
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron transfer induced magnetic ordering of metal-cyanide magnets†
Yulong
Huang
a,
Yong
Hu
a,
Lu
An
a,
Zheng
Li
a,
Jason N.
Armstrong
a and
Shenqiang
Ren
 *abc
*abc
aDepartment of Mechanical and Aerospace Engineering, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA. E-mail: shenren@buffalo.edu
bDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA
cResearch and Education in eNergy, Environment and Water (RENEW) Institute, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA
First published on 28th May 2020
Abstract
After over three decades of intense research effort, molecular cyanide magnets offer the prospect of low-temperature solution growth and stimuli-dependent magnetic properties. Central to the magnetic properties of cyanide compounds, electron transfer and spin coupling of structural cyanide networks are crucial because they control the magnetic ordering characteristics. However, the candidates of magnetically ordered molecules with controlled electron transfer are still very rare. Here we report atomic coordination induced control of electron transfer in solution grown metal-cyanide magnets, where the 2D FeSe layered material can react with the cyanide-chain dependent electron-accepting molecules, 7,7,8,8-tetracyanoquinodimethane and tetracyanoethylene. We demonstrate the electron transfer in Fe:cyanide networks, revealing its striking role in the magnetization and magnetic ordering. This study opens up a new avenue to design coordination magnets with high-temperature magnetic properties.
Introduction
The cooperative magnetism of metal-cyanide molecular magnets arises from the localized spin exchange interaction between metal cations and cyanide radical anions,1 which can be dictated through the control of the cyanide ligand dimension and metal-cyanide stoichiometry ratios.2–4 Therefore, the electron transfer and molecular coupling can be designed to control magnetic hardening of molecular magnets for high Curie temperature.5,6 The molecular magnet, V(TCNE)x (TCNE = tetracyanoethylene) with ferrimagnetic ordering temperature (Tc) beyond 400 K,7 is the first metal-cyanide magnet ordered above room temperature. The abundant organometallic coordination structures render metal–TCNE with metamagnetic, ferro- and photo-electric features to achieve the fundamental spin and charge coupling.8–12 In this context, Fe(TCNE)-based magnets, Fe(TCNE)2 with Tc above 100 K, have been grown by the reaction between FeI2, Fe(CO)5 or FeCl2(NCMe)2 and organic oxidizing agent TCNE.13–15 However, it is challenging to control Fe and TCNE stoichiometry and electron transfer, which play an important role in the spin coupling of Fe–TCNE magnets to determine their magnetic ordering temperature.5The mean field model is suggested to understand the magnetic properties in the insulating magnets, where the ordering temperature Tc could be enhanced by increasing the spin exchange interaction J, number of magnetic neighbors and Curie constants.16 In a ferrimagnetic Fe and cyanide system, Fe(TCNX)y, (TCNX is TCNQ or TCNE, TCNQ = 7,7,8,8-tetracyanoquinodimethane),17 two kinds of molecules show similar coordination with Fe cations. The TCNE molecule has a short distance of two cyano radicals that could increase the spin exchange interaction as a bridge in Fe(TCNX)y. Therefore, spin exchange interaction determined by atomic coordination is a critical factor in improving the Tc of Fe(TCNX)y molecular magnets. Here, we report a solution reaction between 2D FeSe layered material and electron accepting molecules, TCNQ and TCNE, for the understanding of the electron transfer role in the magnetic ordering of metal-cyanide molecular magnets. The substitution reactions of 2D FeSe proceed through electron transfer from Fe to cyanide ligands, leading to Fe(TCNE)y magnets with high Curie temperature, in contrast to M–(TCNE)2 magnets by chemical reactions using metal salts.14,18 Magnetic characterization of these compounds reveals the significance of the stoichiometry and electron transfer in magnetic ordering of such molecular magnets. The magnetic order and electron transfer are influenced by the length of the bridge and stoichiometry ratios between the radical cyanide ligands (TCNQ or TCNE) and Fe(II) cations. Through magnetic susceptibility and valence state analysis, the charge distribution in the Fe–TCNE network is taken into consideration for the mechanistic understanding of electron transfer induced magnetic ordering.
Results
Fig. 1 shows the scheme to control ferrimagnetic exchange coupling order in molecular magnets via the reaction of 2D FeSe and molecular oxidizing agent TCNX (TCNQ and TCNE) cyanide ligands, which exhibit different ligand dimensions. A two-dimensional FeSe (Fig. 1a) layer serves as the iron-based precursor to chemically synthesize molecular Fe(TCNX)y magnets where magnetic order is originated from the spin coupling between Fe(II) cations and TCNX ligands. We reported the strong electronegativity of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N in TCNQ molecules for the synthesis of Fe–TCNQ molecular magnets (Fig. 1b). The TCNE molecule has the same four –CN radicals as TCNQ while a lack of an aromatic carbon ring makes TCNE a shorter chain dimension (Fig. 1c). As an electron acceptor with strong electronegativity, TCNE performs well in the procedure of synthesizing Fe–TCNE molecular magnets by substituting Se in FeSe. The TCNX ligand accepts one electron to form a radical TCNX− oxidation state with an unpaired spin (S = 1/2), distributing over the whole molecule.19 In Fe(TCNX)y molecular magnets, one Fe atom loses two electrons and forms Fe2+ cations with four unpaired spins (S = 2), where the Fe2+ cation coordinates with four –CN radicals in Fe–TCNQ20,21 (Fig. 1d) and six –CN radicals in Fe–TCNE18,22,23 (Fig. 1e). In the layer of Fe–TCNX, the antiferromagnetic spin coupling between the Fe2+ cation (S = 2) and TCNX− (S = 1/2) presents a ferrimagnetic order due to their unbalanced opposing spin numbers. Fig. 1d and e demonstrate the in-plane crystal structure and spin order of the Fe2+ cation where the spin distributions of TCNX are not shown. The Fe(TCNE)y magnet possesses a compact Fe spin network due to its smaller cyanide ligand compared to Fe(TCNQ)y. Under the ligand design rule, Fe(TCNE)y should possess a higher ferrimagnetic ordering temperature due to its shorter ligand connection among spins between Fe2+ cations using TCNE−.13 The magnetization hysteresis (MH) loops at 100 K for Fe(TCNE)y feature ferrimagnetic characteristics (Fig. 1f), which is drastically different from that of the Fe(TCNQ)y system. The paramagnetism of Fe(TCNQ)y at 100 K is consistent with its Curie temperature of 60 K. Because of its relatively short TCNE ligands, larger orbital overlap of unpaired electrons enhances the ferrimagnetic coupling between Fe2+ cations and TCNE− radicals. Thus, Fe(TCNE)y is selected for further studies.
N in TCNQ molecules for the synthesis of Fe–TCNQ molecular magnets (Fig. 1b). The TCNE molecule has the same four –CN radicals as TCNQ while a lack of an aromatic carbon ring makes TCNE a shorter chain dimension (Fig. 1c). As an electron acceptor with strong electronegativity, TCNE performs well in the procedure of synthesizing Fe–TCNE molecular magnets by substituting Se in FeSe. The TCNX ligand accepts one electron to form a radical TCNX− oxidation state with an unpaired spin (S = 1/2), distributing over the whole molecule.19 In Fe(TCNX)y molecular magnets, one Fe atom loses two electrons and forms Fe2+ cations with four unpaired spins (S = 2), where the Fe2+ cation coordinates with four –CN radicals in Fe–TCNQ20,21 (Fig. 1d) and six –CN radicals in Fe–TCNE18,22,23 (Fig. 1e). In the layer of Fe–TCNX, the antiferromagnetic spin coupling between the Fe2+ cation (S = 2) and TCNX− (S = 1/2) presents a ferrimagnetic order due to their unbalanced opposing spin numbers. Fig. 1d and e demonstrate the in-plane crystal structure and spin order of the Fe2+ cation where the spin distributions of TCNX are not shown. The Fe(TCNE)y magnet possesses a compact Fe spin network due to its smaller cyanide ligand compared to Fe(TCNQ)y. Under the ligand design rule, Fe(TCNE)y should possess a higher ferrimagnetic ordering temperature due to its shorter ligand connection among spins between Fe2+ cations using TCNE−.13 The magnetization hysteresis (MH) loops at 100 K for Fe(TCNE)y feature ferrimagnetic characteristics (Fig. 1f), which is drastically different from that of the Fe(TCNQ)y system. The paramagnetism of Fe(TCNQ)y at 100 K is consistent with its Curie temperature of 60 K. Because of its relatively short TCNE ligands, larger orbital overlap of unpaired electrons enhances the ferrimagnetic coupling between Fe2+ cations and TCNE− radicals. Thus, Fe(TCNE)y is selected for further studies.
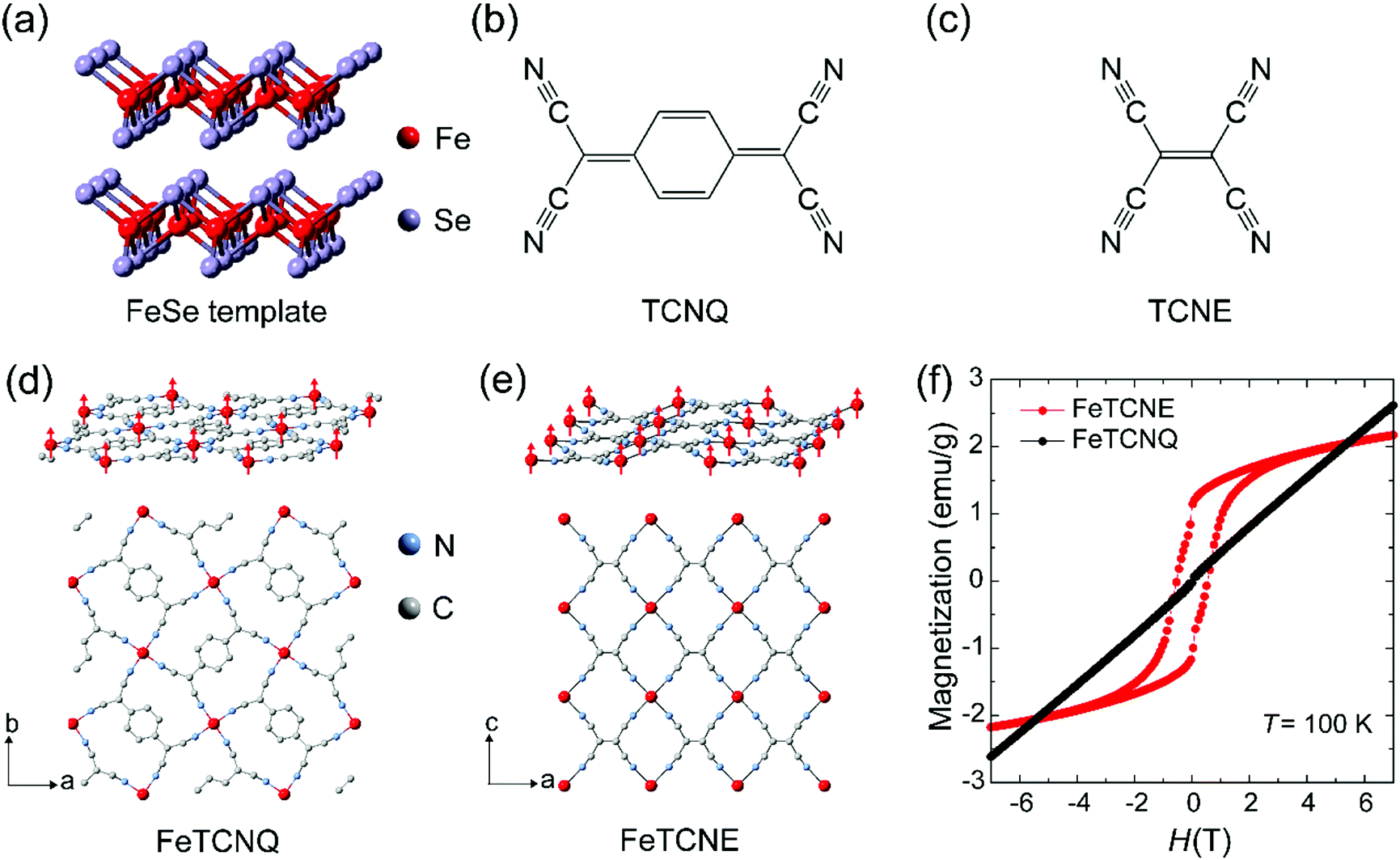 | ||
| Fig. 1 Crystal structures and magnetism of Fe–TCNX (X = Q and E). (a) The FeSe template shows the two-dimensional layered structure where four Se atoms surround Fe as the tetrahedral coordination in each layer. (b and c) Molecular structures of TCNQ and TCNE demonstrate that the molecular length of TCNE (4.189 Å) is smaller than that of TCNQ (7.757 Å). (d and e) Single-layer structures of Fe–TCNX illustrate the in-plane coordination of the Fe atom with cyano radicals. The bindings between layers and solvent molecules are not shown for clarity. Since molecules serve as bridges in the Fe network, all spins on Fe orient ferromagnetically in the ground state. Due to the smaller molecule size, the ferromagnetic coupling in Fe–TCNE with closer exchange interaction is stronger than that in Fe–TCNQ. (f) Magnetic field dependent magnetism of Fe–TCNE shows the large ferromagnetic hysteresis at 100 K above the Curie temperature of Fe–TCNQ of 60 K. | ||
The electron transfer from Fe to TCNE proceeds via the formation of Fe(II) ions and reduced TCNE radicals in a stoichiometry ratio, which leads to the tunability on spin coupling and magnetization of Fe:TCNE controlled under solution-based reaction conditions. The reaction of FeSe and TCNE is thermodynamically driven, while a higher temperature involves an enhanced Fe:TCNE stoichiometry ratio y (Table S1, ESI†) for enhanced ferrimagnetic coupling. Fig. 2a compares the MH data of Fe(TCNE)y samples synthesized at 368 K and 410 K. For the sample synthesized at 410 K, the MH loop features a wide magnetic hysteresis with a coercivity of 7500 Oe at 50 K. When increasing the magnetic field to 3 T, the magnetic hysteresis behavior fades away and the magnetism tends to be paramagnetic. However, once the synthesis temperature decreases to 368 K, the MH hysteresis loop narrows down, in which coercivity reduces to 1200 Oe and shows a kink at around zero magnetic field, in comparison to the sample synthesized at 410 K (Fig. 2a). Therefore, a high reaction temperature proceeds toward the product formation by building up Fe(TCNE)y coordination (Table S1, ESI†).
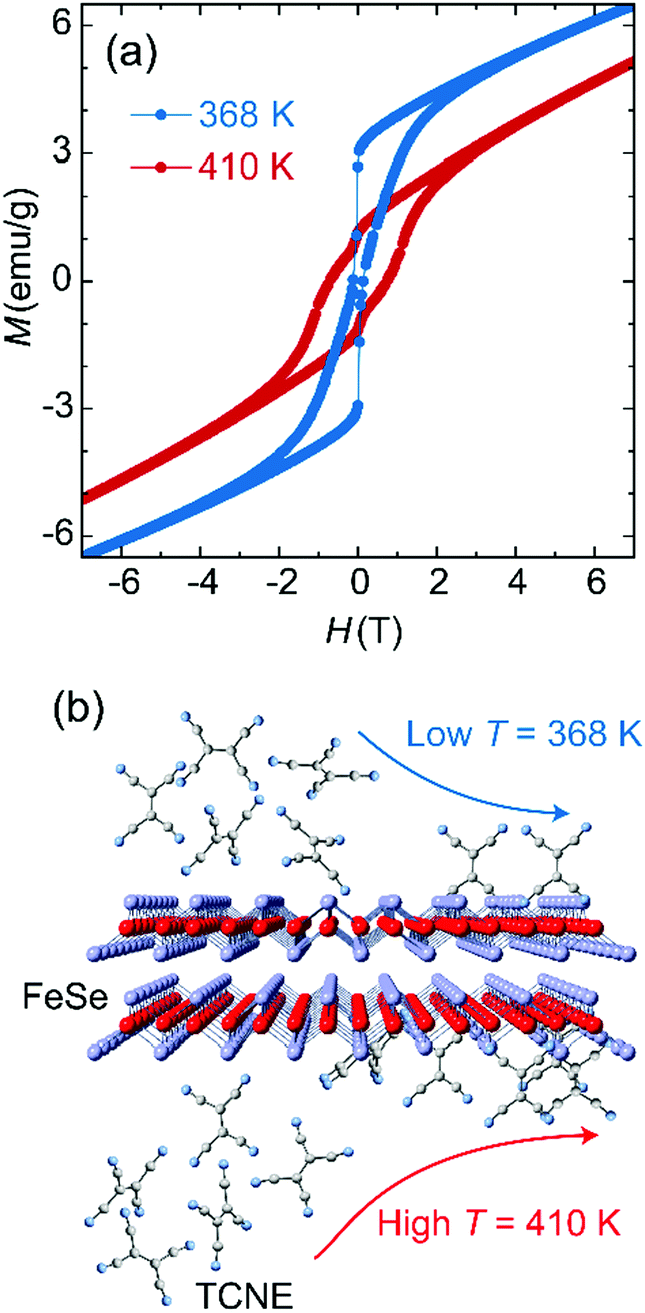 | ||
| Fig. 2 Temperature effects on synthesis of molecular ferromagnet Fe(TCNE)y. (a) Magnetic hysteresis loops of Fe(TCNE)y at 50 K show that higher synthesis temperature improves the coercivity and homogeneity. (b) Reaction progress of FeSe and TCNE could be activated by heat energy. High enough synthesis temperature can improve the substitution reaction to obtain ferrimagnetic Fe(TCNE)y. | ||
In addition, a low reaction temperature leads to a low coercivity that originates from a weak spin coupling interaction. The thermal energy improves the ferrimagnetism by enhancing the exchange interaction via more TCNE ligand connections with Fe atoms. Energy-dispersive X-ray spectroscopy analysis (EDS) on the Fe(TCNE)y sample synthesized at 368 K results in the TCNE stoichiometry of 1.00 (Table S1, ESI†). However, for a sample synthesized at 410 K, the Fe(TCNE)y molecular structure possesses a higher TCNE stoichiometry of 3.75 (Table S1, ESI†) that connects among Fe(II) cations to build up a strong ferrimagnetic coupling. The structure change by the TCNE stoichiometry can also be seen from the X-ray diffraction patterns (Fig. 3 and Fig. S1, ESI†). A wide range of TCNE concentrations in Fe(TCNE)y samples may be attributed to the multiple valence states of TCNE and the mixed stoichiometries under different reaction conditions. The IR spectra of Fe(TCNE)y present multiple absorption peaks at the vCN region (Fig. S2, ESI†). Particularly for the TCNE-rich sample, extra broad absorption at 2329 cm−1 indicates that neutral TCNE molecules24 exist in the Fe(TCNE)y sample. The thermal energy plays a vital role in the substitution reaction for the formation of Fe(TCNE)y. Fig. 2b schematically illustrates the dynamic process of the substitution reaction by TCNE ligands in FeSe. High reaction temperature promotes the reaction for the formation of the Fe(TCNE)y structure, while a sluggish reaction kinetics occurs at low temperature. A lower reaction temperature leads to uncompleted and few substitutions in FeSe that may form the magnetization kink at zero field.
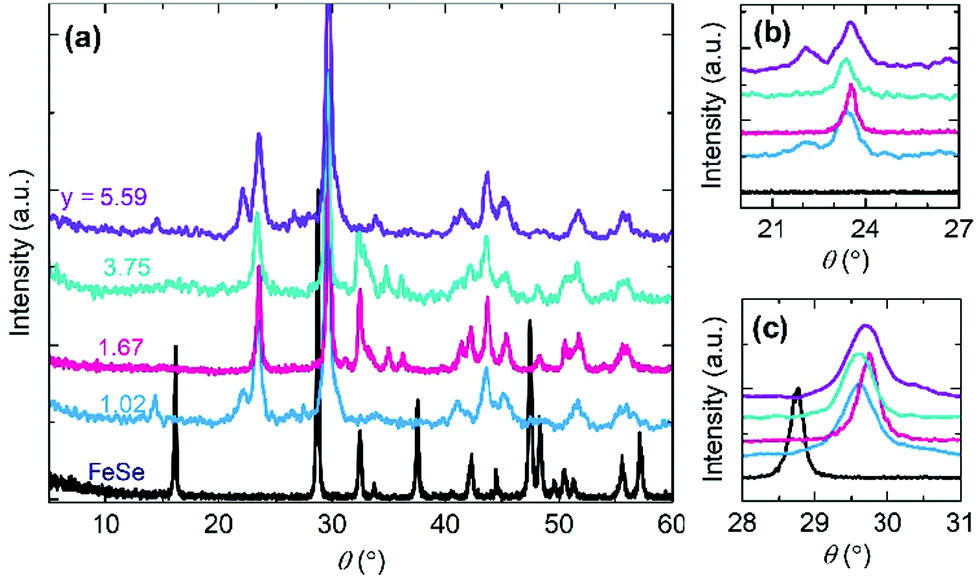 | ||
| Fig. 3 X-Ray diffraction patterns of Fe(TCNE)y synthesized at 410 K by a FeSe template. (a) XRD patterns of the whole range from 5° to 60° present the structure evolution from FeSe to Fe(TCNE)y (y = 1.02, 1.67, 3.75 and 5.59). (b) Enlarged area of XRD patterns from 20° to 27°. (c) Enlarged area of XRD patterns from 28° to 31°. | ||
The electron transfer between Fe and TCNE ligands appears to be an important role in the formation of molecular magnets. The other effective approach to control the electron transfer is to define an appropriate amount of TCNE acceptor in the reaction process. Through controlling the TCNE reactant amount in the reaction solution, the TCNE ratio y in Fe(TCNE)y varies in a large range from 1.02 to 5.59. Fig. 3 shows the X-ray diffraction (XRD) patterns of Fe(TCNE)y synthesized at 410 K with a variable TCNE ratio y. After the substitution reaction, the XRD patterns have considerable changes that a lot of new peaks appear compared to that of the FeSe template (Fig. 3a). At a lower angle, a small peak arises in the Fe(TCNE)1.02 compound indicating a larger crystal lattice than that of FeSe. Meanwhile, most peaks in the XRD patterns of Fe(TCNE)y cannot be tracked from the XRD pattern of FeSe, which could be attributed to the dramatic structure change. Two peaks appear at around 24° in the XRD patterns of Fe(TCNE)y where there should be no peaks for FeSe (Fig. 3b). In addition, the peak of highest intensity in Fe(TCNE)y at around 30° locates at a higher angle than that in FeSe (Fig. 3c). No apparent peak shift law was observed for these Fe(TCNE)y compounds that may result from the inhomogeneity of substitution. The complicated evolution of the XRD patterns of Fe(TCNE)y implies the significant crystal structure change that influences the spin coupling interaction.
The complex magnetic properties of the Fe(TCNE)y products at 100 K (Fig. 4a) reveal the effect of Fe:TCNE stoichiometry ratio y on electron transfer. For a low TCNE stoichiometry (y = 1.02), the MH loop is almost linear and it shows no magnetic hysteresis, suggesting paramagnetic nature. When y = 1.67 and 3.75, as we discussed in Fig. 2, the Fe(TCNE)y network shows a large magnetic hysteresis and remnant magnetization. By further improving the TCNE stoichiometry, the MH loop is linear without hysteresis. The stoichiometry significance can also be observed by the temperature dependent susceptibility (MT) of the as-synthesized Fe(TCNE)y (Fig. 4b). It shows the magnetic susceptibilities of four samples with different TCNE stoichiometry ratios. For the two Fe(TCNE)y samples where the paramagnetic phase is dominated (Fig. 4a), the M–T data show much lower susceptibilities compared to those of ferrimagnetic samples (Fig. S5 and S6, ESI†). Ferrimagnetic Fe(TCNE)y displays a high susceptibility at around 150 K. A small susceptibility upturn at low temperature could be attributed to a possible paramagnetic phase like Se. The tunability of ferromagnetism in Fe(TCNE)y is realized through the adjustment of the quantity of TCNE in the synthesis to engineer FeSe templates at different levels. When the reaction temperature remains constant, more TCNE ligands promote the formation of the Fe(TCNE)y network from a FeSe template. There is a rationale for the critical role of TCNE stoichiometry in the magnetism of Fe–TCNE, where the Se atoms are gradually and partially substituted by TCNE ligands. In order to understand the different reaction stages of FeTCNE, here we propose a magnetic cluster model to picture the magnetic properties after a substitution reaction under different TCNE concentrations. At a low concentration of TCNE, the reaction does not occur uniformly to transform FeSe into Fe(TCNE)y assembly. In this case, magnetic clusters of Fe(TCNE)y form short-range ferrimagnetic order and present weak magnetic behavior (stage I, Fig. 4c). This situation satisfies the sample of y = 1.02 in Fig. 4a that is paramagnetic in nature with nonlinear magnetization at around zero field. The XRD of the sample of y = 1.02 (Fig. 3a) indicates the lattice expansion due to the incidence of TCNE molecules. For the other two samples showing ferrimagnetism, the stoichiometry of the TCNE ligands enables the long-range ferrimagnetic order due to the uniform substitution (stage II and III, Fig. 4d and e). However, the sample of y = 3.75 with highly substituted TCNE ligands results in a slightly disordered coordination among layers in Fe(TCNE)y, leading to the paramagnetic behavior (stage III, Fig. 4e). The XRD pattern of the sample of y = 3.75 (Fig. 3) confirms its disordered feature in the Fe–TCNE network, which could further interrupt the spin lattice and break the ferrimagnetic order (stage IV, Fig. 4f). The strong charm of this work is not only to synthesize ferrimagnetic Fe(TCNE)y of high Tc, but also the various molecular assembly structures by TCNE substitution at different levels. In stage I, only a few TCNE ligands are coordinated in the FeSe framework that is analogous to a molecular heterostructure in different concentrations of TCNE. This hybrid structure may also be the reason for the kink behavior observed in the MH loops of Fe(TCNE)y (Fig. 2a and Fig. S3, S4, ESI†), especially in those samples synthesized at low temperature (Fig. S3, ESI†). Such molecular heterostructures can tune the magnetism by interface and lead to high temperature molecular magnets, which is often contributed to the optimum stoichiometry between TCNE radical anions and metal cations.5
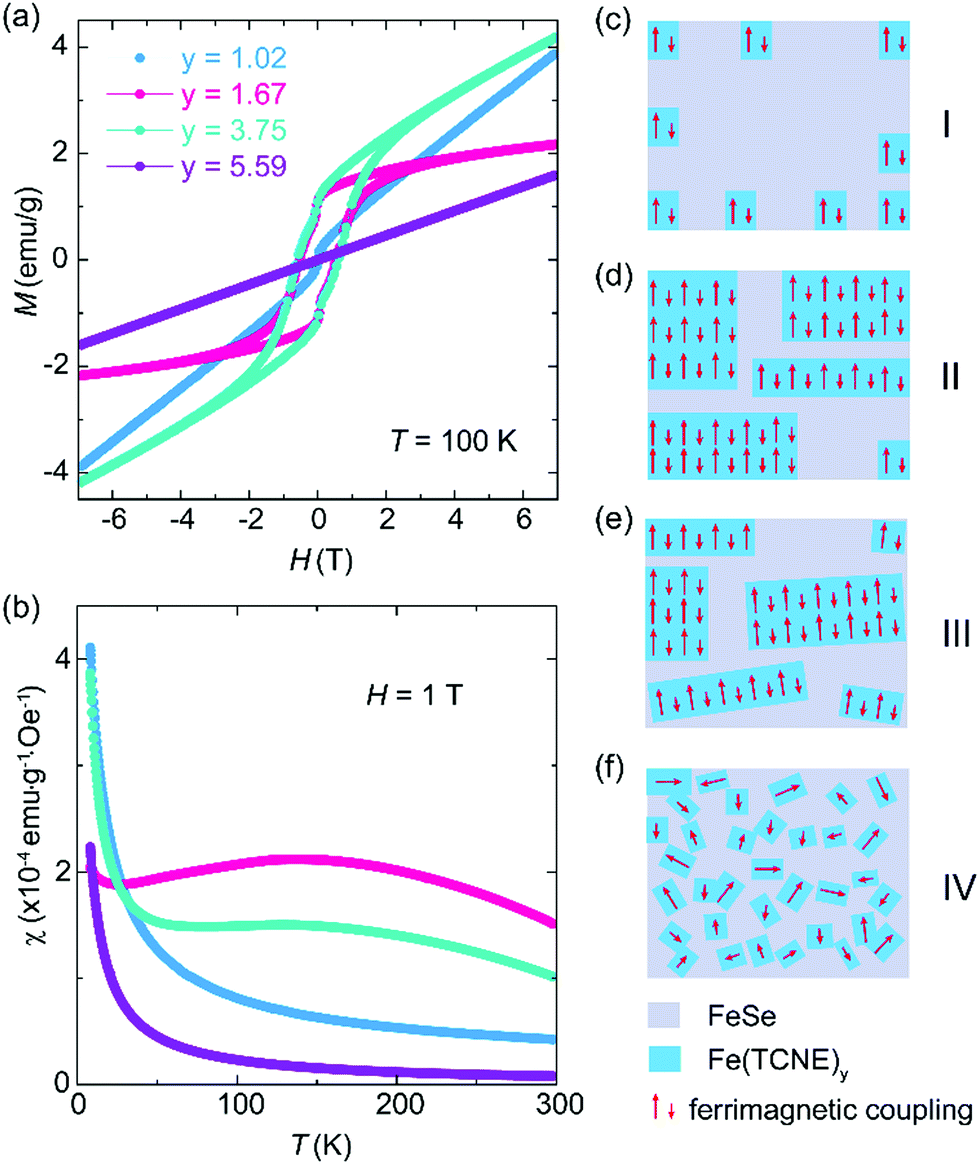 | ||
| Fig. 4 The evolution of the magnetic hysteresis loop of Fe(TCNE)y samples synthesized at 410 K by different TCNE stoichiometry y = 1.02, 1.67, 3.75 and 5.59 which correspond to four stages of spin configuration in Fe(TCNE)y. (a) Too low (y = 1.02) or too high (y = 5.59) TCNE stoichiometry makes a paramagnetic phase, and only appropriate ratios of y = 1.67 and 3.75 result in a ferrimagnetic coupling. (b) The temperature dependent susceptibilities of these four samples in (a) show low (paramagnetic) and high (ferrimagnetic) susceptibilities. (c–f) Schematic figures for different reaction stages of diverse spin configurations according to the TCNE reactant concentration at the synthesis temperature of 410 K. | ||
Conclusions
In conclusion, we design and synthesize a high temperature Fe–cyanide-based molecular magnet via a chemical reaction between 2D FeSe and molecular oxidizing agent TCNX. The shorter TCNE molecules enhance the spin coupling between Fe and cyanide, presenting a higher ordering temperature. The electron transfer induced spin coupling in a molecular magnet enables the design of tunable magnetic interaction by the control of the spin lattice and stoichiometry ratios. The reaction conditions, such as temperature and TCNE concentrations, are vital in tuning the finial structure of Fe(TCNE)y after the substitution reaction that determines the long-range ferrimagnetic coupling and ordering temperature of even possible above room temperature (Fig. S7, ESI†). The selection of the FeSe precursor complex and small cyanide molecules provides a new pathway to synthesize high temperature molecular magnets and molecular interface devices that benefit the potential applications in spin electronics.25Experimental section
Sample preparation
The synthesis of Fe–TCNE was conducted in a 20 ml glass bottle which contains 0.8 mmol FeSe powder, 0.4–6.4 mmol TCNE and 10 ml acetonitrile solvent. All chemicals were sealed in the glass bottle which later on was heated at a synthesis temperature of 368 K or 410 K on a hotplate. A small magnetic stir bar was added into the reaction solution and stirred at 1000 rpm to increase the reaction rate. Three days later, the product was washed with acetonitrile and then centrifuged to condense in the bottom of the centrifuge tube. After the product was dried in a fume hood, the following measurements were taken on those samples.Structural and morphologic characterization
X-Ray diffractions of powder samples were carried out on a Rigaku Ultima IV (40 kV, 44 mA) to characterize the crystal structure evolution due to the TCNE substitution. A Carl Zeiss AURIGA (200 kV) with an Oxford Energy-dispersive X-ray Spectrometer (EDS) was used to collect the surface morphology and element ratios of the samples obtained under different reaction conditions.Magnetic susceptibility and hysteresis loop measurements
Magnetic property measurements were conducted on a Physical Properties Measurement System (PPMS) equipped with a Vibrating Sample Magnetometer (VSM) option. After cooling at zero magnetic field, magnetic susceptibility under different magnetic fields was measured from 8 K to 300 K. Magnetic hysteresis loops were measured at fixed temperatures by sweeping the magnetic field from 7 T to 7 T.Infrared spectra
A Bruker Alpha IR spectrometer with ALPHA-P Platinum ATR module (diamond crystal) is used to obtain transmission ATR-FTIR spectra under an argon atmosphere in a glove box.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering supports S. R. under Award DE-SC0018631 (Organic conductors). Financial support was provided by the U.S. Army Research Office support of S. R. under Award W911NF-18-2-0202 (Materials-by-Design and Molecular Assembly).References
- J. A. Valdez-Moreira, A. E. Thorarinsdottir, J. A. DeGayner, S. A. Lutz, C. H. Chen, Y. Losovyj, M. Pink, T. D. Harris and J. M. Smith, J. Am. Chem. Soc., 2019, 141, 17092–17097 CrossRef CAS PubMed.
- L. M. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325–334 CrossRef CAS PubMed.
- E. B. Vickers, T. D. Selby and J. S. Miller, J. Am. Chem. Soc., 2004, 126, 3716–3717 CrossRef CAS PubMed.
- E. B. Vickers, I. D. Giles and J. S. Miller, Chem. Mater., 2005, 17, 1667–1672 CrossRef CAS.
- R. Jain, K. Kabir, J. B. Gilroy, K. A. Mitchell, K. C. Wong and R. G. Hicks, Nature, 2007, 445, 291–294 CrossRef CAS PubMed.
- J. S. Miller, Mater. Today, 2014, 17, 224–235 CrossRef CAS.
- J. M. Manriquez, G. T. Yee, R. S. McLean, A. J. Epstein and J. S. Miller, Science, 1991, 252, 1415–1417 CrossRef CAS PubMed.
- M. A. Girtu, C. M. Wynn, J. Zhang, J. S. Miller and A. J. Epstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 492–500 CrossRef CAS.
- D. A. Pejakovic, C. Kitamura, J. S. Miller and A. J. Epstein, Phys. Rev. Lett., 2002, 88, 057202 CrossRef PubMed.
- C. Tengstedt, M. P. de Jong, A. Kanciurzewska, E. Carlegrim and M. Fahlman, Phys. Rev. Lett., 2006, 96, 057209 CrossRef CAS PubMed.
- J. W. Yoo, C. Y. Chen, H. W. Jang, C. W. Bark, V. N. Prigodin, C. B. Eom and A. J. Epstein, Nat. Mater., 2010, 9, 638–642 CrossRef CAS PubMed.
- L. Fang, K. D. Bozdag, C. Y. Chen, P. A. Truitt, A. J. Epstein and E. Johnston-Halperin, Phys. Rev. Lett., 2011, 106, 156602 CrossRef PubMed.
- K. I. Pokhodnya, N. Petersen and J. S. Miller, Inorg. Chem., 2002, 41, 1996–1997 CrossRef CAS PubMed.
- J. Zhang, J. Ensling, V. Ksenofontov, P. Gutlich, A. J. Epstein and J. S. Miller, Angew. Chem., Int. Ed., 1998, 37, 657–660 CrossRef CAS PubMed.
- K. I. Pokhodnya, V. Burtman, A. J. Epstein, J. W. Raebiger and J. S. Miller, Adv. Mater., 2003, 15, 1211–1214 CrossRef CAS.
- J. S. Miller and M. Drillon, Magnetism: Molecules to Materials V, Wiley-VCH, 2005, ch. 9, p. 294 Search PubMed.
- J. W. Yoo, V. N. Prigodin, W. W. Shum, K. I. Pokhodnya, J. S. Miller and A. J. Epstein, Phys. Rev. Lett., 2008, 101, 197206 CrossRef PubMed.
- K. I. Pokhodnya, M. Bonner, J. H. Her, P. W. Stephens and J. S. Miller, J. Am. Chem. Soc., 2006, 128, 15592–15593 CrossRef CAS PubMed.
- A. Zheludev, A. Grand, E. Ressouche, J. Schweizer, B. G. Morin, A. J. Epstein, D. A. Dixon and J. S. Miller, J. Am. Chem. Soc., 1994, 116, 7243–7249 CrossRef CAS.
- Y. Ma, Y. Dai, W. Wei, L. Yu and B. Huang, J. Phys. Chem. A, 2013, 117, 5171–5177 CrossRef CAS PubMed.
- X. Zhang, M. R. Saber, A. P. Prosvirin, J. H. Reibenspies, L. Sun, M. Ballesteros-Rivas, H. Zhao and K. R. Dunbar, Inorg. Chem. Front., 2015, 2, 904–911 RSC.
- A. C. McConnell, E. Shurdha, J. D. Bell and J. S. Miller, J. Phys. Chem. C, 2012, 116, 18952–18957 CrossRef CAS.
- J. H. Her, P. W. Stephens, K. I. Pokhodnya, M. Bonner and J. S. Miller, Angew. Chem., Int. Ed., 2007, 46, 1521–1524 CrossRef CAS PubMed.
- W. Kaim and M. Moscherosch, Coord. Chem. Rev., 1994, 129, 157–193 CrossRef CAS.
- L. Guo, X. Gu, X. Zhu and X. Sun, Adv. Mater., 2019, 31, e1805355 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00173b |
| This journal is © The Royal Society of Chemistry 2020 |