
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the block sequence of multi-block oligomer ligands for neutralization of a target peptide†
Hinata
Takimoto‡
,
Sho
Katakami‡
,
Yoshiko
Miura
 and
Yu
Hoshino
*
and
Yu
Hoshino
*
Department of Chemical Engineering, Graduate School of Engineering, Kyushu University, Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: yhoshino@chem-eng.kyushu-u.ac.jp
First published on 30th June 2020
Abstract
A series of block oligomers consisting of the same monomer composition but a different block sequence was prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization to screen high affinity ligands for a toxic peptide. Activity and affinity screening of mono-, di-, and tri-block oligomers revealed that in addition to the localization and combination of functional groups, the sequence of the functional block and end groups are of significant importance to design the ligands that bind and neutralize the peptide.
Affinity ligands recognizing target biomolecules are drawing significant attention regarding applications in disease treatment, diagnosis, and protein purification. These ligands were developed by direct evolution of biomacromolecules such as polypeptides and oligonucleotides through cycles of library generation, screening, and amplification.1,2 However, the application of biomolecular ligands is impeded due to high production costs and low stabilities. Thus, synthetic polymer ligands consisting of versatile functional monomers are of significant interest as a stable and inexpensive substitute for biomolecular ligands.
Radical polymerization is a facile method to prepare stable synthetic polymers, that have a stable C–C backbone, from inexpensive raw materials such as acrylates and acrylamides.3 Schrader et al. reported that the free radical polymerization of optimized combinations of functional monomers leads to their transformation into strong polymeric protein hosts. The optimized functional polymers interact with the surface of target proteins by multi-mode interactions such as electrostatic, hydrophobic, aromatic, and hydrogen bonding.4,5 Shea et al. prepared polymeric nanoparticle ligands via radical and/or imprinting polymerization of combination of functional acrylamides, and employed the product for neutralizing,6–8 purifying,9 and refolding10 of target biomacromolecules.
Recent advances in controlled radical polymerization techniques enabled the synthesis of polymers of well-defined block sequence.11–13 Thus far, researchers have revealed that synthetic polymers produced by living radical polymerization of monomers with various functionalities function as polymer ligands for biomacromolecules.14–16 Haddleton et al. prepared multi-block glycopolymers containing defined block sequences via single electron transfer living radical polymerization and evaluated their binding properties to HIVgp120.14,15 Miura et al. reported that optimizing the size and arrangement of the sugar cluster in glycopolymers facilitates the control of its molecular lectin recognition.16 Effect of topology of glycopolymers on the affinity to target proteins17 and virus18 have also been reported. Perrier et al. reported that multi-block copolymer consisting of negatively charged monomers and nonionic monomers can stabilize fibroblast growth factors depending of molecular weight and charge dispersion.19 We have prepared a small library of di-block oligomer with a uniform number of functional groups and evaluated the interaction with their target peptide.20,21 It was revealed that the multi-modal interaction is important for strong affinity, and the affinity and sequence specificity significantly depends on the number of functionalities. However, the effect of the block sequence of the multiblock copolymers on the interaction with the target biomolecules has not been evaluated.
In this study, we prepared a series of block copolymers consisting of the same monomer composition but different block sequence via reversible addition–fragmentation chain transfer (RAFT) polymerization. The interaction between the polymers and a target peptide was evaluated for the first time. The toxic peptide melittin was selected as model peptide (Fig. 1a). Melittin consists of 26 amino acids and contains consecutive positively charged and hydrophobic sequences. Since melittin shows strong hemolytic activity, it was employed as model target peptide to prepare synthetic polymer ligands working as antidotes.21 To synthesize multi-block polymer ligands binding to melittin for neutralizing its toxicity, we employed negatively charged acrylic acid (AAc) and hydrophobic N-tert-butyl acrylamide (TBAm) as functional monomers (Fig. 1b). N-Isopropylacrylamide (NIPAm) was also selected as the spacer monomer. It has been reported that random ter-oligomers consisting of 3 unit of AAc and 6 unit of TBAm recognize melittin via combination of electrostatic and hydrophobic interaction.21 Density of the functional units have to be diluted by 21 mer NIPAm unit in order to achieve higher binding specificity to melittin.21 Affinity purification experiments revealed that certain sequence of the randomly copolymerized ter-oligomers has stronger affinity to melittin than the as polymerized random oligomers.22
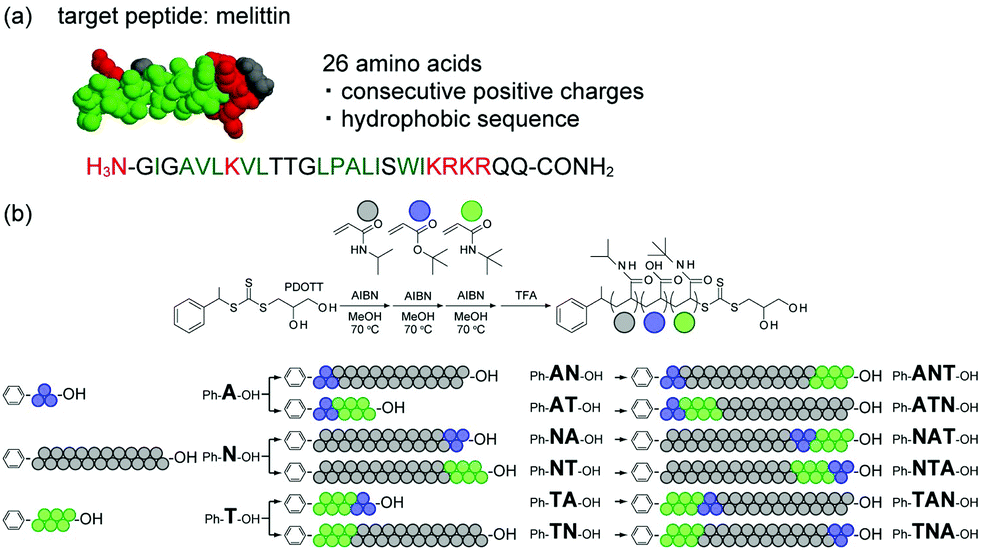 | ||
| Fig. 1 (a) Structure and amino acid sequence of melittin. Positively charged and hydrophobic amino acids are shown in red and green, respectively. (b) Scheme for the preparation of mono-, di-, and tri-block multifunctional ligands. NIPAm, AAc, and TBAm are shown in gray, blue, and green, respectively (NIPAm/AAc/TBAm = 21/3/6). | ||
Three homo oligomers were first polymerized by RAFT polymerization using AAc, TBAm and NIPAm as components (Fig. 1b). Six di-block oligomers were then prepared by using the three homo oligomers as a macro chain transfer agent and one of the remaining two functional monomers as a second component. Six tri-block oligomers were finally prepared using the six di-block oligomers as a macro chain transfer agent and the remaining functional monomer as a third component. The average number of monomer units in the AAc and TBAm blocks were tuned to be 3 mer and 6 mer, respectively; this was because it has been reported that those were the minimum number of functional groups required to interact with the target peptide via combination of multipoint electrostatic and hydrophobic interaction.19 The activity of those oligomers to neutralize melittin hemolysis was evaluated and compared with that of the randomly copolymerized polymers.
For easy purification and analysis of AAc block oligomers, tert-butyl acrylate (tBA), which is AAc protected by a tert-butyl group, is employed for the polymerization instead of AAc.20 The 1-phenylethyl-2,3-propanediolyltrithiocarbonate (PDOTT), which showed a chain transfer efficiency high enough to control the polydispersity index (PDI) of oligomers, was designed and synthesized as chain transfer agent (ESI†). tert-Butyl acrylate (960 mg, 1.5 M, 3 equivalent of PDOTT) was polymerized in degassed methanol (5 mL) at 70 °C for 6 h in the presence of PDOTT (720 mg, 0.5 M, 1 equivalent) using 2,2′-azobis(isobutyronitrile) (AIBN, 82 mg, 0.1 M) as radical initiator to obtain oligo(tBA). The monomer conversion, analyzed by 1H NMR, was 85.2%, which indicates that 2.4 mer of tBA were introduced (Table 1). Residual monomers were removed by extraction (MeOH/heptane). The average molecular weight (Mn, Mw) and the dispersity (PDI) were determined by gel permeation chromatography (GPC) (Fig. 2a). The Mn value was 3.80 × 102 and the PDI value 1.12. Homo-oligomers of NIPAm and TBAm were synthesized in the same manner using 21 and 6 equivalents of each monomer with PDOTT, respectively (ESI,† Table S1). Conversion, GPC, Mn, degree of polymerization (DP) after purification and PDI of oligo(NIPAm) and oligo(TBAm) are shown in Fig. 2 and listed in Table 1 and Table S2 (ESI†).
| Oligomer | Monomer | Monomer feed (1H NMR) | Conversion [%] | Introduction [mer] | M n [×103 g mol−1] | PDI |
|---|---|---|---|---|---|---|
| Ph-A-OH | tBA | 2.8 | 85.2 | 2.4 | 0.38 | 1.12 |
| Ph-N-OH | NIPAm | 21.7 | 97.5 | 21.1 | 3.01 | 1.17 |
| Ph-T-OH | TBAm | 6.6 | 81.8 | 5.4 | 0.68 | 1.27 |
| Ph-AN-OH | NIPAm | 21.4 | 97.2 | 20.8 | 3.56 | 1.18 |
| Ph-AT-OH | TBAm | 7.2 | 98.8 | 7.1 | 1.17 | 1.22 |
| Ph-NA-OH | tBA | 2.4 | 90.4 | 2.2 | 3.18 | 1.17 |
| Ph-NT-OH | TBAm | 5.5 | 97.8 | 5.4 | 3.48 | 1.25 |
| Ph-TA-OH | tBA | 2.7 | 98.4 | 2.6 | 0.85 | 1.34 |
| Ph-TN-OH | NIPAm | 22.0 | 98.8 | 21.8 | 4.03 | 1.24 |
| Ph-ANT-OH | TBAm | 7.2 | 99.2 | 7.2 | 4.59 | 1.25 |
| Ph-ATN-OH | NIPAm | 21.5 | 98.8 | 21.3 | 5.17 | 1.21 |
| Ph-NAT-OH | TBAm | 6.7 | 80.7 | 5.4 | 3.37 | 1.22 |
| Ph-NTA-OH | tBA | 3.2 | 63.0 | 2.0 | 3.57 | 1.17 |
| Ph-TAN-OH | NIPAm | 22.2 | 98.4 | 21.8 | 4.41 | 1.30 |
| Ph-TNA-OH | tBA | 2.5 | 89.7 | 2.3 | 4.58 | 1.22 |
| Ph-R9-OH | tBA, TBAm | 2.80, 6.43 | 96.1, 91.3 | 2.7, 5.9 | 1.11 | 1.15 |
| PH-R30-OH | tBA, NIPAm, TBAm | 2.93, 22.4, 6.4 | 99.0, 97.9, 97.5 | 2.9, 21.9, 6.2 | 4.47 | 1.20 |
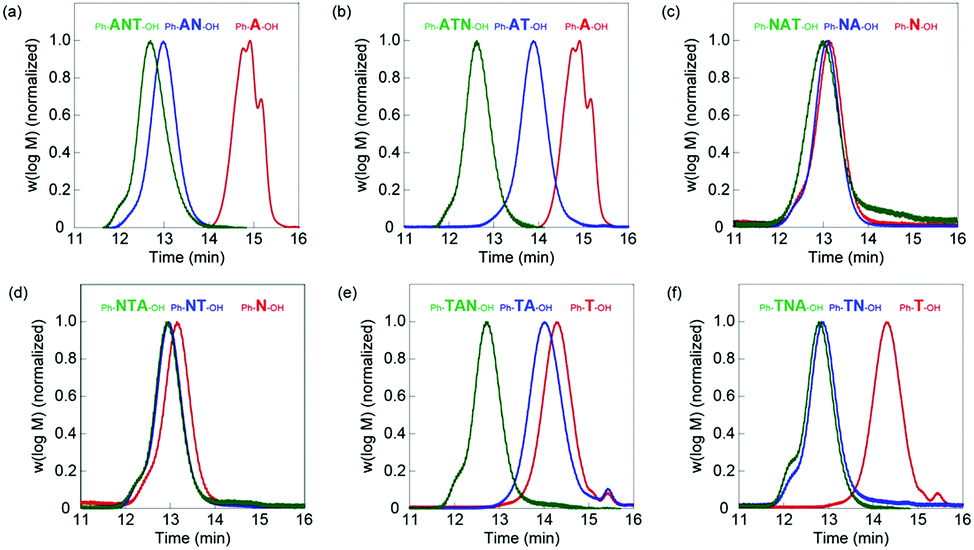 | ||
| Fig. 2 GPC traces for successive block extensions of (a) Ph-ANT-OH, (b) Ph-ATN-OH, (c) Ph-NAT-OH, (d) Ph-NTA-OH, (e) Ph-TAN-OH, and (f) Ph-TNA-OH tri-block oligomers. First, second, and third blocks are shown in red, blue, and green, respectively. | ||
Six di-block oligomers were synthesized using oligo(tBA) (Ph-A-OH), oligo(NIPAm) (Ph-N-OH) and oligo(TBAm) (Ph-T-OH) as a macro chain transfer agent, respectively. Here Ph- and –OH denote phenyl and hydroxyl end groups. NIPAm (848 mg, 1.5 M, 21 equivalent) was polymerized in the presence of Ph-A-OH (210 mg, 0.071 M, 1 equivalent) using AIBN (11 mg, 0.014 M) in degassed methanol (5 mL) at 70 °C for 6 h to prepare oligo(tBA-b-NIPAm) (Ph-AN-OH). The conversion was 97.2% and 20.8 mer of NIPAm were introduced. The residual monomers were removed by dialysis against methanol. The GPC curve shifted to higher molecular weight after polymerization (Fig. 2a). The Mn and PDI values were 3.56 × 103 and 1.18, respectively. Conversion, GPC, Mn, degree of polymerization (DP) after purification and PDI of six di-block oligomers are shown in Fig. 2 and listed in Table 1 and Table S2 (ESI†).
The third blocks were prepared using di-block oligomer as macro chain transfer agent. For example, TBAm (191 mg, 1.5 M, 6 equivalent), the Ph-AN-OH di-block oligomer (734 mg, 0.25 M, 1 equivalent) and AIBN (8 mg, 0.05 M) were dissolved in degassed methanol (1 mL) and heated at 70 °C for 6 h to prepare oligo(AAc-b-NIPAm-b-TBAm) (Ph-ANT-OH). The conversion was 99.2% and 7.2 mer of TBAm were introduced. The Ph-ANT-OH tri-block oligomer was purified by dialysis against methanol. The GPC curve shifted to higher molecular weight (Fig. 2a). The Mn, degree of polymerization (DP) after purification and PDI value were 4.59 × 103 and 1.25, respectively. Conversion, Mn and PDI of the six tri-block oligomers are shown in Fig. 2 and listed in Table 1 and Table S2 (ESI†).
Other di- and tri-block polymers were prepared and purified at similar conditions, shown in ESI† and listed in Table S1. GPC analysis indicated that all polymerizations successfully proceeded as designed (Fig. 2 and Table 1). A series of homo-, di-, and tri-block oligomer ligands with the same composition (A: 2.3 ± 0.3 mer, N: 21.3 ± 0.5 mer, and T: 6.3 ± 0.9 mer) but different block sequence was obtained.
As control samples, random 9 mer oligomers (Ph-R9-OH) consist of 3 mer of AAc and 6 mer of TBAm. Random 30 mer oligomers (Ph-R30-OH) consisting of 3 mer of AAc, 6 mer of TBAm and 21 mer of NIPAm were also prepared and purified at similar conditions, as shown in ESI† and Table S1. The Mn and PDI values of Ph-R9-OH were 1.28 × 103 and 1.15, respectively (Table 1). This indicates that the random oligomers have a molecular weight and composition comparable to that of Ph-AT-OH and Ph-TA-OH, but the functional groups are differently located. The Mn and PDI values of Ph-R30-OH were 4.47 × 103 and 1.20, respectively (Table 1), which indicates that random oligomers with molecular weight and composition comparable to that of tri-block oligomers but with differently localized functional groups were prepared.
Neutralization assay for the hemolytic activity of melittin was performed after the deprotection of the tert-butyl group (ESI† and Fig. S2); the red blood cell lysis behavior of melittin (9 μM) was investigated in the presence of oligomer ligands (60 μM) under physiological conditions (37 °C, 35 mM phosphate buffered saline (150 mM NaCl, pH 7.4), ESI†).
The results for the neutralization activity of homo oligomers are shown in Fig. 3a and indicate little neutralization activity against melittin. The neutralization activity of the di-block oligomers is summarized in Fig. 3b. The Ph-AT-OH and Ph-TA-OH di-block oligomers displayed a moderate neutralization of 39% and 53%, respectively. However, the other four di-block oligomers containing NIPAm-block did not neutralize the hemolytic activity of melittin, although, length of the NIPAm block (21 mer) is longer than the AAc-block (3 mer) and TBAm-block (6 mer). It is suggested that the combination of electrostatic and hydrophobic interaction made the oligomer interact with melittin and neutralize melittin. On the other hand, Ph-R9-OH, the random oligomer with the same number of AAc and TBAm as those of Ph-AT-OH and Ph-TA-OH oligomers, showed only a 29% neutralization activity. The value is lower than that of the Ph-TA-OH oligomers, suggesting that the carboxylic acid and tert-butyl groups have to be localized in an oligomer in such a way that they can effectively interact with melittin, which has a consecutive sequence of positively charged and hydrophobic amino acids.
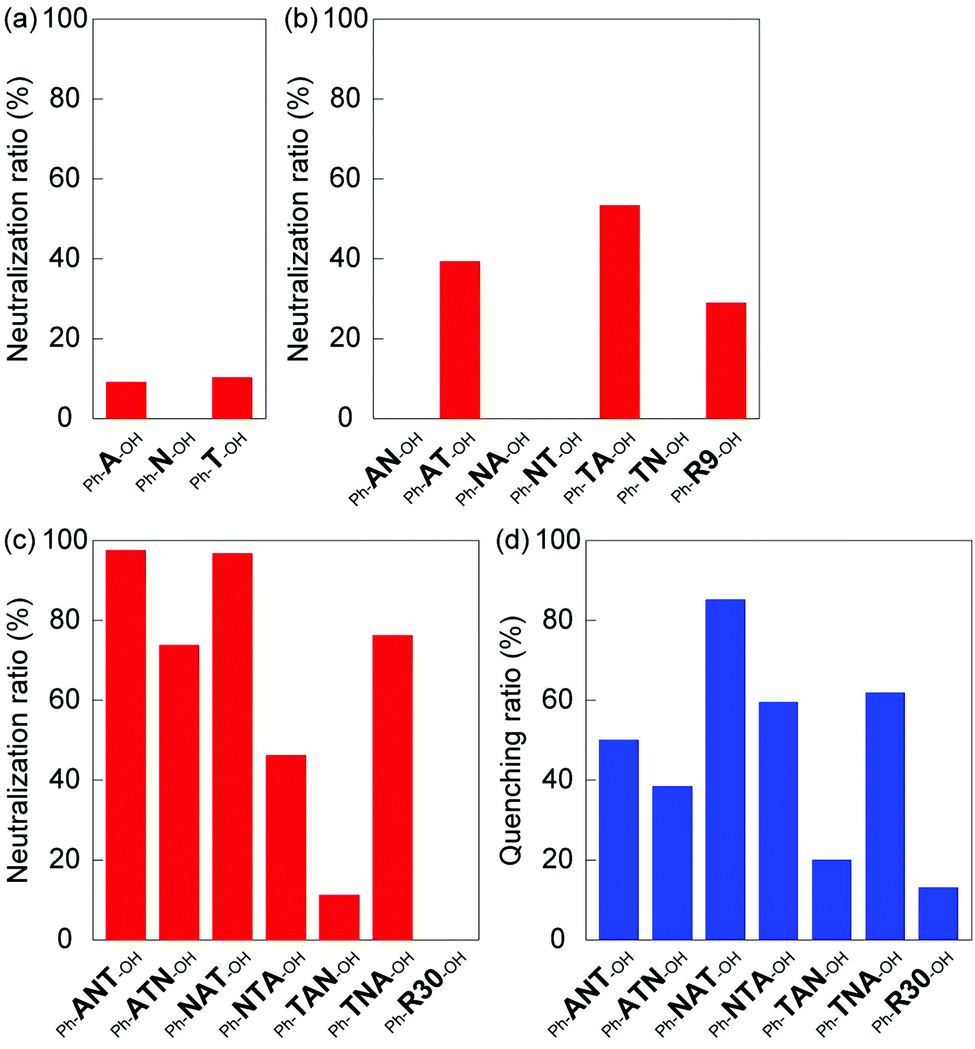 | ||
| Fig. 3 Neutralization of the hemolytic activity of melittin by (a) mono-, (b) di-, and (c) tri-block oligomers, screened by a hemolysis neutralization assay in PBS at 37 °C. (d) Fluorescence quenching by tri-block oligomers in PBS at 37 °C. The oligomer concentration was 60 μM in both experiments. | ||
The neutralization activities of tri-block oligomers were summarized in Fig. 3c. The Ph-ANT-OH, Ph-ATN-OH, Ph-NAT-OH, Ph-NTA-OH and Ph-TNA-OH tri-block oligomers neutralized the hemolytic activity of melittin (98%, 74%, 97%, 46% and 76%, respectively). However, Ph-TAN-OH and Ph-R30-OH did not show neutralization activity. Based on these results, we concluded that the neutralization activity of tri-block oligomer ligands depends on the block sequence, even if the amount and density of functionalities were identical. The position of the long spacer monomer block (oligo(NIPAm)) in tri-block oligomer ligands affected its neutralization activity. Interestingly, although the order of monomer blocks is identical, Ph-NAT-OH and Ph-TAN-OH provided a different neutralization behavior. This result indicate that the location and properties of terminal group have to be optimized in order to maximize the neutralization ability: the higher hydrophobicity of the phenyl group end group as compared to that of the diol group was sufficient to preferably interact with the hydrophobic amino acids of melittin. The long pNIPAm block on the hydrophilic end of Ph-TAN-OH might hindered AAc block to interact with positive charges on melittin. It should be mentioned here again from the result of Ph-R30-OH that the carboxylic acid and tert-butyl groups have to be localized in an oligomer to effectively interact with melittin, which has a consecutive sequence of positively charged and hydrophobic amino acids.
To evaluate the interaction between tri-block oligomer ligands and melittin, the fluorescence emission (340 nm) of melittin (9 μM) was quantified in the presence of tri-block oligomer ligands (60 μM) under physiological conditions (37 °C, 35 mM phosphate buffered saline (150 mM NaCl, pH 7.4), ESI†). The quenching of the tryptophan (Trp-19) fluorescence of melittin, induced by the interaction with oligomer ligands, is summarized in Fig. 3d. The tri-block oligomers Ph-ANT-OH, Ph-ATN-OH, Ph-NAT-OH, Ph-NTA-OH and Ph-TNA-OH, that have high neutralization activity, showed quenching adequately the tryptophan fluorescence of melittin. On the other hand, Ph-TAN-OH and Ph-R30-OH exhibited little fluorescence quenching. These results indicate that hemolytic toxicity of melittin was neutralized by the oligomer ligands as a result of interaction between the peptide and the ligands. Interestingly, the oligomers that showed strongest neutralization activity (such as Ph-ANT-OH) did not always show strongest quenching of tryptophan (Fig. 3c and d). This results indicate that each oligomers neutralized melittin by binding to different sequence on melittin. Sequence specificity was observed in the case of homogeneous di-block oligomers of A and T.20 A di-block hexamer consisting of 4 units of A and 2 units of T recognize negatively charged KRKR sequence on melittin, though, the hexamer consisting of 2 units of A and 4 units of T recognized hydrophobic amino acids including tryptophan next to the KRKR sequence.20
These results demonstrated that the synthetic oligomer ligands, which effectively interact with the target peptide via multi-modal interactions, can be prepared by controlling the block sequence of the multi-functional oligomers. Screening of mono-, di-, and tri-block oligomer ligands revealed that not only the localization and combination of functional groups but also the sequence of the functional block and end groups are of importance to design high affinity ligands that binds and neutralize activity of target biomacromolecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS (23750193 and 20H02789), JST A-STEP (AS2321466 and AS231Z01490D), AMED (JP17efk0108127h), Ogasawara Foundation, Kakihara Foundation, and Kao Foundation for Arts and Sciences.References
- A. D. Ellington and J. W. Szostak, Nature, 1996, 346, 818–822 CrossRef PubMed.
- J. K. Scott and G. P. Smith, Science, 1990, 249, 386–390 CrossRef CAS PubMed.
- (a) J. O’Brien and K. J. Shea, Acc. Chem. Res., 2016, 49(6), 1200–1210 CrossRef PubMed; (b) C. S. Mahon and D. A. Fulton, Nat. Chem., 2014, 6, 665–672 CrossRef CAS PubMed.
- C. Renner, J. Piehler and T. Schrader, J. Am. Chem. Soc., 2006, 128, 620–628 CrossRef CAS PubMed.
- S. J. Koch, C. Renner, X. Xie and T. Schrader, Angew. Chem., Int. Ed., 2006, 45, 6352–6355 CrossRef CAS PubMed.
- (a) M. Nakamoto, D. Zhao, O. R. Benice, S.-H. Lee and K. J. Shea, J. Am. Chem. Soc., 2020, 142, 2338–2345 CrossRef CAS PubMed; (b) K. Yoshimatsu, H. Koide, Y. Hoshino and K. J. Shea, Nat. Protoc., 2015, 10, 595–604 CrossRef CAS PubMed.
- H. Koide, K. Yoshimatsu, Y. Hoshino, S. H. Lee, A. Okajima, S. Ariizumi, Y. Narita, Y. Yonamine, A. C. Weisman and Y. Nishimura, et al. , Nat. Chem., 2017, 9, 715–722 CrossRef CAS PubMed.
- Y. Hoshino, T. Kodama, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2008, 130, 15242–15243 CrossRef CAS PubMed.
- (a) K. Yoshimatsu, T. Yamazaki, Y. Hoshino, P. E. Rose, L. F. Epstein, L. P. Miranda, P. Tagari, J. M. Beierle, Y. Yonamine and K. J. Shea, J. Am. Chem. Soc., 2014, 136, 1194–1197 CrossRef CAS PubMed; (b) M. Liu, R. Huang, A. Weisman, X. Yu, S. H. Lee, Y. Chen, C. Huang, S. Hu, X. Chen, W. Tan, F. Liu, H. Chen and K. J. Shea, J. Am. Chem. Soc., 2018, 140, 6853–6864 CrossRef CAS PubMed.
- M. Nakamoto, T. Nonaka, K. J. Shea, Y. Miura and Y. Hoshino, J. Am. Chem. Soc., 2016, 138, 4282–4285 CrossRef CAS PubMed.
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS; (b) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS; (c) J. S. Wang and K. J. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- (a) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109(11), 5402–5436 CrossRef CAS PubMed; (b) S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS; (c) M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 2603–2614 CrossRef CAS.
- (a) A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed; (b) P. R. Judzewitsch, T.-K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., 2018, 130, 4649–4654 CrossRef; (c) J. L. Grace, E. K. Schneider-Futschik, A. G. Elliott, M. Amado, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, Biomacromolecules, 2018, 19, 4629–4640 CrossRef CAS PubMed.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilsonac and D. M. Haddleton, Polym. Chem., 2014, 5, 3876–3883 RSC.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 52, 4435–4439 CrossRef CAS PubMed.
- K. Jono, M. Nagao, T. Oh, S. Sonoda, Y. Hoshino and Y. Miura, Chem. Commun., 2018, 54, 82–85 RSC.
- L. Liu, F. Zhou, J. Hu., X. Cheng, W. Zhang, Z. Zhang, G. Chen, N. Zhou and X. Zhu, Macromol. Rapid Commun., 2019, 40, 1900223 CrossRef PubMed.
- M. Nagao, T. Matsubara, Y. Hoshino, T. Sato and Y. Miura, Bioconjugate Chem., 2019, 30, 1192–1198 CrossRef CAS PubMed.
- C. Bray, P. Gurnani, E. D. H. Mansfield, R. Peltier and S. Perrier, Biomacromolecules, 2019, 20, 285–293 CrossRef CAS PubMed.
- Y. Hoshino, S. Taniguchi, H. Takimoto, S. Akashi, S. Katakami, Y. Yonamine and Y. Miura, Angew. Chem., Int. Ed., 2020, 59, 679–683 CrossRef CAS PubMed.
- H. Lee, Y. Hoshino, Y. Wada, Y. Arata, A. Maruyama and Y. Miura, J. Am. Chem. Soc., 2015, 137, 10878–10881 CrossRef CAS PubMed.
- Y. Hoshino, S. Shimohara, Y. Wada, M. Nakamoto and Y. Miura, J. Mater. Chem. B, 2020 10.1039/D0TB00849D.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00149j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |