
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanofeatures affect the thermal transitions of polymer thin films: a microcantilever-based investigation
Maximilien
Lopes-Rodrigues
ab,
Didac
Martí-Balleste
a,
Catherine
Michaux
b,
Eric A.
Perpète
b,
Jordi
Puiggalí
 *ac,
Maria M.
Pérez-Madrigal
*ac and
Carlos
Alemán
*ac
*ac,
Maria M.
Pérez-Madrigal
*ac and
Carlos
Alemán
*ac
aDepartament d’Enginyeria Química and Barcelona Research Center for Multiscale Science and Engineering, EEBE, Universitat Politècnica de Catalunya, C/Eduard Maristany, 10-14, 08019, Barcelona, Spain. E-mail: jordi.puiggali@upc.edu; m.mar.perez@upc.edu; carlos.aleman@upc.edu
bLaboratoire de Chimie Physique des Biomolécules, Unité de Chimie Physique Théorique et Structurale (UCPTS), University of Namur, Rue de Bruxelles, 61, 5000 Namur, Belgium
cInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, 08028 Barcelona, Spain
First published on 28th August 2020
Abstract
Nanotechnology is regarded as a promising tool to advance science for a wide range of applications, ranging from nanomedicine, nanoelectronics, imaging and diagnosis. Whilst the unique surface properties of nanostructured materials improve their performance in comparison to conventional materials, little has been done to inspect the correlation between such nanofeatures and the thermomechanical response of nanomaterials. Herein, we report the influence of structural nanofeatures in the glass transition and cold crystallization temperatures (Tg and Tcc) of poly(lactic acid) (PLA) thin films using a technology that detects the microcantilever deflection as a function of temperature. Measurements were conducted on arrays of 8-cantilevers spin-coated with PLA films (thickness of ∼120–200 nm and weight of 6–9 ng) displaying several patterns (compact, nanopored and/or nanoperforated). The Tg increases by 8–13 °C when nanopores and nanoperforations are present, while only the latter affect the Tcc, which increases by ∼6 °C. These phenomena have been attributed (i) the stress of the PLA molecules located at the interface of the pores and perforations, and (ii) to the film-air interface effect, which is associated with the quasi-2D nature of thin films (i.e. those with an aspect ratio of size and thickness greater than 105). On the other hand, the thermomechanical response of PLA thin films loaded with curcumin (CUR) or stiripentol (STP), which formed segregated nanodomains, also differs from that displayed by unloaded PLA films. The size and abundance of CUR and STP nanodomains are directly related to the stress of the PLA chains at the interface and the free volume, which affects the interactions strength and the mobility of polymer molecules (i.e. Tg and Tcc) with respect to neat PLA. Overall, the thermal properties of thin films, which cannot be studied using conventional calorimetric methods, is modified by the presence of nanofeatures. As a consequence, their design needs to be taken into account during the manufacturing of nanomaterials.
Introduction
In the last decades, the utilization of nano- and micro-electromechanical systems (NEMs and MEMs, respectively) has become frequent for characterizing the properties of biological and synthetic macromolecules (i.e. proteins, DNA and polymers) at small scale, due to their high sensitivity and little sample mass requirement (from nano to attograms). For example, these technologies have been applied to study how such large systems can react to pH changes,1,2 wetting and moisture uptake,3,4 swelling and de-swelling,5 the interfacial tension,6 the molecular folding,7,8 and the hydration of self-assembled monolayers,9 among others. Most significantly, NEMs and MEMs can also capture the effect induced by air-sample or liquid-sample interfaces.Consequently, the study at the nanoscale of thermal properties of synthetic polymers using NEMs and MEMs has attracted great interest. For example, Jeon and co-workers examined the glass transition temperature (Tg) of polystyrene, poly(vinyl acetate) and block copolymers using NEMs, which were coated with the polymer solution using inkjet printing.10–12 The heat induced deflection of the NEMs, as a consequence of the changes in volume combined to the elastic properties of the polymers, was used to determine the Tg. Similarly, other authors determined the thermal transitions of ultra-small amounts of semicrystalline poly(lactic acid) (PLA),13 amorphous poly(methylmethacrylate),13 poly(thiophene),14 and semicrystalline poly(propylene azelate).15 Several parameters, including the Tg, the amorphous α and β relaxations, the liquid crystalline transitions of polymers,16 as well as the apparent denaturation temperature (Tad) and other thermal transitions, have been determined for different proteins by means of MEMs technology.8
In recent studies, we developed PLA free-standing nanomembranes (FSNMs) with nanopores crossing the entire thin film thickness to act as a solid support for several applications.17,18 Nanopores were obtained by combining spin-coating and the phase segregation that occurs in immiscible PLA:poly(vinyl alcohol) (PVA) mixtures; subsequently, PVA domains were removed via selective solvent etching.17 Using this procedure, the diameter of the nanoperforations was adjusted for different biomedical applications.17,18
Considering specific applications, quasi-2D (films and membranes), which are characterized by an aspect ratio of size and thickness greater than 105, and 3D (fibres and particles) PLA-based devices are also frequently loaded with drugs for subsequent controlled delivery.19–24 In this case, both the efficiency of the regulation mechanism and the kinetics of the release usually depend on the distribution and organization of the drug into the polymeric scaffold. Therefore, the affinity between the drug and the PLA matrix, which essentially depends on the polarity of the drug and its ability to form polymer–drug specific interactions, as well as the tendency of the drug to organize into nanoaggregates (thus forming nanophases separated from the polymeric matrix) are crucial parameters regulating drug release.
Herein, we report the thermal analysis of different nanostructured PLA samples by means of the cantilever deflection technology. To the best of our knowledge, this is the first time that this technology is used to examine the influence of nanofeatures on the response of the material. More specifically, the thermal responses of nanopored, nanoperforated and drug-loaded PLA thin films have been investigated using a technique named SCALA (SCAnning Laser Analyzer),25,26 which is based on the optical read-out of microcantilevers. We observed that both structural features in the nanometre-length scale, as well as drug–polymer specific interactions, cause important changes in the thermal properties of PLA thin films, which cannot be identified by using conventional calorimetric techniques. Moreover, only sensitive microcantilever-based technologies can deliver relevant information on the evolution with temperature of the polymer–polymer and drug–polymer interactions. Overall, we observed how nanofeatures play an important role in the thermomechanical response of PLA thin films, and thus need to be carefully considered when designing nanomaterials with specific application.
Methods
Materials
PLA 4032D was received from Nupik International (Polinyà, Spain). The residual monomer and D content of such PLA are 0.3% and ∼1.5%, respectively. The main physical properties of the polymer are: density = 1.24 g cm−3; glass transition temperature (Tg) = 55–60 °C; melting temperature (Tm) = 155–170 °C; number and weight average molecular weights (Mn and Mw, respectively) = 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 and 181000 g mol−1, respectively (both determined by gel permeation chromatography); polydispersity index (PDI) = 1.85.
100 and 181000 g mol−1, respectively (both determined by gel permeation chromatography); polydispersity index (PDI) = 1.85.
PVA (87–89% hydrolysed) with Mw = 13000–23000 g mol−1 was purchased from Sigma-Aldrich. Curcumin (CUR) and stiripentol (STP) were purchased from Sigma-Aldrich. 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) was purchased from Apollo Scientific Limit (UK). All reagents were used as received without further purification.
Preparation of thin films
Polymer samples were dissolved in HFIP (10 mg mL−1) for the preparation of PLA and PVA thin films by spin-coating (Spin-coater model: WS-400BZ-6NPP/A1/AR1 Laurell Technologies Corporation). 10 μL of PLA or PVA solution were dropped onto a silicon microcantilevers chip (Fig. 1a), which was fixed on a glass holder for spin-coating at 3500 rpm for 60 seconds. Nanoperforated and nanopored PLA films were obtained using the same procedure but dropping 10 μL of 80:20 and 90:10 PLA:PVA v/v mixtures, which were prepared using PLA (8 and 9 mg mL−1, respectively) and PVA (2 and 1 mg mL−1, respectively) HFIP solutions. Then, chips coated with PLA–PVA films were immersed in Milli-Q water (1 h) to dissolve the PVA nanophase. Finally, CUR- and STP-loaded films were prepared using 80:20 and 90:10 PLA:drug v/v mixtures, which obtained by mixing PLA (8 and 9 mg mL−1, respectively) and the corresponding drug (2 and 1 mg mL−1, respectively) HFIP solutions. Regardless of the composition, 6–9 ng films were obtained. Samples were dried under vacuum for 24 h before analysis.
 | ||
| Fig. 1 (a) Low- and high-magnification optical images of a silicon chip of 8 microcantilevers. (b) Experimental set-up for the thermal analyses conducted with SCALA. | ||
Thermal analyses of microcantilevers
The deflection of the microcantilevers was measured by means of an optical read-out equipment named SCALA (MecWins).25,26 This technique combines the optical beam deflection method and the automated two-dimensional scanning of a single laser beam by voice-coil actuators. The equipment implements a thermal chamber with a thermoelectric cooler and a cartridge heater that control the temperature inside the chamber by flowing thermostatized N2 to the sample, which also avoids its oxidation. Inside the chamber, the holder includes multiple thermometer probes for a better mapping of the temperature. The software (created by MecWins in LabVIEW environment) controls the temperature, acquires data, and sends commands to the hardware.SCALA's experimental set-up consists on a cantilever array that is illuminated by the laser beam and a two-dimensional linear position detector (PSD) that collects the reflected beams (Fig. 1b). Then, TRACKER, an algorithm based on the recognition of reflected intensity patterns, locates and characterizes the cantilever sensors. Hence, such capability allows the user to characterize a single sensor or an array of them in a fully automated process. A change in the local slope at the cantilever surface displaces the reflected laser spot on the PSD, which is denoted S (Fig. 1b). S is used to obtain the cantilever profile along its longitudinal axis, z(x), by integrating:
 | (1) |
Coated cantilevers behave as precise thermometers by exploiting the bimaterial effect,27 which refers to the measurable bending (i.e. differential stress) produced by temperature changes in substrates coated on one side with another material. More specifically, the differential stress in the cantilever is created due to dissimilar thermal expansion coefficients of the silicon substrate and the gold coating. Similarly, coated microcantilevers undergo a deflection, Δz (Fig. 1b), as a result of temperature changes because of the thermal expansion coefficient difference between the coating material of the cantilever and the cantilever itself.11
In this work, monocrystalline Si microcantilever chips containing arrays of eight cantilevers (Micromotive GmbH) were used for the thermomechanical measurements (Fig. 1a). Specifically, the nominal length, width, and thickness of the cantilevers were 500, 90, and 1 μm, respectively. The average deflection curves were obtained using 16 different microcantilevers that were heated from 25 °C to 100 °C at 1 °C min−1.
Profilometry
Film thickness was determined using a Dektak 150 stylus profilometer (Veeco, Plainview, NY) and applying the following optimized conditions: tip radius = 12.5 μm; stylus force = 3.0 mg; scan length = 1 mm; and speed = 100 μm s−1. Measurements were conducted on intentionally scratched films. At least, eighteen measurements were performed using three independent samples for each studied system.Scanning electron microscopy (SEM)
The morphology of the films supported onto silicon sheets was examined using a Focus Ion Beam Zeiss Neon 40 instrument (Carl Zeiss, Germany) equipped with an energy dispersive X-ray (EDX) spectroscopy system and operating at 5 kV. In order to prevent sample charging problems, samples were mounted on a double-sided adhesive carbon disc and sputter-coated with an ultra-thin carbon layer (6–10 nm) The diameter of the perforations was determined using the SmartTiff software from Carl Zeiss SMT Ltd.FTIR spectroscopy
FTIR spectra were recorded on a FTIR Jasco 4100 spectrophotometer by applying 64 scans between 4000 and 600 cm−1 with a resolution of 4 cm−1. Samples were deposited on an attenuated total reflection accessory (Top-plate) with a diamond crystal (Specac model MKII Golden Gate Heated Single Reflection Diamond ATR).Differential scanning calorimetry
Differential Scanning Calorimetry (DSC) experiments were conducted on a PerkinElmer 4000 DSC apparatus calibrated with indium. About 10 mg of each sample were weighed in pressed and punched aluminium crucibles. Nitrogen was used as inert atmosphere at a flow rate of 50 mL min−1. Heating and cooling rates of 10 °C min−1 were applied.Results and discussion
Thermomechanical response of compact PLA and PVA thin films
The thermal response of control non-coated cantilevers was firstly analysed by heating the chips at 1 °C/min from room temperature (25 °C) to 100 °C, and plotting the average deflection against temperature (Fig. 2a). As expected, the deflection of uncoated cantilevers slowly and progressively increases with the temperature, as the deflection of rectangular bare cantilevers of length L grows linearly with the change of temperature (ΔT), according to Δz ∝ L2ΔT.28 However, the deflection reaches a value of only −0.008 nm ± 0.005 at 100 °C, which indicates that changes discussed below for polymer-coated cantilevers are induced by the bimaterial effect (i.e. differentiated thermal expansion/contraction behaviour of the polymer and the silicon cantilever). In this work, all thermal analyses were conducted by heating the sample up to 100 °C, which is not only the denaturation temperature of proteins8 typically used to prepare PLA nanomembranes for biofiltration,18 but also is a reasonable temperature to apply during thermal treatments of drug-loaded PLA devices without melting the polymer. Table 1 compares all the thermomechanical data measured for the studied systems by means of the microcantilever-based technology. | ||
| Fig. 2 Mean deflection curves (obtained using 16 microcantilevers) measured for (a) bare microcantilevers; and microcantilevers coated with (b) PLA and (c) PVA thin films. In all deflection curves, the standard error of the mean is indicated by thick light-grey lines. The ramp of temperature is displayed in the inset of (a). | ||
| System | Nanofeaturea (mean diameter (nm)/drug density (#domains per μm2)) | Thickness roughness (nm) | T g (°C) | T cc (°C) | Other events observed |
|---|---|---|---|---|---|
| a Data shown when applicable. b Thermal event not observed below 100 °C. | |||||
| PLA | None – Compact/Flat film | 154 ± 6/2.5 ± 0.5 | 50 | 70 | Attractive interactions (upward deflection) |
| PVA | None – Compact/flat film | 181 ± 17/1.8 ± 0.4 | 82 | —b | Repulsive interactions (downward deflection); detachment of superficially adsorbed water molecules (55–64 °C) |
| 90-PLA | Nanopores (74 ± 18) | 140 ± 11/90 ± 12 | 58 | 70 | Transition from one regime (downward) to the other (upward) linked to the rearrangement of PLA molecules at the pores |
| 80-PLA | Nanopores (∼45) | 119 ± 9/126 ± 20 | 63 | 76 | Repulsive interactions dominate (downward deflection); |
| Nanoperforations (∼120) (96 ± 29) | Relaxation of PLA molecules located at the film-air interface (48 °C) | ||||
| PLA/CUR(10) | Nanodomains (76 ± 25/4.4) | 155 ± 12/52 ± 29 | 60 | —b | Attractive interactions among PLA increase with temperature (upward deflections) |
| PLA/CUR(20) | Nanodomains (88 ± 22/4.5) | 188 ± 6/32 ± 6 | 47 | 73 | |
| PLA/STP(10) | Nanodomains (99 ± 38/3.3) | 161 ± 9/60 ± 25 | 62 | 89 | |
| PLA/STP(20) | Nanodomains (111 ± 42/3.6) | 180 ± 12/36 ± 8 | 56 | 84 | |
PLA thin films were prepared by spin-coating a HFIP polymer solution onto previously cleaned bare microcantilevers chip. The thickness and roughness of the resulting films were 154 ± 6 nm and 2.5 ± 0.5 nm, respectively, which reveals that they were ultra-flat, as also evidenced by morphological characterization (Fig. 3a). The average deflection curve obtained for compact PLA nanofilms (Fig. 2b) differs from that obtained for bare cantilevers as PLA-coated cantilevers underwent a differential stress caused by the thermal expansion difference between the polymer and the silicon from the microcantilever, which gave rise to a significant net positive (upward) deflection. This observation implies a contraction of the PLA volume, resulting from the reduction of the configurational entropy of PLA chains, which in turn reduces the compressive forces on the silicon side of the cantilever.
 | ||
| Fig. 3 SEM micrographs of thin nanofilms made of: (a) PLA; (b) PVA; (c) nanopored 90-PLA; and (d) nanoperforated 80-PLA. Examples of nanoperforations and nanopores are marked in (d) by red and blue circles, respectively. | ||
The PLA deflection profile shows a subtle slope change at 50 °C, value obtained at the intersection of tangents to the curve, which has been associated with Tg (Fig. 2b). More specifically, the midpoint DIN method was used to define the Tg as is the temperature at which the measured curve is equidistant between the upper and lower tangents. This value is slightly lower than the Tg supplied by the manufacturer (i.e. 55–60 °C), which can be sustained by two explanations. Firstly, the thin nature of the film and the reduced weight of the sample (∼8 ng) favoured the plasticiser behaviour of residual HFIP solvent that remained after the spin-coating process. On the other hand, the decrease in Tg could also be attributed to its well-described variation with the sample degree of crystallinity.29,30 The two fractions that constitute the non-crystalline phase of PLA include a bulk mobile amorphous fraction (MAF) with a Tg of ∼50 °C and a rigid amorphous fraction (RAF) with a higher Tg. Therefore, depending on the crystallization process and the thermal treatment, Tg may vary from 50 °C (e.g. melt quenched samples or obtained from a fast solvent evaporation, as expected for the spin-coating process) to 71 °C (e.g. very slow cooling from the melt or samples submitted to annealing). Furthermore, crystallization in thin PLA polymer films is significantly hindered,31 a feature that leads to a decrease of the RAF fraction and consequently Tg.
Conventional differential scanning calorimetry (DSC) experiments (not shown) demonstrated the absorption of HFIP and its plasticising effect in solvent casted films (thickness around 1 μm), for which a Tg of ∼45 °C was determined. This value progressively increased until a constant Tg of 50 °C was reached after vacuum drying the films at 60 °C for several days. Hence, the lower required amount of trapped molecules and the opportunity of getting rid of careful drying29 both constitute additional advantages of the technique. Moreover, it should be noted that conventional DSC analyses of PLA thin films are completely unfeasible since the preparation of ca. 1250 samples would be needed to reach the necessary 10 mg.
A second change in the slope is detected at 70 °C (Fig. 2c), which has been attributed to the cold crystallization of amorphous domains during the heating process. Typically, the cold crystallization temperature for PLA (Tcc) ranges from 70 °C to 110 °C depending on the molecular weight, D-content, and the previous processing of the sample.32–36 Such a clear detection of the cold crystallization process supports our previous interpretation on the amorphous content of the spin-coated samples.
Considering that nanoperforated PLA thin films were prepared by blending PLA and PVA, and then removing PVA domains by water etching, we decided to complete this section by the study of the thermomechanical response of PVA thin films (Fig. 3b). These films exhibited thickness and roughness of 181 ± 17 nm and 1.8 ± 0.4 nm, respectively. In this case, the profile curve of PVA shows a net negative (downward) average deflection (Fig. 2c), which has been related to the increased repulsive forces on the silicon-coated surface of the microcantilevers. Therefore, the interaction between PVA chains decreases with increasing temperature, whereas the opposite effect was detected for PLA. These features can be explained as follows. The, complex deflection profile recorded for PVA displays an abrupt change in the slope starting at 55 °C, and followed by a straight line until 64 °C. This sharp change in the negative deflection tendency has been associated with the loss of mass produced by the detachment of superficially adsorbed water molecules. Indeed, PVA is a hydrophilic polymer that tends to capture water molecules at the surface. As the vapour pressure of water was low, the release of these superficially adsorbed molecules occurred in the aforementioned interval. The slope then experiences a slight change that allows the determination of Tg at 82 °C, in good agreement with the Tg values described in literature.37–39 Despite its atactic configuration, PVA has a relatively significant crystallinity and, as a consequence, we did not observe any cold crystallization process during heating.
Differences between PLA and PVA have been attributed to their chemical and physical properties. Thus, the methyl group on the carbon backbone of PLA provides a steric impediment to flow but at the same time favours the formation of weak but numerous van der Waals interactions. This particular chemical structure explains the very high and low density of PLA (1.24 g cm−3) in comparison to polyethylene (0.88–0.96 g cm−3) and polyglycolic acid (1.53 g cm−3). Instead, PVA chains present repulsive electrostatic interactions due to a certain ionization degree that, although low, is non-negligible when adsorbed on the surface amorphous silica cantilevers and promotes the formation of polymer coil conformations on the solid substrate.40,41 Furthermore, repulsions increase with the temperature due to the induced conformational changes.
Thermomechanical response of nanopored and nanoperforated PLA thin films
To evaluate the impact of nanofeatures (nanopores and/or nanoperforations) on the thermomechanical response of PLA thin films, nanostructured samples were prepared by the following manufacturing procedure. As PLA and PVA are immiscible polymers, the segregation of the two polymers takes place during the spin-coating process,17 thus yielding spherical PVA nanodomains homogeneously distributed in the resulting PLA film. According to previous work, these PVA regions can be used to create nanofeatures (nanopores or nanoperforations) in PLA thin films by removing them via selective water etching.17,18 More specifically, PVA nanodomains give nanoperforations when their diameter is similar to the entire film thickness, while they lead to nanopores when their diameter is smaller. Moreover, not only do thickness and diameter depend on the operational conditions used during spin-coating (see Methods section), but also on the nature of substrate (i.e. silicon cantilevers in this case). It is worth noting that the combination of phase separation by spin-coating and selective solvent etching causes significant stress at the pore–air and/or perforation–air interfaces. Hereafter, solvent etched PLA thin films derived from spin-coated 90:10 and 80:20 PLA:PVA mixtures onto silicon microcantilevers are denoted 90-PLA and 80-PLA, respectively.
The thickness of 90-PLA and 80-PLA films was determined to be 140 ± 11 and 119 ± 9 nm, respectively, whereas the average diameter of the created nanofeatures was 74 ± 18 and 96 ± 29 nm, respectively. According to such thickness/diameter ratios, the topography of these PLA nanofilms may involve both nanopores and nanoperforations. Actually, the surface of 90-PLA nanofilms exhibits abundant nanopores of very different sizes, but in all cases smaller than ∼100 nm, which are homogeneously distributed across the surface (Fig. 3c). In contrast, the surface of 80-PLA films (Fig. 3d) features a distribution of both nanopores and nanoperforations, with the former more abundant than the latter. In line with previous studies on nanoperforated PLA thin films,17,18 nanoperforations correspond to nanofeatures with diameters of ∼120 nm (some examples are highlighted with red circles in Fig. 3d), while the diameter of nanopores was much smaller, ∼45 nm (blue circles in Fig. 3d). Obviously, the presence of nanofeatures affected the roughness of the films, which increased from 2.5 ± 0.5 nm for neat PLA to 90 ± 12 and 126 ± 20 nm for 90-PLA and 80-PLA, respectively. A more complete structural characterization of the nanoperforations has been described in previous studies.17,18
The thermomechanical response of nanopored PLA (i.e. 90-PLA, Fig. 4a) differs completely from that discussed previously for homogeneous PLA films since two different regimes are clearly identified. The first is defined by the downward profile with negative deflection in the 32–48 °C temperature interval, while the second corresponds to the subsequent ascending profile with positive deflection. The transition from one regime to the other has been ascribed to the rearrangement of PLA molecules at the pores, which were probably stressed at the PLA–PVA interface before the water etching process. Once PVA is removed, the increasing temperature ramp induces the relaxing of such interfacial molecules, thus changing repulsive interactions (negative deflection) into attractive ones (positive deflection). In the second regime, two thermal events are detected at 58 and 70 °C (i.e. change in the curve slope), which have been associated to Tg and Tcc, respectively. Noteworthy, the Tg of 90-PLA films is 8 °C higher than that determined for compact PLA thin films (Tg = 50 °C). According to previous observations,14 this shift originates from the effect of the film-air interface on the interactions between polymer chains. Therefore in nanopored 90-PLA films, the role played by the bulk decreases, whereas the influence of the film-air interface is enhanced. This phenomenon is consistent with the downward deflection in the lower temperature region (Fig. 4a), thus reflecting that interactions at the interface affect both the surface stress and the thermal properties. We also observe that the cold crystallization process in 90-PLA thin films remains unaffected regardless of the presence of nanopores, which suggests that the mobility of relaxed PLA molecules is preserved.
 | ||
| Fig. 4 Mean deflection curves of (a) nanopored 90-PLA and (b) nanoperforated 80-PLA thin films. | ||
On the other hand, the thermomechanical response of 80-PLA films is completely dominated by the nanofeatures (Fig. 4b). In these films, not only was the diameter of the nanopores larger than in 90-PLA films, but also it was similar to the thickness of the film, leading to an assortment of nanopores and nanoperforations. Consequently, both features favoured the effects associated with the film-air interface to the detriment of those derived from bulk PLA. In the deflection profile of 80-PLA films, the continuous negative deflection appears as the most striking characteristic, which indicates that repulsive interactions dominate attractive interactions throughout the whole temperature interval. There are three different possibilities that could explain this observation. The first is related with the effect of the nanoperforations on the thermal expansion coefficient of PLA. However, deeper investigations using modelling tools would be necessary to make this effect understandable. The second is the physical nature of the nanoperforations of 80-PLA, which can be considered as a discontinuity. This could induce stress to the chains located at the boundaries, eliminating the possibility of relaxation when the temperature increases but facilitating the redistribution of the chains. This possibility is consistent with previous results on cantilevers non-uniformly coated with protein aggregates.8 Finally, the third possibility is a combination of the previous two (i.e. a change in the thermal expansion coefficient and the effect of the discontinuities).
Three different thermal events are detected according to changes in the slope of the deflection profile. The first one occurs at 48 °C and could be associated with the relaxation of PLA molecules located at the film–air interface. However, the size and depth of the nanopores is significantly higher in 80-PLA than in 90-PLA and as a consequence, the extent of this relaxation varies, which also explains the continuous negative deflection (Fig. 4b). At higher temperature, two thermal transitions associated to Tg and Tcc are detected at 63 °C and 76 °C, respectively. Again, nanofeatures induce a shift in the transition temperatures towards higher values. Moreover, this effect grows when the stress of PLA molecules located at the interface of the pores and perforations predominate over the film–air interface associated to the quasi-2D nature of the thin film.
Thermomechanical response of drug-loaded PLA thin films
Considering the wide-spread application of PLA-based nanomaterials as drug delivery systems, PLA thin films were loaded with curcumin (CUR; Scheme 1). CUR is a polyphenol soluble in fat but not in water, and displays a wide spectrum of medical properties, including anti-bacterial, anti-viral, anti-protozoal, anti-fungal, anti-inflammatory, as well as anti-cancer activity.42–45 However, the limited stability of CUR molecules in a variety of physical and chemical environments (i.e. heat, pH, light or alkali medium) restricts its practical use. In order to overcome this drawback, researchers encapsulate CUR into drug delivery systems such as PLA, in order to improve its stability and the regulation of its release.46–49 In particular, CUR-loaded PLA have shown good blood compatibility and healing properties.48,49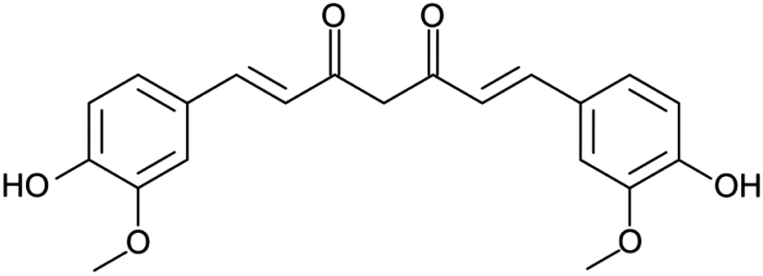 | ||
| Scheme 1 Chemical structure of CUR. | ||
Although CUR is often considered a lipophilic drug, phase segregation occurs upon spin-coating, and CUR separates from PLA by forming well-defined phase-segregated nanodomains (Fig. 5a and b). The resulting morphology derives from the hydrogen bonding capacity of CUR, which promotes the formation of CUR–CUR interactions with respect to CUR–PLA when the solvent rapidly evaporates during the spin-coating process. Films prepared using 90:10 and 80:20 PLA:CUR solutions in HFIP are hereafter denoted as PLA/CUR(10) and PLA/CUR(20), respectively.
 | ||
| Fig. 5 SEM micrographs of (a) PLA/CUR(10), (b) PLA/CUR(20), (c) PLA/STP(10) and (d) PLA/STP(20). Examples of CUR and STP aggregates are marked by red and blue circles, respectively. | ||
The average size of the CUR segregated phases was determined to be 76 ± 25 and 88 ± 22 nm for PLA/CUR(10) and PLA/CUR(20), respectively (Fig. 5a and b). Whilst the size of the domains increases with the amount of CUR, their occurrence per surface unit (i.e. density) is maintained: 4.4 and 4.5 CUR domains per μm2 for PLA/CUR(10) and PLA/CUR(20), respectively. Accordingly, we attributed the changes in the thermal response of CUR-loaded PLA thin films to the increase in the nanodomains size (see below). The thickness of PLA/CUR(10) films, 155 ± 12 nm, is practically identical to that of neat PLA, while that of PLA/CUR(20), 188 ± 6 nm, is slightly higher. Comparing the size of the CUR nanodomains to the thickness of the films suggests that a fraction of CUR experienced phase-segregation across the entire film thickness. Furthermore, we determined a roughness of 52 ± 29 and 32 ± 6 nm for PLA/CUR(10) and PLA/CUR(20), respectively, which is considerably lower than those of nanostructured PLA thin films (i.e. 90-PLA and 80-PLA).
The FTIR spectra of neat CUR, PLA and PLA/CUR(10) were recorded to analyse the nature of the mixture (Fig. 6a). The spectrum of PLA/CUR(20) (available upon request) is practically identical to that of PLA/CUR(10). For PLA, peaks at 1749 cm−1 and 1085 cm−1 correspond to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds, respectively. In addition, peaks related to the asymmetrical vibrations of –CH3 bonds appear at 1455 cm−1 and 1380 cm−1, while the CH–O stretching at 1180 cm−1 is also observed. On the other hand, the spectrum of pure CUR exhibited the characteristic peaks at 1625 cm−1, 1507 cm−1, 1427 cm−1, 1273 cm−1 and 1027 cm−1, which have been associated with the stretching vibrations of CO, CC, olefinic C–H, C–O and C–O–C bonds, respectively. PLA/CUR(10) retains all the characteristic peaks at featured wave numbers, meaning that PLA and CUR remained stable after the manufacturing process, whereas the absence of additional peaks confirms the formation of a physical composite between polymer and drug with no chemical reaction occurring during the spin-coating process.
O and C–O bonds, respectively. In addition, peaks related to the asymmetrical vibrations of –CH3 bonds appear at 1455 cm−1 and 1380 cm−1, while the CH–O stretching at 1180 cm−1 is also observed. On the other hand, the spectrum of pure CUR exhibited the characteristic peaks at 1625 cm−1, 1507 cm−1, 1427 cm−1, 1273 cm−1 and 1027 cm−1, which have been associated with the stretching vibrations of CO, CC, olefinic C–H, C–O and C–O–C bonds, respectively. PLA/CUR(10) retains all the characteristic peaks at featured wave numbers, meaning that PLA and CUR remained stable after the manufacturing process, whereas the absence of additional peaks confirms the formation of a physical composite between polymer and drug with no chemical reaction occurring during the spin-coating process.
 | ||
| Fig. 6 FTIR spectra of: (a) CUR, PLA and PLA/CUR(10); and (b) STP, PLA and PLA/STP(10). | ||
The size of the drug nanodomains affected the thermomechanical behaviour of PLA/CUR(10) and PLA/CUR(20) (Fig. 7a and b); similarly to PLA (Fig. 2c), attractive van der Waals forces between PLA molecules increase with temperature, deflecting the cantilever upwards. The Tg determined for PLA/CUR(10) from the deflection profile is 60 °C, which is 10° higher than for PLA (Fig. 2b). Moreover, the regime associated to cold crystallization was not detected below 100 °C, which indicates that CUR nanodomains also interfere with this process. In contrast, both the glass transition and the cold crystallization thermal events were identified in the deflection profile of PLA/CUR(20). Specifically, Tg and Tcc of PLA/CUR(20) were determined to be 3 °C lower and 3 °C higher with respect to the values for neat PLA.
 | ||
| Fig. 7 Mean deflection curves measured for microcantilevers coated with (a) PLA/CUR(10), (b) PLA/CUR(20), (c) PLA/STP(10) and (d) PLA/STP(20) thin films. | ||
The absence of cold crystallization process in PLA/CUR(10) is consistent with previous works, where bulk PLA microfibers loaded with 5 wt% CUR and were prepared by melt spinning. In comparison to neat PLA, we observe a 20 °C increment in Tcc determined by conventional DSC assays.50 Similarly to electrospinning, melt spinning is a continuous processes, allowing to prepare enough amount of microfibers for DSC evaluation, whereas spin-coating requires the one-by-one preparation of more than a thousand of thin films, making such analysis practically unachievable. Thanks to the thermomechanical response of PLA/CUR(10) and PLA/CUR(20) coated microcantilevers (Fig. 7a and b), it is demonstrated that the shift in Tcc is larger than 30 °C when the polymeric chains and the drug aggregates are confined in thin films. On the other hand, the Tg of electrospun and melt spun PLA/CUR microfibers remains practically unaltered when the drug is loaded in a concentration ≤10 wt%, while it decreases by 4–5 °C when the amount of drug is >10 wt%, on account of the plasticising effect of non-segregated molecules.50,51 Although the Tg reduction is in good agreement with the thermomechanical performance of PLA/CUR(20), the glass transition region of PLA/CUR(10) is largely influenced by the effect of the film thickness (i.e. by the confinement of the polymer chains).
Chen et al.48 reported the lack of PLA–CUR specific interactions (e.g. no hydrogen bonds between the phenolic groups of CUR and the backbone ester groups of PLA backbone) for fibres loaded with ≤5 wt% CUR, which is consistent with our observations (FTIR spectra discussion, Fig. 6a). Accordingly, we associate the changes induced in Tg and Tcc of PLA/CUR thin films to effects induced by CUR nanodomains. Specifically, CUR aggregates (1) stress the PLA chains located at the interface; and (2) enhance the free volume of the polymer matrix, which reduces the strength of attractive polymer–polymer interactions. The amplitude of these effects depend on both the size and the abundance of the drug aggregates. For PLA/CUR(10), the variation in Tg is dominated by the stress of the PLA chains located at the interfaces, which brings rigidity to the chains in the bulk of the film. Contrastingly, drug nanodomains act as a plasticizer in PLA/CUR(20). In this case, the CUR nanodomains are large enough to reduce the energy for molecular motion at the interfaces, which in turn favours PLA mobility at lower temperature.
Overall, regardless of the drug concentration, CUR nanoaggregates alter the glass transition event and hinder the cold crystallization process, with polymer chains increasing their mobility to reach some small amount of crystallization while heating. Indeed, the increase in Tcc for PLA/CUR(10) and PLA/CUR(20) with respect to neat PLA is ascribed to the mobility restrictions imposed by the presence of drug nanodomains.
To corroborate these results, thin films were prepared by loading another drug. Specifically, we have selected stiripentol (STP, (R,S)-(E)-4,4-dimethyl-1-[3,4(methylenedioxy)-phenyl]-1-penten-3-ol, Scheme 2) because it differs from CUR in the amount of hydrogen bonding acceptor and donor groups with CUR/STP ratio of 4/2 and 2/1, respectively. STP is an anticonvulsant drug belonging to the group of aromatic allylic alcohols. It is used in the treatment of epilepsy as it elevates the levels of γ-aminobutyric acid (GABA), a major inhibitory neurotransmitter that regulates electrical activity in the central nervous system.52
 | ||
| Scheme 2 Chemical structure of STP. | ||
Films derived from 90:10 and 80:20 PLA:STP HFIP solutions, are hereafter denoted as PLA/STP(10) and PLA/STP(20), respectively. The thickness/roughness of these films, which were determined to be 161 ± 9 nm/60 ± 25 nm and 180 ± 12 nm/36 ± 8 nm, respectively, follow the same tendency displayed by PLA/CUR. Besides, the drug organizes in segregated nanodomains with an average size/density of 99 ± 38 nm/3.3 STP domains per μm2 and 111 ± 42 nm/3.6 STP domains per μm2 for PLA/STP(10) and PLA/STP(20), respectively (Fig. 5c and d). Accordingly, drug nanodomains are significantly bigger for STP-loaded films than for CUR-loaded films, whereas the density is considerably lower. In the FTIR spectrum of STP (Fig. 6b), the sharp peak at 3551 cm−1 corresponds to the OH group, the broad bands at 2951 and 2866 cm−1 to HCCH and aliphatic C–H, respectively, while the sharp band at 1500 cm−1 is assigned to the stretching vibration of CC. The distribution of peaks in PLA/STP(10) (Fig. 6b) and PLA/STP(20) (available upon request), is nearly identical to that obtained for neat STP and PLA, which indicates the lack of drug–polymer interactions.
Regarding the thermomechanical performance, Tg and Tcc determined for PLA/STP(10) (i.e. 62 °C and 89 °C, respectively) are higher than those obtained for neat PLA (Fig. 7c). The 12 °C increment in Tg is very similar to that obtained for PLA/CUR(10). Again, STP nanoaggregates induce the rigidity of PLA chains confined in thin films. By comparing PLA/STP(10) to PLA/CUR(10) responses, we deduce that the smaller size of STP aggregates is balanced by their abundance, which is significantly lower in CUR-loaded films. Besides, the increase in Tcc with respect to neat PLA (19 °C), which is lower than for PLA/CUR(10), has been associated with the low density of drug nanodomains.
On the other hand, PLA/STP(20) Tg and Tcc (i.e. 56 °C and 84 °C, respectively) are lower than those of PLA/STP(10), but higher than those observed for PLA (Fig. 7d), which is caused by (i) the presence of some plasticizing STP molecules in the amorphous phase, and (ii) the contribution of STP aggregates to the rigidity of the sample. Nevertheless, a slight contrast in the plasticising effect of CUR and STP, as well as differences in the rigidity caused by the presence of agglomerates, are observed by comparing PLA/CUR(20) and PLA/STP(20), possibly ascribed to an increment in the size combined with a reduction in density of the drug nanodomains.
In summary, taking advantage of the sensitivity of the SCALA methodology, we have identified the impact that features in the nanometer-length scale have in the thermomechanical performance of drug loaded PLA thin films. Analysis of these properties using conventional calorimetric methods is virtually impossible since preparation of a huge amount of thin films is needed.
Conclusions
SCALA microcantilever-based technology has been used to investigate the influence of nanofeatures in the thermal response of PLA thin films. Homogeneous and compact PLA and PVA thin films coated onto the cantilevers displayed Tg and Tcc values very similar to those obtained for bulk polymers by conventional DSC analyses. Additional information, such as the variation with temperature of attractive (between PLA chains) and repulsive interactions (between PVA chains), or the release of water molecules from the surface of PVA, was also obtained. However, the presence of physical nanofeatures (i.e. nanopores, nanoperforations and segregated CUR or STP nanodomains) altered the thermal response of PLA thin films, mainly by causing stress in the polymer molecules located at the PLA–air and PLA–drug interfaces. In the case of nanopored and nanoperforated PLA, the significant increase in both Tg and Tcc (with respect to homogeneous and compact PLA thin films) is ascribed to the manufacturing procedure (i.e. phase separation through spin-coating followed by selective solvent etching). For drug-loaded PLA films, the thermal response depends on the size and abundance of the nanodomains, which enhances or reduces the strength of polymer–polymer interactions, thus modifying the mobility of the polymer chains. Consequently, Tg either increases or decreases with respect to neat PLA, while Tcc increases in all studied cases. Therefore, important microscopic information about the influence of nanofeatures on the thermal response of PLA thin films has been obtained through the microcantilever-based technology, which has never been reported before and remains unreachable when using conventional calorimetric methods. Nanofeatures represent a relevant design parameter that affects the performance and, consequently, the application of structured materials at the nanoscale.Conflicts of interest
There is no conflict of interest.Acknowledgements
Authors acknowledge MINECO (RTI2018-098951-B-I00 and RTI2018-101827-B-I00), Agència de Gestió d'Ajuts Universitaris i de Recerca (2017SGR359, 2017SGR373 and FI grant to ML-R) for financial support. Support for the research of C. A. was received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. C. M. and E. A. P. thank the Belgian National Fund for Scientific Research for their research associate and senior research associate positions, respectively.References
- F. Zhou, W. M. Shu, M. E. Welland and W. T. S. Huck, J. Am. Chem. Soc., 2006, 128, 5326–5327 CrossRef CAS.
- T. Chen, D. P. Chang, T. Liu, R. Desikan, R. Datar, T. Thundat, R. Berger and S. J. Zauscher, J. Mater. Chem., 2010, 20, 3391–3395 RSC.
- S. Lenz, S. K. Nett, M. Memesa, R. F. Roskamp, A. Timmann, S. V. Roth, R. Berger and J. S. Gutmann, Macromolecules, 2010, 43, 1108–1116 CrossRef CAS.
- G. M. A. Alves, S. B. Goswami, R. Mansao and A. Boisen, Polym. Test., 2018, 65, 407–413 CrossRef CAS.
- C. Bradley, N. Jalili, S. K. Nett, L. Q. Chu, R. Forch, J. S. Gutmann and R. Berger, Macromol. Chem. Phys., 2009, 210, 1339–1345 CrossRef CAS.
- J. Zhao, R. Berger and J. S. Gutmann, Appl. Phys. Lett., 2006, 89, 033110 CrossRef.
- R. Muñoz, F. Aguilar-Sandoval, L. Bellona and F. Mel, PLoS One, 2017, 12, e0189979 CrossRef.
- M. Lopes-Rodrigues, A. Puiggalí-Jou, D. Martí-Balleste, L. J. del Valle, C. Michaux, E. A. Perpète and C. Alemán, ACS Omega, 2018, 3, 7856–7867 CrossRef CAS.
- A. Cagliani, P. Kosaka, J. Tamayo and Z. J. Davis, Lab Chip, 2012, 12, 2069–2073 RSC.
- N. Jung and S. Jeon, Macromolecules, 2008, 41, 9819–9822 CrossRef CAS.
- N. Jung, H. Seo, D. Lee, C. Y. Ryu and S. Jeon, Macromolecules, 2008, 41, 6873–6875 CrossRef CAS.
- M. Yun, N. Jung, C. Yim and S. Jeon, Polymer, 2011, 52, 4136–4140 CrossRef CAS.
- S. A. M. Bukhari, M. F. Khan, A. Goswami, R. McGee and T. Thundat, RSC Adv., 2017, 7, 8415–8420 RSC.
- O. Ahumada, M. M. Perez-Madrigal, J. Ramirez, D. Curcó, C. Esteves, A. Salvador-Matar, G. Luongo, E. Armelin, J. Puiggali and C. Aleman, Rev. Sci. Instrum., 2013, 84, 053904 CrossRef CAS.
- M. Soccio, G. Luongo, C. Esteves, A. Salvador-Matar, O. Ahumada, D. R. Rueda, M. C. García-Gutiérrez, N. Lotti, A. Munari and T. A. Ezquerra, Appl. Phys. Lett., 2014, 104, 251904 CrossRef.
- M. Marl, P. E. Larsen, V. P. Rangacharya, E. T. Hwu, J. Rantanen, A. Boisen and T. Rades, J. Am. Chem. Soc., 2018, 140, 17522–17531 CrossRef.
- A. Puiggalí-Jou, J. Medina, L. J. del Valle and C. Alemán, Eur. Polym. J., 2016, 75, 552–564 CrossRef.
- A. Puiggalí-Jou, M. M. Pérez-Madrigal, L. J. del Valle, E. Armelin, M. T. Casas, C. Michaux, E. A. Perpète, F. Estrany and C. Alemán, Nanoscale, 2016, 8, 16922–16935 RSC.
- B. Dorj, J. E. Won, O. Purevdorj, K. D. Patel, J. H. Kim, E. J. Lee and H. W. Kim, Acta Biomater., 2014, 10, 1238–1250 CrossRef CAS.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 65, 72–82 CrossRef.
- J. Delmote, L. Teruel-Biosca, J. L. G. Ribelles and G. G. Ferrer, Polym. Degrad. Stab., 2017, 146, 24–33 CrossRef CAS.
- A. L. Gao, F. Liu, Z. Xiong and Q. Yang, J. Colloid Interface Sci., 2017, 505, 49–58 CrossRef CAS.
- J. M. Pang, Y. X. Luan, F. F. Li, X. Q. Cai, J. M. Du and Z. H. Li, Int. J. Nanomed., 2011, 6, 659–665 CrossRef CAS.
- A. Puiggalí-Jou, A. Cejudo, L. J. del Valle and C. Alemán, ACS Appl. Biomater., 2018, 1, 1594–1605 CrossRef.
- L. Lechuga Gómez, V. Álvarez-Sanchéz and F. J. Tamayo De Miguel, US Pat., 7646494B2, 2007 Search PubMed.
- F. J. Tamayo De Miguel, J. Mertens and M. Calleja-Gómez, US Pat., 7978344B2, 2011 Search PubMed.
- R. Berger, Ch Gerber, J. K. Ginzewski, E. Meyer and H. J. Güntherodt, Appl. Phys. Lett., 1996, 69, 40 CrossRef CAS.
- J. R. Barnes, R. J. Stephenson, C. N. Woodburn, S. J. O’Shea, M. E. Welland, T. Rayment, J. K. Gimzewski and Ch. Gerber, Rev. Sci. Instrum., 1994, 65, 3793 CrossRef CAS.
- E. Zuza, J. M. Ugartamendia, A. Lopez, E. Meaurio, A. Lejardi and J. R. Sarasua, Polymer, 2008, 49, 4427 CrossRef CAS.
- A. Sangroniz, A. Chaos, M. Iriarte, J. del Rio, J. R. Sarasua and A. Etxeberria, Macromolecules, 2018, 51, 3923 CrossRef CAS.
- B. D. Fitz, D. D. Jamiolkowski and S. M. Andjelic, Macromolecules, 2002, 35, 5869 CrossRef CAS.
- E. S. Trofimchuk, A. V. Efimov, T. E. Grokhovskaya, N. I. Nikonorova, M. A. Moskvina, N. G. Sedush, P. V. Dorovatovskii, O. A. Ivanova, E. G. Rukhlya, A. L. Volynskii and S. N. Chvalun, ACS Appl. Mater. Interfaces, 2017, 9, 34325–34336 CrossRef CAS.
- C. B. Zhou, H. F. Li, W. Y. Zhang, J. Q. Li, S. Y. Huang, Y. F. Meng, J. D. Christiansen, D. H. Yu, Z. H. Wu and S. C. Jiang, CrystEngCommun, 2016, 18, 3237–3246 RSC.
- R. Androsch, H. M. N. Iqbal and C. Schick, Polymer, 2015, 81, 151–158 CrossRef CAS.
- R. Androsch and M. L. Di Lorenzo, Macromolecules, 2013, 46, 6048–6056 CrossRef CAS.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS.
- J. Rault, R. Gref, H. Ping, O. T. Nguyen and J. Neel, Polymer, 1995, 36, 1655–1661 CrossRef CAS.
- E. Fathi, N. Atyabi, M. Imani and Z. Alinejad, Carbohydr. Polym., 2011, 84, 145–152 CrossRef CAS.
- A. Stavropoulou, K. G. Papadokostaki and M. Sanopoulou, J. Appl. Polym. Sci., 2004, 93, 1151–1156 CrossRef CAS.
- M. Wisniewska, I. Ostolska, K. Szewczuk-Karpisz, S. Chibowski, K. Terpiłowski, V. M. Gun’ko and V. I. Zarko, J. Nanopart. Res., 2015, 17, 12 CrossRef.
- M. Wisniewska, Cent. Eur. J. Chem., 2012, 10, 1236–1244 CAS.
- D. Karunagaran, R. Rashmi and T. R. Kumar, Curr. Cancer Drug Targets, 2005, 5, 117–129 CrossRef CAS.
- C. H. Hsu and A. L. Cheng, Adv. Exp. Med. Biol., 2007, 595, 471–480 CrossRef.
- B. B. Aggarwal and B. Sung, Trends Pharmacol. Sci., 2009, 30, 85–94 CrossRef CAS.
- P. Anand, A. B. Kunnumakkara, C. Sundaram, K. B. Harikumar, B. Sung, S. T. Tharakan, K. Misra, I. K. Priyadarsini, K. N. Rajasekharan and B. B. Aggarwal, Biochem. Pharmacol., 2008, 76, 1590–1611 CrossRef CAS.
- D. Chopra, L. Ray, A. Dwivedi, S. K. Tiwari, J. Singh, K. P. Singh, H. N. Kushwaha, S. Jahan, A. Pandey, S. K. Gupta, R. K. Chaturvedi, A. B. Pant, R. S. Ray and K. C. Gupta, Biomaterials, 2016, 84, 25–41 CrossRef CAS.
- A. Puiggalí-Jou, P. Micheletti, F. Estrany, L. J. del Valle and C. Alemán, Adv. Healthcare Mater., 2017, 6, 1700453 CrossRef.
- Y. Chen, J. Lin, Y. Wan, Y. Fei, H. Wang and W. Gao, Fibers Polym., 2012, 13, 1254–1258 CrossRef CAS.
- T. T. T. Nguyen, C. Ghosh, S.-G. Hwang, L. Dai Tran and J. S. Park, J. Mater. Sci., 2013, 48, 7125–7133 CrossRef CAS.
- I. S. S. Sharifah, A. A. B. Qairol, H. N. Azlina and M. K. Nor Khairusshima, Procedia Eng., 2017, 184, 544–551 CrossRef CAS.
- G. Perumal, S. Pappuru, D. Chakraborty, A. M. Nandkumar, D. K. Chand and M. Doble, Mat. Sci. Eng., C, 2017, 76, 1196–1204 CrossRef CAS.
- J. L. Fisher, Neuropharmacology, 2009, 56, 190–197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |