
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu atomic clusters on N-doped porous carbon with tunable oxidation state for the highly-selective electroreduction of CO2†
Jin
Gao
a,
Hui
Wang
a,
Kun
Feng
a,
Chensheng
Xiang
b,
Huibo
Wang
a,
Huihui
Qi
a,
Yang
Liu
*a,
He
Tian
 *b,
Jun
Zhong
*a and
Zhenhui
Kang
*a
*b,
Jun
Zhong
*a and
Zhenhui
Kang
*a
aInstitute of Functional Nano and Soft Materials Laboratory (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 199 Ren’ai Road, Suzhou 215123, P. R. China. E-mail: yangl@suda.edu.cn; jzhong@suda.edu.cn; zhkang@suda.edu.cn
bCenter of Electron Microscopy, State Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, Zhejiang, China. E-mail: hetian@zju.edu.cn
First published on 11th August 2020
Abstract
Efficient catalysts for the electrochemical CO2 reduction reaction (CO2RR) are highly required for solving the environmental crisis. However, the product selectivity and the catalyst durability for CO2RR are still big problems. Here, we synthesized ultra-small Cu atomic clusters (ACs) dispersed on an N-doped porous carbon (NPC) support (Cu ACs/NPC) for efficient and highly selective CO2RR to CO. The Cu ACs/NPC catalyst can achieve a maximum faradaic efficiency (FE) of 93.2% for CO production at a low potential of −0.5 V (vs. RHE), without any deactivation during 24 h of electrolysis. To the best of our knowledge, the Cu ACs/NPC catalyst, with high selectivity, low overpotential and excellent stability, represents one of the best Cu-based catalysts ever reported for CO2RR to CO. The favorable structure of the Cu ACs on the support means that they have large surface active sites to enhance CO2 adsorption for excellent performance. Moreover, in situ X-ray absorption spectroscopy (XAS) reveals that the Cu ACs/NPC has a tunable oxidation state, which can be easily switched between air-stable Cu2+ and highly reductive Cu0 for efficient CO2RR. A combination of favorable structure and tunable oxidation state thus results in high activity towards the CO2RR.
1 Introduction
The wide utilization of fossil fuels produces an enormous amount of carbon dioxide, leading to environmental crises such as global warming.1–3 Removing the extra CO2 from the environment is now an urgent problem. One of the most compelling approaches towards the problem is to convert greenhouse CO2 into valuable chemicals.4 Electrochemical CO2 reduction reaction (CO2RR) to CO is an important energy conversion process, which is the initial step for the synthesis of more complex hydrocarbon fuels.5,6 However, the reaction still suffers from several bottlenecks, such as a high overpotential resulting from the thermodynamic stability of CO2 molecules, low selectivity for the target product due to competitive side reactions (e.g., the hydrogen evolution reaction (HER)), and the poor durability of the catalysts.7–9 Developing an active and robust electrocatalyst with high selectivity for the CO2RR is still crucial for realizing practical industrial application.Many efficient CO2RR electrocatalysts, such as different metals or metal oxides, have been studied.10–18 Among the various types of catalysts, Cu as the most promising candidate can catalyse CO2 into a wide range of products, including C1 species (CO, CH4, HCOOH), C2 species (C2H4, C2H5OH), and even C3 species (C3H7OH).19,20 However, such products are typically hard to separate, resulting in low selectivity. The overpotential to produce an expected product is also very large and the catalyst stability is not so good. Many related factors have been investigated to optimize the catalytic activity and selectivity of Cu-based catalysts for the CO2RR, such as particle size,21 morphology,22,23 crystal facets,24 oxidation states,25 and the alloying of Cu with other metals.26 The catalytic size effect is of particular interest. For instance, Kanan et al. synthesized a Cu catalyst with 100–1000 nm diameters, achieving a maximum faradaic efficiency (FE) towards CO of around 40% at −0.5 V (vs. RHE).27 Rulle et al. also investigated the catalytic activity and selectivity of Cu nanoparticles (NPs) ranging from 2 to 15 nm, in which hydrocarbon selectivity was increasingly suppressed for nanoscale Cu surfaces.21 Therefore, better CO2RR performance might be observed for Cu samples with a particle size of smaller than 2 nm. Although some Cu-based single atom catalysts have been reported, to date there have been limited studies on ultra-small Cu clusters for the CO2RR. The understanding of the working mechanism with the ultra-small Cu clusters, especially that in the in situ working state, is also an open question that needs to be clearly addressed.
In this work, we synthesized ultra-small Cu atomic clusters (of less than 1 nm) dispersed on an N-doped porous carbon support for efficient and highly selective CO2RR to CO. The sample was prepared using a facile pyrolysis method assisted by a zeolitic imidazole framework (ZIF).28–30 ZIFs have been widely used to synthesize transition metal catalysts supported on nitrogen-doped porous carbon (TM/N–C) materials, which show large amounts of active sites with a unique structure.31–34 Here, Cu atomic clusters (ACs) on an N-doped porous carbon support (Cu ACs/NPC) exhibit high selectivity with a maximum CO FE of 93.2% at a very low potential of −0.5 V (vs. RHE). The corresponding CO partial current density also reached −5.09 mA cm−2, which is a 46-fold and 1159-fold enhancement when compared to that for Cu NPs on an N-doped porous carbon support (Cu NPs/NPC) and bulk Cu on an N-doped porous carbon matrix (bulk Cu/NPC), respectively. The Cu ACs/NPC catalyst also shows excellent durability, with a stable current density and a constant CO FE during 24 h of electrolysis.
Synchrotron radiation based X-ray absorption spectroscopy (XAS) tests indicated that the Cu atomic clusters in Cu ACs/NPC have a high Cu2+ oxidation state with Cu–O or Cu–N bonds, which can thus be anchored on the NPC support. However, in situ XAS experiments revealed that the active sites in the CO2RR process switched to Cu0 clusters under the applied potentials, which can effectively reduce CO2 to CO with their favorable low oxidation state. When the potential was removed, the Cu clusters quickly recovered to their oxidized state as a stable form in air for excellent durability. Compared to Cu-based single atom catalysts with typical stable Cu2+ states, the Cu ACs/NPC catalyst has a tunable oxidation state, which can be easily switched between air-stable Cu2+ and highly reductive Cu0 for efficient CO2RR. The favorable structure of the ultra-small Cu clusters on the NPC support has large surface active sites to enhance the CO2 adsorption, and the tunable oxidation state of the Cu clusters can be easily switched to a low Cu0 state for the efficient reduction of CO2 to CO. A combination of the favorable structure and the tunable oxidation state thus resulted in the excellent CO2RR performance.
2 Experimental
2.1 Materials
Zinc acetate (Zn(CH3COO)2·2H2O, 99.0%), zinc chloride (ZnCl2, 98.0%), 2-methylimidazole (2-MeIM, 99%), copper chloride (CuCl2·2H2O, 99.0%), sodium hydroxide (KOH, 85.0%), potassium bicarbonate (KHCO3, 99.5%), potassium chloride (KCl, 99.5%), and ethanol (EtOH, 99.8%) were used in this study without further purification. All reagents above were purchased from Sinopharm Chemical Reagent Co. Ltd (China). Carbon dioxide (99.999%) and nitrogen (99.999%) were purchased from Airgas, and Nafion perfluorinated resin solution (5 wt%) was purchased from Adamas-Beta®; Nafion®212 proton exchange membrane and Toray Carbon Paper (TGP-H-60) were purchased from Alfa Aesar. Deionized water (purified using a MilliQ-MilliRho system, ≥18 MΩ cm−1) was used to prepare all aqueous solutions and to rinse samples and glassware.2.2 Instruments
The crystal structure of the target was measured by X-ray diffraction (XRD) using an X’Pert-ProMPD (Holland) D/max-γAX-ray diffractometer with Cu Kα radiation (λ = 0.154178 nm). The morphologies of the samples were characterized by scanning electron microscopy (SEM, ZEISS MERLIN Compact) and transmission electron microscopy (TEM, FEI-Tecnai F20). The Fourier-transform infrared (FTIR) spectra of the samples were recorded on a Hyperion spectrophotometer (Bruker) in the scan range of 400–4000 cm−1 using a standard KBr pellet technique. N2/CO2 sorption analysis was conducted using Micromeritics ASAP 2050 apparatus at 77 and 273 K, using Barrett–Emmett–Teller (BET) calculations for the surface areas. X-ray photoelectron spectroscopic (XPS) spectra were obtained using a KRATOS Axis ultra-DLD X-ray photoelectron spectrometer equipped with a monochromatised Mg Kα X-ray source (hν = 1283.3 eV). Raman scattering spectra were recorded with a laser excitation wavelength of 633 nm. The electrocatalysis reactions were tested on a Model CHI 760E workstation (CH Instruments, Chenhua, Shanghai, China). XAS experiments were performed at the Shanghai Synchrotron Radiation Facility (SSRF, beamline 14 W) and the National Synchrotron Radiation Laboratory (NSRL, XMCD beamline). The obtained gas phase composition and liquid products were detected by gas chromatograph (PERSEE, G5) with a flame ionization detector (FID) and by 1H nuclear magnetic resonance spectroscopy (Bruker AVANCE III 600), respectively.2.3 Fabrication of copper atomic clusters supported on an N-doped porous carbon material (Cu ACs/NPC)
A ZnO@ZIF-8 precursor was synthesized as shown in the ESI.† One sixth of the ZnO@ZIF-8 product was put into a 100 mL beaker, then 40 mL of a CuCl2 solution (2 mmol) was added and the reaction mixture was stirred for 40 min at room temperature in order for the salt solution to be completely adsorbed. Then, the sample was centrifuged and dried under vacuum at 50 °C for 6 h. The sample was put in a tube furnace and heated to 900 °C for 2 h under a stream of N2 to yield the Cu ACs/NPC. The Cu content was measured to be 0.2 wt% based on induced coupled plasma mass spectrometry (ICP-MS) analysis.2.4 Electrochemical measurements
The electrochemical experiments were performed using a typical three-electrode cell with the catalysts deposited on the working electrode. The experiments were performed in 0.5 M KHCO3 solution under CO2 and N2 atmospheres, respectively. Gas chromatography (GC) and nuclear magnetic resonance (NMR, Bruker AVANCE III 600) spectroscopy were used to detect the gaseous products and the liquid products, respectively. Details are shown in the ESI.†3 Results and discussion
3.1 Characterization of the catalysts
SEM and TEM images reveal the morphology of Cu ACs/NPC catalyst (Fig. S1, ESI† and Fig. 1a). No obvious particles can be observed due to the small size of the Cu in the Cu ACs/NPC. As a contrast, the images of Cu NPs/NPC and bulk Cu/NPC show clear Cu NPs (mean size of around 20 nm) and bulk spinous Cu (length of around 400 nm), respectively (Fig. S2, ESI†). The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of the Cu ACs/NPC clearly demonstrate the existence of Cu clusters on the N-doped porous carbon support (Fig. 1b and Fig. S3, ESI†). The clusters are very small (less than 0.5 nm) and randomly dispersed on the support. We cannot assign the light points to Cu single atoms since some of them are obviously larger than one single Cu atom. Thus, we labelled them as “Cu atomic clusters”. Some of them gather together similar to nanoparticles but each of the clusters can be well separated. Energy-dispersive X-ray spectroscopy (EDS) analysis further revealed that Cu and N were dispersed on the whole structure (Fig. S4, ESI†). ICP-MS was used to measure the loading of Cu in Cu ACs/NPC, with a value of 0.2% recorded (Table S1, ESI†). It should be noted that the XRD pattern (Fig. 1c) of Cu ACs/NPC displays two broad diffraction peaks at 2θ = 23.5° and 43.0°, which are similar to that of the NPC support for the (002) and (100) crystal planes (JCPDS card no. 34-0567). No characteristic peaks for Cu can be observed in the pattern of Cu ACs/NPC due to its low content and poor crystallinity. For comparison, additional Cu metal peaks at 43.3°, 50.4° and 74.1° were observed in the Cu NPs/NPC and bulk Cu/NPC patterns, in good agreement with the TEM images.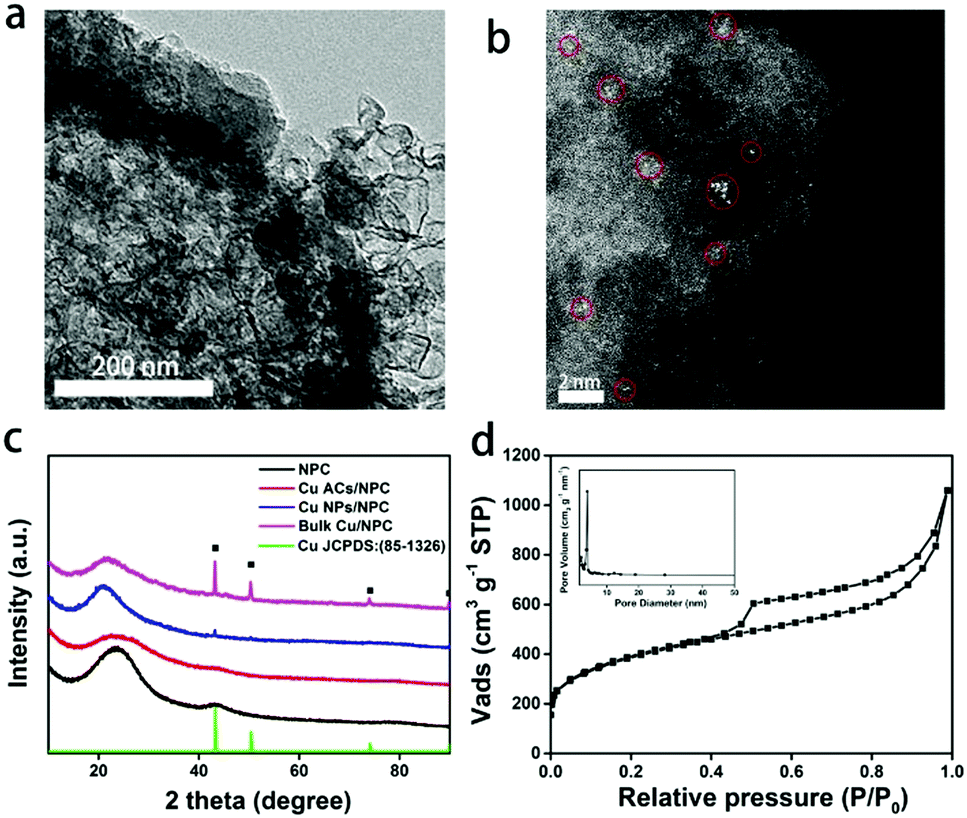 | ||
| Fig. 1 Structural characterization of Cu ACs/NPC. (a) TEM and (b) HAADF-STEM images of Cu ACs/NPC. (c) XRD patterns of NPC (black trace), Cu ACs/NPC (red trace), Cu NPs/NPC (blue trace), bulk Cu/NPC (pink trace) and standard Cu metal (green trace). (d) N2 adsorption–desorption isotherms and pore size distribution (inset) of Cu ACs/NPC. | ||
BET measurements were used to explore the surface areas of the three samples. Among them, Cu ACs/NPC shows the largest BET surface area of 1329.4 m2 g−1, obtained from the N2 adsorption isotherm (Fig. 1d and Fig. S5, ESI†). The inset of Fig. 1d displays the pore size distribution, which further demonstrates the mesoporous structure of the catalyst. A porous structure can facilitate electrolyte diffusion and reactant/product mass transfer in the electrocatalytic process.34 Surface functional groups of the catalysts were also investigated using FTIR spectroscopy (Fig. S6, ESI†), in which several peaks were observed, suggesting that there are abundant surface groups upon which to anchor the Cu clusters. Raman spectra were also used to evaluate the graphitization degree of the NPC (Fig. S7, ESI†). In the Raman data, two broad peaks at around 1325 and 1594 cm−1 were assigned as defective sp3 carbon (D band) and graphitic sp2 carbon (G band), respectively.35,36 The relative ratios of the D to G band (ID/IG) values were found to be all around 1, suggesting similar graphitization degrees of the three catalysts.
To examine the electronic structure of Cu ACs/NPC, XPS measurements were employed. Fig. S8 (ESI†) shows the XPS spectra of various samples (NPC, Cu ACs/NPC, Cu NPs/NPC and bulk Cu/NPC). The XPS survey spectrum of Cu ACs/NPC confirmed the presence of the C, N, O and Cu elements. The presence of N in all the samples confirmed the successful N-doping in NPC. It is worth noting that the content of nitrogen decreased with an increase in the Cu content. The doped N stabilized CO2 in the initial stages of the catalytic process, which is favorable for better CO2RR performance.37 In the high-resolution N 1s spectrum of Cu ACs/NPC (Fig. S8g, ESI†), two major peaks at 398.4 and 401.0 eV can be observed, which are assigned to pyridinic N and graphitic N.37 As shown in Fig. 2a (also in Fig. S8e, ESI†), the high-resolution Cu 2p spectrum of Cu ACs/NPC is dominated by a main peak at 933.5 eV for Cu2+, with a weak feature at 935.0 eV for the presence of Cu+.37 It is reasonable for Cu ACs/NPC to be in the Cu2+ state due to the ultra-small size of the Cu clusters in air, which are easily oxidized and can be easily anchored on the NPC support. In Fig. S8i and m (ESI†), the Cu NPs/NPC and bulk Cu/NPC catalysts also show a Cu2+ oxidation state due to their surface oxidation. However, according to the XRD data, the Cu NPs/NPC and bulk Cu/NPC catalysts mainly contain Cu metal.
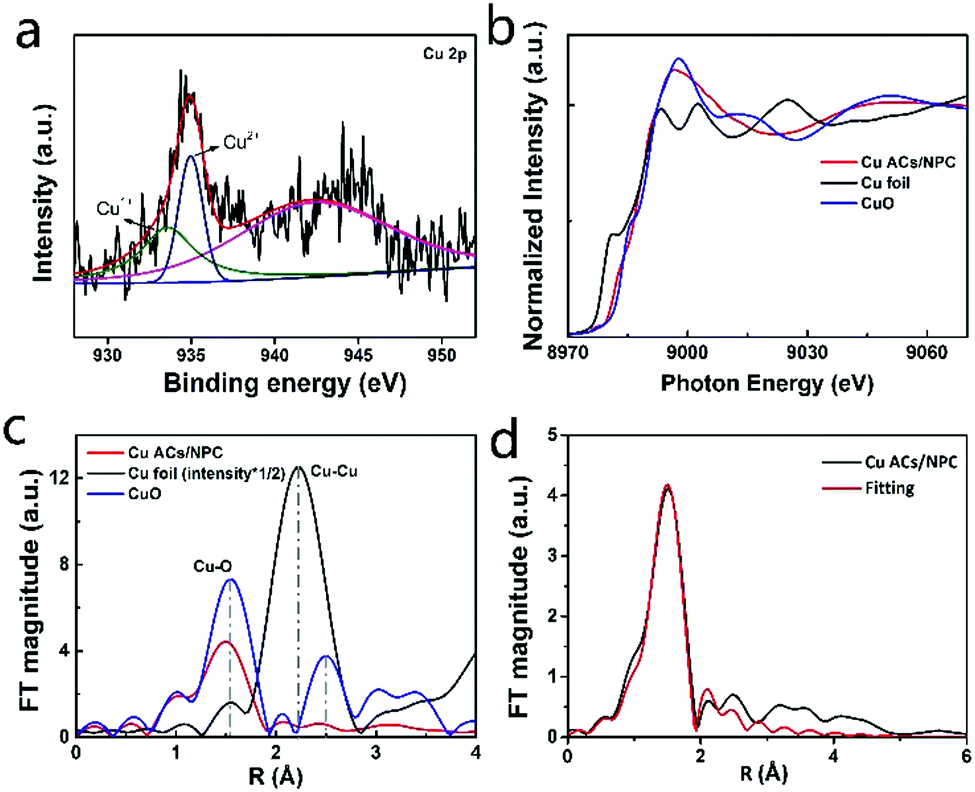 | ||
| Fig. 2 Electronic structural characterization of Cu ACs/NPC. (a) Cu 2p XPS spectra of Cu ACs/NPC. (b) Cu K-edge XAS spectra of Cu foil, CuO and Cu ACs/NPC. (c) Fourier transform curves of the EXAFS data of Cu foil, CuO and Cu ACs/NPC at Cu K-edge. (d) Fitting results of the EXAFS data for Cu ACs/NPC. | ||
Synchrotron radiation based X-ray absorption spectroscopy was also applied to investigate the electronic structure of Cu atoms in Cu ACs/NPC. The XAS spectra of Cu ACs/NPC, the reference Cu foil, and the CuO samples, are shown in Fig. 2b. The spectrum of Cu ACs/NPC is very similar to that of CuO, suggesting a Cu2+ oxidation state, in good agreement with the XPS results. The corresponding extended X-ray absorption fine structure (EXAFS) spectra are also displayed in Fig. 2c. The curve for Cu ACs/NPC shows a main peak at around 1.5 Å, which can be attributed to Cu–O or Cu–N bonds.37 The formation of Cu–O or Cu–N bonds can maintain the stability of the Cu clusters in air and anchor them onto the NPC support. It should be noted that the second shell Cu–Cu configuration (as that for CuO) can only be very weakly identified, strongly confirming the small size of the Cu atomic clusters in Cu ACs/NPC. The EXAFS data was also well fitted using Cu–O bonds and the results are shown in Fig. 2d and Table S2 (ESI†).
3.2 Electrochemical performance and the mechanism
The electrochemical reduction of CO2 was explored in a standard three-electrode system. Cyclic voltammetry (CV, Fig. S9, ESI†) and linear sweep voltammetry (LSV, Fig. 3a) were performed in CO2-saturated and N2-saturated 0.5 M KHCO3 solution to evaluate the catalytic activity of Cu ACs/NPC. The LSV curve measured under a CO2 atmosphere indicates an onset potential of around −0.2 V (vs. RHE), representing a low overpotential of 90 mV. Moreover, the current density of Cu ACs/NPC in CO2-saturated KHCO3 electrolyte shows an obvious increase compared with that in N2-saturated solution (from 7.24 to 13.43 mA cm−2 at −0.65 V vs. RHE). The significant growth in current density can be ascribed to the CO2 reduction reaction as well as the hydrogen evolution reaction. To further analyze the performance of Cu ACs/NPC, the electrolysis of CO2 at controlled potentials was tested in a gastight H-type cell separated by a Nafion 212 membrane. The gas-phase and liquid-phase products after testing were analyzed using GC and 1H NMR. For the Cu ACs/NPC catalyst, only H2 and CO were found during the entire electrolysis process, while no liquid products were observed in the studied potential range. As shown in Fig. 3b, the faradaic efficiencies (FEs) of CO and H2 for Cu ACs/NPC were measured in the potential range from −0.4 to −1.2 V (vs. RHE). It is noted that the FE of CO can reach 79.7% at −0.4 V (vs. RHE), corresponding to a very low overpotential of 0.29 V. Moreover, the CO2-to-CO conversion started to increase and exhibit a maximum CO FE of 93.2% at −0.5 V (vs. RHE). The high CO FE for the CO2RR is comparable to the best catalytic values for Cu-based materials reported in the literature (Table S3, ESI†), especially at a very low potential of −0.5 V (vs. RHE).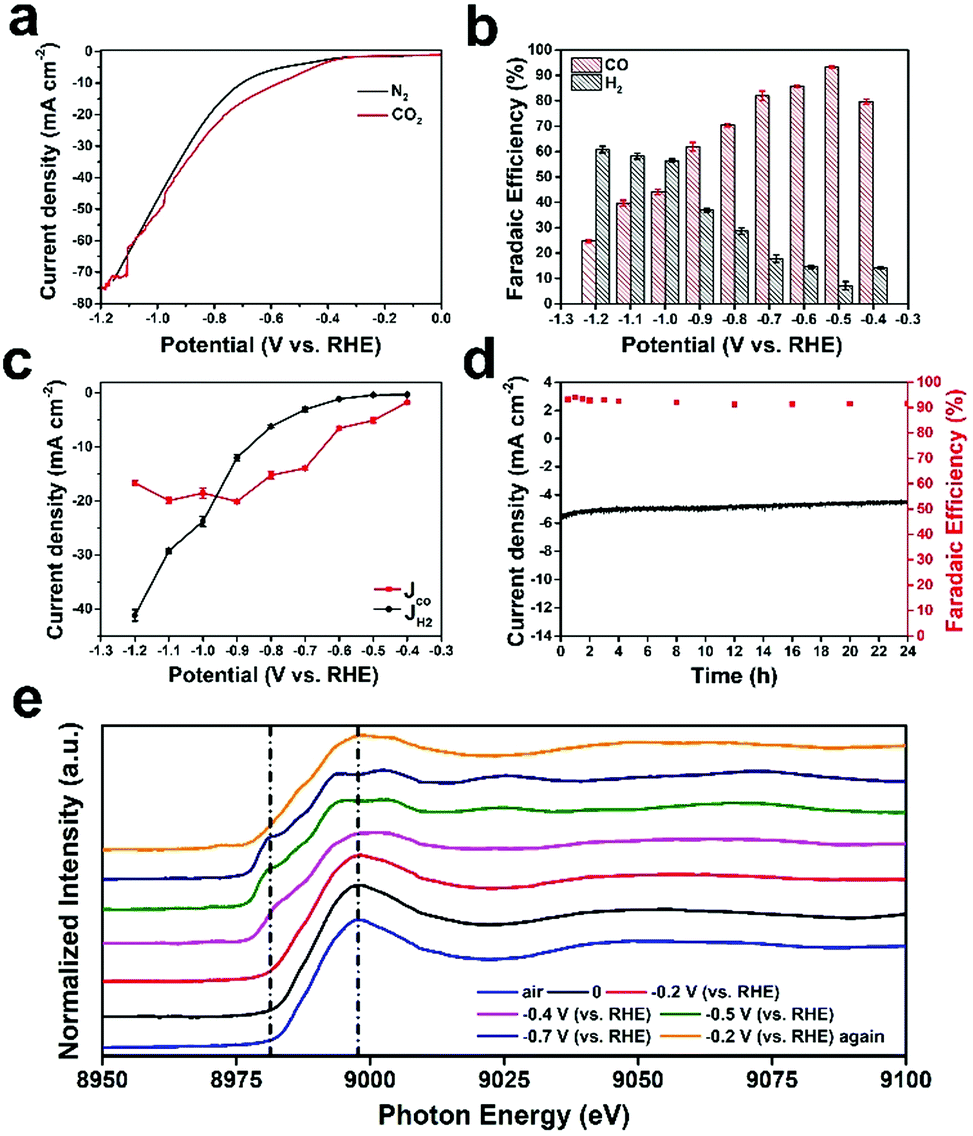 | ||
| Fig. 3 CO2RR performance and in situ characterization of Cu ACs/NPC. (a) LSVs for Cu ACs/NPC in CO2-saturated and N2-saturated 0.5 M KHCO3 electrolyte at a rate of 20 mV s−1. (b) Faradaic efficiencies of CO and H2vs. the applied potentials, catalyzed by Cu ACs/NPC. (c) Partial current density of CO (JCO) and H2 (JH2) vs. the applied potentials. (d) Stability curves of Cu ACs/NPC for the CO2RR at −0.5 V (vs. RHE). FEs for CO vs. time (right axis) and total current density vs. time (left axis). (e) In situ Cu K-edge XAS spectra of Cu ACs/NPC at different applied potentials. | ||
At more negative potentials, the CO FE declined slowly and reached ∼61.9% at −0.9 V (vs. RHE). The FE for H2 showed the opposite tendency, first dropping from 14.1% (−0.4 V vs. RHE) to 7.2% (−0.5 V vs. RHE), and then increasing slowly. The partial current densities of CO (JCO) and H2 (JH2) (Fig. 3c) were calculated from the data of total current density and FE (Fig. 3b), which showed that the CO partial current density of Cu ACs/NPC was 5.09 mA cm−2 at −0.5 V (vs. RHE) and increased to a maximum value of 20.08 mA cm−2 at −0.9 V (vs. RHE) in the applied potential range. Durability tests were also performed to further explore the catalytic properties of Cu ACs/NPC (Fig. 3d). During 24 h of electrolysis at −0.5 V (vs. RHE), the current density was maintained at around −5.3 mA cm−2 and the CO FE stayed at around 92% without any obvious change, indicating the increased stability of the Cu ACs/NPC catalyst. In addition, the Cu ACs/NPC catalyst was characterized by XRD, XPS and TEM after long-term testing (Fig. S10, ESI†). Compared with the original Cu ACs/NPC before electrolysis, the crystal structure, oxidation state and morphology were well retained, which further confirmed the excellent stability of Cu ACs/NPC during the CO2RR. To the best of our knowledge, the Cu ACs/NPC catalyst, with high selectivity, low overpotential and excellent stability, may represent one of the best Cu-based catalysts ever reported for the CO2RR to CO (Table S3, ESI†).
Although the Cu ACs/NPC catalyst can maintain its structure after the CO2RR reaction, the real catalytic sites during the reaction are still unclear. In situ XAS experiments at various potentials were thus performed to probe the working mechanism of Cu ACs/NPC during the CO2RR process. Fig. 3e shows the Cu K-edge XAS spectra of Cu ACs/NPC at different potentials. Among them, the curve in air (blue line) represents the ex situ XAS spectrum of Cu ACs/NPC, and the curve with a potential at 0 V (black line) represents the in situ XAS spectrum of Cu ACs/NPC in solution but without any applied potential. The curve in air is almost identical to that at 0 V, suggesting the negligible influence from the reaction solution. The curves at potentials from −0.2 to 0.7 V (vs. RHE) represent the in situ XAS spectra of Cu ACs/NPC during the real CO2RR process. The spectrum at −0.2 V (vs. RHE) is also similar to that in air or at 0 V, which shows a Cu2+ oxidation state and is in good agreement with the electrochemical performance (the onset potential).
However, when the reaction obviously started at −0.4 V (vs. RHE), the main XAS peak showed a clear shift to a lower energy position and a pre-edge peak at around 8980 eV was also observed, which is an indication of the presence of Cu0. Moreover, when a potential was further applied at −0.5 V (vs. RHE) for the highest CO FE, the main XAS peak also split into two features with the presence of a prominent pre-edge peak, which is almost the same as that for Cu foil (it is not Cu2O due to the spectral shape).37 The curve at −0.7 V (vs. RHE) was also very similar to that at −0.5 V (vs. RHE). The XAS spectra recorded at high potentials during the CO2RR strongly suggest that the active sites in the Cu ACs/NPC catalyst are metallic Cu (Cu0), which is significantly different from the Cu2+ oxidation state in the original sample. This is also reasonable since metallic Cu has a low oxidation state, which is favourable for reducing CO2 molecules. Interestingly, when the applied potential returned to the onset potential of −0.2 V (vs. RHE), the XAS spectrum also recovered to show a XAS spectral shape for Cu2+, with the disappearance of the pre-edge peak and a single main feature. The catalyst also fully recovered after the reaction, according to the XRD, XPS and TEM results shown in Fig. S10 (ESI†). Thus, the Cu ACs/NPC catalyst shows interesting properties, in which a high Cu2+ oxidation state maintains the stability of the ultra-small clusters in air as well as allowing them to bond to the support to anchor the catalyst, and this oxidation state can also be easily switched to a low Cu0 oxidation state to reduce CO2 molecules under the applied potentials. Different from the stable structure in single atom catalysts (such as Cu–N4 or Cu–O), which will be destroyed and then aggregate with the reduced state (Cu0) (a bond is needed to keep the isolated single atom state),37 the Cu ACs/NPC catalyst has a tunable oxidation state and can maintain its small size, with a favorable cluster morphology. Thus, the Cu ACs/NPC catalyst shows excellent CO2RR performance with good selectivity and long-term stability, as shown in Fig. 3.
Control experiments with the similar NPC support but different Cu catalysts were also performed to understand the working mechanism. The detailed synthesis processes for Cu NPs/NPC and bulk Cu/NPC are shown in the ESI.† The electrochemical measurements were performed under the same experimental conditions (Fig. S11, ESI†). Both Cu NPs/NPC and bulk Cu/NPC show low activities in CO2-saturated 0.5 M KHCO3 electrolyte than that in N2-saturated 0.5 M KHCO3, suggesting low CO2RR activity. A comparison of the LSV curves for Cu ACs/NPC, Cu NPs/NPC, bulk Cu/NPC and the NPC support in CO2-saturated KHCO3 electrolyte is also shown in Fig. S11c (ESI†), revealing the much lower catalytic activities of Cu NPs/NPC and bulk Cu/NPC, with low current densities. This result strongly suggests that the Cu ACs/NPC catalyst with ultra-small particle size has the best catalytic performance. The NPC support also shows a very low catalytic performance, suggesting that the support alone does not contribute much to the CO2RR.
Fig. 4 shows more comparisons of the catalysts with different Cu sizes. In Fig. 4a and b, it can be seen that the Cu ACs/NPC presents the highest current density among the three samples and shows the lowest onset potential of around −0.2 V (vs. RHE). The CO FEs and CO partial current densities for the three catalysts were also measured in the range of −0.4 to −1.2 V (vs. RHE) (Fig. 4c–e). In Fig. 4c, the Cu ACs/NPC catalyst exhibits the highest CO FEs (24.71–93.21%) compared with those for Cu NPs/NPC (1.34–21.99%) and bulk Cu/NPC (0.8–6.17%). The maximum CO FEs and the corresponding CO partial current densities are highlighted in Fig. 4d. The Cu ACs/NPC shows an obviously enhanced selectivity for CO production, with a maximum CO FE of 93.2% at −0.5 V (vs. RHE), which is much higher than that for the other two catalysts. The complete CO and H2 FE data for the Cu NPs/NPC and bulk Cu/NPC with partial current densities are also presented in Fig. S12 (ESI†). As shown in Fig. 4e and Fig. S13 (ESI†), the CO partial current density (normalized by the geometric electrode area) and the mass specific partial current density (normalized by the Cu content, see the Supplementary Notes, ESI†) of Cu ACs/NPC exhibit the highest values among the three samples. Notably, the CO partial current density for Cu ACs/NPC can reach up to −5.09 mA cm−2 at −0.5 V (vs. RHE), which is a 46-fold and 1159-fold enhancement in comparison with those for Cu NPs/NPC and bulk Cu/NPC, respectively. The calculated turnover frequency (TOF, see the Supplementary Notes, ESI†) of Cu ACs/NPC is also a high value of 4425 h−1 at −0.5 V (vs. RHE), much higher than those of Cu NPs/NPC and bulk Cu/NPC (Fig. 4f). These results strongly suggest the Cu ACs/NPC catalyst with ultra-small size can benefit the CO2RR performance.
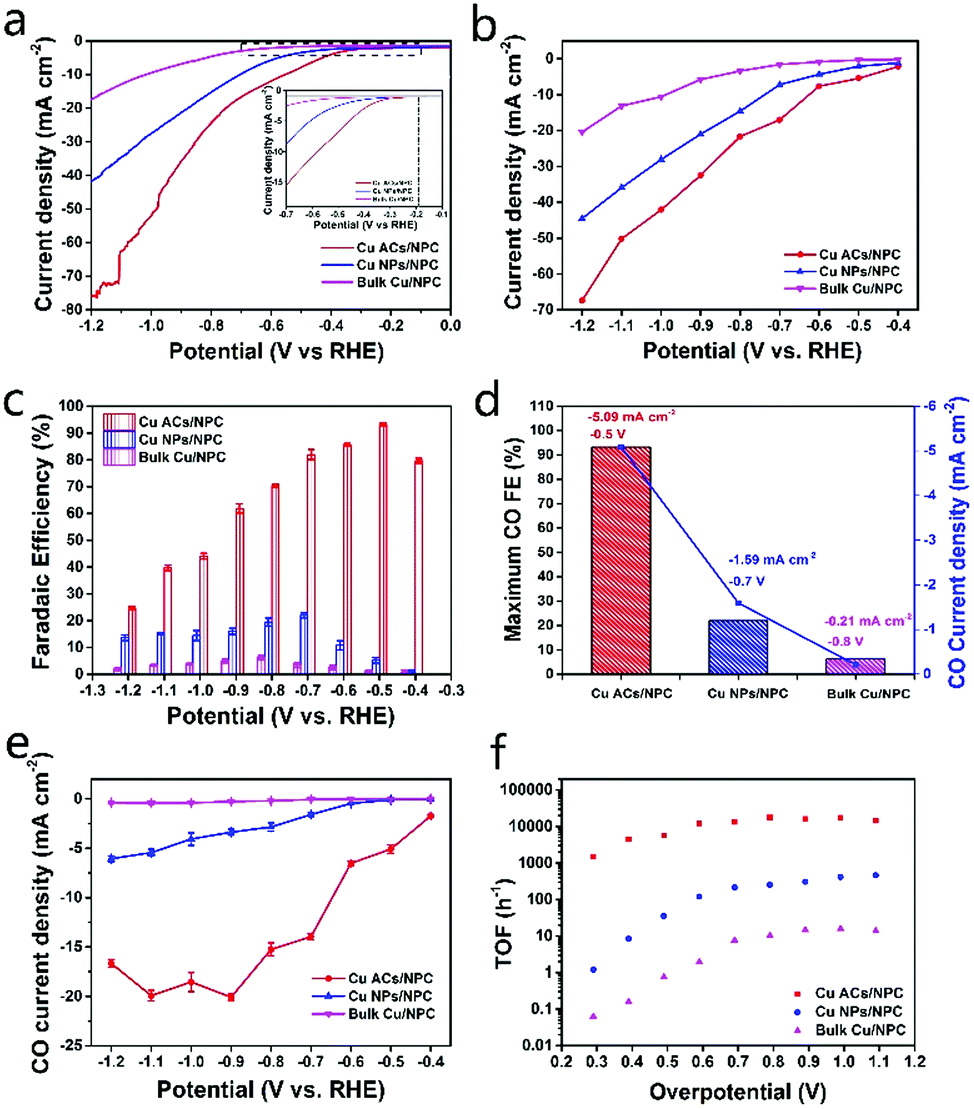 | ||
| Fig. 4 Performance comparison of Cu ACs/NPC, Cu NPs/NPC and bulk Cu/NPC for the electroreduction of CO2 to CO. (a) LSV curves in CO2-saturated 0.5 M KHCO3 electrolyte at a rate of 20 mV s−1. The inset stresses the curves within the range from −0.1 to −0.7 V (vs. RHE). (b) Total current densities and (c) FEs of CO at various applied potentials. (d) Maximum CO FEs (left axis) and the corresponding CO partial current density (right axis). (e) CO partial current densities and (f) TOF values at various applied potentials. | ||
The electrochemically active surface areas (ECSA, see the Supplementary Notes, ESI†) of the three catalysts were also investigated using electrochemical capacitance measurements. The ECSA of Cu ACs/NPC is much larger than those of Cu NPs/NPC and Bulk Cu/NPC (Fig. 5a and Fig. S14, Table S4, ESI†). A larger surface area can also contribute to better CO2RR performance, as shown in Fig. 4. Electrochemical impedance spectroscopy (EIS) was implemented to explore the CO2RR kinetics. Cu ACs/NPC shows the lowest electron-transfer impedance among the three catalysts (Fig. S15, ESI†), indicating the much faster electron-transfer capacity than that of the other two control samples during the CO2RR process.
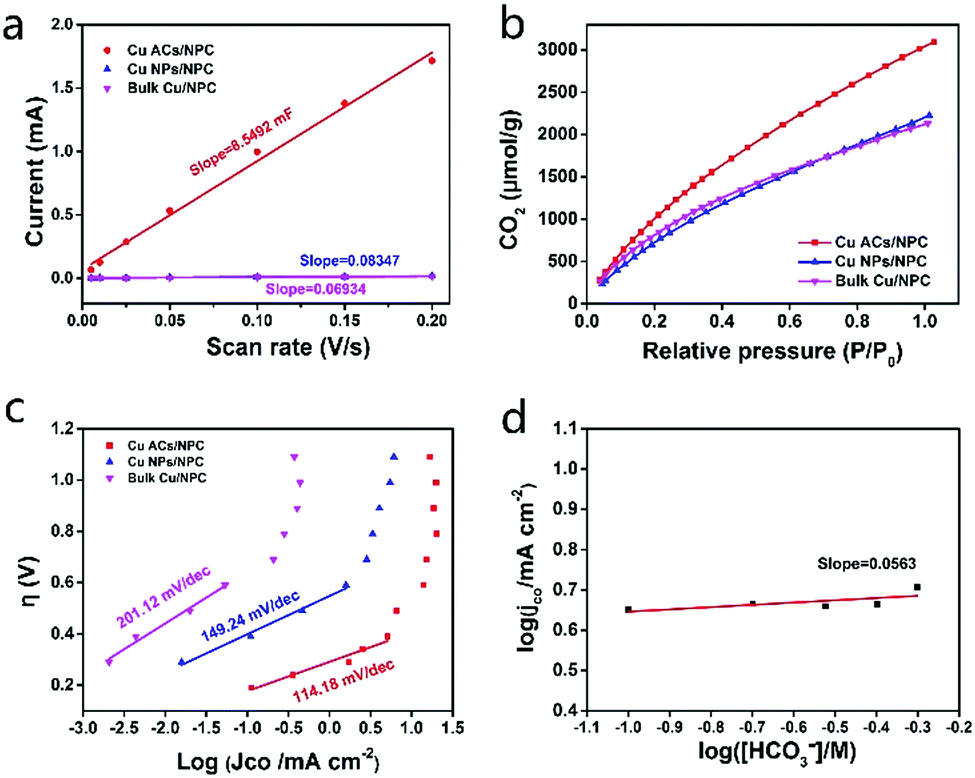 | ||
| Fig. 5 Investigation of the catalytic mechanisms. (a) Charging current differences plotted as a function of the scan rates. The linear slope was used to estimate Cdl for Cu ACs/NPC, Cu NPs/NPC and bulk Cu/NPC. (b) CO2 adsorption isotherms and (c) Tafel plots (overpotential vs. JCO) of the three catalysts. (d) Effect of HCO3− concentration on the CO partial current density at a constant potential of −0.5 V (vs. RHE) for Cu ACs/NPC. | ||
According to the literature, CO2 adsorption is an essential step for further CO2RR.37 Interestingly, the Cu ACs/NPC catalyst appears to adsorb more CO2 at room temperature than the other two samples, which may originate from the size effect of Cu clusters and their high surface area (Fig. 5b and Fig. S5, ESI†). Tafel plot (overpotentials vs. the logarithm of JCO) for Cu ACs/NPC, Cu NPs/NPC and bulk Cu/NPC are presented in Fig. 5c. The Tafel slope of Cu ACs/NPC at low overpotential is 114.18 mV per decade, much smaller than those of Cu NPs/NPC (149.24 mV per decade) and bulk Cu/NPC (201.12 mV per decade), indicating faster kinetics for CO production. The large Tafel slope at higher overpotential might be attributed to a mass transport limitation. Constant-potential electrolysis for Cu ACs/NPC was also performed in different HCO3− solutions with concentrations from 0.5 to 0.1 M. A plot of log(JCO) vs. log([HCO3−]) with a slope of 0.0563 indicates zero-order dependence on HCO3− concentration (Fig. 5d, also see the Supplementary Notes, ESI†), suggesting that the donation of protons from HCO3− is not the rate-limiting step for the CO2RR on the surface of Cu ACs/NPC. This work does not consider the proton from H2O due to the higher pKa of H2O compared with that of HCO3−. From the Tafel plot and the HCO3− concentration analysis, it can thus be concluded that the single electron reduction of CO2 to generate the adsorbed “CO2−*” intermediate is the rate-determining step for the entire CO2RR process. Thus, the excellent CO2RR properties of Cu ACs/NPC can also be partly attributed to abundant surface active sites that stabilize the CO2− intermediate to improve the catalytic activity. The combination of ultra-small Cu clusters and a NPC support with large surface active sites can lead to a favorable structure for CO2 adsorption, and the tunable oxidation state of Cu can be easily switched to a low Cu0 state for the efficient reduction of CO2 to CO with a small cluster structure. Thus, the Cu ACs/NPC catalyst shows high selectivity, low overpotential and excellent stability for the CO2RR, meaning that it might be used for practical applications in the future.
4 Conclusions
In summary, we synthesized Cu clusters dispersed on N-doped porous carbon using a facile method assisted by the pyrolysis of ZIF-8. Compared with Cu NPs and bulk Cu, the Cu clusters show admirable activity and selectivity for the CO2RR, in which the FE for CO can reach up to 93.2% at a low potential of −0.5 V (vs. RHE), accompanied by a high current density of −5.09 mA cm−2. Furthermore, the Cu ACs/NPC catalyst exhibits a superior durability over at least 24 h of electrolysis for the CO2RR. The present Cu ACs/NPC catalyst, with high selectivity, low overpotential and excellent stability, might represent one of the best Cu-based catalysts ever reported for the CO2RR to CO. The favorable structure of the Cu atomic clusters on NPC means that this material has large surface active sites that enhance the CO2 adsorption. In situ XAS measurements revealed that Cu ACs/NPC has a tunable oxidation state, which can be easily switched between air-stable Cu2+ and highly reductive Cu0 for efficient CO2RR. A combination of a favorable structure and tunable oxidation state thus results in the excellent CO2RR performance of the material. This work also provides a promising path for the design of non-noble-metal element based catalysts for efficient CO2RR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National MCF Energy R&D Program (2018YFE0306105), the Innovative Research Group Project of the National Natural Science Foundation of China (51821002), the National Natural Science Foundation of China (21771132, 51725204, U1932211, 51972216, 52041202), the Natural Science Foundation of Jiangsu Province (BK20190041), the Users with Excellence Program of Hefei Science Center CAS (2019HSC-UE002), the Key-Area Research and Development Program of GuangDong Province (2019B010933001), the Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project.Notes and references
- S. H. Schneider, Science, 1989, 243, 771–781 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- J. Gao, C. Zhu, M. Zhu, Y. Fu, H. Huang, Y. Liu and Z. Kang, ACS Sustainable Chem. Eng., 2019, 7, 3536–3543 CrossRef CAS.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- C. H. Lee and M. W. Kanan, ACS Catal., 2015, 5, 465–469 CrossRef CAS.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- Q. Wu, J. Gao, J. Feng, Q. Liu, Y. Zhou, S. Zhang, M. Nie, Y. Liu, J. Zhao, F. Liu, J. Zhong and Z. Kang, J. Mater. Chem. A, 2020, 8, 1205–1211 RSC.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- K. Klingan, T. Kottakkat, Z. P. Jovanov, S. Jiang, C. Pasquini, F. Scholten, P. Kubella, A. Bergmann, B. Roldan Cuenya, C. Roth and H. Dau, ChemSusChem, 2018, 11, 3449–3459 CrossRef CAS PubMed.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS PubMed.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- B. Y. Guan, X. Y. Yu, H. B. Wu and X. W. D. Lou, Adv. Mater., 2017, 29, 1703614 CrossRef PubMed.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- H. Zhong, J. Wang, Y. Zhang, W. Xu, W. Xing, D. Xu, Y. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- J. Wang, G. Han, L. Wang, L. Du, G. Chen, Y. Gao, Y. Ma, C. Du, X. Cheng, P. Zuo and G. Yin, Small, 2018, 14, 1704282 CrossRef PubMed.
- Y. Ye, F. Cai, H. Li, H. Wu, G. Wang, Y. Li, S. Miao, S. Xie, R. Si, J. Wang and X. Bao, Nano Energy, 2017, 38, 281–289 CrossRef CAS.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, 1903668 CrossRef CAS PubMed.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2020, 30, 1907658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00433b |
| This journal is © The Royal Society of Chemistry 2020 |