
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent poly(dendrimer)s for the detection of explosives†
Kinitra L.
Hutchinson
,
Dani M.
Stoltzfus
,
Paul L.
Burn
 * and
Paul E.
Shaw
*
* and
Paul E.
Shaw
*
Centre for Organic Photonics & Electronics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: p.burn2@uq.edu.au; p.shaw3@uq.edu.au
First published on 26th June 2020
Abstract
Three poly(dendrimer)s and their corresponding monomers have been studied for the detection of trace quantities of nitro-based explosives. The poly(dendrimer) structures consist of side-chain conjugated triphenylamine-based chromophores attached to a non-conjugated norbornenyl-derived polymer backbone, with the polymers prepared using a ring opening metathesis polymerisation. The conjugation length, steric bulk, and surface groups of the chromophores were varied to explore their effects on sensing performance. Solution-based Stern–Volmer (SV) measurements were conducted on the materials to investigate the quenching responses to the nitro-aliphatic taggant, 2,3-dimethyl-2,3-dinitrobutane (DMNB), and the nitroaromatic analyte, 2,4-dinitrotoluene (DNT). The SV measurements showed a general trend of an increase in the solution quenching response (observed for both analytes) when going from the monomer to the corresponding poly(dendrimer). Furthermore, the poly(dendrimer) that had the smallest and least sterically encumbered chromophore was found to have the largest response to both DNT and DMNB. Combination of time-resolved and steady-state SV measurements revealed that for the poly(dendrimer)s the quenching by DNT was dominated by a dynamic mechanism, whereas for DMNB it was roughly split between instantaneous and dynamic quenching.
Introduction
Trace detection of explosives and related compounds is crucial to countering terrorism and providing homeland security. In particular, there is demand for in-field methods that can detect vapours of nitro-based chemicals such as 2,4,6-trinitrotoluene (TNT) and its by-product 2,4-dinitrotoluene (DNT), as well as nitrated explosive taggants like 2,3-dimethyl-2,3-dinitrobutane (DMNB).1–4 Detectors based on standard spectroscopic methods such as mass spectrometry, infrared spectrometry, terahertz spectrometry, and Raman spectroscopy can be selective and sensitive, but are often expensive, power hungry and cumbersome, and hence less amenable to field use.5–8 Fluorescence-based detectors can sense trace levels of explosives and taggants and can be incorporated into robust and compact devices.9–11 Hence, they are a promising technology for developing handheld detectors for explosives.The fluorescent sensing material is the key component in a vapour detector and largely determines the performance of the device. In the case of nitro-containing analytes the sensing mechanism is oxidative fluorescence quenching. Oxidative fluorescence quenching is achieved by designing the sensing material such that when photoexcited, the excited electron is transferred to a high electron affinity nitro-based analyte.11 Previous studies have explored the potential of linear conjugated polymers,2,12 dendrimers,11 and small molecules,12 as well as aggregate induced emission,13 for the detection of nitro-based explosives and taggants. Small molecule-based sensing materials have included anthracene, pyrene, carbazole, fluoranthene, and triphenylamine moieties within the sensing chromophore.11,14–16 However, one of the limitations of small molecule solution-based quenching is that no signal amplification is observed as the interaction of one analyte molecule can only quench the fluorescence of one chromophore due to localisation of the exciton. In contrast, it has been reported that conjugated polymers such as poly(phenylethynylene) (PPE) and poly(1,4-phenylenevinylene) (PPV) can exhibit an amplified quenching response in solution due to exciton diffusion along the conjugated polymer backbone.17–20 Similar amplification of the quenching response has been demonstrated for dendrimeric sensing materials. For example, Cavaye et al. showed that an adamantyl-centred dendrimer composed of four phenylbifluorene-based chromophores capped with first generation biphenyl dendrons containing 2-ethylhexyloxy surface groups had an amplified quenching response in solution with one analyte quenching multiple chromophores.21–23
In this manuscript we introduce a family of poly(dendrimer)s as a new class of materials to explore the role of exciton diffusion and analyte–sensor interactions for detection of explosives in solution. The poly(dendrimers) are composed of conjugated chromophore side-chains and a norbornenyl-derived polymer backbone (P1–3, Fig. 1), which were synthesised using a ring opening metathesis polymerisation.24 The chromophores of the poly(dendrimer)s were all triphenylamine centred, as this moiety has been previously used in materials designed to detect nitro-based explosives,25–27 and in some cases selective detection has been demonstrated.25 The poly(dendrimer)s differ with respect to the conjugated unit attached to the triphenylamine centre. The simplest poly(dendrimer), P1, had a fluorenyl moiety connected to two of the phenyl rings of the triphenylamine. P2 is similar to P1 but has bifluorenes rather than fluorene units attached, whilst P3 contains first generation biphenyl dendrons with 2-ethylhexyloxy surface groups in place of the second fluorene units of P2. Thus, the poly(dendrimer) structures allow comparison of the effect of chromophore size (P1versusP2) and steric hindrance (P1versusP3) on the photophysical properties of the poly(dendrimer)s and their solution -based sensing response. We describe the synthesis and characterisation of the poly(dendrimer)s, and the mechanism and efficiency of the solution quenching response with two nitro-containing analytes, namely DMNB and DNT. In particular, we explore whether a non-conjugated polymer with conjugated side-chain chromophores can exhibit an amplified quenching response in solution in a similar manner to conjugated polymers and multi-chromophoric dendrimers.
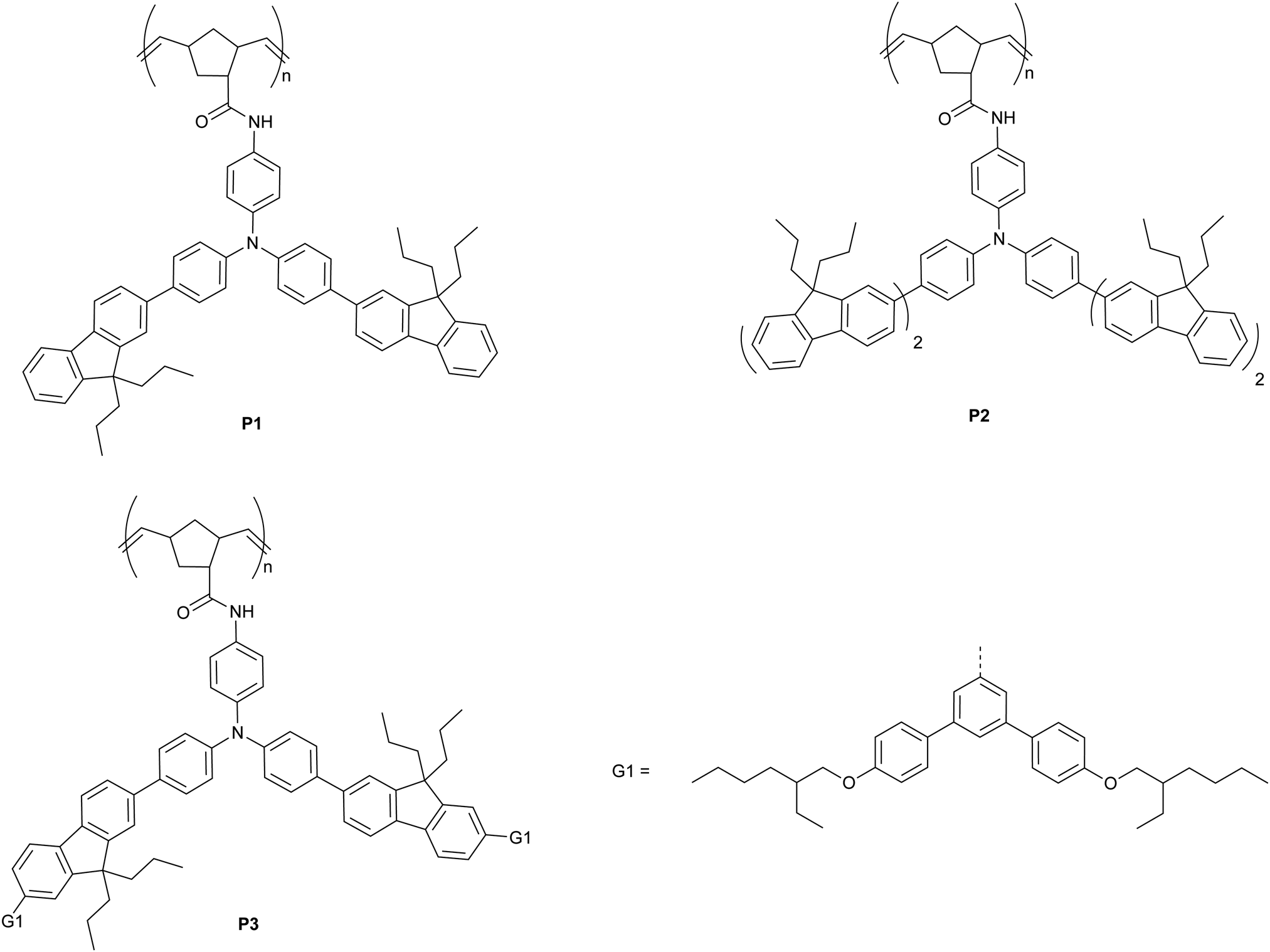 | ||
| Fig. 1 Chemical structures for materials P1–3. | ||
Results and discussion
Synthesis and physical characterisation
The synthetic pathways to the three poly(dendrimer)s are illustrated in Scheme 1 and all follow the same general strategy (see ESI† for experimental details). In the first step, the requisite boronic acid was coupled under Suzuki conditions with 4-bromo-N-[4-bromophenyl]-N-[4-nitrophenyl]aniline to give the nitro-focussed dendrons 4–6 in yields of order 90%. The nitro compounds were then reduced using hydrogen and palladium on charcoal to give the corresponding amines. However, we found that the amines were prone to oxidation and decomposition and hence were used without purification to form their respective amides by reaction with (1S,4R)-bicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid (97% exo) to give M1, M2, and M3 in yields of 27%, 35%, and 35%, respectively. The monomers (M1–M3) were then polymerised using the Grubbs third generation catalyst to give the polymers in yields by weight of 95%, 68%, and 65% for P1, P2 and P3, respectively. Table 1 summarises the poly(dendrimer) molecular weights and dispersities. We found that the more sterically encumbered monomers, M2 and M3, required a higher catalyst loading (5 mol%) than M1 (1 mol%) and this is reflected in the molecular weights of the poly(dendrimer)s.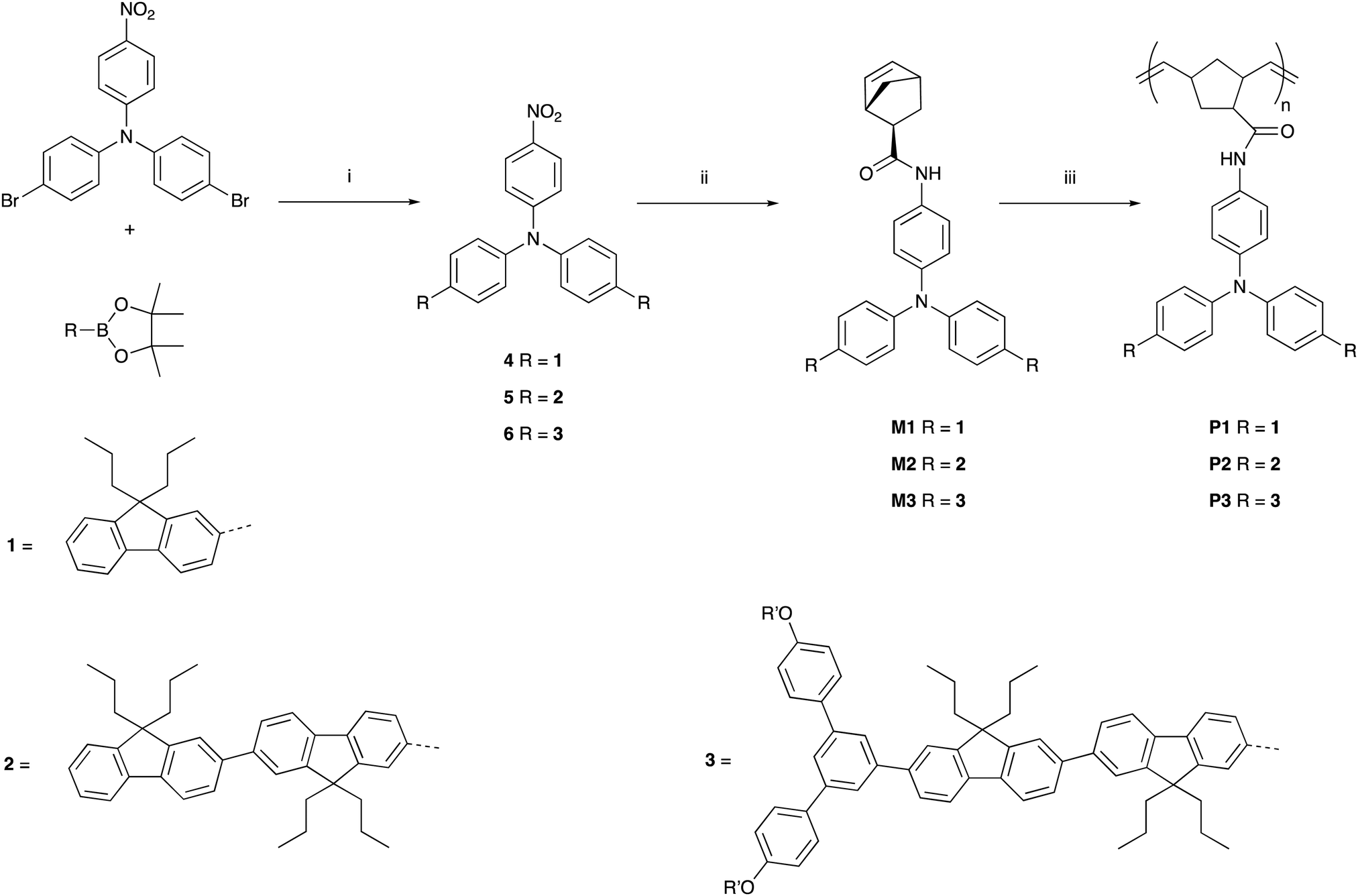 | ||
| Scheme 1 Reactions and conditions (i) Pd(PPh3)4, aq. K2CO3, PhMe, Ar, heat; (ii) Pd/C (10% w/w), H2, CHCl3, EtOH, r.t. then (1S,4R)-bicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid (97% exo), DCC, DMAP, DCM, r.t.; (iii) Grubbs third generation catalyst (1 mol% or 5 mol%), THF, r.t. R′ = 2-ethylhexyl. | ||
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n
n
|
w
|
Đ | Catalyst ratio (mol%) | |
|---|---|---|---|---|
| P1 | 1.4 × 105 | 1.9 × 105 | 1.4 | 1 |
| P2 | 1.8 × 104 | 2.4 × 104 | 1.3 | 5 |
| P3 | 2.5 × 104 | 3.5 × 104 | 1.4 | 5 |
Photophysical and electronic properties
The solution absorption and photoluminescence (PL) spectra of the monomers and polymers are shown in Fig. 2. It can be seen that the absorption spectra for each monomer and polymer pair were essentially the same. It can also be observed that the onset of the absorption of M2/P2 and M3/P3 were at longer wavelengths than M1/P1, which is consistent with extension of the chromophore conjugation length by the extra fluorene moiety on each arm for the former and the dendron on the latter. The red shift in the absorption onset and peak maximum was mirrored in the PL spectra, with M2/P2 and M3/P3 having emission at longer wavelengths than M1/P1. An interesting feature of the PL spectra is that the poly(dendrimer)s all had a larger Stokes shift compared to their corresponding monomers, which is advantageous for sensing as it reduces the inner filter effect and attenuation of the emission. The exact reason for the larger apparent Stokes shift is unclear but may be due to stabilisation of the excited state through interaction with neighbouring ground-state chromophores along the polymer backbone.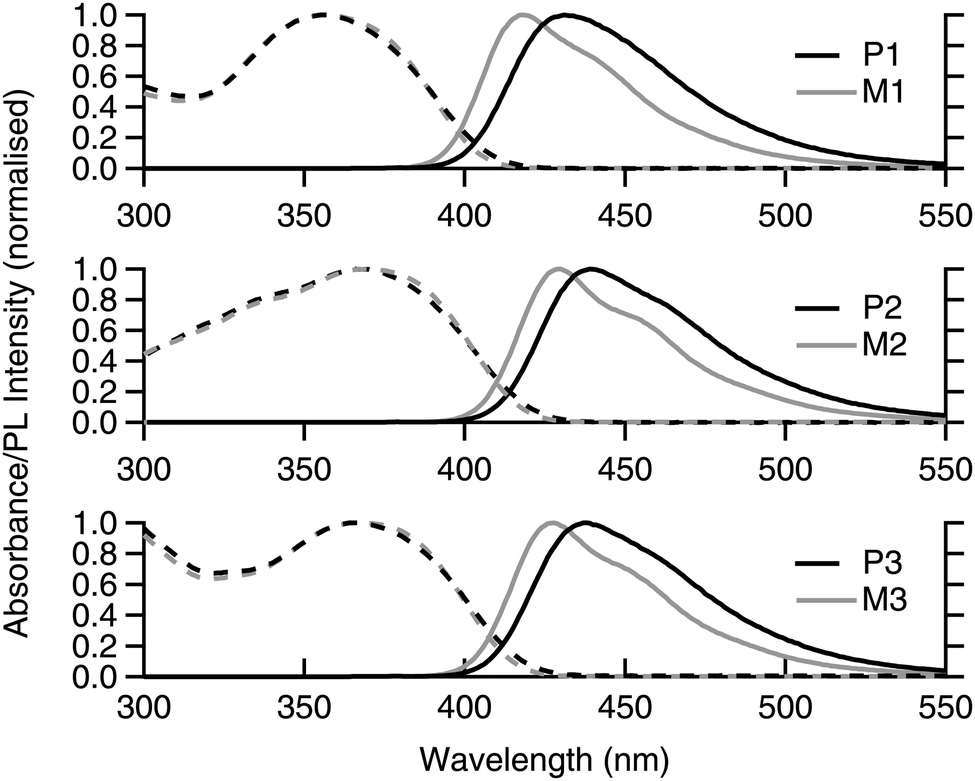 | ||
| Fig. 2 Solution (toluene) absorption (dashed lines) and emission (solid lines) spectra of monomers M1–3 (grey) and poly(dendrimers) P1–3 (black). | ||
To further probe the photophysical properties of the materials we measured their PL quantum yields (PLQYs) and lifetimes, with the results summarised in Table 2. The first point to note is that the PLQY values of the monomers and poly(dendrimer)s are all high and suitable for use as materials for oxidative fluorescence-based quenching measurements. The monomers all have similar PLQY values with the poly(dendrimer)s on average having slightly lower values than their corresponding monomers. The fact that the PLQY values of the poly(dendrimer)s are slightly lower than those of the monomers is consistent with there being weak intra-polymer inter-chromophore interactions. This analysis is supported by the fact that the PL lifetimes of the poly(dendrimer)s are longer than those of the corresponding monomers with their radiative rates being slightly decreased.
| PLQY (%) | τ 0 (ns) | K R (×108 s−1) | K NR (×108 s−1) | |
|---|---|---|---|---|
| a The error on the lifetime measurement is the standard deviation from multiple independent measurements. | ||||
| M1 | 66 ± 7 | 1.17 ± 0.04 | 5.7 ± 0.6 | 2.9 ± 0.8 |
| P1 | 59 ± 6 | 1.34 ± 0.02 | 4.5 ± 0.5 | 3.1 ± 0.5 |
| M2 | 71 ± 7 | 0.90 ± 0.01 | 7.9 ± 0.8 | 3.3 ± 0.8 |
| P2 | 66 ± 7 | 1.32 ± 0.01 | 5.0 ± 0.5 | 2.6 ± 0.5 |
| M3 | 70 ± 7 | 0.97 ± 0.01 | 7.3 ± 0.7 | 3.1 ± 0.7 |
| P3 | 57 ± 6 | 1.03 ± 0.04 | 5.5 ± 0.6 | 4.2 ± 0.8 |
To determine whether a material is suitable for detecting nitro-based materials by oxidative fluorescence quenching it is important to know the relative electron affinities. We have used cyclic voltammetry to determine the solution oxidation potentials of the materials. All the materials were found to undergo chemically reversible oxidations with the E1/2oxs summarised in Table 3. Within experimental error the oxidation potentials of the materials were the same at around 0.2–0.3 V. Chemically reversible reductions for all the materials could not be observed and so the electron affinities of the monomers and poly(dendrimers)s were estimated from the oxidation potential and optical gap in the following manner. The ionisation potentials were calculated from the E1/2s of the first oxidation relative to the ionisation potential of ferrocene. The optical gap was estimated from the absorption and PL spectra using a previously reported method.28 Finally, the electron affinity was calculated by subtracting the optical gap from the ionisation potential. The key point from these calculations was that the electron affinities of all the materials were sufficiently smaller than those reported for DMNB and DNT (see Table 3) by more than the ∼0.3–0.5 eV typically reported for exciton binding energies,29 and hence the materials would be expected to detect the analytes by oxidative fluorescence quenching.
Characterisation of the quenching response
To characterise and quantify the quenching efficiencies of the materials in solution, Stern–Volmer measurements were performed. The nitro-containing analytes chosen for this study were DNT and DMNB, which are both real-world targets for detection. DNT is a nitroaromatic by-product of TNT synthesis, which is more volatile and, thus, easier to detect than TNT.31 DMNB is a widely used nitro-aliphatic taggant that is added to most commercially available plastic explosives. Therefore, these analytes provide insight into whether the polymers interact with aliphatic and aromatic analytes.For steady-state measurements, the Stern–Volmer equation is given by
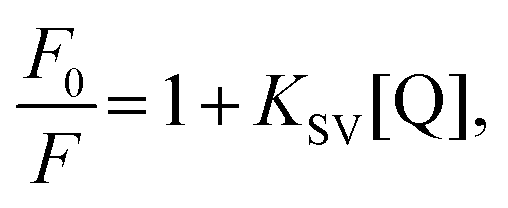 | (1) |
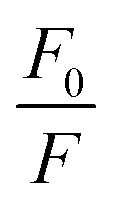 versus [Q] is linear, the value of KSV can be determined from fitting to the data. However, the details of the quenching mechanism, cannot be determined from steady-state measurements alone.
versus [Q] is linear, the value of KSV can be determined from fitting to the data. However, the details of the quenching mechanism, cannot be determined from steady-state measurements alone.
Steady-state quenching experiments were performed on optically dilute (absorbance of ∼0.1 at the excitation wavelength) toluene solutions of M1–3 and the corresponding poly(dendrimer)s P1–3. Given that the chromophores in the poly(dendrimer) were not in conjugation, the concentration of the chromophores in solution was essentially the same between each monomer and poly(dendrimer) pair. Hence, the stoichiometric ratio of analyte to chromophore was to first order the same for monomer and poly(dendrimer) solutions at each analyte concentration. The excitation wavelength selected for each sensor material and analyte combination was chosen to enable photo-excitation of the sensing material while minimising parasitic absorption by the analyte. While the molar extinction coefficient of the analyte was relatively low at the excitation wavelength the fact that it was present at relatively high concentrations led to attenuation of the excitation. Furthermore, the sensing material and analyte absorb a fraction of the emission from the sensing material, resulting in a greater than expected reduction in PL intensity with analyte concentration than would be expected from quenching alone. We have corrected the data for both of these effects following a previously reported method22 to avoid the resulting overestimation of KSV.
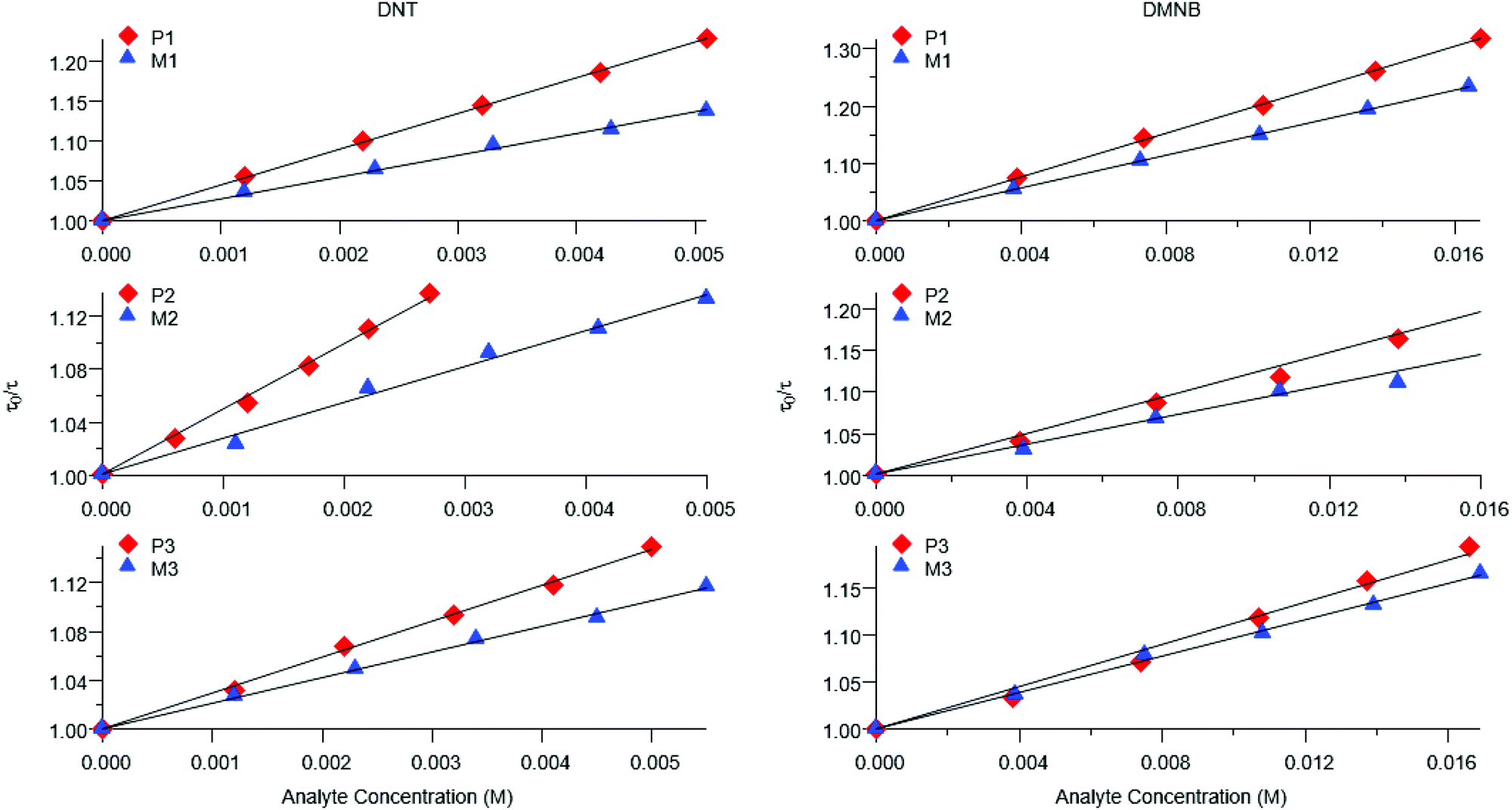
Representative plots for the steady-state Stern–Volmer measurements of the monomers and poly(dendrimer)s with DNT and DMNB are shown in Fig. 3 and the values of the Stern–Volmer constants are summarised in Table 4. The Stern–Volmer constants were of similar magnitude to those previously reported for sensing materials containing triarylamine moieties.27,32 DMNB has a lower electron affinity than DNT (see Table 3) and being a nitro-aliphatic compound does not have the same capability as DNT for π–π interactions. These characteristics typically result in reduced quenching efficiency and lower KSV values for DMNB. All the materials except for P2 showed a linear relationship between 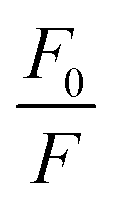 and [Q] at the concentrations used. The plot for P2 and DMNB showed a slight downward curvature, which is indicative of a saturation of the response. Saturation can occur when if some of the chromophores in the system are in accessible to the analyte, for example, if some of the chromophores are buried due to the conformation of the polymer backbone.28
and [Q] at the concentrations used. The plot for P2 and DMNB showed a slight downward curvature, which is indicative of a saturation of the response. Saturation can occur when if some of the chromophores in the system are in accessible to the analyte, for example, if some of the chromophores are buried due to the conformation of the polymer backbone.28
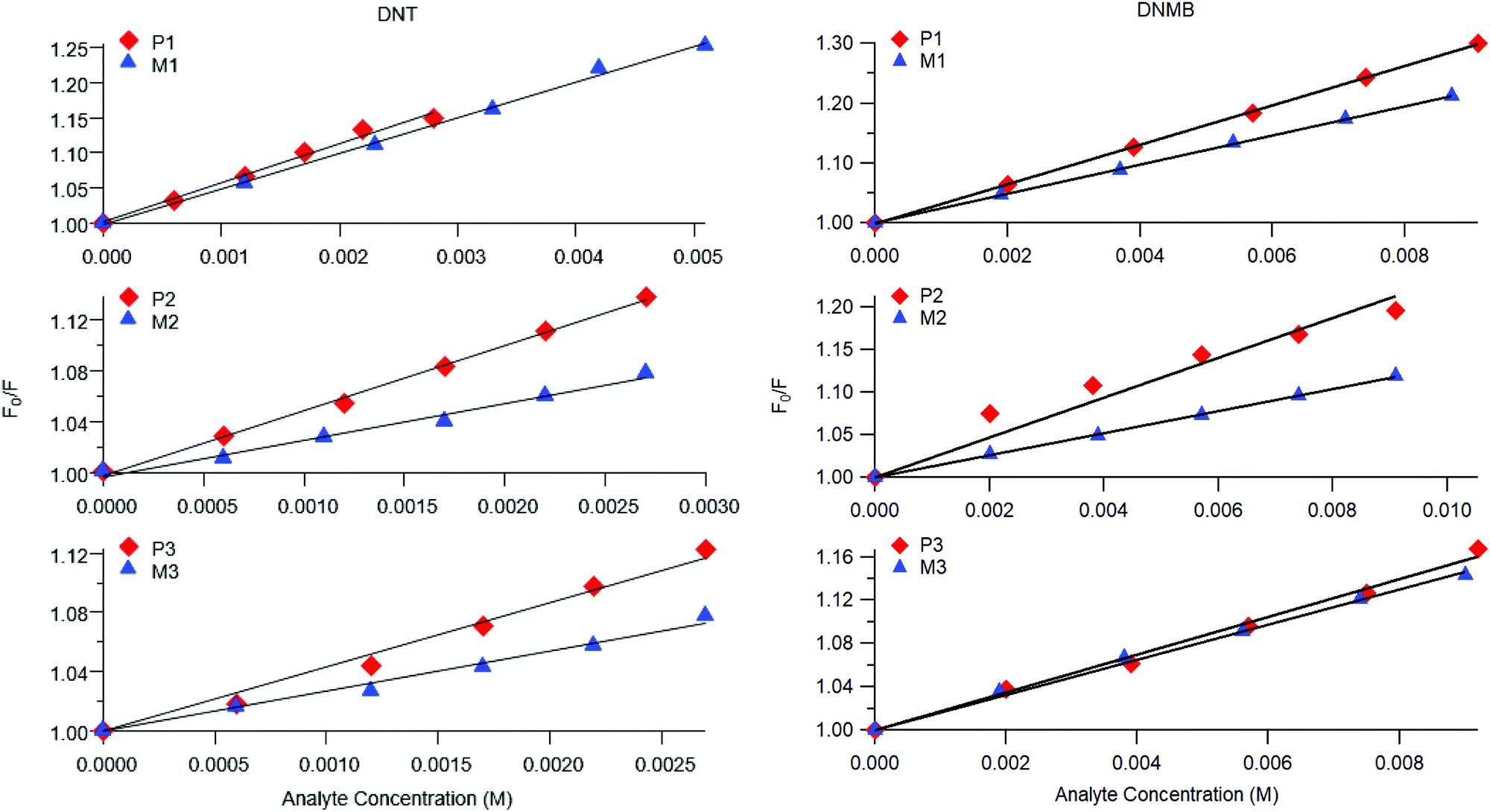 | ||
| Fig. 3 Representative steady-state Stern–Volmer plots for poly(dendrimer)s P1–3 (red diamonds) and corresponding monomer M1–3 (blue triangles) with DNT (left) and DMNB (right). | ||
| DNT | DMNB | |||
|---|---|---|---|---|
| K SV (M−1) | K D (M−1) | K SV (M−1) | K D (M−1) | |
| M1 | 50 ± 3 | 27 ± 3 | 24 ± 1 | 14 ± 1 |
| M2 | 27 ± 3 | 27 ± 3 | 13 ± 1 | 10 ± 1 |
| M3 | 27 ± 6 | 21 ± 2 | 17 ± 1 | 10 ± 1 |
| P1 | 62 ± 6 | 45 ± 5 | 32 ± 1 | 19 ± 2 |
| P2 | 49 ± 5 | 35 ± 4 | 23 ± 1 | 12 ± 1 |
| P3 | 43 ± 2 | 29 ± 3 | 20 ± 1 | 11 ± 1 |
At first glance it can be seen in Table 4 that the Stern–Volmer constants for the poly(dendrimer)s are all higher than their respective monomers for each of the analytes. However, to gain deeper insight into the quenching mechanisms of the materials, time-resolved Stern–Volmer measurements were also performed with the results shown Fig. 4. The PL lifetime of a material is affected by processes that result in quenching of the singlet exciton following photoexcitation. If the quenching is instantaneous, perhaps due to the binding of an analyte to the photoexcited chromophore, then the PL intensity is reduced with no change in the PL lifetime. In solution-based measurements on single chromophore systems, a change in the PL lifetime corresponds to quenching of the excited state as a result of a collisional interactions with the analyte. In polymeric systems, there are two mechanisms that can change the lifetime. A collisional interaction, as observed in single chromophore systems, could lead to quenching of the singlet exciton or the exciton could migrate between adjacent chromophores until it reaches a chromophore to which an analyte is bound and is quenched. Since there are multiple processes that can lead to the decrease in the PL lifetime of the singlet exciton for the poly(dendrimer)s we define these effects as “dynamic quenching” rather than “collisional quenching”. Given that dynamic quenching encompasses processes that depopulate the excited state, the lifetime in the absence (τ0) and presence (τ) of the quencher leads to the following Stern–Volmer relation
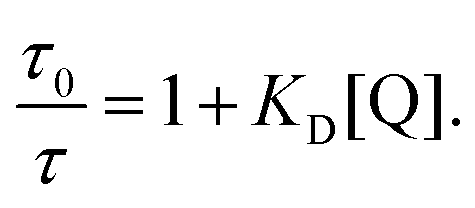 | (2) |
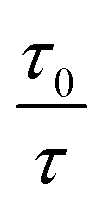 versus [Q] that is a straight line has the slope equal to the dynamic quenching constant KD. At low analyte concentrations the contribution from instantaneous quenching, KI, can be estimated from KSV = KI + KD.
versus [Q] that is a straight line has the slope equal to the dynamic quenching constant KD. At low analyte concentrations the contribution from instantaneous quenching, KI, can be estimated from KSV = KI + KD.
 | ||
| Fig. 4 Representative PL lifetime Stern–Volmer plots for M1–3 (blue triangles) and corresponding poly(dendrimer)s P1–3 (red diamonds) for DNT (left) and DMNB (right). | ||
The measured steady-state Stern–Volmer constant and the dynamic quenching constants (from the time-resolved PL measurements) for the monomers and poly(dendrimer)s with DNT and DMNB are summarised in Table 4. Fig. 5 shows the relative proportions of dynamic and instantaneous quenching for each monomer and poly(dendrimer) with DNT and DMNB. The KSV values show that for the monomers there is an overall decrease in the quenching response to DNT when going from M1 to M3, which is driven by a reduction in the instantaneous quenching component. This suggests that there is stronger binding of DNT with M1 than M2 and M3. When going from a monomer to the corresponding poly(dendrimer) there was an increase in the KSV value with DNT, which is similar to the behaviour reported for linear conjugated polymers, where large enhancements in Stern–Volmer constants were observed in the polymer relative to a monomeric chemosensor.18 From the time-resolved PL measurements the contribution from dynamic quenching was found to increase in going from the monomers to the poly(dendrimer)s, which was also accompanied with an increase in the instantaneous quenching contribution in going from M2 to P2 and M3 to P3. The lower KSV values of P2 and P3 than P1 are primarily due to the lower dynamic quenching constants and suggests that intra-polymer exciton transfer for P2 and P3 is less efficient. This could be due to differences in polymer conformation with the latter materials having bulkier side-chains. Alternatively, it could be due to a reduction in collisional interactions due to the lower molecular weights of P2 and P3 relative to P1.
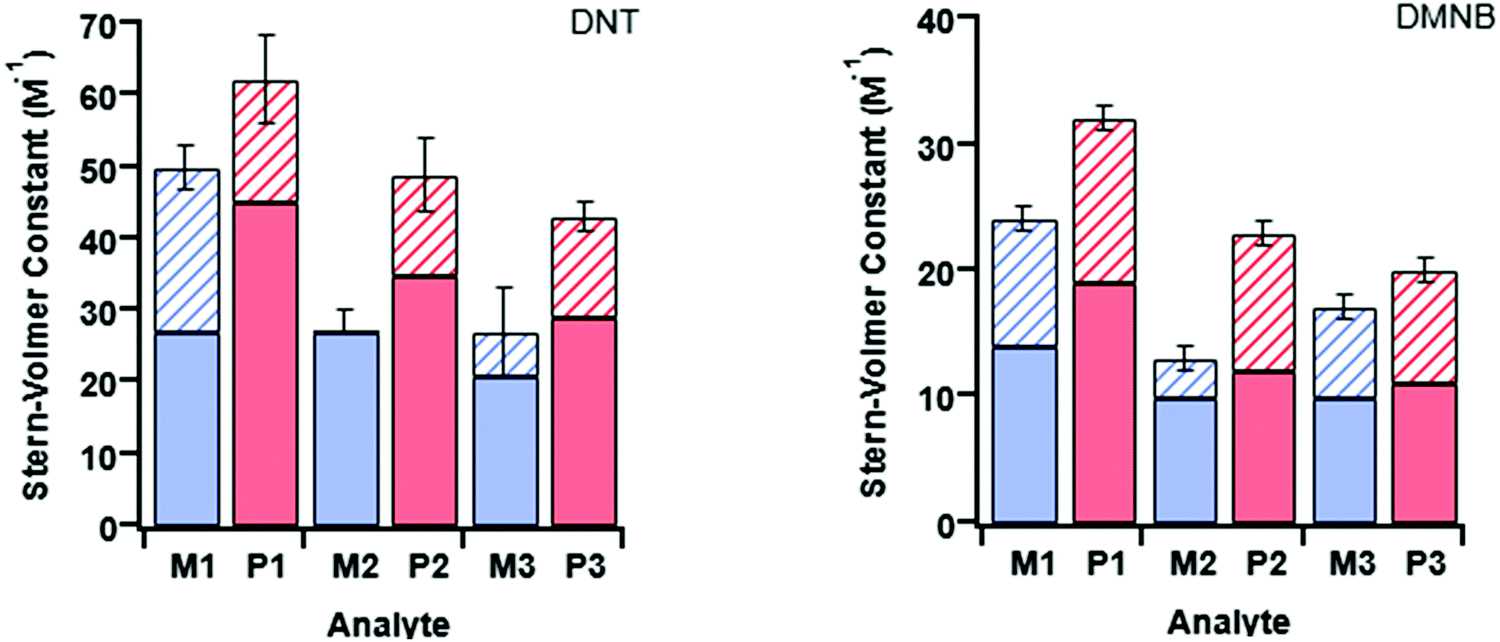 | ||
| Fig. 5 Summary of the Stern–Volmer constants for M1–3 (in blue) and P1–3 (in red) with DNT (left) and DMNB (right). On each plot there are three pairs of bars corresponding to each monomer/polymer pair. The solid-fill corresponds to the dynamic component of quenching (KD) with the hatched part corresponding to the instantaneous quenching contribution (KI). The total height of the bar is the measured Stern–Volmer constant (KSV). | ||
The monomer responses to DMNB were found to be generally smaller than that of the corresponding poly(dendrimer)s. Furthermore, the KSVs for the monomers and poly(dendrimer)s for DMNB were all lower than DNT, which is consistent with its smaller electron affinity and the lack of an aromatic unit that encourages π–π interactions. Interestingly, the poly(dendrimer)s all exhibited significant instantaneous quenching, particularly P2 and P3 when compared to their monomers and previously reported triphenylamine-centred dendrimers. That is, the DMNB is binding more strongly with the poly(dendrimer)s, which could be due to adjacent dendritic side-chains along the polymer backbone acting in concert to bind the DMNB.
Conclusions
Three dendritic monomers and their corresponding poly(dendrimer)s with variations within the triphenylamine-based sensing chromophore were synthesised and their sensing ability for nitro-based analytes determined. Of particular interest was the effect of varying the chromophores on sensing performance in solution, as well as the effect of going from the monomer (single chromophore) to poly(dendrimer) (multiple chromophores). Stern–Volmer measurements confirmed that the fluorescence of the monomer/polymer pairs was quenched by DNT and DMNB, indicating that these are candidate sensing materials for the detection of nitro-based explosives and taggants. It was found that the bulkier monomers (M2 and M3) had smaller quenching responses than M1 for both analytes. The Stern–Volmer constants of the poly(dendrimer)s were roughly 50% larger than their corresponding monomers. This enhancement is consistent with an amplification effect in solution and was attributed to a combination of increased binding of the analyte to the poly(dendrimer)s and/or intramolecular exciton diffusion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. E. S. was supported by an Advance Queensland Research Fellowship. P. L. B. is an Australian Research Council Laureate Fellow (FL160100067). This work was performed in part at the Queensland node of the Australian National Fabrication Facility (ANFF-Q) – a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers.References
- S. Shoaee, S. S. Y. Chen, H. Cavaye, A. R. G. Smith, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, Sens. Actuators, B, 2017, 239, 727–733 CrossRef CAS.
- Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- J. P. Agrawal and R. D. Hodgson, Organic Chemistry of Explosives, Wiley, 2007 Search PubMed.
- J. Akhavan, Chemistry of Explosives, Royal Society of Chemistry, 2004 Search PubMed.
- K. E. Brown, M. T. Greenfield, S. D. McGrane and D. S. Moore, Anal. Bioanal. Chem., 2016, 408, 35–47 CrossRef CAS PubMed.
- W. R. de Araujo, T. M. G. Cardoso, R. G. da Rocha, M. H. P. Santana, R. A. A. Munoz, E. M. Richter, T. Paixao and W. K. T. Coltro, Anal. Chim. Acta, 2018, 1034, 1–21 CrossRef CAS PubMed.
- C. Chen, D. Jiang and H. Li, Anal. Chim. Acta, 2019, 1077, 1–13 CrossRef CAS PubMed.
- S. Singh, J. Hazard. Mater., 2007, 144, 15–28 CrossRef CAS PubMed.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- F. Akhgari, H. Fattahi and Y. M. Oskoei, Sens. Actuators, B, 2015, 221, 867–878 CrossRef CAS.
- P. E. Shaw and P. L. Burn, Phys. Chem. Chem. Phys., 2017, 19, 29714–29730 RSC.
- M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- H. Zhu, M. H. Chua, B. Z. Tang and J. Xu, Polym. Chem., 2019, 10, 3822–3840 RSC.
- A. M. Sanjuán, J. A. Reglero Ruiz, F. C. García and J. M. García, React. Funct. Polym., 2018, 133, 103–125 CrossRef.
- L. Mokalled, M. Al-Husseini, K. Y. Kabalan and A. El-Hajj, Int. J. Appl. Sci. Eng. Res., 2014, 5, 337–350 Search PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- T. M. Swager and J. H. Wosnick, MRS Bull., 2002, 27, 446–450 CrossRef CAS.
- S. W. G. Thomas, J. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- S. W. Thomas III, J. P. Amara, R. E. Bjork and T. M. Swager, Chem. Commun., 2005, 4572–4574 RSC.
- M. Guo, O. Varnavski, A. Narayanan, O. Mongin, J.-P. Majoral, M. Blanchard-Desce and T. Goodson III, J. Phys. Chem. C, 2009, 113, 4763–4771 CrossRef CAS PubMed.
- H. Cavaye, P. E. Shaw, A. R. G. Smith, P. L. Burn, I. R. Gentle, M. James, S.-C. Lo and P. Meredith, J. Phys. Chem. C, 2011, 115, 18366–18371 CrossRef CAS.
- H. Cavaye, P. E. Shaw, X. Wang, P. L. Burn, S.-C. Lo and P. Meredith, Macromolecules, 2010, 43, 10253–10261 CrossRef CAS.
- H. Cavaye, A. R. Smith, M. James, A. Nelson, P. L. Burn, I. R. Gentle, S. C. Lo and P. Meredith, Langmuir, 2009, 25, 12800–12805 CrossRef CAS PubMed.
- W.-Y. Lai, M. N. Balfour, J. W. Levell, A. K. Bansal, P. L. Burn, S.-C. Lo and I. D. W. Samuel, Macromolecules, 2012, 45, 2963–2971 CrossRef CAS.
- N. Niamnont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel and M. Sukwattanasinitt, Chem. Commun., 2013, 49, 780–782 RSC.
- G. Vamvounis, P. E. Shaw and P. L. Burn, J. Mater. Chem. C, 2013, 1, 1322–1329 RSC.
- Y. Geng, M. A. Ali, A. J. Clulow, S. Fan, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, Nat. Commun., 2015, 6, 8240 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 7017–7018 CrossRef CAS.
- P. D. Barata and J. V. Prata, Supramol. Chem., 2013, 25, 782–797 CrossRef CAS.
- R. Meyer, J. Köhler and A. Homburg, Explosives, Wiley-VCH & Co. KGaA, Weinheim, 2007 Search PubMed.
- D. A. Olley, E. J. Wren, G. Vamvounis, M. J. Fernee, X. Wang, P. L. Burn, P. Meredith and P. E. Shaw, Chem. Mater., 2011, 23, 789–794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00249f |
| This journal is © The Royal Society of Chemistry 2020 |