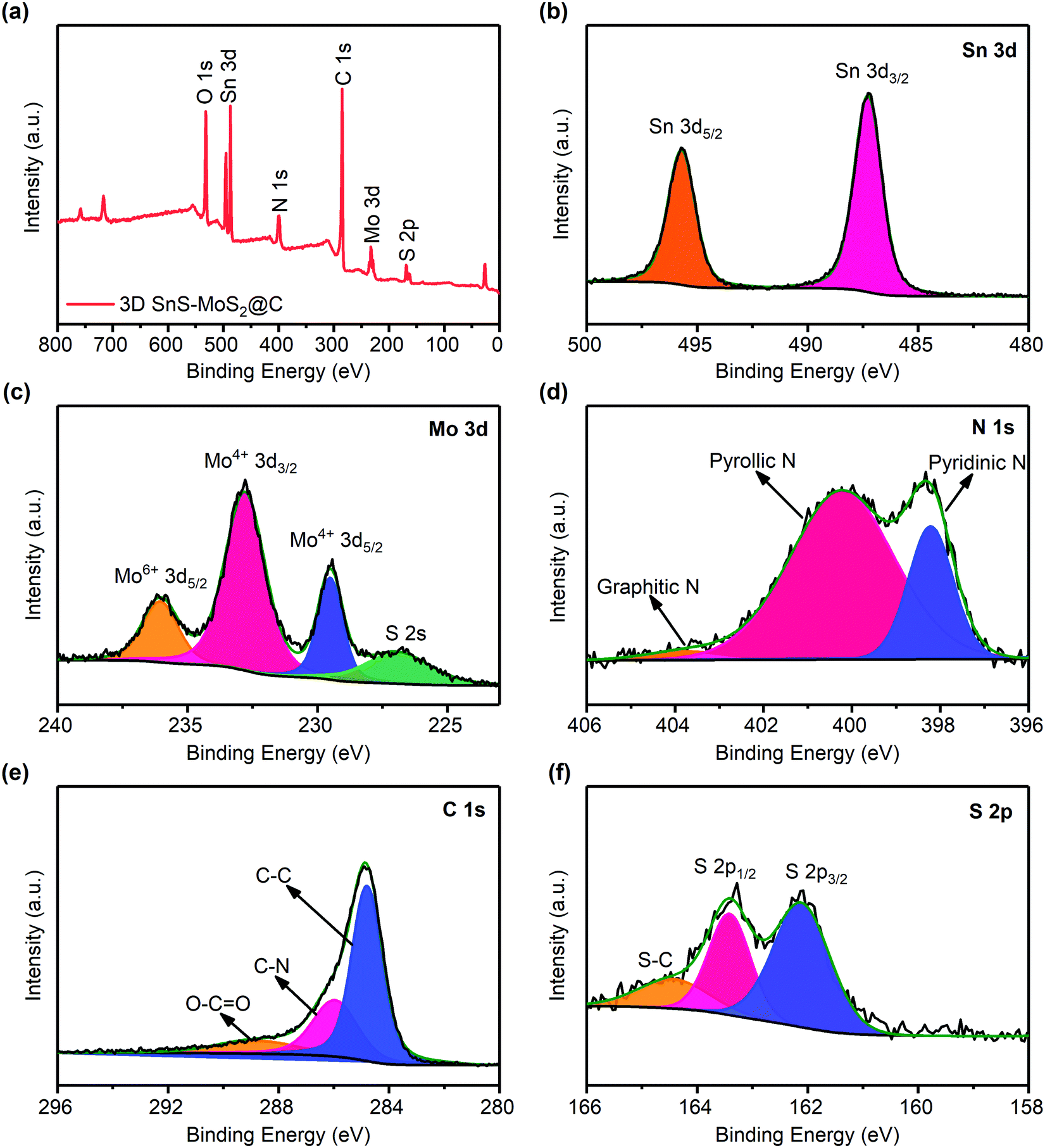 | ||
| Fig. 5 (a) XPS survey spectrum of 3D SnS–MoS2@C. XPS spectra of Sn 3d (b), Mo 3d (c), N 1s (d), C 1s (e), and S 2p (f). | ||
Fig. 5e shows the XPS spectrum of C 1s. The peaks around 284.8, 286.0, and 288.6 eV could be assigned to C–C, C–N, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. Recent studies suggest that heteroatom doping of carbon materials could effectively promote the electronic conductivity, leading to an improved rate performance.54,55 The high-resolution XPS spectrum of S 2p reveals two peaks near 162.2 and 163.4 eV, which originate from S2−, and the peak near 164.5 eV is associated with S–C (Fig. 5f).56
O, respectively. Recent studies suggest that heteroatom doping of carbon materials could effectively promote the electronic conductivity, leading to an improved rate performance.54,55 The high-resolution XPS spectrum of S 2p reveals two peaks near 162.2 and 163.4 eV, which originate from S2−, and the peak near 164.5 eV is associated with S–C (Fig. 5f).56
Electrochemical performance
CV tests were first conducted to study the electrochemical behavior of 3D SnS–MoS2@C (Fig. 6a). During the first cathodic scan, the broad peak at 0.81 V could be ascribed to the lithiation of SnS into Li2S and Sn, which is disguised by the simultaneous irreversible SEI formation. This broad peak disappeared during the subsequent scans, suggesting the formation of a stable SEI film during the first reduction. We notice that a new cathodic peak at 1.3 V evolved during the subsequent cycles, which could be assigned to the lithiation of SnS after the formation of the stable SEI.26,34 The broad peak at 0.31 V could be assigned to the reversible conversion process: MoS2 + 4Li + 4e− ↔ Mo + 2Li2S.34 The broad peak near 0.1 V could result from the reversible alloying process between Sn and Li, namely Sn + xe− + xLi+ ↔ LixSn (0 ≤ x ≤ 4.4).57 A series of oxidation peaks between 0.2 and 0.8 V could be observed, which is ascribed to the sequential de-alloying of LixSn and the conversion of Mo and Li2S to MoS2 and Li+.57 Two broad peaks around 1.8 and 1.34 V could be assigned to the conversion of SnS to Sn and Li2S.58,59 Significantly, the redox peaks of the subsequent CV curves almost coincide with each other, which suggests the superior stability and reversibility of 3D SnS–MoS2@C. Fig. 6b presents the first galvanostatic voltage profiles of the three SnS-based composites at 200 mA g−1. Two distinct plateaus could be observed during the discharge process. The first discharge plateau located between 0.8 and 1.0 V corresponds to the lithiation of SnS, forming Sn and amorphous Li2S, accompanied with the irreversible SEI formation. The second plateau between 0.2 and 0.5 V could be associated with the alloying reaction of Sn with Li. The 3D SnS–MoS2@C anode demonstrates a superior stability and reversibility over the 3D SnS@C and SnS@C anodes upon repeated charge and discharge at low rates (Fig. S4 and S5, ESI†). The first charge process delivered a gravimetric capacity of 887.1 mA h g−1 at 200 mA g−1. Remarkably, a high capacity of 861.8 mA h g−1 was maintained over 200 cycles with a high capacity retention of 97.1% and CEs close to 100%. Although 3D SnS@C shares the same porous architecture with 3D SnS–MoS2@C, its cycling stability and capacity retention are much lower than those of 3D SnS–MoS2@C. The enhanced reversibility and stability of 3D SnS–MoS2@C over 3D SnS@C originate from the reduced kinetic barrier of Li2S oxidation as well as the mitigated polysulfide shuttling as discussed earlier.35–42 To evaluate the rate performance of the three SnS-based anodes, galvanostatic cycling tests with current densities in the range from 100 to 4000 mA g−1 were performed (Fig. 6c). The performance of the 3D SnS–MoS2@C electrode is significantly improved over the SnS@C and 3D SnS@C counterparts. The achieved gravimetric capacities of all the three samples decreased with the increasing rates. When the applied current was increased to 4000 mA g−1, the gravimetric capacities of the SnS@C and 3D SnS@C electrodes were found to be 30 and 200 mA h g−1, respectively. Remarkably, the specific capacity of the 3D SnS–MoS2@C composite stabilized around 533.7 mA h g−1 at 4000 mA g−1and recovered to 880.8 mA h g−1 when the applied current was reduced to 200 mA g−1. The weight ratio of the active materials not only greatly influenced the performance of the electrodes but also directly determined their energy density. We notice that 70 wt% of 3D SnS–MoS2@C is relatively lower compared to those of commercialized LIBs. Cycling of the 3D SnS–MoS2@C electrode with 90 wt% active materials was performed (Fig. S6, ESI†). Both electrodes shared similar high CEs close to 100% while the electrodes with 90 wt% active material suffered from more severe capacity fading than the ones with lower active material content. Further optimization of the electrode structural design is needed to improve the performance of the 3D SnS–MoS2@C electrodes. The outstanding rate performance of the 3D SnS–MoS2@C composites could be ascribed to the following two aspects: (1) the pore size lies between 100 nm and 1 μm which greatly reduces the diffusion path of Li+ and a large number of microspores enlarge the electrode/electrolyte contact area, which is beneficial for the rate performance of the anode26,60 and (2) the catalytic effect of Mo on the oxidation of Li2S as well as its ability to regulate the conversion of polysulfide during de-lithiation.35–42 The rate capability of 3D SnS–MoS2@C was compared with that of the other two SnS-based composites at 1000 mA g−1 (Fig. 6d). During the first 50 cycles, the gravimetric capacities of both 3D SnS@C and SnS@C were sharply decreased to less than 200 mA h g−1, whereas 3D SnS–MoS2@C demonstrated stable cycling of more than 500 cycles with a gravimetric capacity of 651.2 mA h g−1 and an average CE of 99.75%. The difference in the rate performance between 3D SnS–MoS2@C and SnS@C was investigated by monitoring their morphology evolution during cycling. The 3D porous structure remained well-preserved while the structures of 3D SnS@C and SnS@C were shattered after 50 cycles (Fig. S7, ESI†). Galvanostatic cycling tests of the full cell configuration with the 3D SnS–MoS2@C anode and the commercial LiFePO4 cathode were conducted at 100 mA g−1 (Fig. S8, ESI†). A gravimetric capacity of 485 mA h g−1 could be achieved with an average CE of 97.5% after 100 cycles. We notice that the initial CE is as low as 60%, which could be associated with the irreversible SEI formation on the 3D SnS–MoS2@C anode during the first charging process. The practical application of the full cell was also evaluated by powering programmed light-emitting diode (LED) fans. Three 3D SnS–MoS2@C/LiFePO4 full cells connected in series could power the LED fans for several minutes (Fig. S9 and Video, ESI†).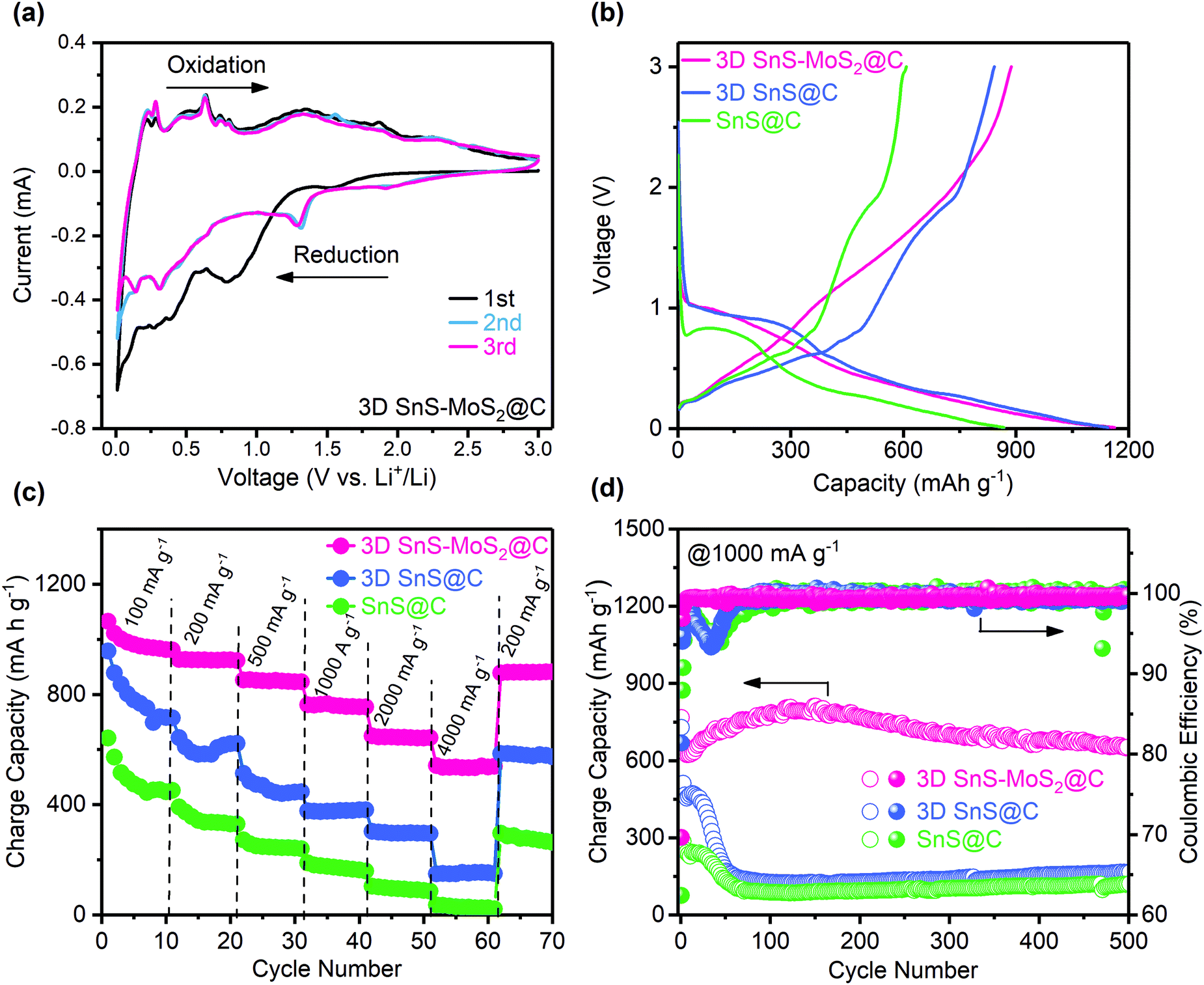 | ||
| Fig. 6 Electrochemical characterization of the SnS-based electrodes. (a) Cyclic voltammetry (CV) of 3D SnS–MoS2@C at 0.1 mV s−1. (b) Voltage versus capacity curves of the three SnS-based electrodes at 200 mA g−1. Rate capability (c) and long-term cycling performance (d) of the three SnS-based electrodes. | ||
Electrochemical impedance spectroscopy was conducted to probe the resistance evolution of the three SnS-based anodes before and after 50 cycles (Fig. S10, ESI†). The total resistance of the 3D SnS@C and SnS@C electrodes gradually increases during cycling which might be related to their structure instability during repeated lilthiation and de-lithiation. In contrast, the total resistance of the 3D SnS–MoS2@C electrode decreased after cycling which could be ascribed to the improved wettability between the electrode and the electrolyte. The high-frequency semicircle could be ascribed to the charge transfer, and the low-frequency slope could be assigned to the Li+ diffusion behavior. As can be seen from the fitting results (Table S1, ESI†), the total resistance consists of three components: charge transfer resistance Rct, film resistance Rf, and the Warburg diffusion resistance W. Among all the three anodes, SnS@C exhibits the largest film resistance, which reflects the formation of a thick and unstable SEI film between the material and the electrolyte. The 3D SnS–MoS2@C composite has the smallest Rf, suggesting the formation of a stable SEI over a unique three-dimensional porous structure. Both the Warburg impedance and charge transfer resistance of 3D SnS–MoS2@C are smaller than those of the other samples, which agrees well with the rate performance results (Fig. 6c)
To differentiate the surface capacitance from the electrochemical reactions, CV tests of the 3D SnS–MoS2@C electrode with the scan rates varying from 0.1 to 2.0 mV s−1 were conducted (Fig. 7a). The current response at a specific voltage can be divided into two parts: (1) the current contribution of the surface capacitance and (2) the diffusion-controlled current contribution. The corresponding relationship is described as follows:61
| i(v) = k1v + k2v1/2 = avb | (1) |
| logi(v) = loga + blogv | (2) |
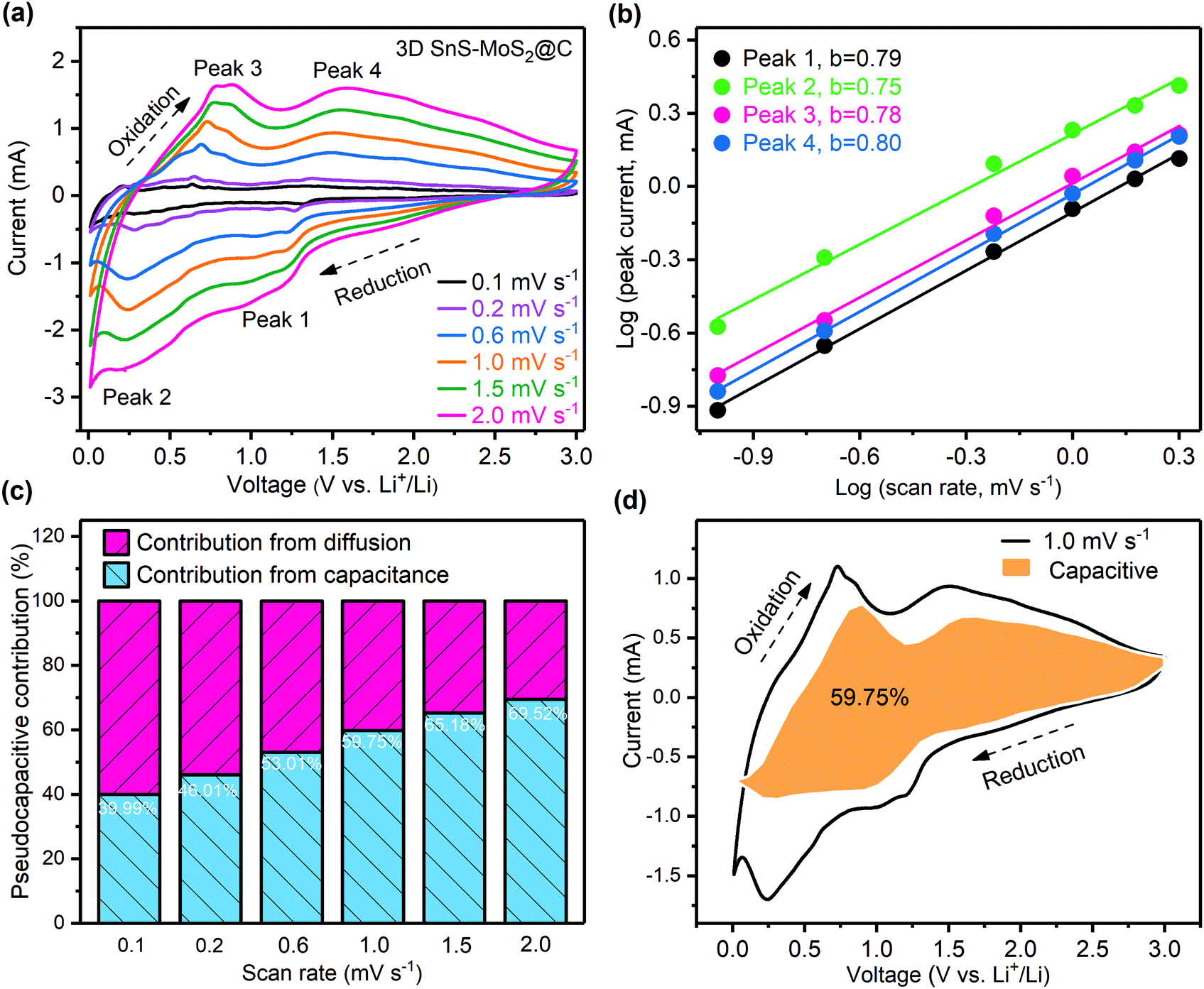 | ||
| Fig. 7 (a) CV scans of the 3D SnS–MoS2@C anode at varying scan rates from 0.1 to 2.0 mV s−1. (b) Linear correlation between the logarithm of the peak currents and the logarithm of the scan rates. (c) Contribution of the pseudo-capacitive process at different scan rates depicted by bar charts. (d) Current contribution of the pseudo-capacitive process (orange area) during the CV tests at 1.0 mV s−1. | ||
Conclusions
In summary, a rationally designed highly porous 3D SnS–MoS2@C composite was successfully synthesized using a facile and mild procedure. The 3D SnS–MoS2@C composite achieves a superior capacity of 861.8 mA h g−1 after 200 cycles at 200 mA g−1. In addition, the as-prepared ternary composite demonstrates an outstanding rate performance and a long lifespan as well. A remarkable capacity of 651.2 mA h g−1 was maintained after 500 cycles at 1000 mA g−1. This facile and effective method of preparing highly porous electrodes could potentially be applied to other metal sulfide-based electrodes.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21805193 and 51673117), the Science and Technology Innovation Commission of Shenzhen (JCYJ20180305125319991, JCYJ20170818093832350, JCYJ20170818100112531, JSGG20170824112840518, JCYJ20180507184711069, JCYJ20170818101016362, and JCYJ20170817094628397), the Key R&D Programme of Guangdong Province (2019B010941001 and 2019B010929002), and the China Postdoctoral Science Foundation (2017M622786, 2019M650212, and 2017M622787).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 Search PubMed.
- C. Kim, G. Song, L. Luo, J. Y. Cheong, S.-H. Cho, D. Kwon, S. Choi, J.-W. Jung, C.-M. Wang, I.-D. Kim and S. Park, ACS Nano, 2018, 12, 8169–8176 CrossRef.
- M.-S. Balogun, M. Yu, Y. Huang, C. Li, P. Fang, Y. Liu, X. Lu and Y. Tong, Nano Energy, 2015, 11, 348–355 CrossRef.
- W. Li, X. Li, J. Liao, B. Zhao, L. Zhang, L. Huang, G. Liu, Z. Guo and M. Liu, Energy Environ. Sci., 2019, 12, 2286–2297 RSC.
- W. Li, J. Liao, X. Li, L. Zhang, B. Zhao, Y. Chen, Y. Zhou, Z. Guo and M. Liu, Adv. Funct. Mater., 2019, 29, 1903638 CrossRef.
- W. Li, J. Yu, J. Wen, J. Liao, Z. Ye, B. Zhao, X. Li, H. Zhang, M. Liu and Z. Guo, J. Mater. Chem. A, 2019, 7, 16785–16792 RSC.
- X. Xia, S. Deng, S. Feng, J. Wu and J. Tu, J. Mater. Chem. A, 2017, 5, 21134–21139 Search PubMed.
- H. Liang, J. Ni and L. Li, Nano Energy, 2017, 33, 213–220 CrossRef.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- C. Wang, L. Wu, H. Wang, W. Zuo, Y. Li and J. Liu, Adv. Funct. Mater., 2015, 25, 3524–3533 Search PubMed.
- Y. Xu, S. Zheng, H. Tang, X. Guo, H. Xue and H. Pang, Energy Storage Mater., 2017, 9, 11–30 CrossRef.
- J.-M. Liang, L.-J. Zhang, D.-G. XiLi and J. Kang, Rare Met., 2020, 1–14 Search PubMed.
- Y.-F. Huo, N. Qin, C.-Z. Liao, H.-F. Feng, Y.-Y. Gu and H. Cheng, J. Cent. South Univ., 2019, 26, 1416–1425 CrossRef.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- L. Zhao, X. Yu, J. Yu, Y. Zhou, S. N. Ehrlich, Y.-S. Hu, D. Su, H. Li, X.-Q. Yang and L. Chen, Adv. Funct. Mater., 2014, 24, 5557–5566 CrossRef.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 RSC.
- W. Qiu, J. Xia, H. Zhong, S. He, S. Lai and L. Chen, Electrochim. Acta, 2014, 137, 197–205 CrossRef.
- J. Xiao, X. Wang, X.-Q. Yang, S. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840–2846 CrossRef.
- J. Wang, H. He, Z. Wu, J. Liang, L. Han, H. L. Xin, X. Guo, Y. Zhu and D. Wang, J. Power Sources, 2018, 392, 193–199 Search PubMed.
- L. Ma, W. Zhang, L. Wang, Y. Hu, G. Zhu, Y. Wang, R. Chen, T. Chen, Z. Tie, J. Liu and Z. Jin, ACS Nano, 2018, 12, 4868–4876 CrossRef.
- F. Jing, Q. Lv, J. Xiao, Q. Wang and S. Wang, J. Mater. Chem. A, 2018, 6, 14207–14214 Search PubMed.
- K. Yang, Q. Guo, H. Li, X. Hao, Y. Ma, M. Yang, T. Zhai, S. V. Savilov, V. V. Lunin and H. Xia, J. Power Sources, 2018, 402, 340–344 CrossRef.
- P. He, Y. Fang, X.-Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 12202–12205 CrossRef CAS.
- P. Xue, N. Wang, Y. Wang, Y. Zhang, Y. Liu, B. Tang, Z. Bai and S. Dou, Carbon, 2018, 134, 222–231 CrossRef.
- X. Hu, J. Chen, G. Zeng, J. Jia, P. Cai, G. Chai and Z. Wen, J. Mater. Chem. A, 2017, 5, 23460–23470 RSC.
- Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486–1491 CrossRef.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef.
- L. Wang, Y. Wang and Y. Xia, Energy Environ. Sci., 2015, 8, 1551–1558 RSC.
- D. Guan, J. Li, X. Gao and C. Yuan, RSC Adv., 2015, 5, 58514–58521 RSC.
- S. Chen, K. Xing, J. Wen, M. Wen, Q. Wu and Y. Cui, J. Mater. Chem. A, 2018, 6, 7631–7638 RSC.
- S. H. Choi and Y. C. Kang, Nano Res., 2015, 8, 1595–1603 CrossRef CAS.
- Q. Pan, F. Zheng, X. Ou, C. Yang, X. Xiong and M. Liu, Chem. Eng. J., 2017, 316, 393–400 CAS.
- Z. Xing, G. Tan, Y. Yuan, B. Wang, L. Ma, J. Xie, Z. Li, T. Wu, Y. Ren, R. Shahbazian-Yassar, J. Lu, X. Ji and Z. Chen, Adv. Mater., 2020, 32, e2002403 Search PubMed.
- Y.-Z. Wang, X.-Y. Shan, D.-W. Wang, Z.-H. Sun, H.-M. Cheng and F. Li, Joule, 2018, 2, 1278–1286 CAS.
- X. Li, J. Zai, S. Xiang, Y. Liu, X. He, Z. Xu, K. Wang, Z. Ma and X. Qian, Adv. Energy Mater., 2016, 6, 1601056 CrossRef.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 CAS.
- Y. Wei, Z. Kong, Y. Pan, Y. Cao, D. Long, J. Wang, W. Qiao and L. Ling, J. Mater. Chem. A, 2018, 6, 5899–5909 CAS.
- P. T. Dirlam, J. Park, A. G. Simmonds, K. Domanik, C. B. Arrington, J. L. Schaefer, V. P. Oleshko, T. S. Kleine, K. Char, R. S. Glass, C. L. Soles, C. Kim, N. Pinna, Y. E. Sung and J. Pyun, ACS Appl. Mater. Interfaces, 2016, 8, 13437–13448 CAS.
- M. D. Walle, K. Zeng, M. Zhang, Y. Li and Y.-N. Liu, Appl. Surf. Sci., 2019, 473, 540–547 CrossRef CAS.
- P. Han, S. H. Chung and A. Manthiram, ACS Appl. Mater. Interfaces, 2018, 10, 23122–23130 CAS.
- L. Sygellou, Appl. Surf. Sci., 2019, 476, 1079–1085 Search PubMed.
- X. Li, R. Liu, C. Xu, Y. Bai, X. Zhou, Y. Wang and G. Yuan, Adv. Funct. Mater., 2018, 28, 1800064 Search PubMed.
- T. Zhang, L.-B. Kong, M.-C. Liu, Y.-H. Dai, K. Yan, B. Hu, Y.-C. Luo and L. Kang, Mater. Des., 2016, 112, 88–96 CAS.
- L. Kumari, Y.-R. Ma, C.-C. Tsai, Y.-W. Lin, S. Y. Wu, K.-W. Cheng and Y. Liou, Nanotechnology, 2007, 18, 115717 CrossRef.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Adv. Energy Mater., 2012, 2, 970–975 CAS.
- Z. Deng, H. Jiang, Y. Hu, Y. Liu, L. Zhang, H. Liu and C. Li, Adv. Mater., 2017, 29, 1603020 Search PubMed.
- A. Yanilmaz, A. Tomak, B. Akbali, C. Bacaksiz, E. Ozceri, O. Ari, R. T. Senger, Y. Selamet and H. M. Zareie, RSC Adv., 2017, 7, 28383–28392 CAS.
- Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S. Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B. Anikeeva, T. I. Buryakov and E. N. Tkachev, Carbon, 2009, 47, 1922–1929 CrossRef CAS.
- Y. Qiu, X. Zhang and S. Yang, Phys. Chem. Chem. Phys., 2011, 13, 12554–12558 RSC.
- Y. Zhao, Z. Wang, X. Zhao, X. Wang, K.-H. Lam, F. Chen, L. Zhao, S. Wang and X. Hou, Energy Fuels, 2020, 34, 10188–10195 CAS.
- F. Ma, Y. Wan, X. Wang, X. Wang, J. Liang, Z. Miao, T. Wang, C. Ma, G. Lu, J. Han, Y. Huang and Q. Li, ACS Nano, 2020, 14, 10115–10126 CAS.
- Y. Jiang, M. Wei, J. Feng, Y. Ma and S. Xiong, Energy Environ. Sci., 2016, 9, 1430–1438 RSC.
- X. Zhou, Y. Zhong, M. Yang, M. Hu, J. Wei and Z. Zhou, Chem. Commun., 2014, 50, 12888–12891 CAS.
- Q.-Y. Li, Q.-C. Pan, G.-H. Yang, X.-L. Lin, Z.-X. Yan, H.-Q. Wang and Y.-G. Huang, J. Mater. Chem. A, 2015, 3, 20375–20381 CAS.
- Q. Pan, F. Zheng, Y. Wu, X. Ou, C. Yang, X. Xiong and M. Liu, J. Mater. Chem. A, 2018, 6, 592–598 CAS.
- S. Zhang, G. Wang, Z. Zhang, B. Wang, J. Bai and H. Wang, Small, 2019, 15, 1900565 Search PubMed.
- Y. Fang, Y. Lv, R. Che, H. Wu, X. Zhang, D. Gu, G. Zheng and D. Zhao, J. Am. Chem. Soc., 2013, 135, 1524–1530 CAS.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00493f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |