
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic properties of {Mo3S4}-based complexes with regard to the hydrogen evolution reaction and application to PEM water electrolysis†
Joumada Al
Cheikh
a,
Rosa
Zakari
ab,
Amit C.
Bhosale‡
ac,
Angel
Villagra
a,
Nathalie
Leclerc
b,
Sébastien
Floquet
 b,
Prakash C.
Ghosh
c,
Alireza
Ranjbari
a,
Emmanuel
Cadot
b,
Pierre
Millet
a and
Loïc
Assaud
*a
b,
Prakash C.
Ghosh
c,
Alireza
Ranjbari
a,
Emmanuel
Cadot
b,
Pierre
Millet
a and
Loïc
Assaud
*a
aEquipe de Recherche et d’Innovation en Electrochimie pour l’énergie (ERIEE), Institut de Chimie Moléculaire et des Matériaux d’Orsay (ICMMO), UMR CNRS 8182, Université Paris-Saclay, 91405 Orsay, France. E-mail: loic.assaud@universite-paris-saclay.fr
bILV, CNRS, Université de Versailles Saint Quentin, Université Paris-Saclay, Avenue des Etats Unis, 78035 Versailles, France
cDepartment of Energy Science and Engineering, IIT Bombay, Mumbai – 400076, India
First published on 12th May 2020
Abstract
We report on the synthesis and the electrochemical characterization of two [Mo3S4]4+ nanoclusters, [Mo3S4(acacbut)3]Cl and [Mo3S4–Pd(acacbut)3]Cl. Their electrochemical properties have been measured in solution in organic and in aqueous media, by cyclic voltammetry, linear sweep potential voltammetry and chrono-coulometry at controlled potential. The different redox couples have been identified. Their electrochemical activity with regard to the hydrogen evolution reaction (HER) has been determined by dropwise addition of controlled amounts of formic acid in acetonitrile. The turn over frequency of the two compounds has been determined to compare their HER activity. Then the two compounds have been functionalized onto two different carbonaceous substrates (graphene and carbon black) of practical interest for applications in water electrolysis cells. The polarization curves and Tafel plots have been measured in 0.1 M H2SO4 aqueous solutions. Finally, the two complexes have been implemented at the cathode of a PEM water electrolysis cell. The polarization curves have been measured and compared to those obtained with metallic platinum under similar operating conditions.
1. Introduction
Large amounts of molecular hydrogen (H2) are used in the chemical industry for a quite diverse set of applications, including gasoline refining, the synthesis of nitrogenous fertilizers, metal reduction, and many others.1–6 Hydrogen is also considered as the most suitable energy vector of the future, for the large scale storage of renewable electricity. Due to the increase in the world population and the decrease in fossil fuel reserves, some forecasts anticipate a strong increase in demand for hydrogen in the coming years. Today, most of the hydrogen is produced either by steam reforming of methane (natural gas) (according to: CH4 + 2H2O → 4H2 + CO2), or by cracking of hydrocarbons.7,8 However, these industrial processes directly contribute to global warming because they release CO2 (a greenhouse gas) which is released into the atmosphere. The production of hydrogen by electrolysis of water from renewable energy sources is seen as a key technology that could help decarbonize the energy sector.9 Among a bunch of water splitting processes, water electrolysis can produce H2 in a cost-affordable and efficient way. The electrical consumption of water electrolyzers operating at temperatures and pressures close to ambient is of the order of 4.5 to 5.0 kWh Nm−3 (the thermodynamic requirement under the conditions standard is 3.55 kWh Nm−3, which gives a conversion efficiency of 70 to 80%). PEM (polymer electrolyte membrane) technology10 is not as mature as alkaline technology, but it is considered as the most promising water electrolysis technology for large scale deployments, due to some specific advantages such as an ability to operate in the multi A cm−2 range and to provide grid services.11 However, a current limitation of PEM technology comes from its cost. Because of the strong acidity of the polymer electrolyte, only platinum group metals (PGMs) can be used as electrocatalysts for both the hydrogen (HER) and oxygen (OER) evolution reactions. Unsupported or carbon-supported Pt nanoparticles are used at the cathode for the HER and unsupported IrO2 particles are used at the anode for the OER. Significant cost reductions can be expected by replacing these PGMs with cheaper transition metals or their oxide derivatives.In this paper, we report on the HER properties of two non-PGM thiomolybdate catalysts that could be used in acidic media at the cathode of PEM water electrolysis cells in place of platinum (2H+ + 2e− → H2).12–22 The HER is of course only half of the problem since a similar cost problem is found on the anode side for the OER. However, it is a much more challenging task to find non-PGM alternatives to IrO2 and the discussion reported here is restricted to the HER on the cathode side of the cell. A successful replacement of Pt would already be a very significant step forward and a game-changing result. The challenge is to identify innovative materials with a sufficient level of activity to minimize the HER overpotential and a sufficient chemical stability in strongly acidic media.23 Platinum, either in the form of unsupported nanoparticles, or in the form of nanoparticles supported by an electron-conducting carbonaceous substrate of large surface area, is the reference material against which platinum-free alternatives must be benchmarked.24–29 Besides its chemical stability in acidic environments, the key feature of platinum (which explains its high HER electroactivity) is the weak energy of the M–H bond that forms during the reaction. Hence, H2 can evolve rapidly and the HER kinetics is usually limited by electronic proton discharge. Electrocatalysts based on transition metals (such as Co, Ni and Fe) are Earth abundant and much cheaper than platinum but they are usually insufficiently stable in acidic environments under open-circuit conditions and form M–H bonds of larger energy. Hydrogen adatoms tend to accumulate at the surface of the electrode during the reaction and limit the kinetics. In spite of such limitation, they are found as active centers in some natural systems such as enzymes (hydrogenase and nitrogenase), and therefore bio-inspiration can help the chemical designer to find viable configurations for his processes. It has for example already been reported24,25 that some non-PGM chemical compounds (e.g., carbides and sulfides) exhibit electronic structures close to that of Pt and can, for that reason, be considered as potential alternatives to Pt for the HER. The high HER efficiency of the MoSx species (either in the form of amorphous polymers or MoS2 crystallites) was attributed to the presence of edge sulfur atoms in MoS2, comparable to some extent to those found in the active sites of enzymatic systems.30 It is worth to mention here from a historical perspective that the identification of the active sites within the MoS2 nanocrystallites is an old story that was initiated by Kazstelan et al. in the 1980's in the context of the HDS oil refinery.31 The Kazstelan's model, supported by experimental studies and theoretical considerations, showed that sulphur atoms lining the edges of the MoS2 crystallites were active towards the hydrogen dissociation reaction.32,33 In agreement with the principle of microreversibility used in catalysis,34 the molybdenum–sulphur based catalysts were also found active for the reverse hydrogen oxidation reaction (HOR) and the initial hypotheses were revised in the recent years to explain the hydrogen formation from electro- or photo-catalysis HER processes.35–41 By increasing the number of terminal active sulphur atoms in MoSx systems, it was expected to improve the HER activity. For instance, Kibsgaard et al. reported that molecular [Mo3S13]2− clusters containing intrinsically high ratio of terminal sulphur/molybdedum complexes is an adequate and model HER catalytic site.42 Such structures are also highly stable in acidic media and comparable to those of PGMs such as Pt.43 This striking HER behaviour has been attributed to the distinctive structural arrangement found in [Mo3S13]2−, which exhibits diverse kinds of sulfur ligands distributed as terminal or bridging disulfido and sulfido groups.44 In addition to this, the [Mo3S13]2− anion has been reported to be also highly HER-efficient and stable when used as co-catalyst in electrophotocatalytic devices.45 More generally, molecular molybdenum sulphide species have been extensively studied and considered as versatile chemical platform allowing to develop multifunctional catalytic materials, such as metal-substituted derivatives or coordination complexes, specifically designed with regard to the applied process.46–52
In order to further investigate the HER properties of such class of compounds, we report herein on the development of {Mo3S4}-based molecular HER catalysts, taking benefit of the well-known chemical properties of the aqua cluster [Mo3S4(H2O)9]4+, used as precursor in this work.53 As illustrated in Fig. 1, the nine aquo ligands attached to the Mo centers are labile enough to be exchanged by a large variety of multidentate ligands, while the three bridging sulfido groups are able to coordinate zero-valent metal centers such as Ni, Pd, Pt to form heterometallic derivatives as reported by Uriel and Llusar in their review.47
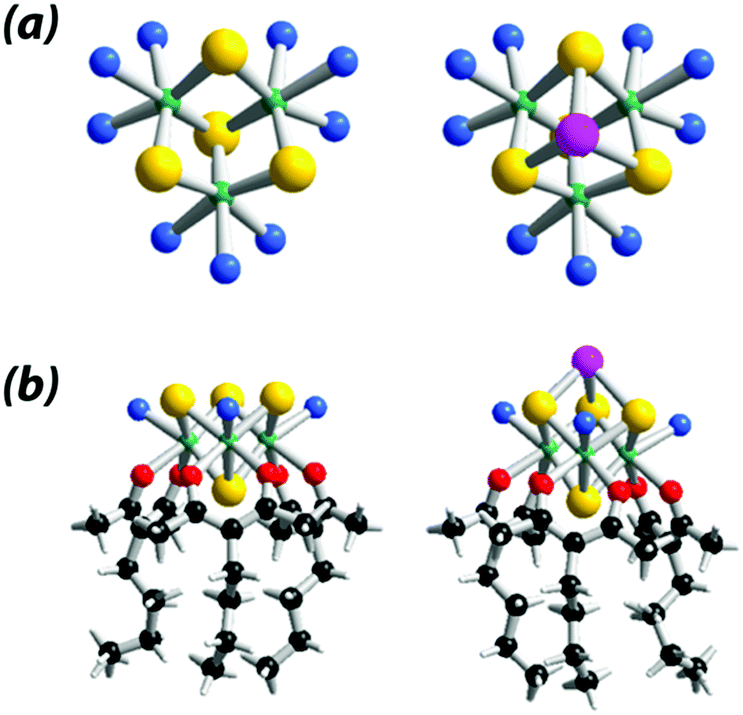 | ||
| Fig. 1 Structural models of the {Mo3S4}-based arrangements. (a) Aquo clusters {Mo3S4(H2O)9}4+ (left) and {Mo3S4Pd(H2O)9}4+ (right) used as precursors;47 (b) 3-butylpentane-2,4-dione (Acac-But) coordination complexes, [Mo3S4(acacBut)3(H2O)3]+ (left) and [Mo3S4Pd(acacBut)3(H2O)3]+ (right). Color code: yellow = S, green = Mo, pink = Pd, red = O, blue = H2O, black = C, white = H. | ||
The coordination properties of the {Mo3S4} cluster are very versatile. They can be used to develop strategies for catalyst's immobilization onto surfaces, or for tuning physical–chemical properties of the {Mo3S4} core according to the redox potential of the semi-reaction of interest.54 Herein, we report on {Mo3S4}-based hybrid electrocatalyst using alkyl β-dikenotate (noted acac-R) as bidentate ligands. The HER electrocatalytic performances of this hybrid catalyst have been investigated in both homogeneous and heterogenous phase, with the goal to design the first {Mo3S4}-based PEM (proton exchange membrane) cathode for water electrolysis. In addition, we also report on the heterometallic cuboidal derivative {Mo3S4Pd}, to show that addenda metal atoms can alter the catalytic properties of the hybrid catalyst. Palladium is an excellent HER catalyst, sometimes used in place of Pt in water electrolysis cells, and its incorporation into the {Mo3S4} structure could potentially enhance the HER electroactivity of these complexes using only tiny Pd amounts (down to the μg cm−2 scale instead of the 0.5 mg cm−2 range in the current state of art).
2. Experimental
2.1. Chemicals
The chemicals used for the synthesis were purchased from Aldrich Chemicals or Acros Chemicals and used as-received without further purification. All solvents were of reagent grade quality, and were also used without further purification. The [Mo3S4]4+ cationic cluster was prepared in the form of (NH4)4[Mo3S4(H2O)9]Cl6·3H2O, as described elsewhere.55 The complex [Mo3S4(acacBut)3(H2O)3]Cl·H2O (where HacacBut corresponds to 3-butylpentane-2,4-dione), denoted hereafter Mo3S4(acacbutyl)3 for short was synthetized as previously reported and characterized by elemental analysis, ESI-MS, EDX and FT-IR (see Fig. S1–S5, ESI† for more details).562.2. Synthesis procedures
2.3. Physico-chemical characterizations
Fourier transform infrared (FT-IR) analysis was used to confirm the formation of target compounds. The spectra were recorded on a 6700 FT-IR Nicolet spectrophotometer, using the diamond ATR technique. The spectra were recorded on non-diluted compounds in the 400–4000 cm−1 range. ATR correction was applied. EDX measurements were made using a JEOL JSM 5800LV apparatus. Elemental analyses (C, H, N, S) were performed by the analytical service of the French CNRS at Gif-sur-Yvette, France. 1H NMR solution spectra were recorded at 25 °C using a Bruker Avance 400 MHz spectrometer equipped with a 5 mm BBI probe head and operated at a magnetic field strength of 9.4 T. Scanning electron micrographs were acquired using a Zeiss Supra 55 VP variable pressure scanning electron microscope with field emission gun (SEM-FEG), equipped with energy dissipative X-ray spectroscopy (EDS) for chemical microanalysis. UV-vis spectra were recorded in transmission mode, using a Cary 60 spectrometer from Agilent Technologies. Electrospray Ionization Mass Spectrometry (ESI-MS) spectra were collected using a Q-TOF instrument supplied by WATERS. For the analysis, the samples were solubilized in water at a concentration of 10−4 M and were introduced into the spectrometer via an ACQUITY UPLC WATERS system whilst a Leucine Enkephalin solution was co-injected via a micro-pump as internal standard. In addition to the EDX and ESI-MS data reported above, in order to support the structural model of the synthesized electrocatalysts, FT-IR, TGA and NMR spectra have been plotted in ESI,† Fig. S1–S5.2.4. Electrochemical measurements
The electrochemical measurements were carried out in a conventional three-electrode cell, equipped with a gas bubbling system. A glassy carbon disk (0.07 cm2) polished with 100% wool clothings (Struers Co.) of different sizes (1 μm, 3 μm and 6 μm) and three diamond pastes of the same sizes were used as working electrode. A droplet of 2 μl of electrocatalyst mixture was drop-casted at the surface of the glassy carbon electrode (the mixture consisted in 4 mg of electrocatalytic compounds dissolved in 200 μl of dichloromethane). A carbon plate of large surface area was used as counter electrode to avoid Pt etching and cathode contamination. Three cycles were recorded systematically. The potential of the working electrode was measured against a saturated calomel reference electrode (SCE) (E0SCE = +242 mV vs. NHE). A PZG402 (Radiometer) potentiostat was used for the electrochemical characterization. Freshly distilled acetonitrile (ACN) was used as solvent and 0.1 M tetrabutylammonium perchlorate (TBAP) in ACN was used as supporting electrolyte in the experiments. The solutions were de-aerated thoroughly for at least 45 minutes by argon bubbling and then an argon cloud was left over to prevent air contamination. Experiments were performed at ambient laboratory temperature. Note that systematic electrochemical measurements were performed on a Pt disk of 0.07 cm2 electrode used as a reference sample.2.5. PEM water electrolysis measurements
The membrane-electrode assemblies (MEAs) were prepared by spraying alcoholic suspensions of catalyst particles over Nafion 117 membranes, using an ink printer (SONO-TEK) equipped with an ultrasonic Syringe. A PEM laboratory cell (circular shape, 7 cm2 active area) was used for measuring electrochemical performances. The cell was made of two titanium plates equipped with a network of flow fields for water circulation and collection of biphasic liquid–gas mixtures. Two circular rubber sealants were placed at the periphery to void leaks. A porous titanium disk made of sintered titanium particles (∼50% open porosity) was used on both sides of the MEAs as porous transport layers (PTLs). Thermostatic deionized (milliQ grade) water was pumped on both sides during operation. Measurements were made at atmospheric pressure. The IV curves were recorded using a DC power source (XHR 20–50 DC power supply from XANTEREX). The methodology used for measuring these IV curves has been described elsewhere.57,58 In order to prepare the catalytic ink, 163 mg of Mo3S4(acacbutyl)3 (respectively Mo3S4Pd(acacbutyl)3), 82 mg of Vulcan and 565 μL of Nafion® in 25 mL of water/ethanol (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) were mixed together.
:50) were mixed together.
3. Results and discussion
3.1. Electrochemical redox characterization
The chloride salts of the two cationic complexes investigated in this study, [Mo3S4(H2O)3(acacbutyl)3]+ (denoted [1] in the followings) and [Mo3S4Pd(H2O)3(acacbutyl)3]+ (denoted [2]), were found both soluble in acetonitrile (ACN). Their redox characteristics were determined by cyclic voltammetry, using 0.1 M TBAP as supporting electrolyte. Results are reported in Fig. 2a and b respectively. The cyclic voltammograms (CVs) of Fig. 2 reveal the presence of several oxidation and reduction redox transitions. Peak potentials are sufficiently well separated to identify the different redox couples. Regarding complex [1] (the one with three molybdenum metal centers), three different monoelectronic redox waves were observed on the cyclic voltamograms of Fig. 2a along the cathodic scan:55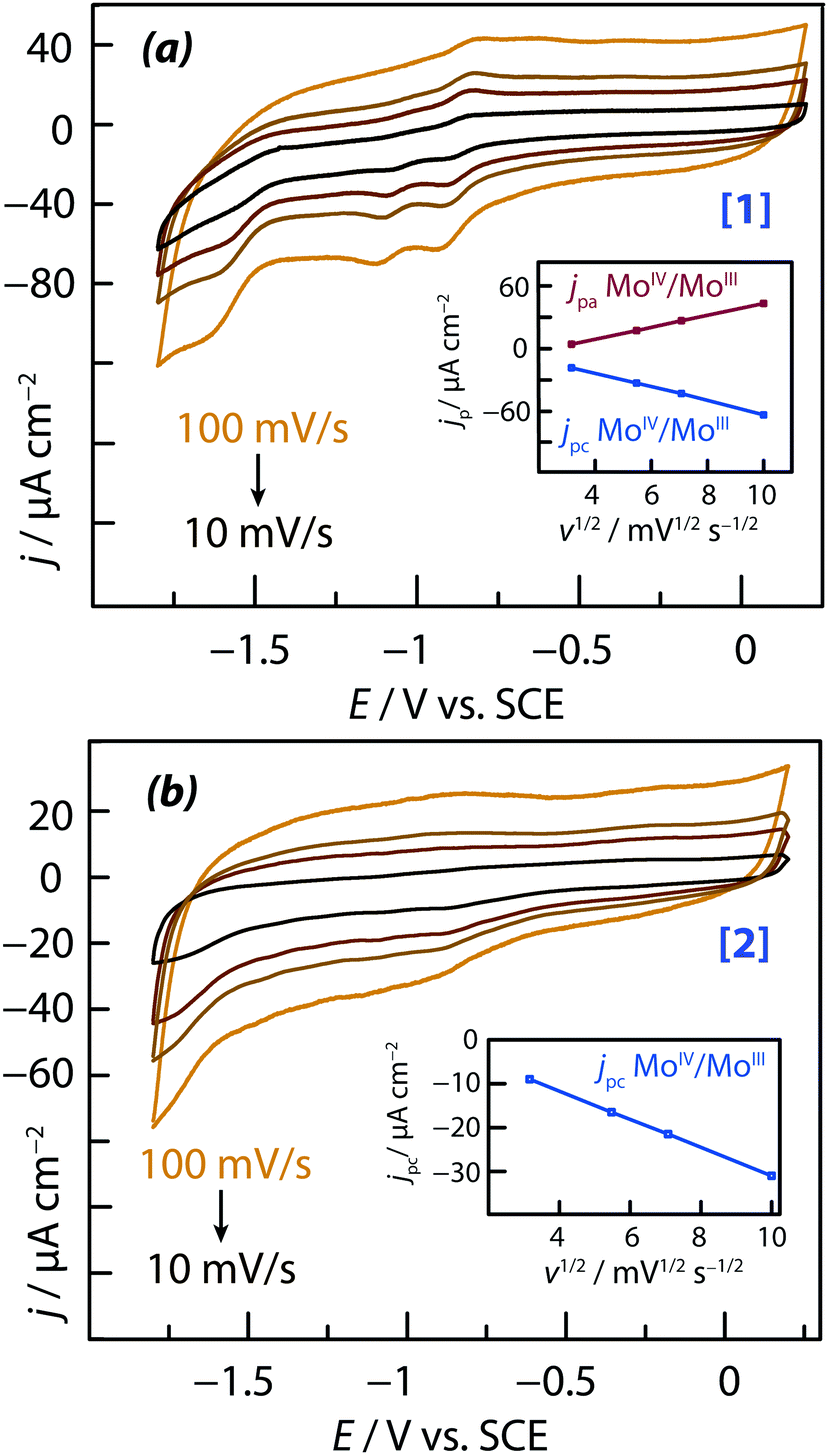 | ||
| Fig. 2 Cyclic voltammograms of 0.2 mM thiomolybdate solutions in ACN at 10, 30, 50 and 100 mV s−1: (a) [Mo3S4(H2O)3(acacbutyl)3]+; (b) [Mo3S4Pd(H20)3(acacbutyl)3]+. Inserts: Plots of the anodic and cathodic peak current densities as a function the square root of the potential scan rate. | ||
• the first monoelectronic wave was found to be almost reversible and was attributed to the MoIV3 ↔ MoIIIMoIV2 redox transition; the potential of the peak current during the cathodic scan was measured at Epc = −920 mV vs. SCE and the potential of the associated anodic peak current during the anodic scan was measured at Epa = −840 mV vs. SCE).
• the second monoelectronic wave was found to be irreversible; the potential of the peak current during the cathodic scan was measured at Epc = −1120 mV vs. SCE) and was attributed to the MoIIIMoIV2 → MoIII2MoIV redox transition.
• the third monoelectronic wave was also found to be irreversible; the potential of the peak current during the cathodic scan was measured at Epc = 1650 mV vs. SCE and was attributed to the irreversible reduction of the remaining MoIV according to: MoIII2MoIV → MoIII3.
Regarding complex [2] (the one with a palladium metal centers), two irreversible electronic transfers were observed (Fig. 2b). The potentials of the peak currents along the cathodic scan were shifted towards either upper or lower potential values compared to those of complex [1]. The first cathodic peak was measured at a potential of Epc = −880 mV vs. SCE (against Epc = −920 mV for complex [1]), corresponding to a +40 mV potential shift. The reduction of the first molybdenum center was easier in complex [2] than in complex [1], but at the same time the process became irreversible. It has to be noted that the second transfer observed on complex [1] was not observed on complex [2]. The third transfer was localized at a cathodic potential of Epc = −1720 mV vs. SCE (against Epc = −1650 mV for complex [1]), corresponding to a −70 mV potential shift. The reduction of the last molybdenum center was found to be more difficult in complex [2] than in complex [1]. Plots of peak currents versus the square root of the potential scan rate (inserts of Fig. 2a and b) were linear in shape. This is an indication of mass transport (diffusion-controlled) kinetics.
3.2. The HER electroactivity with complexes dissolved in solution
The HER activity of complexes [1] and [2] dissolved in acetonitrile was determined electrochemically by adding controlled amounts of protons to the solution. Several proton sources (acids) were tested in order to identify those in the contact of which the two complexes are sufficiently stable to perform the experiments. Formic acid was selected as the most appropriate one because complexes [1] and [2] were found fairly stable in formic acid/acetonitrile solutions. This was demonstrated by UV-vis spectroscopy: the UV-vis spectra of both compounds were recorded in the presence of increasing amounts of HCOOH and were found to remain mostly unchanged overnight while the acid concentration was increased (see ESI,† Fig. S6). In such media, the possible de-coordination of the acacbutyl ligands and the {Mo3S4} core did not occur. The CVs measured on complexes [1] and [2] in the presence of HCOOH showed characteristic plateau-shaped HER waves at large negative potentials (Fig. 3a and b). Since the working electrode used for these measurements was a stationary (i.e. non-rotating) electrode, plateau currents were not totally flat but mean plateau current values could be determined for each CV. The current density of the plateau currents was found proportional to the HCOOH (proton) concentration, suggesting that kinetic mass transport limitations (i.e. the diffusion-controlled of proton to the interface) are taking place. A correlation was also observed between the potential of the HER onset and the redox state of the compounds: proton reduction starts once the core metallic electro-active centers of the complexes are reduced. In order to analyze the reaction mechanism and to determine the reaction orders of these electro-catalytic processes, a series of experiments were performed with varying amounts of catalyst and acid in the acetonitrile solution. Measures were made in the negative potential range, where the kinetics of electrons transfer (ET) is fast and where plateau currents are observed. It was first assumed that the HER is of the first order with respect to the catalyst concentration, and of the first order with respect to the concentration of acid (i.e. the total reaction order is of two). Such assumption is justified because the HER is a simple reaction involving the transfer of only one electron.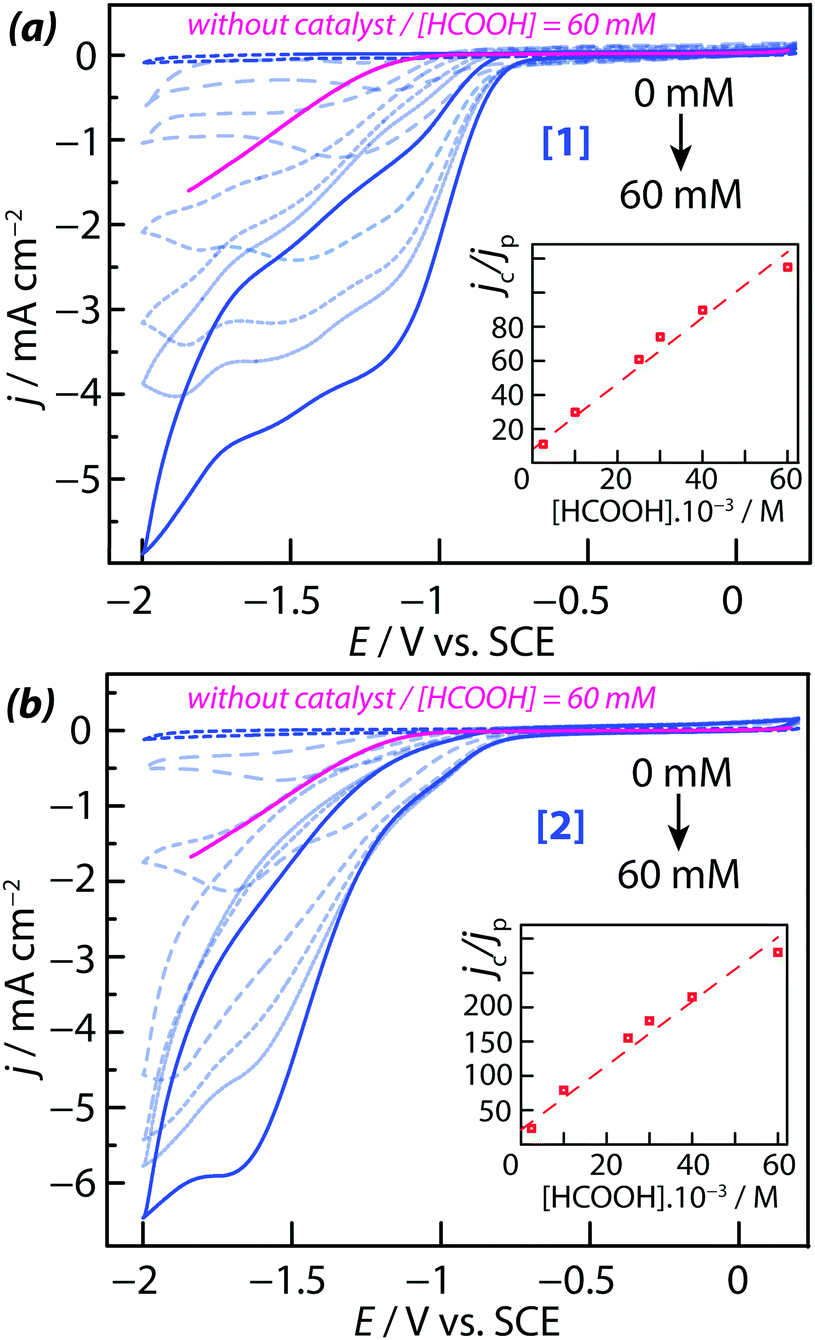 | ||
| Fig. 3 Cyclic voltammograms (CVs) for 0.2 mM acetonitrile solutions of the complexes [Mo3S4(H2O)3(acacbutyl)3]+ = [1] (a) and [Mo3S4Pd(H2O)3(acacbutyl)3]+ = [2] (b), in the absence and the presence of formic acid HCOOH. Inserts show a plot of jc/jpversus HCOOH concentration for 0.2 mM acetonitrile solutions of the both complexes. The electrolyte was ACN + 0.1 M TBAP. The reference electrode was a saturated calomel electrode (SCE). The scanning rate was ν = 50 mV s−1. | ||
In such case, the plateau current (ic) is related to the proton and catalyst concentrations by eqn (1):26,27
| ic = nFAD0.5k0.5[cat][Heq+] | (1) |
 | (2) |
| Catalyst | k 0 (103 mol−2 l2 s−1) | D (10−10 cm2 s−1) | Catalytic efficiency (%) |
|---|---|---|---|
| [1] | 218 | 17 | 5950 |
| [2] | 1284 | 30 | 3281 |
The HER rate constant of the Pd-complex [2] was found to be 6 times larger than the HER rate constant of Pd-free complex [1]. A similar trend was observed for the diffusion coefficient D. A comparison of the current densities measured with 60 mM of formic acid at E = −1 V vs. SCE (before the onset of the plateau currents) show that a larger current density is obtained with [1] instead of [2]. The electrocatalytic effect of the two complexes was clearly demonstrated by comparing the CVs with and without the two complexes in 60 mM acid solutions (in Fig. 3, the forward scan of the cyclic voltammetry recorded without catalyst is highlighted in pink color). In the presence of catalyst [1] or [2], the overpotential required for proton reduction was decreased. Without catalyst in solution, the HER onset was measured at −1 V vs. SCE whereas in the presence of [1] and [2], it was measured at −750 ± 30 mV vs. SCE. Therefore, the HER overpotential was decreased by 260 ± 30 mV for [Mo3S4(H2O)3(acacbutyl)3]+ [1] and [Mo3S4Pd(H2O)3(acacbutyl)3]+ [2].
As can be seen from Fig. 4a, the consumption of charge for the formation of molecular hydrogen during electrolysis (the working electrode was a bare glassy carbon electrode and the counter-electrode a glassy carbon plate of large surface) without electrocatalyst in solution was less than that of the presence of the thiomolybdic complexes. The long-term stability of the {M–S} complexes [1] and [2], in organic medium, with 60 mM HCOOH, at a constant potential of −1.5 V vs. SCE, was investigated by chronoamperometry. During the 4 h long experiment, a total charge of 450 C, 550 C and 345 C was passed in the solution containing complex [1], [2] and without complex, respectively (Fig. 4a). These values correspond to a faradaic yield (the hydrogen content was determined by gas chromatography) of 90 ± 10% (with thiomolybdate compounds) and 70% (without catalyst). Based on these results, it can be concluded that in the definition of the catalyst turnover number (TON) and turnover frequency (TOF), the number of moles of catalyst that are actually involved are those contained in the reaction layer rather than those in the bulk solution. The maximum turnover frequencies (TOFmax) obtained from these data are 785 and 4622 mol H2 per mol catalyst for [1] and [2], respectively (Fig. 4b). Again, a linear correlation was observed for both complexes between the plateau current and the acid concentration, suggesting that the electrochemical reactions are of first order with respect to HCOOH concentration. The electrocatalytic performances of the two compounds can be quantitatively compared by calculating their turnover frequency. For each catalyst, the turnover frequency is related to the HER overpotential by eqn (3):60
 | (3) |
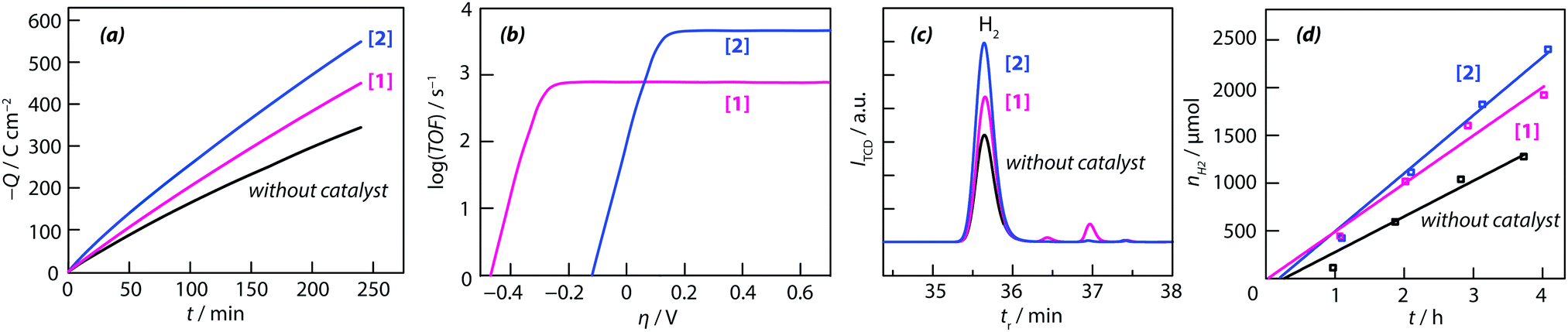 | ||
| Fig. 4 (a) Chronocoulombic experiments using 0.2 mM acetonitrile solutions with and without catalytic complexes: [1] = [Mo3S4(H2O)3(acacbutyl)3]+ and [2] = [Mo3S4Pd(H2O)3(acacbutyl)3]+. Insert shows a linear sweep voltammogram for bulk electrolysis at −1.5 V vs. SCE of 60 mM HCOOH at a glassy carbon electrode in the absence and in the presence of the both catalysts. The electrolyte was ACN + 0.1 M TBAP. The reference electrode was a saturated calomel electrode (SCE). (b) Shows a plot of turnover frequency vs. overpotential for both catalysts (TOFmax and E1/2 have been determined for 60 mM HCOOH concentration using the above eqn (3)), (c) and (d) represent evolution of hydrogen production by gas chromatography analysis. [Mo3S4(H2O)3(acacbutyl)3]+ = [1] and of [Mo3S4Pd(H2O)3(acacbutyl)3]+ = [2]. | ||
3.3. The HER electroactivity of the two complexes after their functionalization on a surface
The two compounds of interest were physisorbed at the surface of two different carbonaceous substrates (Vulcan and graphene) of interest for practical applications. Fig. 5 shows SEM micrographs of the resulting structures and the corresponding chemical microanalyses performed by energy-dissipative spectroscopy. Fig. 5a shows that the bare Vulcan electrode (without catalyst) contains spherical carbon particles (mean diameter ≈ 100 nm). No other element was detected by EDS (insert of Fig. 5a). Fig. 5b and c show the SEM micrographs of Vulcan-based electrodes after addition of [1] and [2] in the ratio 3:7 wt%, respectively. Some crystals of the complexes are observed. The EDS spectra revealed the presence of Mo and Pd, as expected. Fig. 5d shows the SEM micrograph of a bare graphene electrode (without catalyst). The graphene sheets are homogeneously dispersed over the whole surface of the electrode. Fig. 5e and f show the micrographs of graphene-containing electrodes after addition of [1] or [2] in the ratio 1:1 wt%, respectively. SEM micrographs underline an homogeneous graphene/complex mixture on large-scale. The corresponding EDS spectra (inserts) reveal the presence of Mo and Pd, as expected.
 | ||
| Fig. 5 SEM micrographs of glassy carbon electrode modified with Vulcan (a), with Vulcan/[1] (in the ratio 3:7) (b) and with Vulcan/[2] (in the ratio 3:7) (c). Glassy carbon electrode modified with graphene (d), with graphene/[1] (in the ratio 1:1) (e) and with graphene/[2] (in the ratio 1:1) (f). | ||
The HER activity of these electrodes was measured in 0.1 M H2SO4 aqueous solution (pH = 0.7). Results are reported on Fig. 6a and b. All four samples exhibit excellent HER catalytic activity, as evidenced by the Tafel slopes (Fig. 6a) and an early onset of H2 evolution (on Fig. 6b). Compared to bare Vulcan, the HER overpotential has been reduced by 682 mV with [1]/Vulcan and 823 mV with [2]/Vulcan (70:30 wt%). A similar improvement of the HER activity was observed on graphene-containing samples (50:50 wt%), but the HER overvoltage was less reduced than with Vulcan (300 mV and 262 mV with [2]/graphene and [1]/graphene, respectively). Such difference between Vulcan-containing and graphene-containing samples was attributed to a better physisorption of the complexes on Vulcan. We noted that Vulcan, used a catalyst support, could enhance the stability of the electrocatalytic complex leading to a larger-scale hydrogen generation using this type of electrode. Moreover, one can see from the cyclic voltammogram of Fig. 6b that using the Vulcan-based complex electrodes, the overpotential is decreased, getting closer to the one measured on a Pt electrode. The complex–substrate interaction can strongly influence the stability of the complex which is of major importance for electrolysis applications. The carbon substrates used for the tests are conventional materials, commonly used in PEM technology. The type of interaction between the complex and the substrate is central. Using the Pd-containing catalyst, it was not possible to determine the formation of palladium metal nanoparticles and therefore the activity is attributed to complex [2]. Results show that for a given potential, higher complex loadings yield higher current densities. Tafel plots (i.e. plots of the HER overpotential against the logarithm of the current density) with units of millivolts per decade of current density are shown in Fig. 6a. The cathodic Tafel slope related to complexes [1] and [2] was found to progressively decrease by addition of increasing amounts of catalyst, corresponding to an increasing HER electroactivity.
 | ||
| Fig. 6 (a) Tafel slopes and (b) linear voltammograms, with and without Vulcan, with and without graphene, with and without [1] or [2] at a glassy carbon electrode. The electrolyte was 0.1 M H2SO4 in water, pH 0.7. Scanning rate: 50 mV s−1. | ||
Fig. 6b displays the polarization curves measured on the drop-casted [1] and [2] for HER featured by a very significant HER activity. Compared to the electrocatalytic activity of Vulcan/[2] films, an onset potential of −124 mV vs. RHE, a Tafel slope of −150 mV dec−1 and a current density of 30 mA cm−2 at an overpotential of −67 mV (with respect to a smooth planar Pt electrode) were measured. However, the drop-casted [2] films mixed with graphene showed highly efficient electrocatalytic activity for HER with a higher onset potential of −439 mV vs. RHE, a Tafel slope of −117 mV dec−1 and a current density of 30 mA cm−2 at an overpotential of −382 mV with respect to a planar Pt electrode. On the basis of the kinetic principle of HER, a Tafel slope means that HER mechanism at a drop-casted MoxSy films might be Volmer–Heyrovsky mechanism with Heyrovsky reaction as a rate-determining step.61
3.4. Application to PEM water electrolysis
The two coordination complexes ([1] and [2]) have been tested as HER electrocatalysts at the cathode of a PEM water electrolysis cell. Practical details are provided in the experimental section. The loading of [1] or [2] at the electrode was 2.33 mg cm−2. The IV curves measured at 70 °C are shown in Fig. 7. The reference curve measured with Pt (loading = 1 mgPt cm−2) at the cathode is shown for comparison. It has to be noted that considering [2], the amount of Pd loading corresponds to 0.24 mgPd cm−2, four times less than Pt on the reference sample. No Pd is present in [1]. All experiments were made using the same membrane (Nafion® 117), the same anode (IrO2, 2 mg cm−2), and the same experimental cell to facilitate the comparison of the results. Performance differences are therefore attributed to the different cathode catalysts only. It has to noted that individual experimental datapoints plotted in Fig. 7 were recorded by chronoamperometry at low scan rate (1 mV s−1). The cell voltage was sampled at a variable frequency (fast at low current density to acquire the envelope and slower after in the linear region). For Pt, the iV curve is smooth over the entire current range. For complexes [1] and [2] there are some small fluctuations over the entire current density range. Mass-transport limitation are observed at elevated current densities (for j > 600 mA cm−2 for complex [1] and for j > 800 mA cm−2 for complex [2]). This is attributed to a lack of HER catalytic sites. Care should be taken in the analysis of these performances to take into account the activity of the catalytic phases and the engineering of the catalyst layers which usually require a complex optimization process. These layers are basically a dispersion of two powdered components (carbon black and active phase) in a Nafion® matrix. Proportions and quality of the dispersions also contribute to the performance level. Each of the three IV curves has been fitted using a model described elsewhere.57,62 In this model, the cell voltage is obtained by summing the HER, OER and ohmic cell voltage contributions. Experimental curves were fitted by changing only the HER exchange current density since the same membrane and anode were used as described earlier. Fit parameters are compiled in Table 2 (ja0 and jc0 are the anodic and cathodic exchange current densities, and raf, rcf are the anodic and cathodic roughness factors). It can be recalled here that the roughness factor rf is a dimensionless number obtained by taking the ratio of the total surface area to the geometrical surface area of the MEA. This is a useful parameter used to take into account the geometry of large surface area electrodes such as the ones used here with powdered catalysts. The product of the exchange current density with the roughness factor give an apparent exchange current density noted j0*. The same j0* value can be obtained by the product of different j0 (true electrochemical activity) and rf (geometrical factor) values.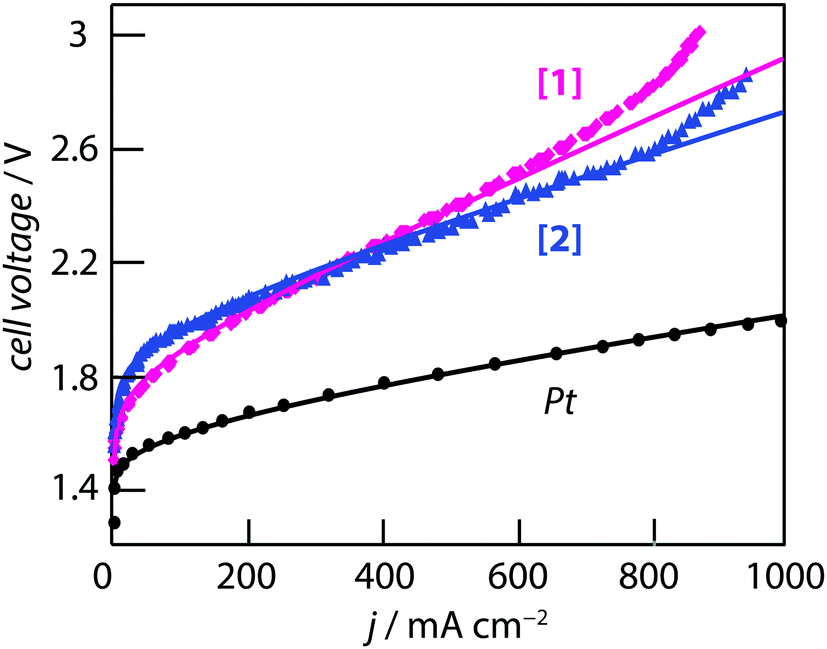 | ||
| Fig. 7 iV curves measured at 70 °C on a 7 cm2 MEA with 2 mg cm−2 IrO2 at the anode and a cathode containing: 1 mg cm−2 of Pt nanoparticles, 2.33 mg cm−2 of [1] and 2.33 mg cm−2 of [2], respectively. | ||
| Cat. |
|
j a0 (A cm−2) | r af (adim) | j c0 (A cm−2) | r cf (adim) |
|---|---|---|---|---|---|
| Pt | 325 | 5 × 10−6 | 100 | 1 × 10−3 | 70 |
| [1] | 1000 | 5 × 10−6 | 100 | 4.3 × 10−7 | 70 |
| [2] | 700 | 5 × 10−6 | 100 | 1.1 × 10−8 | 70 |
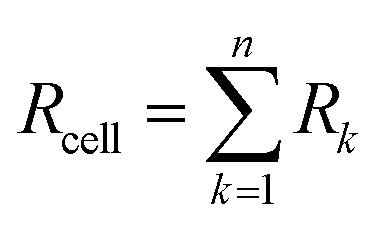
On the OER side, an exchange current density of 5 × 10−6 A cm−2 and a roughness factor of 100 (yielding an apparent current density of (j0*)a = 5 × 10−4 A cm−2) is commonly obtained with IrO2. The cell resistance (Rcell) is the summation of the individual resistances of the membrane resistance, electronic and contact resistances of the different cell components, and also the resistance of the catalyst layers which have been determined by electrochemical impedance spectroscopy. The experimental IV curves were fitted using the same roughness factor on the HER side since in all three cases this is a carbon powder containing particles of the active phase. Fig. 7 and the data of Table 2 show that, assuming a constant roughness factor, the main difference between the three curves comes for the exchange current density at the cathode. Compared to Pt, the j0 value obtained for [Mo3S4(H2O)3(acacbutyl)3]+ is ∼2320 times less than Pt and the j0 value obtained [Mo3S4Pd(H2O)3(acacbutyl)3]+ is ∼91000 times less than Pt. As a result, the IV curves are shifted upwards to higher cell voltage values. At 50 mA cm−2 (a current density for which the ohmic drop due to the cell resistance is less than 50 mV), the HER overvoltage is increased by ∼250 mV with [1] and by ∼350 mV with complex [2] compared to Pt. These results are consistent with those of Fig. 6a, except that the performance measured with the Pd-containing compound are slightly less efficient.
4. Conclusions
We report on the synthesis and electrochemical characterization of two {Mo3S4}-based complexes, i.e. [Mo3S4(H2O)3(acacbutyl)3]+ and [Mo3S4Pd(H2O)3(acacbutyl)3]+. Their properties with regard to the hydrogen evolution reaction (HER) have been measured in aqueous media. The electrochemical behavior of Pd-free [1] and Pd-containing [2] complexes in acetonitrile has been investigated by cyclic voltammetry, linear potential and chrono-coulometry at controlled potential. Their electrochemical activity with regard to the HER has been evidenced by dropwise addition of controlled amounts of HCOOH in acetonitrile. The HER activity of the two compounds has been compared by calculating their corresponding turn over frequency. The higher TOFmax was obtained with [2]. The two compounds have been subsequently functionalized onto two different carbonaceous substrates of practical interest for applications: graphene and carbon black. The polarization curves and Tafel plots have been measured in 0.1 M H2SO4 aqueous solutions. The electrodes modified with Mo3S4 drops exhibit a high HER activity with an onset potential at −124 mV vs. RHE and a Tafel slope of −150 mV dec−1. The potential at which a current density of 30 mA cm−2 was passed in 0.1 M H2SO4 (pH = 0.7) was found to be −0.21 V vs. RHE on Pt, −0.31 V vs. RHE on [2] + Vulcan, and −0.57 V vs. RHE on [1] + Vulcan. Finally, the two complexes have been implemented at the cathode of PEM water electrolysis cells in place of platinum. The polarization curves have been measured and compared to those obtained with platinum. The HER overvoltage was increased by ∼250 mV with [1] and by ∼350 mV with complex [2] compared to Pt. Although the catalytic system based on {MoS} electrocatalysts is not fully ready for large-scale implementation in industrial applications, these results show they are good candidates to replace Pt for the HER in PEM water electrolysis technology but optimization is still required. Finally, it is worth noting that Mo resources are about 1.1 × 10−4% in the Earth crust whereas they are of 3.7 × 10−6% for Pt.63Abbreviations
| ACN | Acetonitrile |
| CV | Cyclic voltammetry |
| EDS-EDX | Energy dissipative X-ray spectroscopy |
| ESI-MS | Electrospray ionization mass spectrometry |
| ET | Electron transfer |
| FT-IR | Fourier transform infrared spectroscopy |
| HER | Hydrogen evolution reaction |
| MEA | Membrane electrode assembly |
| OER | Oxygen evolution reaction |
| PEM | Polymer electrolyte membrane/proton exchange membrane |
| PGM | Platinum group metal |
| PTL | Porous transport layer |
| SCE | Saturated calomel electrode |
| SEM | Scanning electron micrograph |
| TOF | Turnover frequency |
| TON | Turnover number |
Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
This work has been supported by the French National Research Agency (ANR) (ANR-15-CE06-0002-03, CHALCOCAT, ANR-11-IDEX-0003-02 and CHARMMMAT ANR-11-LABX-0039 as part of the “Investissements d’Avenir” program). We also thank CNRS, and Universities of Versailles Saint Quentin and Paris-Saclay as well as the IRS MOMENTOM (Idex Université Paris-Saclay), for financial support.References
- S. Asnavandi and C. Zhao, ACS Sustainable Chem. Eng., 2017, 5(1), 85 CrossRef.
- W. Gao, Y. Q. Shi, Y. F. Zhang, L. Z. Zuo, H. Y. Lu, Y. P. Huang, W. Fan and T. X. Liu, ACS Sustainable Chem. Eng., 2016, 4(12), 6313 CrossRef CAS.
- K. Kawashima, M. Hojamberdiev, H. Wagata, K. Yubuta, K. Domen and K. Teshima, ACS Sustainable Chem. Eng., 2017, 5(1), 232 CrossRef CAS.
- X. D. Yan, L. H. Tian, S. Atkins, Y. Liu, J. Murowchick and X. B. Chen, ACS Sustainable Chem. Eng., 2016, 4(7), 3743 CrossRef CAS.
- Z. Ren, X. Xu, X. Wang, B. Gao, Q. Yue, W. Song, L. Zhang and H. Wang, J. Colloid Interface Sci., 2016, 468, 313 CrossRef CAS PubMed.
- H. Liu, D. Chen, Z. Wang, H. Jing and R. Zhang, Appl. Catal., B, 2017, 203, 300 CrossRef CAS.
- L. Ziani and A. haker, Int. J. Hydrogen Energy, 2016, 41(27), 11842 CrossRef CAS.
- A. R. Martins, L. S. Carvalho, P. Reyes, J. M. Grau and M. D. C. Rangel, Mol. Catal., 2017, 429, 1 CrossRef CAS.
- Z. Pu, S. Wei, Z. Chen and S. Mu, Appl. Catal., B, 2016, 196, 193 CrossRef CAS.
- D. Bessarabov and P. Millet, in PEM water Electrolysis, ed. B. G. Pollet, Elsevier, 1st edn, 2018, vol. 1 and 2, ISBN: 978-0-12-811145-1; ISBN 978-0-08-102830-8 Search PubMed.
- L. Allidières, A. Brisse, P. Millet, S. Valentin and M. Zeller, Int. J. Hydrogen Energy, 2019, 44, 9670 CrossRef.
- D. Akyüz, B. Keskin, U. Şahintürk and A. Koca, Appl. Catal., B, 2016, 188, 217 CrossRef.
- S. Reddy, R. Du, L. Kang, N. Mao and J. Zhang, Appl. Catal., B, 2016, 194, 16 CrossRef CAS.
- L. Wang, Y. Li, M. Xia, Z. Li, Z. Chen, Z. Ma, X. Qin and G. Shao, J. Power Sources, 2017, 347, 220 CrossRef CAS.
- R. Li, L. Yang, T. Xiong, Y. Wu, L. Cao, D. Yuan and W. Zhou, J. Power Sources, 2017, 356, 133 CrossRef CAS.
- Y. Feng, H. Zhang, Y. Guan, Y. Mu and Y. Wang, J. Power Sources, 2017, 348, 246 CrossRef CAS.
- Z. Deng, J. Wang, Y. Nie and Z. Wei, J. Power Sources, 2017, 352, 26 CrossRef CAS.
- T. Li, K. Niu, M. Yang, N. K. Shrestha, Z. Gao and Y.-Y. Song, J. Power Sources, 2017, 356, 89 CrossRef CAS.
- L. Yang, W. Zhou, D. Hou, K. Zhou, G. Li, Z. Tang, L. Li and S. Chen, Nanoscale, 2015, 7(12), 5203 RSC.
- Y. N. Wang, S. H. Ma, Q. C. Li, Y. Zhang, X. J. Wang and X. J. Han, ACS Sustainable Chem. Eng., 2016, 4(7), 3773 CrossRef CAS.
- J. Al Cheikh, A. Villagra, A. Ranjbari, A. Pradon, M. Antuch, D. Dragoe, P. Millet and L. Assaud, Appl. Catal., B, 2019, 250, 292 CrossRef CAS.
- M. Antuch, A. Ranjbari, S. A. Grigoriev, J. Al-Cheikh, A. Villagrá, L. Assaud, Y. Z. Voloshin and P. Millet, Electrochim. Acta, 2017, 245, 1065 CrossRef CAS.
- X. Weng, Q. Liu, A.-J. Wang, J. Yuan and J.-J. Feng, J. Colloid Interface Sci., 2017, 494, 15 CrossRef CAS PubMed.
- Y. Luo, D. Huang, M. Li, X. Xiao, W. Shi, M. Wang, J. Su and Y. Shen, Electrochim. Acta, 2016, 219, 187 CrossRef CAS.
- J. Y. Kim, S. Han and J. H. Bang, Mater. Lett., 2017, 189, 97 CrossRef CAS.
- J. M. Saveant, Chem. Rev., 2008, 108, 2348 CrossRef CAS PubMed.
- J. P. Bigi, T. E. Hanna, W. Hill Harman, A. Changa and C. J. Chang, Chem. Commun., 2010, 46, 958 RSC.
- W. Zhou, J. Lu, K. Zhou, L. Yang, Y. Ke, Z. Tang and S. Chen, Nano Energy, 2016, 28, 143 CrossRef CAS.
- S. Schlicht, L. Assaud, M. Hansen, M. Licklederer, M. Bechelany, M. Perner and J. Bachmann, J. Mater. Chem. A, 2016, 4, 6487 RSC.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127(15), 5308 CrossRef CAS PubMed.
- S. Kasztelan, Langmuir, 1990, 6, 590 CrossRef CAS.
- S. Kasztelan, C. R. Acad. Sci., Ser. II, 1993, 317, 171 CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15(3), 364 CrossRef CAS PubMed.
- O. Weisser and S. Landa, Sulfide Catalysts: Their Properties and Applications, Pergamon Press, New-York, 1973 Search PubMed.
- S. L. Qiu, W. Y. Xing, X. W. Mu, X. M. Feng, C. Ma, R. K. K. Yuen and Y. Hu, ACS Appl. Mater. Interfaces, 2016, 8(47), 32528 CrossRef CAS PubMed.
- L. Yang, W. Zhou, J. Lu, D. Hou, Y. Ke, G. Li, Z. Tang, X. Kang and S. Chen, Nano Energy, 2016, 22, 490 CrossRef CAS.
- W. Zhou, D. Hou, Y. Sang, S. Yao, J. Zhou, G. Li, L. Li, H. Liu and S. Chen, J. Mater. Chem. A, 2014, 2(29), 11358 RSC.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29 CrossRef CAS.
- C.-B. Ma, X. Qi, B. Chen, S. Bao, Z. Yin, X.-J. Wu, Z. Luo, J. Wei, H.-L. Zhang and H. Zhang, Nanoscale, 2014, 6(11), 5624 RSC.
- J. Lv, M. Yang, T. Liang, S. Ken and M. Hideo, Chem. Phys. Lett., 2017, 678, 212 CrossRef CAS.
- X. Zhang, P. Ding, Y. Sun, Y. Wang, J. Li and J. Guo, Mater. Lett., 2017, 197, 41 CrossRef CAS.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6(3), 248–253 CrossRef CAS.
- D. Yue, T. Zhang, M. Kan, X. Qian and Y. Zhao, Appl. Catal., B, 2016, 183, 1 CrossRef CAS.
- A. Müller, S. Sarkar, R. G. Bhattacharyya, S. Pohl and M. Dartmann, Angew. Chem., Int. Ed. Engl., 1978, 17, 535 CrossRef.
- D. Yue, Z. Zhang, Z. Tian, T. Zhang, M. Kan, X. Qian and Y. Zhao, Catal. Today, 2016, 274, 22 CrossRef CAS.
- T. F. Jaramillo, J. Bonde, J. Zhang, B.-L. Ooi, K. Andersson, J. Ulstrup and I. Chorkendorff, J. Phys. Chem. C, 2008, 112(45), 17492 CrossRef CAS.
- R. Llusar and S. Uriel, Eur. J. Inorg. Chem., 2003, 1271 CrossRef CAS.
- J. Á. Pino-Chamorro, A. L. Gushchin, M. J. Fernández-Trujillo, R. Hernández-Molina, C. Vicent, A. G. Algarra and M. G. Basallote, Chem. – Eur. J., 2015, 21, 2835 CrossRef CAS PubMed.
- B. Keita, S. Floquet, J. F. Lemonnier, E. Cadot, A. Kachmar, M. Benard, M. M. Rohmer and L. Nadjo, J. Phys. Chem. C, 2008, 112(4), 1109 CrossRef CAS.
- A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, J. Fize, V. Artero, O. David, E. Magnier, B. Pegot and E. Cadot, Dalton Trans., 2013, 42(14), 4848 RSC.
- J. Kristensen, J. D. Zhang, I. Chorkendorff, J. Ulstrup and B. L. Ooi, Dalton Trans., 2006, 3985 RSC.
- Y. D. Hou, B. L. Abrams, P. C. K. Vesborg, M. E. Bjorketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen, O. Hansen, J. Rossmeisl, S. Dahl, J. K. Norskov and I. Chorkendorff, Nat. Mater., 2011, 10(6), 434 CrossRef CAS PubMed.
- H. Akashi, T. Shibahara and H. Kuroya, Polyhedron, 1990, 9(14), 1671 CrossRef CAS.
- B. Seger, K. Herbst, T. Pedersen, B. Abrams, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Electrochem. Soc., 2014, 161(12), H722 CrossRef.
- T. Shibahara, M. Yamasaki, G. Sakane, K. Minami, T. Yabuki and A. Ichimura, Inorg. Chem., 1992, 31, 640 CrossRef CAS.
- D. Fu, B. Fabre, G. Loget, C. Meriadec, S. Ababou-Girard, E. Cadot, N. Leclerc-Laronze, J. Marrot and Q. de Ponfilly, ACS Omega, 2018, 3, 13837 CrossRef CAS PubMed.
- P. Millet, ECS Trans., 2016, 75(14), 28 Search PubMed.
- EU Harmonised Polarisation Curve Test Method for Low Temperature Water Electrolysis, ed. T. Malkow, A. Pilenga and G. Tsotridis, EUR 29182 EN, JRC104045, Publications Office of the European Union, Luxembourg, 2018, ISBN 978-92-79-90388-6 Search PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235 CrossRef CAS.
- R. Kucernak and C. Zalitis, J. Phys. Chem. C, 2016, 120, 10721 CrossRef.
- S. A. Grigoriev, M. S. Mamat, K. A. Dzhus, G. S. Walker and P. Millet, Int. J. Hydrogen Energy, 2011, 36, 4143 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, FT-IR, ATG, UV-visible spectra. See DOI: 10.1039/d0ma00138d |
| ‡ Present address: Department of Hydro and Renewable Energy, Indian Institute of Technology Roorkee, Uttarakhand, India. |
| This journal is © The Royal Society of Chemistry 2020 |