
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal organic framework-derived porous Fe2N nanocubes by rapid-nitridation for efficient photocatalytic hydrogen evolution†
Zhixing
Cheng‡
ab,
Ali
Saad‡
ab,
Samira
Adimi‡
ab,
Haichuan
Guo‡
ab,
Siqi
Liu‡
 *ab,
Tiju
Thomas‡
*c and
Minghui
Yang‡
*ab
*ab,
Tiju
Thomas‡
*c and
Minghui
Yang‡
*ab
aNingbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo 315201, P. R. China. E-mail: liusiqi@nimte.ac.cn; myang@nimte.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology Madras, Adyar, Chennai 600036, Tamil Nadu, India. E-mail: tijuthomas@iitm.ac.in
First published on 24th June 2020
Abstract
Transition metal nitrides are promising substitutes for noble metal catalysts in photocatalytic hydrogen evolution reaction. Here, we use the cubic metal organic framework (MOF) Prussian blue, which has been rarely explored for nitride preparation. A rapid nitridation process is used for obtaining porous iron nitride (Fe2N) nanoparticles. The phase-pure Fe2N catalyst reported largely maintains the cubic geometry and specific surface area of the parent MOF. Fe2N is sensitized using Eosin-Y for boosting photocatalytic hydrogen evolution. Hydrogen production rates close to ∼14.5 mmol g−1 h−1 are observed, which are considered desirable. This is reasonable since the nitride phase has high electronic conductivity and low electrocatalytic H2-generation overpotential. Calculations indicate that the electronic conductivity of Fe2N is due to Fe-3d states. This work opens up new possibilities for the production of porous nitride photocatalysts for efficient hydrogen evolution.
Introduction
Transition-metal nitrides (TMNs) represent an important category of materials with interesting optoelectronic, catalytic, electrochemical, and structural functions.1–5 In particular, the Pt-group like properties of TMNs lead to significant binding and hence strong adsorption to hydrogen. These characteristics make them promising in the field of catalytic hydrogen production.6–12 However, there are challenges associated with the synthesis of TMNs. Notably, the traditional synthesis of TMNs involves high temperatures and pressures. The products thus formed tend to have larger particle sizes, and hence are rarely suitable for catalytic purposes.6,8,13–15 Furthermore, conventional ammonothermal and nitridation techniques pose significant challenges to scalability. It is very important to industrial applications. However currently, due to synthetic developments, TMN materials can be synthesized in the nano-regime with different morphologies.6,8,16–19 Currently, both (i) low temperature, rapid and high yield methods of preparation and (ii) nanosizing are imperative for the eventual success of TMNs in catalysis and related areas. In particular, there is value in developing methods that enable the production of porous 3D TMNs using soft chemical approaches. Clearly, the development of such techniques merits further attention.Metal–organic frameworks (MOFs) present an opportunity to address the synthetic requirements mentioned above. Thus far MOFs that consist of metal ions/clusters and coordinated organic linkers have found uses in applications such as gas storage/separation, catalysis, sensing and drug delivery.20–23 Importantly, their high porosity and large specific surface area, as well as tunable pore size and morphology make them excellent sacrificial templates for fabricating porous MOF-derived functional materials.24–26 For example, various MOF-derived porous carbon, metal oxides, metal sulfides, metal phosphates and their nanocomposites have been reported.27–31 In these cases, MOFs have been used as both the precursor and template for the synthesis of catalysts.32 Although ammonia treatment of MOF-derived metal/carbon nanocomposites can yield porous metal nitride/carbon composites, pure metal nitrides derived from MOFs with porous structures have been rarely reported. This is primarily due to the ease of collapse of the pore structures associated with MOFs at high reaction temperatures; this is especially the case with long reaction times.33,34
As a subclass of MOFs, Prussian blue (PB) created from the supramolecular assembly of iron components with N-containing organic ligands has attracted considerable interest. This is due to the diverse architectures and morphologies it offers.35 In fact, there exist reports on topotactic transformation for the fabrication of various nanomaterials derived from PB or PB analogues. Materials obtained using this approach include porous metal phosphates, sulfides, carbides, oxides, etc.36–39 However, PB-derived porous metal nitrides have not been explored extensively. Previously we reported the synthesis of PB-derived metal nitrides, which yielded nanoparticles without substantial porosity.8 Therefore, a novel synthesis process is highly desirable for achieving MOF-derived porous metal nitrides which nearly retain the specific structure of the parent precursor; this would be highly desirable for catalytic applications.
In this work, the PB precursors are first oxidized and then subjected to rapid nitridation to obtain porous Fe2N nanocubes. The nanocubes obtained are phase pure. The process used for heating is essential for obtaining the material. Using a slip furnace for pre-heating and rapid cooling also reduces the reaction time. The samples thus obtained are sensitized using Eosin-Y (EY) in photocatalytic hydrogen evolution reaction. PB-derived porous Fe2N nanocubes are synthesized while maintaining the pattern and structure of the parent MOF precursor. The performance of cube-like Fe2N is duly rationalized using density functional theory (DFT) based calculations and its metallic nature is also duly elaborated. The superior metallic conductivity of Fe2N boosts the separation and migration of charge carriers excited from EY. Besides, the porous structure of PB-derived Fe2N nanocubes contributes to the high density of active sites, which in turn yields desirable kinetics for photocatalytic hydrogen evolution. The optimal Fe2N/Eosin-Y system exhibits excellent photocatalytic hydrogen evolution performance with a H2 generation rate at 14.5 mmol g−1 h−1.
Experimental section
Reagents
All chemicals are of analytical grade and are obtained from commercial suppliers and used without further purification. Potassium hexacyanoferrate(II) trihydrate {K4[Fe(CN)6]·3H2O, AR}, hydrochloric acid (HCl, AR) and polyvinylpyrrolidone K30 {(C6H9NO)n, GR} are purchased from Sinopharm Chemical Reagent Co., Ltd. Triethanolamine (TEOA, C6H15NO3, AR) is purchased from Aladdin Chemical Co., Ltd. Eosin Y disodium salt (C20H6Br4Na2O5, RG) is purchased from Adamas Reagent, Ltd.Synthesis of photocatalysts
Subsequently, 10 mg of the oxide obtained is moved to the middle of the tube. The tube is placed in a rail tube furnace. After that, the air is pumped out and the tube is filled with ammonia. This is repeated thrice, to avoid oxygen contamination. When the temperature approaches 400 °C, the sample is allowed to soak for ∼10 min for confirming the stability of the temperature. The reaction time is 30 min, then the Fe2N is produced. Then the sample obtained is used for measurements.
Characterization
X-ray powder diffraction (XRD) measurements are performed using a powder X-ray diffractometer (Rigaku Miniflex 600) with Cu-Kα radiation (λ = 1.54178 Å). The 2θ range is set from 10° to 80° and the measurement rates are kept at 1° min−1. The morphological structure and microstructures of the samples are characterized using scanning electron microscopy (SEM) (FE-SEM, Hitachi S4800, Japan) and transmission electron microscopy (TEM) (JEOL model JEM 2100 EX instrument) measurements. In addition, high-resolution TEM, selected area electron diffraction (SAED) and elemental mapping measurements are also performed using the same TEM instrument.Photocatalytic H2 evolution measurements
Photocatalytic H2 evolution measurements are carried out in a Pyrex top-irradiation vessel connected to a glass closed gas circulation system. The temperature of the reactant solution is maintained at 5 °C through the flow of cooling water during the reaction. A 300 W xenon lamp (PLS-SXE300C, Perfectlight) is used as the visible light source. The irradiation area is 18.1 cm2 and the incident light power density is 304 mW cm−2. Typically, 5 mg of the catalyst and 80 mg of Eosin-Y are dispersed in 80 mL of a 10% (v/v) triethanolamine–water (TEOA–H2O) solution by ultrasonication for 15 min.The cyclic stability of the catalyst is tested as follows and the initial reaction amount of Eosin-Y is 20 mg. After the first run, under visible light irradiation, the photocatalytic system is thoroughly degassed. Then 20 mg of fresh EY, 2 mL of TEOA and 15 mL of deionized water are added without any other treatments like centrifugation and washing (pH = 11). Subsequently, the thoroughly degassed system is irradiated again using a 300 W Xe lamp. In controlled experiments, the pH values of the reaction solution are adjusted by the addition of hydrochloric acid (HCl) or sodium hydroxide (NaOH).
Results and discussion
As shown in Scheme S1 (ESI†), Prussian blue (PB)-derived Fe2N nanocubes are synthesized via a two-step process, i.e. oxidation followed by rapid-nitridation. In brief, PB is first synthesized via a hydrothermal method.40 Then, PB precursors are heated in air at 350 °C for 1 h to form Fe2O3. Finally, the as-prepared Fe2O3 is treated in an NH3 atmosphere at 400 °C for 30 min. Actually, this is the rapid nitridation step that yields Fe2N. The typical Rietveld refined XRD patterns of the as-prepared samples are shown in Fig. 1 and Fig. S1 (ESI†). The PB precursor, the iron oxide that derived from it, and the corresponding nitride all belong to the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Fdm and Pbcn space groups, respectively. The detailed results can be seen in Table S1 (ESI†); there is no indication of oxygen or nitrogen vacancies. Hence the occupancies for oxygen and nitrogen sites are fixed at 1.0. Notably, for the iron oxide sample, the yellowish-brown sample suggests that Fe3+ makes up the majority of the compounds. Refinement shows that there are obvious vacancies at the iron sites (the site occupancy of Fe is only about 0.6–0.7 as shown in Table S1b, ESI†). However, the Fe occupancy of the iron nitride sample is 0.9398 (Table S1c, ESI†), which is consistent with the simple model of the fully nitrogenated Fe2N.
m, Fdm and Pbcn space groups, respectively. The detailed results can be seen in Table S1 (ESI†); there is no indication of oxygen or nitrogen vacancies. Hence the occupancies for oxygen and nitrogen sites are fixed at 1.0. Notably, for the iron oxide sample, the yellowish-brown sample suggests that Fe3+ makes up the majority of the compounds. Refinement shows that there are obvious vacancies at the iron sites (the site occupancy of Fe is only about 0.6–0.7 as shown in Table S1b, ESI†). However, the Fe occupancy of the iron nitride sample is 0.9398 (Table S1c, ESI†), which is consistent with the simple model of the fully nitrogenated Fe2N.
 | ||
| Fig. 1 (a) Rietveld refined XRD pattern and (b) SEM images of Fe2N, with the inset showing the cube size in high magnification. (c) Low magnification TEM image, (d) HRTEM images and (e–h) the elemental mapping images of Fe2N. | ||
The morphology of the as-prepared samples has been tested by electron microscopy (EM) images. As shown in Fig. S2 (ESI†), PB exhibits uniform cube-like morphology with a smooth surface. Its average size is found to be ∼0.53 μm. After the oxidation process, Fe2O3 retains the cube-like morphology while its size shrinks to ∼0.47 μm. This size shrinkage is due to the oxidation of the ligands in PB. As shown in Fig. 1, further nitridation of Fe2O3 nanocubes leads to the formation of Fe2N with a rougher surface and marginally smaller size (∼0.43 μm). TEM analysis clearly indicates the coarse surface associated with Fe2N cubes. In addition, the lattice fringes in HRTEM of Fe2N indicate an interplanar distance of ∼0.26 nm. This corresponds to the (100) crystalline planes of Fe2N, which confirms the XRD results. Furthermore, elemental mappings (Fig. 1e–h) confirm the homogeneous distribution of Fe and N contents in the binary Fe2N sample.
It is important to note that the oxidation step plays a critical role in forming Fe2N nanocubes by retaining the morphology of PB. The oxidation process plays an important role in enabling the contraction of the nanocubes to a certain extent so that the collapse happens in the nitridation process. As shown in Fig. S3 (ESI†), although pure phase metal nitride is observed, direct rapid-nitridation of PB leads to collapsed Fe2N particles. In addition, the purity and morphology of PB-derived Fe2N samples are also heavily influenced by the nitridation time and temperature.
X-ray photoelectron spectroscopy (XPS) is employed to investigate the surface compositions and chemical states of the samples. As shown in Fig. S4 (ESI†), both Prussian blue and Fe2O3 exhibit an ionic iron state in Fe 2p spectra, which is in agreement with previous reports.41–46 For the Fe2N sample, the peaks centered at 710.68 eV and 724.28 eV correspond to Fe 2p3/2 and Fe 2p1/2 of metallic Fe.47,48 Besides, the peaks centered at 713.71 eV and 727.31 eV corresponding to the Fe 2p3/2 and Fe 2p1/2 of Fe3+ suggest the occurrence of surface oxidation in Fe2N.49,50 This is in agreement with other reported metal nitrides.51–53 The N 1s spectrum of the Fe2N sample can be divided into three peaks at 396.13 eV, 397.84 eV and 399.48 eV, respectively, which in turn can be assigned to metal nitride and N–H.51–53 The FT-IR spectra of the samples are displayed in Fig. S5 (ESI†). The characteristic peak at 2090 cm−1 is assigned to the Fe(CN)64− group in the molecules of Prussian Blue according to previous work.54 The dominant band at 592 cm−1 is a characteristic of α-Fe2O3.55,56 The peaks at 1048, 1348 and 1540 are due to the C–O bond.55,57,58 For the Fe2N sample, the peaks at 1374 cm−1 and 628 cm−1 correspond to N–O and Fe–O–H bonds, respectively.59–61 All the results mentioned above demonstrate the successful synthesis of Fe2N.
The N2 adsorption–desorption isotherms and pore structure parameters of Prussian blue and Fe2N samples are summarized in Fig. S6 and Table S2 (ESI†). The specific surface area of PB is 5.74 m2 g−1, which is consistent with previous reports.35 It can be clearly seen that the porous structure of PB is retained during oxidation and rapid-nitration processes. In fact, the surface area of the Fe2N sample (10.12 m2 g−1) is larger than that of PB. This is due to roughness that is brought about during the processing stages.
The photocatalytic hydrogen evolution activities of the Fe2N sample under different conditions are investigated using Eosin-Y dye and triethanolamine (TEOA) as the photosensitizer and the electron donor, respectively. As can be seen in Fig. S7a (ESI†), with an increase in the mass of Eosin-Y, the amount of evolved hydrogen increases until it reaches a maximum value (14.5 mmol g−1 h−1), which corresponds to the maximum value of Eosin-Y of 80 mg (16 times the mass of catalyst). This is consistent with the fact that the dye is indeed sensitizing and provides sufficient electrons for the reaction. This is actually consistent with the literature.62 Furthermore, the increase of dye concentration improves the adsorption of Eosin-Y on the Fe2N surface, which increases the hydrogen evolution rate.62 Beyond the optimal dye loading, there is a reduction in the activity. This is likely due to the light-shielding effect.63,64 It means that beyond a certain optimal concentration, Eosin-Y exists in a free state and it quenches the reaction at the active sites. This also prevents sufficient light absorption in the reaction system.
Fe2N exhibits better activity for hydrogen evolution reaction (HER) while pH = 11, when compared to the situation wherein pH is set to 9. The observed trends are given in Fig. S7b (ESI†). Clearly, Fe2N is well suited for hydrogen evolution reaction in an alkaline condition. Fig. 2a compares the hydrogen production activity of Prussian blue, Fe2O3 and Fe2N photocatalyst under irradiation over 2 h of reaction with the usage of 80 mg of Eosin-Y, at pH = 11. The activity of Fe2N (14.5 mmol g−1 h−1) is over three times higher than that of Prussian blue (4.3 mmol g−1 h−1) and Fe2O3 (4.7 mmol g−1 h−1). The very favorable photocatalytic activity of Fe2N is aided by the high conductivity associated with the nitride phase.65 In this alkaline condition (pH = 11), the HER activity of Fe2N is comparable to most of the previously reported photocatalytic Eosin-Y sensitized systems (Table S3, ESI†). Additionally, turnover numbers (TON) of 57.8 and turnover frequencies (TOF) of 0.016 s−1 are obtained when 5 mg of Fe2N is used. This also proves the considerable catalytic ability of Fe2N.
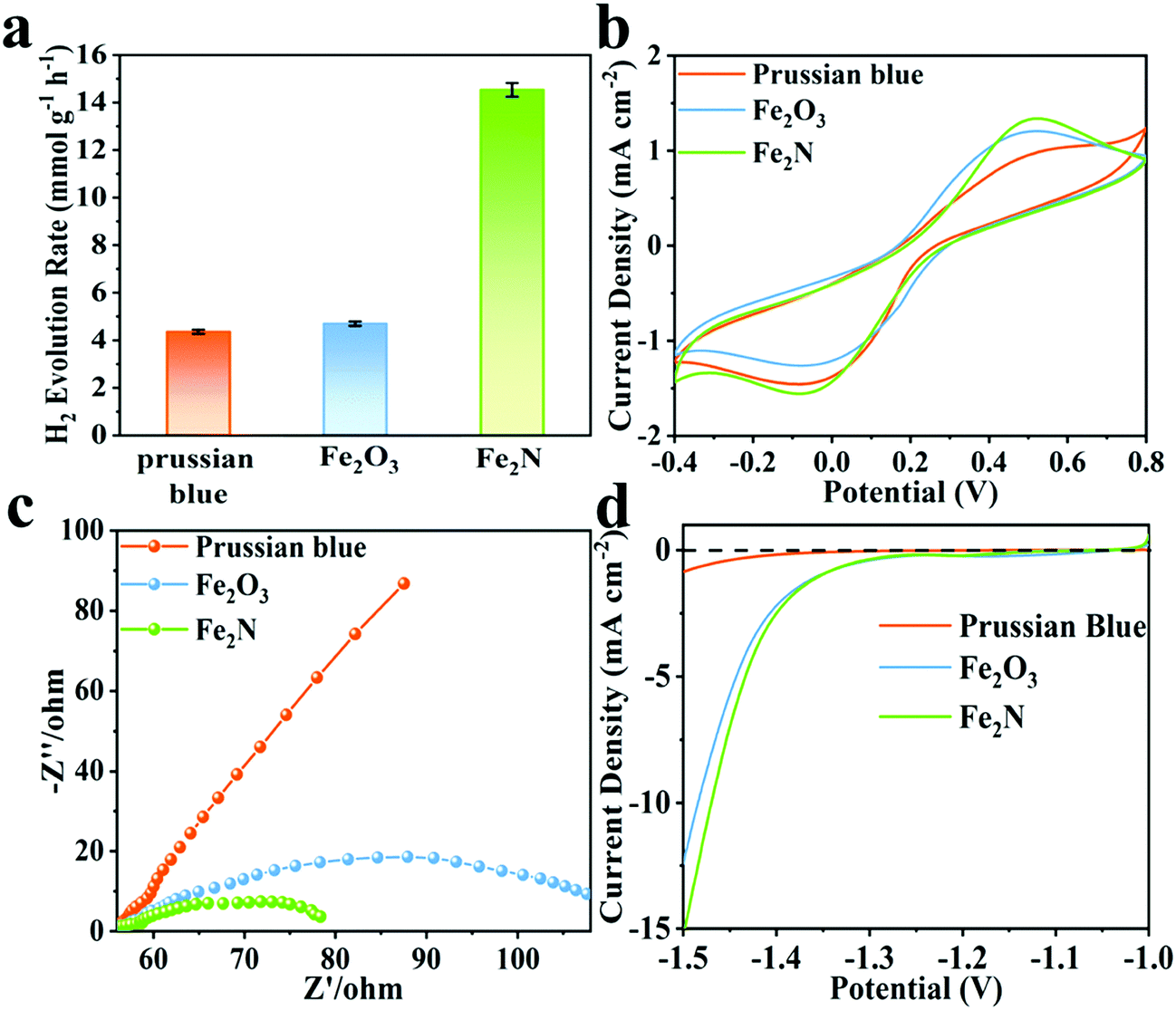 | ||
| Fig. 2 (a) Comparison of activity among Prussian blue, Fe2O3 and Fe2N under optimal conditions, (b) cyclic voltammograms, (c) EIS Nyquist plots and (d) polarization curves for the three samples. | ||
From Fig. S7c (ESI†), it is clear that in all cases, Eosin-Y and sacrificial agents are necessary for the HER process. In addition to the photocatalytic activity, stability has also been tested through several reaction cycles. As can be seen in Fig. S7d (ESI†), Fe2N shows slightly decreased activity. This is likely due to the adsorption of the samples on the magnetic stirrer, which in turn leads to a decrease in the effective interfacial area between catalyst and solution. Moreover, controlled experiments with prolonging reaction times have been tested. As shown in Fig. S8 (ESI†), the synthesized sample exhibits pure Fe2N phase with nanoparticle morphology. The photoactivity of the Fe2N nanoparticles has also been tested. As shown in Fig. S8 (ESI†), the H2 evolution rate of Fe2N nanoparticles is 12.26 mmol g−1 h−1, which is lower than that of Fe2N nanocubes. This demonstrates the advantages of the morphology and pore structure of the cubic Fe2N sample to a certain extent.
Fig. 2b displays the cyclic voltammograms (CV) of Prussian blue, Fe2O3 and Fe2N with obvious cathodic and anodic peaks. In CV measurements, the electrodes and the electrolyte are identical. Hence the current density is only related to the electron transfer rate of the electrode materials.66–69 The current density obtained over the Fe2N electrode is higher than that obtained over Prussian blue and Fe2O3; this is again an indicator of Fe2N's ability to enable the transfer of charge carriers required for catalysis. Electrochemical impedance spectroscopy (EIS) is carried out and the corresponding Nyquist plots of different samples are given (Fig. 2c). It is observed that the radius of the Fe2N sample is smaller than that of Prussian blue and Fe2O3. This actually is consistent with earlier inferences drawn from CV; Fe2N indeed has more efficient charge transfer capabilities.70,71Fig. 2d shows the polarization curves; it is clear that Fe2N improves the current density in a similar potential range. Once again this indicates that the transition metal nitride promotes the H2 reduction reaction.72 This is reasonable since the nitride phase has higher electronic conductivity and lower electrocatalytic H2-generation overpotential than the two other samples. Furthermore, specific capacitance performance is measured in a three-electrode cell system (Fig. S9, ESI†). Fe2N has a much larger curve than the other two samples, illustrating better electron accepting abilities.
The optimized structure of Fe2N and the calculated partial density of states (PDOS) are presented in Fig. 3a and b, respectively. The theoretical calculation results shown in Fig. 3b and Fig. S10 (ESI†) clearly illustrates the metallic character of the material, illustrating the existence of free electrons, which are responsible for the good conductivity of Fe2N.73 Considering the DOS plot, while there is p–d hybridization between Fe and N in the energy range of −9 to −5.8 eV, the material conductivity mostly arises from the Fe-3d distributions around the Fermi level. The conductivity of transition metal nitride is not only proved by calculation of density of states, but also suggested by reported research works.74–78
 | ||
| Fig. 3 (a) Optimized structural representation of Fe2N; iron and nitrogen atoms are shown in blue and silver colors respectively. The metallic character of the material is obvious due to the zero band-gap of the plot. (b) Total electron density of states (DOS) projected on iron and nitrogen atoms. The Fermi level is set at zero. (c) Proposed photocatalytic hydrogen production mechanism over the Eosin-Y sensitized Fe2N under visible light irradiation. | ||
The mechanism of photocatalytic H2 production enabled through the Eosin-Y sensitized Fe2N system is proposed in Fig. 3c. First, the molecules absorb photons from visible light and form the singlet excited state EY1*. Subsequently, a more stable three excited state EY3* is formed through an intersystem crossing (ISC). In the presence of TEOA as an electron donor, EY3* is reduced to EY−˙. This ion has a strong reduction ability.62 The electrons of EY−˙ are conducted into the surfaces of Fe2N owing to its metallic-like properties. A reduction reaction occurs here which results in the production of H2. At the same time, reduced Eosin-Y dye molecules return to the ground state. Because of the considerable ability of dye adsorption and electron acceptor behavior of the Fe2N structure, the recombination of photo-generated charges and holes is limited.62 Therefore, the efficiency of photocatalytic hydrogen production is improved.
Conclusion
In conclusion, MOF derived porous Fe2N has been synthesized using two-step oxidation followed by a rapid-nitridation technique. The MOF based on Prussian blue (PB), which is made from the supramolecular assembly of iron components with N-containing organic ligands, has been employed. Usage of this MOF ensures that the nitride samples retain the shape of the precursor while showing only a nominal shrinkage during the chemical transformation (hence indicating a topotactic transformation). The preparation process is both quick and rather chimie douce (soft chemical). Fe2N samples thus obtained have a high specific surface area, and offer several exposed active sites for photocatalytic activity. Fe2N is sensitized using Eosin-Y for boosting photocatalytic hydrogen evolution. Hydrogen production rates close to ∼14.5 mmol g−1 h−1 are observed, which are considered desirable. The superior conductivity of Fe2N offers enhanced electron transfer performance between the catalyst and photosensitizer, and also promotes electron-hole separation, hence yielding the observed hydrogen production rates. Calculations show that free electrons in Fe2N, responsible for its conductivity, arise primarily because of Fe-3d. Overall, this work provides a new means of producing MOF derived TMN materials, which in conjunction with suitable dyes offer high-efficiency and low-cost avenues for making photocatalysts for hydrogen production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Plan (Grant No. 2016YFB0101205), Key Program of the Chinese Academy of Sciences (Grant No. KFZD-SW-320), Natural Science Foundation of China (Grant No. 21471147), and Opened Fund of the State Key Laboratory on Integrated Optoelectronics (Grant No. IOSKL2017KF08M). M. Yang would like to thank the Ningbo 3315 program. Tiju Thomas thanks Department of Science and Technology, GoI, for support via projects titled Solar Fuels: DST Solar Energy Harnessing Center (IITM) and Materials for Energy storage.References
- X. Li, Z. Ao, J. Liu, H. Sun, A. I. Rykov and J. Wang, ACS Nano, 2016, 10, 11532–11540 CrossRef CAS PubMed.
- M. Chhowalla and H. E. Unalan, Nat. Mater., 2005, 4, 317–322 CrossRef CAS PubMed.
- Y. Yuan, Y. Zhou, H. Shen, T. Thomas, J. Wang, C. Wang, J. Wang and M. Yang, ACS Appl. Energy Mater., 2018, 1, 6774–6780 CrossRef CAS.
- Y. Gu, S. Chen, J. Ren, Y. A. Jia, C. Chen, S. Komarneni, D. Yang and X. Yao, ACS Nano, 2018, 12, 245–253 CrossRef CAS PubMed.
- Y. Yuan, L. Yang, B. He, E. Pervaiz, Z. Shao and M. Yang, Nanoscale, 2017, 9, 6259–6263 RSC.
- S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946–1956 CrossRef CAS.
- A.-M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- W. Qi, S. Liu, F. Li, H. Jiang, Z. Cheng, S. Zhao and M. Yang, Catal. Sci. Technol., 2019, 9, 2571–2577 RSC.
- D. Gao, J. Zhang, T. Wang, W. Xiao, K. Tao, D. Xue and J. Ding, J. Mater. Chem. A, 2016, 4, 17363–17369 RSC.
- Z. Sun, H. Chen, L. Zhang, D. Lu and P. Du, J. Mater. Chem. A, 2016, 4, 13289–13295 RSC.
- H. Chen, D. Jiang, Z. Sun, R. M. Irfan, L. Zhang and P. Du, Catal. Sci. Technol., 2017, 7, 1515–1522 RSC.
- X. Yu, Z. Zhao, D. Sun, N. Ren, L. Ding, R. Yang, Y. Ji, L. Li and H. Liu, Chem. Commun., 2018, 54, 6056–6059 RSC.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- A. M. Alexander, J. S. J. Hargreaves and C. Mitchell, Top. Catal., 2012, 55, 1046–1053 CrossRef CAS.
- R. Michalsky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- W. Chen, james T. Muckerman and E. Fujita, ChemComm, 2013, 49, 8896 RSC.
- R. Fang, R. Luque and Y. Li, Green Chem., 2017, 19, 647–655 RSC.
- M. Lan, R. Guo, Y. Dou, J. Zhou, A. Zhou and J. Li, Nano Energy, 2017, 33, 238–246 CrossRef CAS.
- H. Bin Wu, B. Yu Xia, L. Yu, X. Yu and X. Wen (David) Lou, Nat. Commun., 2015, 6, 6512 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- Y. Lv, W. Zhan, Y. He, Y. Wang, X. Kong, Q. Kuang, Z. Xie and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 4186–4195 CrossRef PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O’Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- T. Toyao, M. Fujiwaki, K. Miyahara, T.-H. Kim, Y. Horiuchi and M. Matsuoka, ChemSusChem, 2015, 8, 3905–3912 CrossRef CAS PubMed.
- X. Kang, H. Liu, M. Hou, X. Sun, H. Han, T. Jiang, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 1080–1084 CrossRef CAS PubMed.
- Y. Xu, X. Lv, Y. Chen and W. Fu, Catal. Commun., 2017, 101, 31–35 CrossRef CAS.
- H. Liu, S. Zhang, Y. Liu, Z. Yang, X. Feng, X. Lu and F. Huo, Small, 2015, 11, 3130–3134 CrossRef CAS PubMed.
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS.
- Y. Chen, R. Zhang, L. Jiao and H. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- D. D. Vaughn, J. Araujo, P. Meduri, J. F. Callejas, M. A. Hickner and R. E. Schaak, Chem. Mater., 2014, 26, 6226–6232 CrossRef CAS.
- Y. Zhu, G. Chen, X. Xu, G. Yang, M. Liu and Z. Shao, ACS Catal., 2017, 7, 3540–3547 CrossRef CAS.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- J. Wang, C. F. Tan, T. Zhu and G. W. Ho, Angew. Chem., Int. Ed., 2016, 128, 10482–10486 CrossRef.
- C. W. Abney, K. M. L. Taylor-Pashow, S. R. Russell, Y. Chen, R. Samantaray, J. V. Lockard and W. Lin, Chem. Mater., 2014, 26, 5231–5243 CrossRef CAS.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu and X. W. (David) Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- L. Zhang, H. Bin Wu, S. Madhavi, H. H. Hng and X. Wen (David) Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed.
- M. Xia, X. Zhang, T. Liu, H. Yu, S. Chen, N. Peng, R. Zheng, J. Zhang and J. Shu, Chem. Eng. J., 2020, 394, 124923 CrossRef CAS.
- X. W. He, L. D. Tian, M. T. Qiao, J. Z. Zhang, W. C. Geng and Q. Y. Zhang, J. Mater. Chem. A, 2019, 7, 11478–11486 RSC.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- J. A. R. Guivar, E. A. Sanches, F. Bruns, E. Sadrollahi, M. A. Morales, E. O. Lopez and J. Litterst, Appl. Surf. Sci., 2016, 389, 721–734 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handb. X-ray Photoelectron Spectrosc., Phys. Electron., 1992 Search PubMed.
- P. Mills and J. L. Sullivan, J. Phys. D: Appl. Phys., 1983, 16, 723–732 CrossRef CAS.
- D. Liang, H. Zhang, X. Ma, S. Liu, J. Mao, H. Fang, J. Yu, Z. Guo and T. Huang, Mater. Energy, 2020, 17, 100433 Search PubMed.
- S. Sultan, J. N. Tiwari, J. H. Jang, A. M. Harzandi, F. Salehnia, S. J. Yoo and K. S. Kim, Adv. Energy Mater., 2018, 8, 1801002 CrossRef.
- Z. Chen, Y. Li, L. Lei, S. Bao, M. Wang, H. Liu, Z. Zhao and M. Xu, Catal. Sci. Technol., 2017, 7, 5670–5676 RSC.
- Y. Lou, J. Liu, M. Liu and F. Wang, ACS Catal., 2020, 10, 2443–2451 CrossRef CAS.
- Z. Cheng, A. Saad, H. Guo, C. Wang, S. Liu, T. Thomas and M. Yang, J. Alloys Compd., 2020, 838, 155375 CrossRef CAS.
- Y. Wang, C. Xie, D. Liu, X. Huang, J. Huo and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18652–18657 CrossRef CAS PubMed.
- Z. Liu, H. Tan, J. Xin, J. Duan, X. Su, P. Hao, J. Xie, J. Zhan, J. Zhang, J. J. Wang and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 3699–3706 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd edn, 2004 Search PubMed.
- M. M. Rafi, K. S. Z. Ahmed, K. P. Nazeer, D. S. Kumar and M. Thamilselvan, Appl. Nanosci., 2015, 5, 515–520 CrossRef CAS.
- V. Barron and J. Torrent, J. Colloid Interface Sci., 1996, 177, 407–410 CrossRef CAS.
- X. F. Ma, P. R. Chang, J. W. Yang and J. G. Yu, Carbohydr. Polym., 2009, 75, 472–478 CrossRef CAS.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, J. Photochem. Photobiol., A, 1989, 48, 161–169 CrossRef CAS.
- X. Zhang, Q. Yan, J. Li, J. Zhang and Z. Cai, Materials, 2018, 11, 139 CrossRef PubMed.
- M. A. Fernandez de la Ossa, M. Torre and C. Garcia-Ruiz, Adv. Mater. Sci. Res., 2012, 7, 201–220 Search PubMed.
- S. Zhang, W. Wu, X. Xiao, J. Zhou, F. Ren and C. Jiang, Nanoscale Res. Lett., 2011, 6, 1–9 Search PubMed.
- D. Liu, Z. Jin and Y. Bi, Catal. Sci. Technol., 2017, 7, 4478–4488 RSC.
- J. Moser and M. Grátzel, J. Am. Chem. Soc., 1984, 106, 6557–6564 CrossRef CAS.
- X. Hao, Z. Jin and G. Lu, Chem. Lett., 2016, 45, 116–118 CrossRef CAS.
- L. Chen, H. Huang, Y. Zheng, W. Sun, Y. Zhao, P. S. Francis and X. Wang, Dalton Trans., 2018, 47, 12188–12196 RSC.
- B. Weng, Q. Quan and Y. Xu, J. Mater. Chem. A, 2016, 4, 18366–18377 RSC.
- C. Han, Z. Chen, N. Zhang, J. C. Colmenares and Y. Xu, Adv. Funct. Mater., 2015, 25, 221–229 CrossRef CAS.
- M. Yang, B. Weng and Y. Xu, J. Mater. Chem. A, 2014, 2, 1710–1720 RSC.
- X. Pan, Y. Zhao, S. Liu, C. L. Korzeniewski, S. Wang and Z. Fan, Appl. Mater. Interfaces, 2012, 4, 3944–3950 CrossRef CAS PubMed.
- N. Zhang and Y. Xu, Chem. Mater., 2013, 25, 1979–1988 CrossRef CAS.
- B. Weng and Y. Xu, Appl. Mater. Interfaces, 2015, 7, 27948–27958 CrossRef CAS PubMed.
- M. Yang, C. Han and Y. Xu, J. Phys. Chem. C, 2015, 119, 27234–27246 CrossRef CAS.
- Z. Chen, J. Wang and C. Li, J. Alloys Compd., 2013, 575, 137–144 CrossRef CAS.
- Y. Yuan, Y. Zhou, H. Shen, S. A. Rasaki, T. Thomas, J. Wang, C. Wang, J. Wang and M. Yang, ACS Appl. Energy Mater., 2018, 1, 6774–6780 CrossRef CAS.
- Y. Gu, S. Chen, J. Ren, Y. A. Jia, C. Chen, S. Komarneni, D. Yang and X. Yao, ACS Nano, 2018, 12, 245–253 CrossRef CAS PubMed.
- Y. Yuan, L. Yang, B. He, E. Pervaiz, Z. Shao and M. Yang, Nanoscale, 2017, 9, 6259–6263 RSC.
- L. Chen, H. Huang, Y. Zheng, W. Sun, Y. Zhao, P. S. Francisand and X. Wang, Dalton Trans., 2018, 47, 12188–12196 RSC.
- H. Yang, S. Luo, X. Li, S. Li, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 18499–18508 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section (electrochemical measurements and computational methodology) and characterization (synthesis flows, XRD, SEM, TEM, BET, photocatalytic properties and comparison). See DOI: 10.1039/d0ma00074d |
| ‡ All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |