
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentrated aqueous dispersions of low-defect few-layer thick graphene using surface active ionic liquid for enhanced enzyme activity†
Gagandeep
Singh‡
a,
Manvir
Kaur‡
a,
Gurbir
Singh
ab,
Komal
Arora
a,
Manpreet
Singh
a,
Bilal A.
Sheikh
 a and
Tejwant S.
Kang
*a
a and
Tejwant S.
Kang
*a
aDepartment of Chemistry, University Grants Commission (UGC) Centre for Advanced Studies-II, Guru Nanak Dev University, Amritsar-143005, Punjab, India. E-mail: tejwant.chem@gndu.ac.in; tejwantsinghkang@gmail.com; Tel: +91-183-2258802 ext. 3291
bDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 11th July 2020
Abstract
Low energy ultrasonic assisted aqueous phase exfoliation of defect free graphene (D/G = 0.19) in a high yield (3.0 mg ml−1) is achieved using benzimidazolium based surface active ionic liquid (SAIL) appended with an ester functionalized alkyl chain. The obtained dispersions are found to be stable against agglomeration or sedimentation for at least 15 days, owing to coulombic repulsions offered by electron deficient π-conjugated SAIL ions adsorbed onto graphene. The exfoliated few-layer thick graphene sheets (2 layers 53% and 3–5 layers 20%) offer a high surface area in conjunction with high concentration, which is exploited for the efficient loading of an enzyme, cellulase, via hydrophobic interactions. The graphene appended cellulase is found to exhibit a remarkable increase in the enzyme activity (13-fold) as compared to cellulase in the buffer. It is expected that the present results would act as a platform for the synthesis and utilization of new SAILs for graphene exfoliation, stabilization, and functionalization required to widen the application arena of graphene.
Introduction
Since its introduction, graphene,1 owing to its unique physicochemical properties,2–8 has attracted great interest for a wide array of applications.9–11 One such application is the use of graphene as a support for enzyme loading owing to its large surface area. In past, different enzymes such as lipase,12,13 horseradish peroxidase,14,15 and cellulase16,17 have been successfully immobilized onto graphene oxide (GO) via electrostatic12,15,16 or covalent interactions13,14 due to the presence of functional groups on GO; however, graphene has not been investigated for the same purpose. The immobilization of enzymes onto GO has been marked by improved thermal13,15 and solvent stability of enzyme along with reusability.16 Such immobilized enzymes retains enzymatic activity, however, a decrease in the enzyme activity has been reported as compared to the bare enzyme, particularly in the case of enzymes adsorbed onto GO via a covalent linkage.18 Alterations in the secondary (2°) or tertiary (3°) structure of enzymes and hence the deformation of the active site of an enzyme at the surface of GO could be a reason for decreased enzymatic activity.14 On the other hand, the enzyme adsorbed physically on GO has been reported to exhibit higher activity as compared to that tethered covalently.14It is conceived that defect-free graphene instead of GO would offer a more suitable surface with the high surface area for enzyme loading via physical-adsorption exploiting hydrophobic interactions. Such weaker interactions are not expected to alter the microenvironment of the active site of an enzyme. Further, the aqueous solutions of well-dispersed single-layered or a few-layered thick graphene would result in a higher surface area required for efficient enzyme loading. The high stability of graphene dispersion against agglomeration and phase separation is another critical aspect to achieve the best enzyme activity as agglomeration and phase separation of enzyme-loaded graphene would decrease the concentration of enzyme dispersed in an aqueous medium, hence decreasing the enzyme activity. Therefore, new eco-friendly and cost-effective methods for the exfoliation of very thin defect-free graphene in an aqueous medium with high dispersion stability and subsequent enzyme loading, where enzymes show enhanced activities, should be devised to enhance the application arena of graphene.
In past, different methods1,19–23 have been employed for the preparation of graphene from graphite; however, these methods have certain limitations. The ultrasonic-assisted exfoliation of graphene in organic solvents of appropriate surface energy is another route that produces high quality graphene,24–29 but this process is limited by the high toxicity of organic solvents. Ionic liquids (ILs), an emerging class of green solvents, have also been used for graphene exfoliation30,31 but they confront the problem of high cost when used neat. Therefore, the best solvent for the liquid phase exfoliation of graphene is water, where the surface energy is lowered by the addition of surfactants that not only helps in exfoliation but also assists in the long-term stabilization of the colloidal solutions of graphene against agglomeration.32–39 Although this method seems to be most appropriate for the mass production of graphene, a low concentration (<0.5 mg ml−1) of exfoliated graphene in aqueous-conventional surfactant solutions is the major drawback.
Herein, we report the direct exfoliation of graphene in an aqueous medium up to high concentrations (3.0 mg ml−1), which exhibits long-term stability towards agglomeration, using ester-functionalized benzimidazolium based surface active IL (SAIL) (Scheme 1).
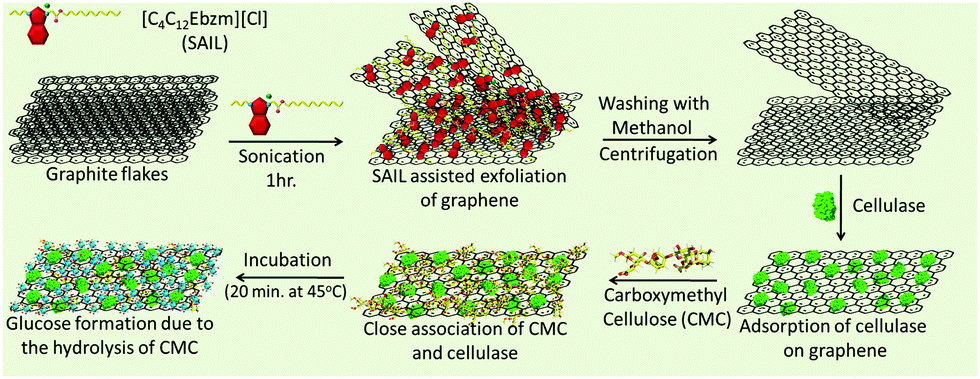 | ||
| Scheme 1 SAIL-assisted exfoliation of graphene and loading of cellulase on exfoliated graphene. | ||
The better surface active properties and designer nature of SAILs40–48 over conventional ionic surfactants prompted us to evaluate the graphene exfoliation efficiency and stability of the colloidal dispersions of graphene in aqueous SAILs. The ease of the synthetic procedure40 of SAIL would be beneficial in scaling-up the process required for the mass-production of graphene in a cost-effective way. The employed SAIL also exhibits a low critical aggregation concentration (cac)40 of ≈1.4 mmol L−1 in an aqueous medium, which also would add to the economy of the exfoliation process as the maximum exfoliation of graphene has been found to be achieved around cac of conventional ionic surfactants.33 The choice of the benzimidazolium cationic head group in SAIL is governed by its planner and π-conjugated electron deficient nature, which would lead to the efficient adsorption of SAIL onto graphene surface via π–π interactions supplemented by hydrophobic interactions, required for the desirable exfoliation of graphene. Further, H-bonding prone ester-moiety is expected to anchor with graphene sheets while retaining the hydrating water, which not only would help in exfoliation but would also add to the colloidal stability. In this way, the present study represents a new and efficient low energy ultrasonic-assisted method for the mass production of highly concentrated, relatively defect-free few-layered graphene in an aqueous medium exhibiting desired colloidal stability for different applications. The exfoliated graphene is utilized as a support for the loading of cellulase, where a 13-fold increase in an enzyme activity is observed as compared to the bare enzyme. At first, we shall discuss the efficiency of the exfoliation process and the quality of exfoliated graphene, followed by its utilization as support for cellulase loading and role in enhanced enzyme activity.
Result and discussion
The detailed aspects of graphene exfoliation and characterization are provided in the Experimental section (Annexure S1, ESI†). The absorption spectra of graphene (Fig. 1A) dispersion were measured and found to be flat and featureless as expected for quasi-2D materials.49 The absorption coefficient, α, which is an indicator of dispersion stability, is found to be 747 ml mg−1 m−1, which is in good agreement with that reported previously.50 The concentration of exfoliated graphene in aqueous supernatant was calculated by the careful measurement of the filtered mass of graphene, after drying, accounting for the mass of residual SAIL. The amount of exfoliated graphene was found to increase with the increase in the concentration of SAIL up to cac, which decreases thereafter (Fig. 1B). At concentrations much lower or higher than cac, the dispersions were found to be phase-separated just after 24 hours (Fig. S1, ESI†). As shown in Fig. 1C, at optimum [SAIL] = 1.0 mM, a graphene dispersion of 3.0 mg ml−1 is obtained. It is important to mention that the cac of SAIL is found to be shifted from 1.4 mM to 1.0 mM in the presence of graphene. During the exfoliation of graphene in an aqueous medium, the hydrophobic surface of graphene sheets are in great demand for the adsorption of SAIL moieties. Relatively higher concentration of SAIL on the graphene surface as compared to that present in bulk resulted in the advancement of the self-assembly of SAIL similar to that reported in literature.51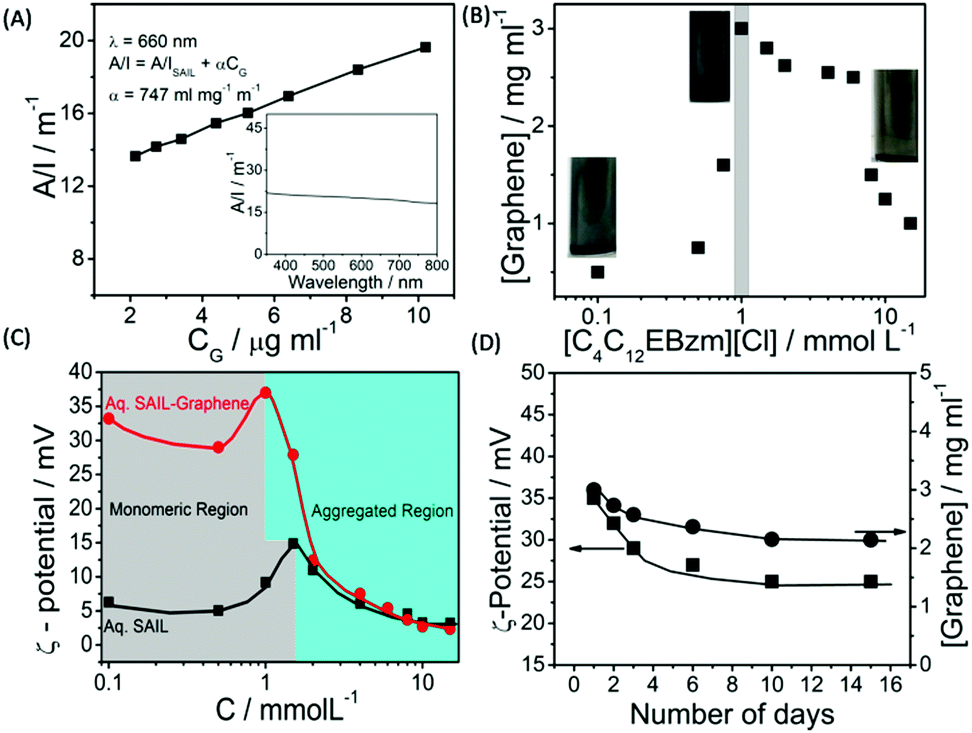 | ||
| Fig. 1 Absorbance per unit length at 660 nm as a function of graphene concentration (CG) (Inset shows the absorbance spectra of graphene dispersion) (A); effect of the concentration of SAIL on the extent of graphene exfoliation (B); the variation of ζ-potential in aqueous SAIL solutions in the absence and presence of graphene (C) and the variation of ζ-potential and residual content of exfoliated graphene in aqueous dispersions as a function of time (D) at 298.15 K. As per the law of mass action, the number of surfactant monomers below cac in a solution is equal to cac,52 the increase in the number of which increases the extent of penetration in graphene layers and hence greater exfoliation is observed. | ||
The obtained concentration of graphene is much higher than that reported for conventional ionic surfactants (0.1–0.5 mg ml−1).33–39 The content of exfoliated graphene is higher than that obtained using large ionic aromatic molecules (>0.1 mg ml−1)53–57 and then that obtained (0.1–1 mg ml−1) for the exfoliation of GO expanded by the intercalation of H2SO4/HNO3.58 It has been shown that surfactants having ionic groups attached to an electron deficient π-conjugated unit through alkyl spacers exhibit high tendency to exfoliate (1.2–5.0 mg ml−1) graphene depending on the centrifugation rate.59 The used SAIL is highly efficient in promoting the exfoliation of graphene without oxidation in high concentrations in an economical manner. This is attributable to the presence of π–π interactions between graphene and aromatic benzimidazolium ring supplemented by the hydrophobic and electrostatic forces of interaction with graphene.
Further, the similarity in concentration of SAIL at which maximum graphene exfoliation is observed to that where the maximum magnitude of ζ-potential is observed (Fig. 1C) confirms the electrostatic repulsion between adsorbed SAILs onto graphene as the driving force behind colloidal stability. Owing to the flexible nature provided by the ester moiety,44 SAIL could adopt varying orientations for an optimal contact with graphene, resulting in efficient exfoliation. Further, the ester-group is expected to retain hydrating water molecules along with SAIL via H-bonding to provide colloidal stability to exfoliated graphene.
Thus, formed graphene dispersions after centrifugation ([SAIL] = 1.0 mM, [CG] = 3.0 mg ml−1) were tested for stability against agglomeration and sedimentation. The formed graphene–SAIL dispersions were found to be stable for at least 15 days against agglomeration as suggested by the negligible change in the concentration of graphene in dispersion (Fig. 1D). However, initially, the content of dispersed graphene decreases by ≈17% from 3.0 mg ml−1 to 2.5 mg ml−1 for 2 days. Further, ζ-potential, which is an important parameter to monitor the colloidal stability, is found to be +35 mV, which is much above than the colloidal stability limit of +25 mV. It is to be noted that the ζ-potential of the graphene dispersion is found to be more than twice of that observed in the absence of graphene at the SAIL concentration where maximum exfoliation takes place (Fig. 1D). This shows the efficient adsorption of SAIL ions on to the graphene surface, required for efficient exfoliation and colloidal stability (Scheme 1).
The exfoliated graphene was characterized via powder X-ray diffraction, Raman spectroscopy, transmission electron microscopy (TEM), and atomic force microscopy (AFM) techniques. An efficient exfoliation of graphene is suggested via XRD measurements, where in the case of exfoliated graphene only a broad band is observed as compared to a sharp diffraction peak at 26.5° in the case of graphite (Fig. 2A). Fig. 2B shows the Raman spectra obtained for exfoliated graphene sheets. The presence of G (1580 cm−1) and D (1350 cm−1) bands is assigned to the sp2-hybridized nature of carbon of graphene. The appearance of a relatively stronger D-band in graphene as compared to that of graphite indicates the presence of edge or basal defects; however, a lower intensity ratio of D/G bands (D/G = 0.190) suggests the smaller extent of these defects33,60 as compared to that observed in the presence of sodium cholate (D/G = 0.57, on average).34 The shift and shape of the 2D band of graphene is peculiar of a few-layer thick (<5 layers) graphene,33,59 while the shift in the G-band attributes to the change in the electronic structure of graphene due to the π–π interactions between graphene and aromatic benzimidazolium ring.
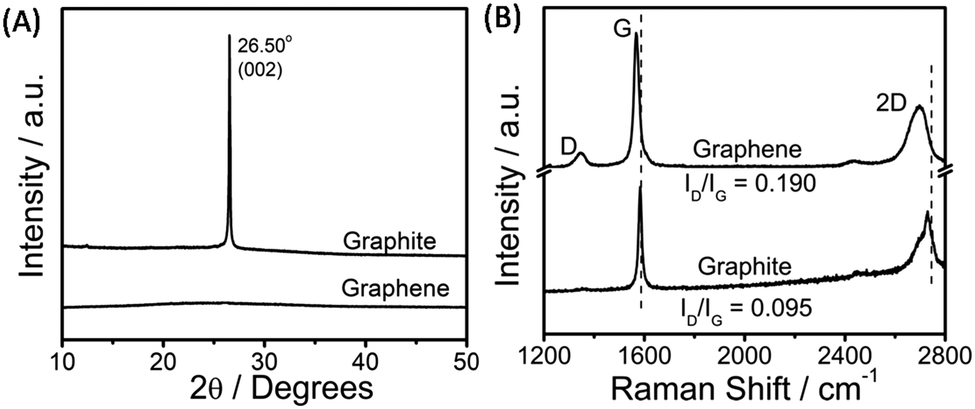 | ||
| Fig. 2 X-ray diffraction pattern (A) and the obtained Raman spectra (B) for pristine graphite and exfoliated graphene. | ||
Fig. 3(A–D) shows the TEM images of exfoliated graphene flakes of different thickness along with selected area electron diffraction (SAED) pattern. High resolution TEM image (Inset Fig. 3A and C) of very thin graphene flakes suggest the presence of 3–4 layered graphene. The observed SAED (Fig. 3D) pattern is peculiar of the hexagonally arranged lattice of graphene, where the higher intensity of the second diffraction layer as compared to the first one suggests the exfoliation of two/three layered thick graphene.60
 | ||
| Fig. 3 TEM images of exfoliated graphene as long flakes (A); smaller but few-layer thick flake (B); high resolution TEM images near the edge of a few-layer graphene flake showing the number of graphene layers (C); and SAED pattern of exfoliated graphene flakes (D). The inset of (A) shows the number of graphene layers as observed at the edge of an exfoliated graphene flake. | ||
To statistically analyze the size distribution of graphene flakes, AFM measurements (Fig. 4A–C) have been carried out after depositing the graphene on mica sheets. Graphene sheets with a lateral size of 50–500 nm have been obtained with a mean lateral size of ≈150 nm. The analysis of AFM images (Fig. 4B and C) suggests that ≈45% of the flakes comprised two layers, exhibiting a thickness of 0.8–1.2 nm. 42% of the analyzed flakes are found to be few-layered thick with a thickness of 1.2–3.0 nm.
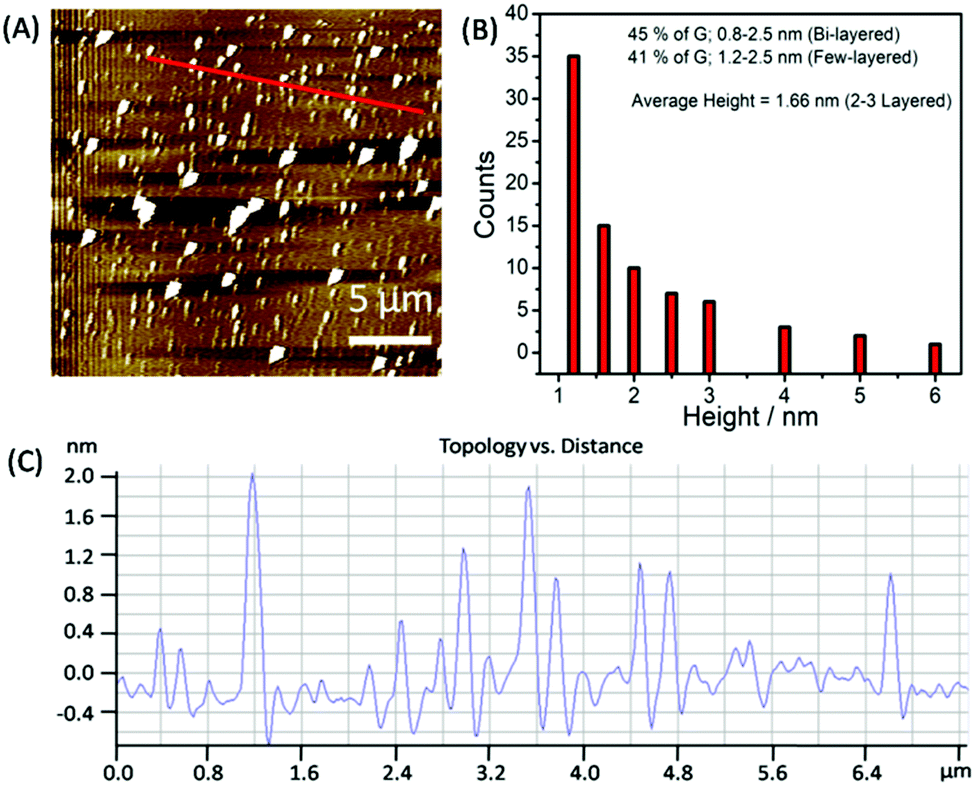 | ||
| Fig. 4 AFM image of the exfoliated graphene flakes (A); the statistical analysis of the obtained height profile (B) and surface topology vs. the distance profile of exfoliated graphene flakes (C) corresponding to the red line shown in (A). | ||
To probe the mechanism of the SAIL-mediated exfoliation of graphene, high disperse-ability and colloidal stability, steady-state fluorescence, 2D 1H–1H NMR measurements, and computational investigations were performed. A decrease in the intensity of inherent fluorescence exhibited by SAIL40 with an increase in the content of dispersed graphene is observed (Fig. S2, ESI†). This is attributed to the interactions of the π-conjugated electron-deficient benzimidazolium head group of SAIL aided by its planer nature with the electron-rich and planner surface of graphene. The appearance of 2D 1H–1H NOESY cross-peaks between protons of the cationic head group and hydrophobic alkyl chain of SAIL in the presence of graphene, which otherwise are absent suggests their adsorption at the graphene surface (Fig. S3, ESI†). The calculated absorption free energy of SAIL onto the surface of graphene comes out to be −13.76 kcal mol−1, which comes out to be close to that observed for the naphthalene diimide-based ionic surfactant having a carboxylate ionic head group and more than that of conventional ionic surfactants.59 This suggests a high affinity of SAIL towards the graphene surface. Both the reported surfactants59 and SAIL used in this work have the π-conjugated electron-deficient system; however, the extent of conjugation in case of SAIL is less, even then, an appreciable absorption energy has been observed. This is attributable to the presence of the relatively flexible ester moiety near the ionic head group by virtue of which the ionic head group could adopt different orientations for efficient absorption at the graphene surface. The retention of H-bonded water with the H-bonding prone ester moiety is expected to retard agglomeration.
Further, SAIL was removed from exfoliated graphene via the method mentioned in Annexure S1, ESI,† and the removal of SAIL has been confirmed via the thermogravimetric analysis (Fig. S4, ESI†). Thus, obtained graphene is used as nano-support for the loading of cellulase, and enzyme activity of the loaded cellulase is investigated. Cellulase has been immobilized on graphene using a cellulase buffer solution at pH 4.8. The TEM image shown in Fig. 5A suggests the adsorption of cellulase on graphene, which is also supported via AFM measurements (Fig. S5 and S6, ESI†). The presence of lattice planes having a distance of 0.261 nm indicates the crystalline nature of cellulase adsorbed on graphene with an interplanar distance of 0.34 nm (Fig. S7A, ESI†).
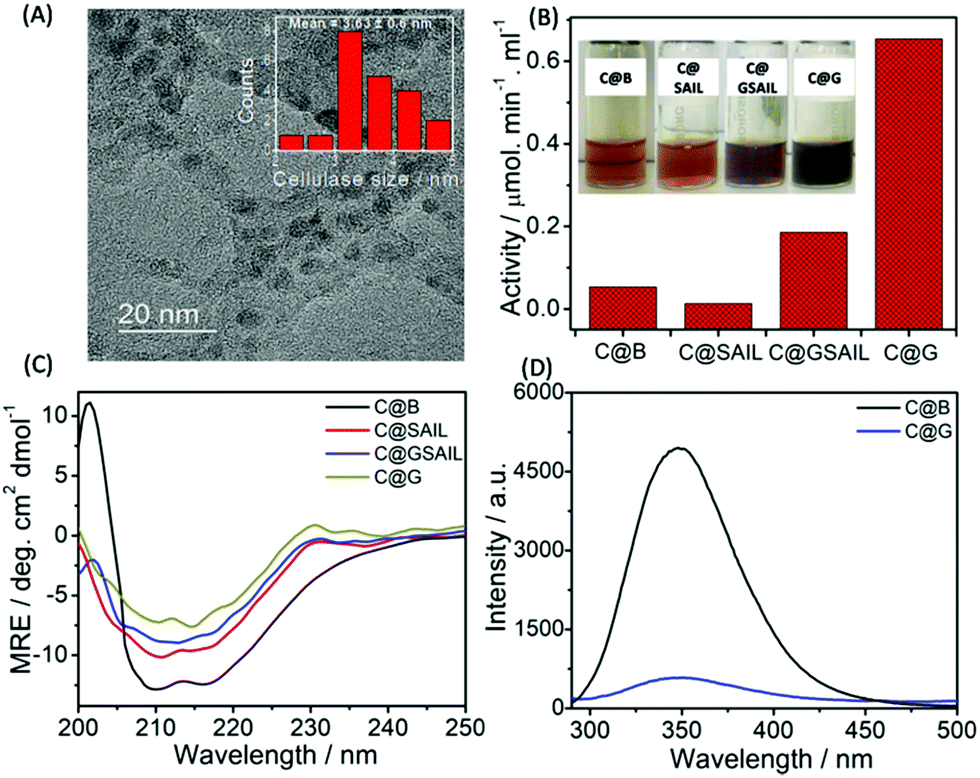 | ||
| Fig. 5 TEM image of C@G (A); activity of cellulase towards CMC adsorbed on washed graphene, on graphene with SAIL, in the buffer and in the SAIL aqueous medium (B); far UV CD spectra of native cellulase, cellulase adsorbed on graphene, on graphene with SAIL and in the SAIL aqueous medium (C); and intrinsic fluorescence of native cellulase and cellulase adsorbed on washed graphene (D). (Inset of A shows the histograms showing the mean particle size of cellulase along with standard deviation.) | ||
The functional stability of cellulase adsorbed onto the surface of graphene (C@G) is probed by measuring its enzyme activity towards carboxymethyl cellulose (CMC) (Annexure S1 and S2, ESI†) and is compared with that observed in the case of cellulase dispersed in the buffer (C@B), in aqueous SAIL solution (C@SAIL) and in the aqueous dispersions of SAIL and graphene (C@GSAIL) (Fig. 5B). The enzyme activity is found to be remarkably enhanced in the case of C@G (13-fold), whereas it enhances appreciably in the case of C@GSAIL (3.4-fold) as compared to that observed in the case of C@B. On the other hand, a decrease in the enzyme activity is observed in the case of G@SAIL.
A 13-fold increase in the enzyme activity in the case of C@G is assigned to favorable conformational changes in secondary (2°) structure of cellulase, which opens up the active sites of enzyme resulting in enhanced enzyme activity. The conformational changes in cellulase in the form of C@G are confirmed from far-UV CD measurements (Fig. 5C). The 2°-structure of cellulase mainly comprises α-helices, β-sheets and, turns, and the bands present at the −θ209 and −θ217 corresponds to the presence of α-helices and β-sheets, respectively.61 The alterations in the 2°-structure of cellulase in the case of C@G are evidenced by a decrease in the magnitude of MRE values corresponding to −θ209 and −θ217 along with the change in the shape of CD spectra owing to the unfolding of cellulase. Further, a drastic decrease in the inherent fluorescence intensity of cellulase, which mainly arises from 7 Tryptophan residues present at various sites of enzyme,62 is observed when cellulase is appended with graphene (Fig. 5D). This indicates the destabilization of the 3°-structure of cellulase owing to hydrophobic interactions with graphene in the case of C@G, which exposes the Trp residues towards a relatively more polar environment. Two each of the Trp residues are present in the active site tunnel of cellulase and near the catalytic site, which provides hydrophobic stacking interactions for glycosyl units and are essential for substrate loading.63 Therefore, it is natural to assume that the unfolding of cellulase modifies the active as well as the catalytic site of an enzyme for efficient substrate binding and enzymatic activity. The fluorescence bands of cellulase and SAIL overlap with each other in the case of C@SAIL and C@GSAIL (Fig. S8, ESI†). Therefore, it was difficult to discuss the structural stability of cellulase in presence of SAIL using fluorescence measurements. The relatively lesser increase in the enzyme activity in the case of C@GSAIL is assigned to the presence of SAIL. Similar to earlier reported SAIL–protein systems, SAIL is expected to interact not only with cellulase but also with the substrate (CMC)64via electrostatic and hydrophobic interactions. The interactions of SAIL in C@SAIL with cellulase results in structuring rather than unfolding of cellulase marked by an increase in the magnitude of MRE values, corresponding to −θ209 and −θ217 with the decreased content of turns. This results in the decreased enzyme activity as compared to that observed in buffer (Fig. 5B). In the case of C@GSAIL, there work two forces, one is the hydrophobic interactions of cellulase with the graphene surface, which favors increased enzyme activity and the other is structuring of the enzyme via interactions with SAIL, which disfavors the enzyme activity. The dominance of the graphene effect in the case of C@GSAIL results in increased enzyme activity but to a relatively lesser extent as compared to C@G.
Further, the colloidal stability of the C@GSAIL dispersion has been found to be at least 15 days due to the presence of electrostatic repulsions between SAIL ions adsorbed on graphene. However, colloidal stability reduces to three days in the absence of SAIL (C@G). The observed stability of the C@G dispersion could be assigned to the presence of highly hydrated cellulase at the surface of graphene. Even though C@G dispersion possesses a lower colloidal stability, the enzymatic activity of cellulase in C@G is retained, up to 41% of its original activity, after a period of 15 days (Fig. S9, ESI†). It is important to mention that the enzyme activity decreases both in C@G as well as in C@B with time. The observed activity is still higher than that observed for C@B. Hence, the prepared C@G would lay a new platform to establish systems with enhanced enzymatic activity. In this way, a new approach for the efficient exfoliation of defect-free graphene in high concentration for achieving remarkably enhanced enzyme activity is developed.
Conclusions
Surface active ionic liquid (SAIL) having a positively charged benzimidazolium based aromatic head group appended with an ester-functionalized alkyl chain is used to achieve highly stable and concentrated aqueous dispersion of few-layer thick graphene. In comparison to conventional surfactants, the exfoliation of graphene with the used SAIL is better even at a very low concentration of SAIL. Further, the formed defect-free graphene is used as an enzyme-immobilizing substrate for the adsorption of cellulase that exhibits 13-fold increase in the catalytic activity as compared to native enzyme in buffer. This is assigned to the structuring of the active site of enzyme when adsorbed physically onto the surface of graphene. It is anticipated that the content of graphene, colloidal stability and exclusive monolayer formation may be achieved by a decisive choice of cation, anion and functionalization over the alkyl chain of SAILs. Therefore, the present study along with previous reports on the use of ILs65,66 and surfactants32–36 for graphene exfoliation would offer a new platform for exploiting SAILs in an aqueous medium in conjunction with other non-conventional methods such as hydrothermal method67 for the efficient exfoliation and stabilization of a defect-free single-layer graphene in an aqueous medium.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by DST, Govt. of India wide project number SB/FT/CS-057/2013. Gurbir, Manvir, Komal and Manpreet are thankful to CSIR and UGC, Govt. of India, respectively for Fellowship. The infrastructure facility provided under the UPE, PURSE and CAS grant for this work is highly acknowledged.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- Y. Wang, Y. Huang, Y. Song, X. Zhang, Y. Ma, J. Liang and Y. Chen, Nano Lett., 2009, 9, 220 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem. Int. Ed., 2010, 49, 3014 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS.
- L. Dai, Acc. Chem. Res., 2013, 46, 31 CrossRef CAS PubMed.
- W. Xu, Z. Fu, G. Chen, Z. Wang, Y. Jian, Y. Zhang, G. Jiang, D. Lu, J. Wu and Z. Liu, Nat. Commun., 2019, 10, 2684 CrossRef PubMed.
- S. Hermanová, M. Zarevúcká, D. Bouša, M. Pumera and Z. Sofĕr, Nanoscale, 2015, 7, 5852 RSC.
- Y. Zhang, J. Zhang, X. Huang, X. Zhou, H. Wu and S. Guo, Small, 2012, 8, 154 CrossRef CAS PubMed.
- F. Zhang, B. Zheng, J. Zhang, X. Huang, H. Liu, S. Guo and J. Zhang, J. Phys. Chem. C, 2010, 114, 8469 CrossRef CAS.
- A. A. Gokhale, J. Lu and I. Lee, J. Mol. Catal. B: Enzym., 2013, 90, 76 CrossRef CAS.
- N. Srivastava, M. Srivastava, V. K. Gupta, P. W. Ramteke and P. K. Mishra, Bioresour. Technol., 2018, 270, 337 CrossRef CAS PubMed.
- J. Xu, Z. Sheng, X. Wang, X. Liu, J. Xia, P. Xiong and B. He, Bioresour. Technol., 2016, 200, 1060 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS.
- W. Yang, G. Chen, Z. Shi, C. C. Liu, L. Zhang, G. Xie, M. Cheng, D. Wang, R. Yang, D. Shi, K. Watanabe, T. Taniguchi, Y. Yao, Y. Zhang and G. Zhang, Nat. Mater., 2013, 12, 792 CrossRef CAS PubMed.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem. Int. Ed., 2010, 49, 9336 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460 CrossRef CAS PubMed.
- U. Khan, H. Porwal, A. O'Neill, K. Nawaz, P. May and J. N. Coleman, Langmuir, 2011, 27, 9077 CrossRef CAS.
- A. O’Neill, U. Khan, P. N. Nirmalraj, J. Boland and J. N. Coleman, J. Phys. Chem. C, 2011, 115, 5422 CrossRef.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis and A. K. Stubos, Small, 2009, 5, 1841 CrossRef CAS PubMed.
- E. Y. Choi, W. S. Choi, Y. B. Lee and Y. Y. Noh, Nanotechnology, 2011, 22, 365601 CrossRef PubMed.
- X. Wang, P. F. Fulvio, G. A. Baker, G. M. Veith, R. R. Unocic, S. M. Mahurin, M. Chi and S. Dai, Chem. Commun., 2010, 46, 4487 RSC.
- D. Nuvoli, L. Valentini, V. Alzari, S. Scognamillo, S. B. Bon, M. Piccinini, J. Illescasd and A. Mariani, J. Mater. Chem., 2011, 21, 3428 RSC.
- L. Guardia, M. J. Fernández-Merino, J. I. Paredes, P. Solís-Fernández, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 2011, 49, 1653 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS PubMed.
- M. Lotya, P. J. King, A. U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155 CrossRef CAS PubMed.
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288 CrossRef CAS.
- S. Lin, C. J. Shih, M. S. Strano and D. Blankschtein, J. Am. Chem. Soc., 2011, 133, 12810 CrossRef CAS.
- S. De, P. J. King, M. Lotya, A. O'Neill, E. M. Doherty, Y. Hernandez, G. S. Duesberg and J. N. Coleman, Small, 2010, 6, 458 CrossRef CAS PubMed.
- K. Zhang, L. Mao, L. L. Zhang, H. S. On Chan, X. S. Zhao and J. Wu, J. Mater. Chem., 2011, 21, 7302 RSC.
- K. Nawaz, M. Ayub, M. B. Khan, A. Hussain, A. Q. Malik, M. B. K. Niazi, M. Hussain, A. U. Khan and N. Ul-Haq, Nanomater. Nanotechnol., 2016, 6, 14 CrossRef.
- G. Singh, M. Kaur, M. Drechsler and T. S. Kang, Chem. Commun., 2018, 54, 2432 RSC.
- H. Wang, L. Zhang, J. Wang, Z. Li and S. Zhang, Chem. Commun., 2013, 49, 5222 RSC.
- S. Ghosh, C. Ghatak, C. Banerjee, S. Mandal, J. Kuchlyan and N. Sarkar, Langmuir, 2013, 29, 10066 CrossRef CAS PubMed.
- G. Singh, G. Singh and T. S. Kang, J. Phys. Chem. B, 2016, 120, 1092 CrossRef CAS PubMed.
- G. Singh, R. Kamboj, V. S. Mithu, V. Chauhan, T. Kaur, G. Kaur, S. Singh and T. S. Kang, J. Colloid Interface Sci., 2017, 496, 278 CrossRef CAS PubMed.
- M. Blesic, M. Swadzba-Kwasny, J. D. Holbrey, J. N. Canongia Lopes, K. R. Seddon and L. P. Rebelo, Phys. Chem. Chem. Phys., 2009, 11, 4260 RSC.
- B. Dong, N. Li, L. Zheng, L. Yu and T. Inoue, Langmuir, 2007, 23, 4178 CrossRef CAS.
- S. Mandal, J. Kuchlyan, S. Ghosh, C. Banerjee, N. Kundu, D. Banik and N. Sarkar, J. Phys. Chem. B, 2014, 118, 5913 CrossRef CAS PubMed.
- H. Wang, J. Wang, S. Zhang and X. Xuan, J. Phys. Chem. B, 2008, 112, 16682 CrossRef CAS PubMed.
- D. S. L. Abergel and V. I. Fal’ko, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 155430 CrossRef.
- S. Wang, M. Yi and Z. Shen, RSC Adv., 2016, 6, 56705 RSC.
- C. M. Wijmans and P. Linse, J. Phys. Chem., 1996, 100, 12583 CrossRef CAS.
- M. J. Rosen, Surfactants and Interfacial Phenomena, Wiley, New York, 2nd edn, 1989 Search PubMed.
- D. Parviz, S. Das, H. S. T. Ahmed, F. Irin, S. Bhattacharia and M. J. Green, ACS Nano, 2012, 6, 8857 CrossRef CAS PubMed.
- X. An, T. Simmons, R. Shah, C. Wolfe, K. M. Lewis, M. Washington, S. K. Nayak, S. Talapatra and S. Kar, Nano Lett., 2010, 10, 4295 CrossRef CAS PubMed.
- A. Ghosh, K. V. Rao, S. J. George and C. N. Rao, Chem. – Eur. J., 2010, 16, 2700 CrossRef CAS PubMed.
- D. W. Lee, T. Kim and M. Lee, Chem. Commun., 2011, 47, 8259 RSC.
- S. Sampath, A. N. Basuray, K. J. Hartlieb, T. Aytun, S. I. Stupp and J. F. Stoddart, Adv. Mater., 2013, 25, 2740 CrossRef CAS PubMed.
- V. Panwar, A. Chattree and K. Pal, Phys. E, 2015, 73, 235 CrossRef CAS.
- L. Zhang, Z. Zhang, C. He, L. Dai, J. Liu and L. Wang, ACS Nano, 2014, 8, 6663 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- P. Bharmoria, M. J. Mehta, I. Pancha and A. Kumar, J. Phys. Chem. B, 2014, 118, 9890 CrossRef CAS PubMed.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443 RSC.
- A. Nakamura, T. Tsukada, S. Auer, T. Furuta, M. Wada, A. Koivula, K. Igarashi and M. Samejima, J. Biol. Chem., 2013, 288, 13503 CrossRef CAS PubMed.
- G. Singh, G. Singh and T. S. Kang, Phys. Chem. Chem. Phys., 2018, 20, 18528 RSC.
- C. Wang, Y. Chen, K. Zhuo and J. Wang, Chem. Commun., 2013, 49, 3336 RSC.
- M. Kaur, G. Singh, K. Damarla, G. Singh, H. Wang, J. Wang, V. K. Aswal, A. Kumar and T. S. Kang, Phys. Chem. Chem. Phys., 2019, 22, 169 RSC.
- S. Yang, K. Zhuo, Z. Zhang, J. Liu, G. Bai and J. Wang, J. Phys. Chem. C, 2019, 123, 27969 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00159g |
| ‡ G. S. and M. K. contributed equally to this research work. |
| This journal is © The Royal Society of Chemistry 2020 |