
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn automated microfluidic system for efficient capture of rare cells and rapid flow-free stimulation†
Philip
Dettinger
a,
Weijia
Wang
a,
Nouraiz
Ahmed
a,
Yang
Zhang
a,
Dirk
Loeffler
 a,
Tobias
Kull
a,
Martin
Etzrodt
a,
Claudia
Lengerke
b and
Timm
Schroeder
*a
a,
Tobias
Kull
a,
Martin
Etzrodt
a,
Claudia
Lengerke
b and
Timm
Schroeder
*a
aDepartment of Biosystems Science and Engineering, ETH Zurich, Basel, Switzerland. E-mail: timm.schroeder@bsse.ethz.ch
bDepartment of Biomedicine, University of Basel, Basel, Switzerland
First published on 16th October 2020
Abstract
Cell fates are controlled by environmental stimuli that rapidly change the activity of intracellular signaling. Studying these processes requires rapid manipulations of micro-environmental conditions while continuously observing single cells over long periods of time. Current microfluidic devices are unable to simultaneously i) efficiently capture and concentrate rare cells, ii) conduct automated rapid media exchanges via diffusion without displacing non-adherent cells, and iii) allow sensitive high-throughput long-term time-lapse microscopy. Hematopoietic stem and progenitor cells pose a particular challenge for these types of experiments as they are impossible to obtain in very large numbers and are displaced by the fluid flow usually used to change culture media, thus preventing cell tracking. Here, we developed a programmable automated system composed of a novel microfluidic device for efficient capture of rare cells in independently addressable culture chambers, a custom incubation system, and user-friendly control software. The chip's culture chambers are optimized for efficient and sensitive fluorescence microscopy and their media can be individually and quickly changed by diffusion without non-adherent cell displacement. The chip allows efficient capture, stimulation, and sensitive high-frequency time-lapse observation of rare and sensitive murine and human primary hematopoietic stem cells. Our 3D-printed humidification and incubation system minimizes gas consumption, facilitates chip setup, and maintains stable humidity and gas composition during long-term cell culture. This approach now enables the required continuous long-term single-cell quantification of rare non-adherent cells with rapid environmental manipulations, e.g. of rapid signaling dynamics and the later stem cell fate choices they control.
Introduction
Extracellular stimuli control cell fates like cell cycle progression, death, differentiation or lineage choice.1–6 These stimuli, like cytokines, engage specific intracellular signalling pathways and can rapidly elicit varying responses depending on e.g. signal strength, combinatorics and temporal encoding, or on the type or current state of the receiving cell.1,5,7–10 Individual stimuli usually activate multiple intracellular signalling pathways, and different stimuli leading to different cell fate choices often, surprisingly, activate overlapping pathways.1,5 The specific combination of activated pathways may thus encode the relevant cellular response like activation of a specific set of target genes.11 In addition, it recently became clear that signalling pathways are not only inactive or active with different strengths, but also show different dynamics of activity over time.1,8,10–12 Activation can be e.g. sustained, transient or oscillatory.13,14 These dynamics can be specific for the receiving cell type, and for e.g. the type, concentration or temporal encoding of stimulation.1,5,7–9 This extends the parameter space in which cells can compute their reaction to stimuli, and different signalling activity dynamics can indeed lead to different cellular fates.8,10,12Understanding the dynamics of these signalling pathway responses to cytokines requires the continuous quantification of large numbers of individual cells before, during, and after cytokine stimulation.4,15,16 Continuous time-lapse imaging utilizing environmentally controlled microscope setups can enable this in combination with in vitro fate mapping assays.3,17–19 However, producing rapid temporally and quantitatively defined cytokine changes in standard cell culture vessels presents unsolved challenges. Time consuming and slow manual media exchange can obscure fast responses of signalling pathways occurring in minutes, and displace non-adherent cells thus preventing single-cell tracking.
Large-scale integrated microfluidics (LSIM)20–22 enable cell culture while automating otherwise labour intensive interventions during complex experiments.7,21 However, available designs suffer from limitations in effective media exchange, manufacturing complexity, cell survival, capacity for cytokine multiplexing, and cell capture efficiency. Miniaturized flow chambers present in many microfluidic systems struggle to culture rare non-adherent cells at sufficient density because direct flow of media during cell seeding and culture washes cells away.23,24 Cell trapping structures as an alternative to flow chambers can rapidly stimulate non-adherent cells without identity loss but require extremely large numbers of cells for effective single cell trapping,25,26 which prevents work with rare and valuable primary cells. Diffusion-based media exchange systems can slowly apply stimulations to cells.24,27 However, it is difficult to capture rare cell types in these systems, and their diffusion-based media exchange is too slow for studying fast responding signalling networks. Additionally, the complex manufacturing requirements for these systems preclude their use by most researchers.27,28 Microfluidic devices intended for rare or non-adherent cells, rapid stimulation, or sensitive and efficient long-term time-lapse microscopy, respectively, have been developed.24,27,29–31 However, no single device combines all these solutions in the required way.
Here, we present an easy-to-assemble two-layer, valve-controlled microfluidic chip that efficiently captures individual cells, and allows stimulation sequences by rapid diffusion-based media exchange during sensitive time-lapse imaging of rare non-adherent cells without cell displacement. In combination with novel 3D printed incubation and humidification equipment and an intuitive graphic user interface, our system enables long-term culture of non-adherent murine and human hematopoietic stem and progenitor cells (HSPCs) to study fast activation dynamics of many previously unapproachable biological systems like the extracellular signal-regulated kinase (ERK) in response to cytokines.
Results
Design of a microfluidic chip for rapid diffusion-based media exchange of rare non-adherent cells
We devised a valve-controlled two-layer microfluidic chip that concentrates rare cells in selected cell culture chambers and permits rapid media exchange via diffusion without displacement of non-adherent cells (https://bsse.ethz.ch/csd/Hardware/C4F4.html). Users can individually combine 16 separate media inputs controlled by a multiplexed network of flow channels. Control of flow rates into the main body of the device occurs by programmatically opening and closing sequences of valves on the chip to induce pumping or by externally adjusting pressure on flow layer liquids. A multiplexed tree of channels addresses 32 separate chamber units (Fig. 1A). Each chamber unit contains two culture chambers connected to one flow chamber (W × L × H: 625 × 150 × 25 μm) via a network of capillaries (10 × 3 × 4 μm) for diffusion-based media exchange and cell capture (Fig. 1B and S1†). These culture chamber dimensions are optimized for fast and uniform diffusion-based media exchange, while providing sufficient space for colony expansion due to cell proliferation during long-term culture. Three independent valves present before each chamber unit direct flow into the culture chambers during cell trapping or to flow chambers during cell feeding or stimulation (Fig. 1B). During cell seeding, media containing the cells flows through culture chambers into the flow chambers via the capillary network (Fig. 1C). Even small cells like murine HSCs (<10 μm cell diameter) are effectively captured and concentrated (Fig. 1C and 2A). During cell culture, media flowing through the flow chamber diffuses through these capillaries into the 2 adjacent culture chambers by Fick's law (Fig. 1D). Media rapidly exchanges by diffusion due to the small dimensions of the culture chambers (Fig. 1E). Importantly, a network of capillaries before and after each chamber unit prevents fluid flow pulses induced by the opening and closing of adjacent valves by providing flow resistance between the central flow chamber of each culture chamber and the rest of the chip (Fig. 1B). This avoids unintended jumps in media flow and the resulting cell displacement or change in diffusion speed.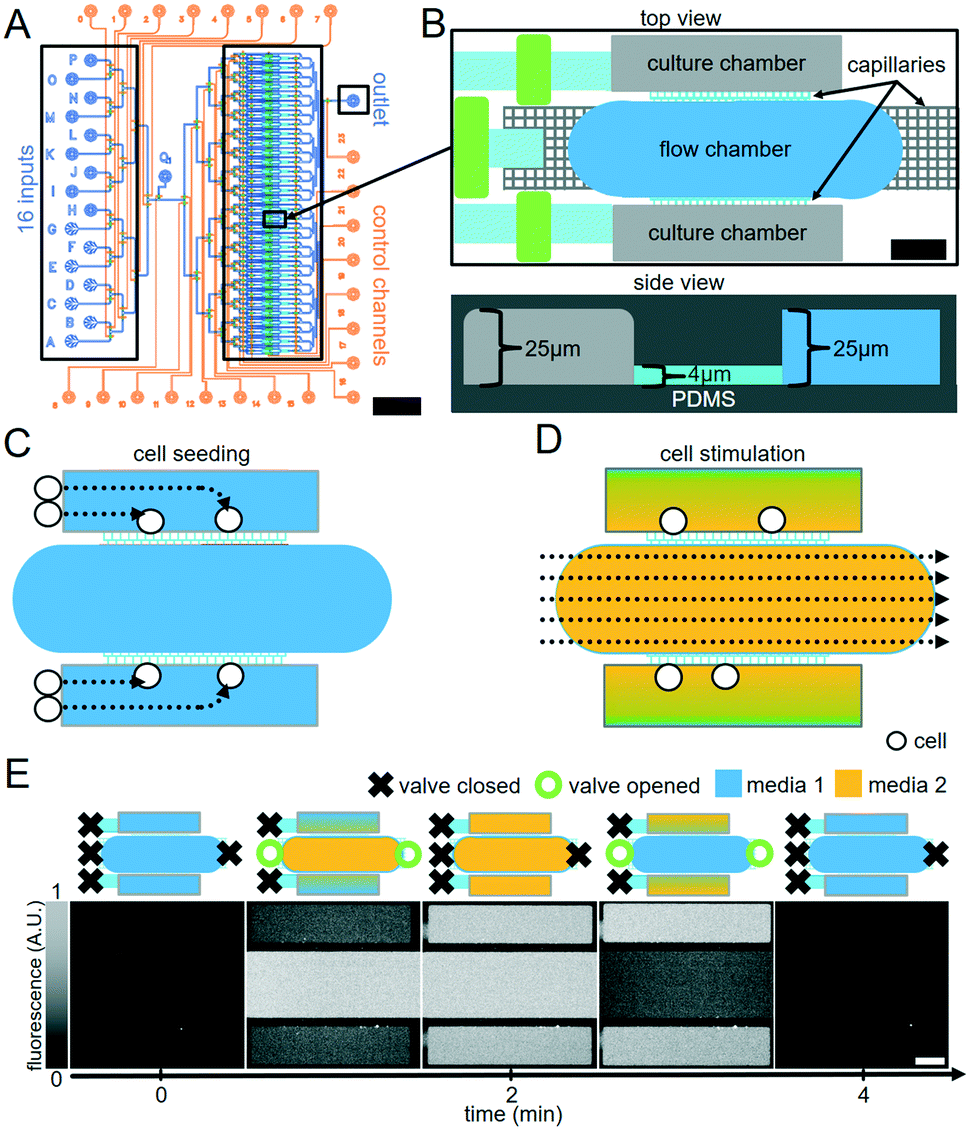 | ||
| Fig. 1 Cell capture and stimulation chip design and operation. (A) The two-layer PDMS device layout with 32 independent chamber units. Orange: control lines operating valves; blue: flow lines for media exchange and cell loading. 16 independent inputs on left, 1 outlet on right. Scale bar = 3 mm. (B) (Top) one of 32 chamber units viewed from above. 2 culture chambers flank a shared flow chamber. 3 PDMS membrane valves (green) control flow into culture chambers and flow chambers. Diffusion between flow chamber and culture chambers via capillaries (light blue) establishes equilibrium. A 3 μm wide 4 μm tall capillary network (grey) prevents bulk fluid flow during valve opening and closing. Scale bar = 100 μm. (Bottom) chambers viewed from side. 25 μm tall culture chambers are separated from 25 μm tall flow chambers by a 3 μm wide 4 μm tall capillary network. (C) Cell capture. Cell-containing medium passes through the culture chambers to the flow chamber and capillaries retain cells in culture chambers. (D) Diffusion-based cell stimulation. Culture chamber medium is changed via diffusion across capillaries from the central flow chamber. (E) Visualization of diffusion speed by microscopy. (Top) valves settings at different states of media pulsing. (Bottom) microscopy images of FITC–dextran fluorescence in chambers. Switching to media with or without 40 kDa FITC–dextran (300 μM) in the flow chamber leads to fast diffusion-based FITC–dextran addition to or removal from culture chambers. Scale bar = 100 μm. | ||
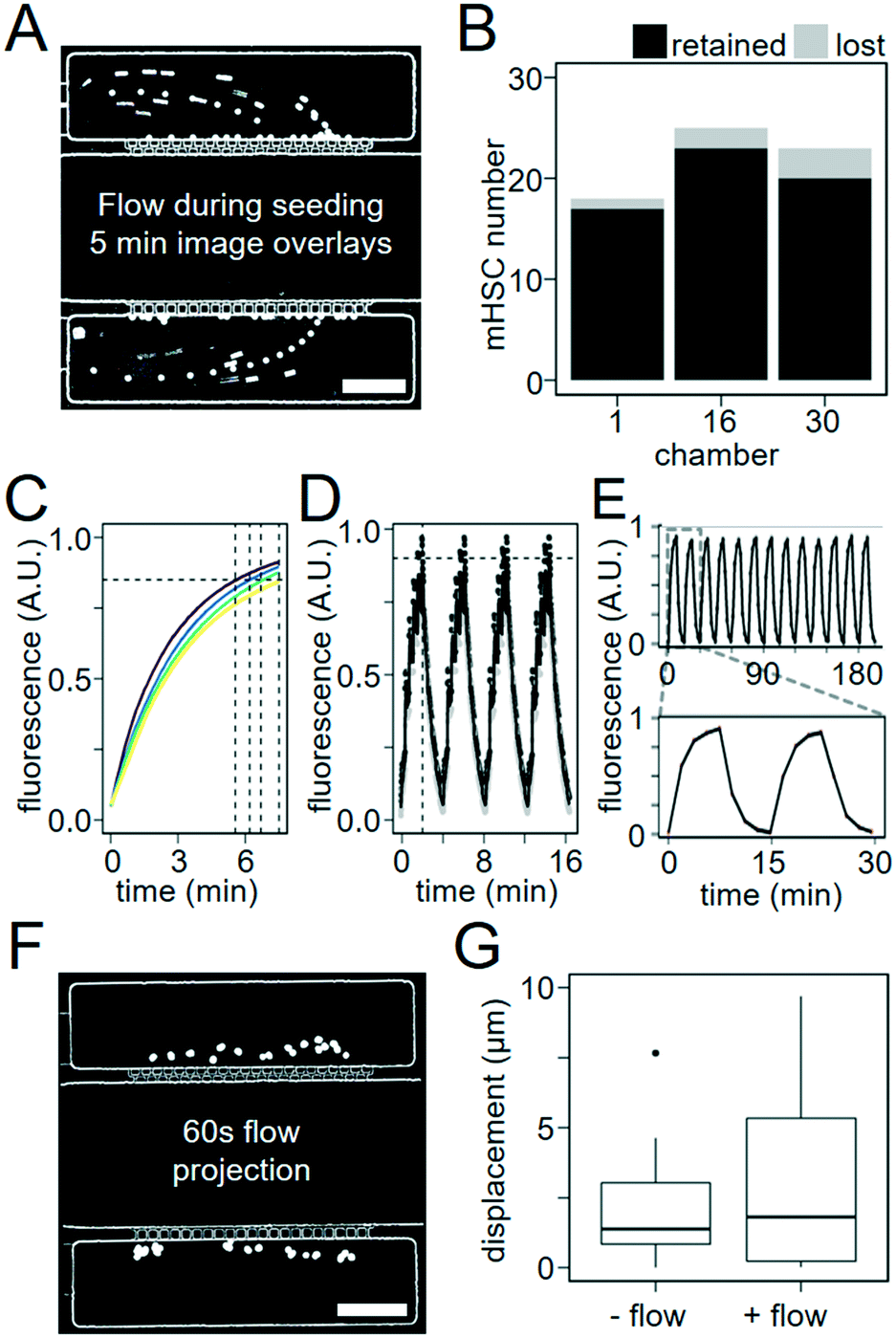 | ||
| Fig. 2 Capillary structures enable robust small cell capture and fast diffusion-based media exchange. (A) 10 μm polystyrene beads are trapped inside culture chambers by 3 μm sieve-like capillary structures. Minimum projection of recorded bead locations over 5 min of flow imaged at a 1 s interval. Scale bar = 100 μm. (B) 88–95% of murine HSCs were captured during 15 minutes of seeding (2 experiments). (C) Molecules with molecular weights comparable to commonly used cytokines rapidly diffuse into cell culture chambers. 20 kDa (purple), 40 kDa (blue), 70 kDa (green), and 150 kDa (yellow) FITC–dextran (300 μM) reach 85% saturation (horizontal line) in culture chambers during perfusion from the flow chambers after 5.5, 6.2, 6.7, and 7.5 min, respectively (n = 4 experiments). (D) Rapid 4 min oscillating FITC–dextran (40 kDa) patterns created by diffusion of pulsed flow steps in flow chambers. Mean fluorescence was imaged every 1 s, adjacent to capillary networks (black) and adjacent to outer edge of chambers (grey), and reliably reached 90% of maximum fluorescence (horizontal line) on a two minute interval (vertical line) when media was applied in 10 s pulses (e.g. 10 s flow, 10 s no flow). (E) Consistent 15 minute FITC–dextran oscillations can be maintained over hours. (F) Minimal displacement in culture chambers during diffusion-based media exchange (no cell path trailing as in A). Image overlays of 10 μm polystyrene beads during 60 s of continuous flow in flow chamber. Scale bar = 100 μm. (G) Median displacement during 60 s of flow is less than 3 μm, similar to displacement due to Brownian motion (p = 0.032, two-tailed t-test, 2 experiments, n = 35 & 36). | ||
Culture media auto-fluorescence hampers detection of weak fluorescent signals. For optimal sensitivity of fluorescence imaging, we reduced the height of culture chambers to 25 μm, thus minimizing the media columns contributing to background fluorescence (Fig. 1B). This minimizes background fluorescence, which proved crucial for the detection of typically weak fluorescent signals.
Robust cell capture by capillary networks
To test cell-trapping efficacy, we loaded culture chambers with 40 μL PBS containing only about 600 polystyrene beads with 10 μm diameter, 40 μL media with 600 murine HSCs, or 40 μL media with 1900 human HSCs. We directed these particles into culture chambers by pressurizing their respective vessels and opening valves leading into the desired culture chambers, through the flow chambers at 0.14 μL min−1 (estimated from media volume consumed), and then to the waste outlet. The capillary networks prevented both cells and polystyrene beads from passing due to their size and rigidity (Fig. 2A and B). We quantified capture efficiency by time-lapse microscopy. Polystyrene beads and HSPCs arriving at the capillaries were captured with 100% and 88–95% efficiency, respectively (Fig. 2A and B and Movie S1 and S2†). While smaller capillary width would increase cell retention, it would decrease diffusion efficiency and chip production reliability. Several rounds of optimization led to the current design as the optimal combination. Additionally, to quantify the capture efficiency per cell entering the chip, we sorted 8700 UCB hHSCs, imaged their capture in 3 chambers per chip, before counting cells remaining in the seeding vessel and waste product. We seeded 3 chips with 1875, 1900, and 4900 hHSCs, respectively. 7%, 71% and 20% of cells initially present in the seeding vessel were captured, respectively, while no cells were detected in the waste. The remaining cells were lost in the tubing connecting the seeding vessel and the culture chambers. While cell loss at the world-to-chip interface thus isn't eliminated, our chip design allows capturing up to 70% of all cells leaving the seeding tube. This is much better than what can typically be achieved, and an important prerequisite for use with precious primary stem cells available only in very low numbers. While further development of the interface between seeding tube and microfluidic channels may further improve cell loss, the incorporation of industrial fittings into our device complicated production beyond what could be accomplished by inexperienced users.32 In our hands, the current design therefore represents the optimized compromise between cell loss and ease of use.Rapid diffusion-based culture media exchange
A flow rate of 0.14 μL min−1 media flow in flow chambers (estimated from total used media volume) was used for all following experiments. To visualize and quantify the speed of diffusion-based media exchange, we used FITC-conjugated dextrans with 20, 40, 70, or 150 kDa molecular weights resembling typical molecular weights of different cytokines. These dextrans required 5.5, 6.2, 6.7 and 7.5 min to reach 85% replacement, respectively (Fig. 2C). We then generated 4 min oscillations in culture chamber media composition by flowing PBS with and then without 300 μM 40 kDa FITC–dextran for 2 min each in flow chambers. During these 2 min intervals, media was pulsed with 10 s on/10 s off cycles. We quantified the change in fluorescence in the culture chambers over time. We were able to generate oscillating stimulations with period lengths as short as 4 min and replace 90% of culture chamber volumes over a time period of 2 min (Fig. 2D and S2).† Reproducible oscillations could be maintained for hours (Fig. 2E and Movie S3†).We next assessed whether fluid flow within flow chambers would displace non-adherent objects in culture chambers. Non-adherent 10 μm polystyrene beads were loaded into culture chambers, and displacement of beads was measured every 1 s over 60 s of microscope stage movement with and without flow through the flow chambers (Movie S4†). The mean displacement of beads was 3.2 ± 1.0 μm with flow, similar to 2 ± 0.6 μm without flow (Fig. 2F and G). The difference between median particle displacements for beads with or without flow was approximately 1 μm, which is unproblematic for robust cell tracking. Our approach can thus be used for time-lapse microscopy with reliable single-cell tracking (Movie S5†).
Long-term culture of sensitive cells
Initially, HSPCs cultured in our chip and several other previously published PDMS chips24,33,34 failed to divide normally (data not shown). PDMS devices are sensitive to changes in osmotic balance caused by media evaporation and creating a sealed enclosure for them is challenging due to the many tubes required to control them.24,35 In order to effectively maintain humidity and gas composition within our enclosure, we designed a temperature-controlled air humidifier (https://bsse.ethz.ch/csd/Hardware/3DPHumidifier.html) and a sealed incubation chamber with dedicated slots for tube organization (https://bsse.ethz.ch/csd/Hardware/3DPChipEnclosure.html) (Fig. S3 and S4†). They can be produced using a common desktop 3D printer, inexpensive laser-cut parts, and an affordable temperature control circuit, and are compatible with other standard-sized microfluidic chips. To reduce evaporation due to gas exchange, we minimized the gas-containing volume within the enclosure. During HSPC culture, we used only 1 mL of mixed gas per hour, and consistently measured humidity levels greater than 99% over 3 days during all experiments (Fig. S5†).To compare the chip and 3D printed enclosure's capacity to maintain demanding cell cultures for extended periods to a standard culture device, we seeded 10–20 murine HSCs in each of 20 culture chambers of 3 chips and 20 wells of 3 standard 384 well plates (Greiner). We monitored cell proliferation continuously in each device by time-lapse-imaging for 3 days, with an automated feeding cycle of 30 min in the chip. No change in mean time to first divisions between 384 well plates (32.9 ± 2.0 h) and chips (34.7 ± 2.7 h) were detectable (p = 0.286, n = 3, two-tailed t-test, 3 experiments) (Fig. 3B). Similarly, cell division rates over 72 h were comparable between 384 well plates and chips, with mean doubling times of 20.7 h and 19.1 h, respectively as calculated from the slope of the log2 transform of hourly cell counts (p = 0.228, two-tailed t-test, 3 experiments) (Fig. 3A and B and S6, and Movie S6†). Finally, total cell number expansion after 72 h were comparable between well plates and chips, with an average of 11.5 and 11.6 progeny from each input cell, respectively (p = 0.993, Welch two sample t-test, 3 experiments), also demonstrating comparable cell survival. Thus, even sensitive and difficult to culture primary HSPCs can be cultured in the chip throughout long-term time lapse imaging experiments and without changed proliferation kinetics.
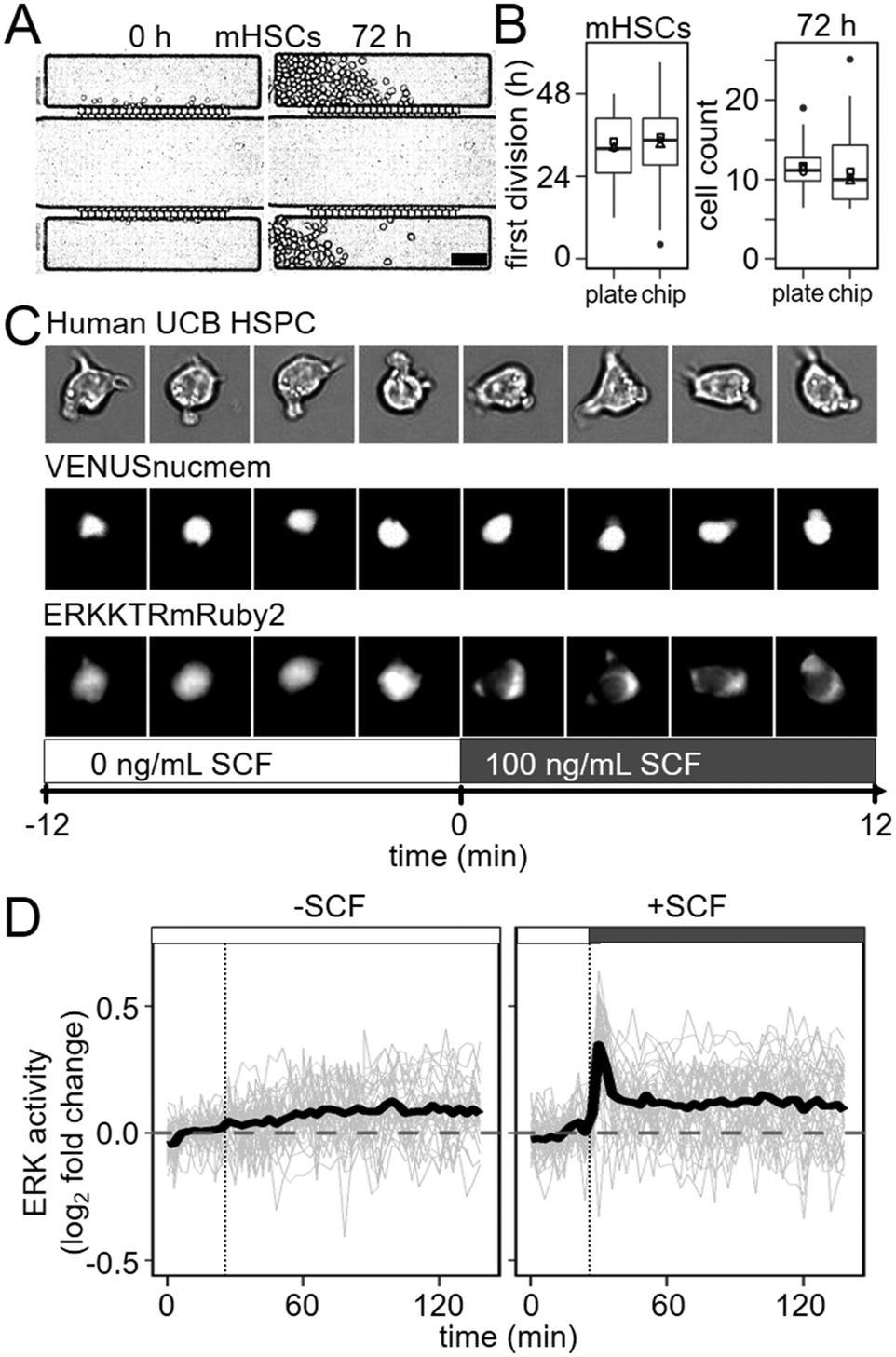 | ||
| Fig. 3 The chip cultures mouse HSCs and enables quantification of fast intracellular dynamics in human HSCs. (A) Non-adherent murine HSCs can be cultured and monitored over 3 days. Cell density at start (left) and after 72 h (right) of culture. Scale bar = 100 μm. (B) Normal behaviour of murine HSCs cultured in the chip. Murine HSCs cultured in our chip and a 384 well plate for 72 hours showed comparable proliferation kinetics. Mean time to first division (left) occurred at 32.9 and 34.7 hours, respectively (p = 0.286, Welch two sample t-test, 3 experiments) while mean cell counts per input cell in the chip and 384 well plate were 11.5 and 11.6 (p = 0.993, Welch two sample t-test, 3 experiments). Symbols indicate mean for each experiment. (C) ERKKTR rapidly exports from human HSC nucleus upon ERK pathway activation. (D) Rapid and transient dynamics of ERK activity in hHSCs after SCF stimulation can be induced and quantified. ERK response peaks after 3 to 6 minutes before returning to an elevated baseline level. | ||
Diffusion-based stimulation is fast enough to detect rapid signalling dynamics changes in human HSPCs
To confirm that this is sufficient to detect and quantify even very fast dynamics on the scale of minutes in rare and difficult to culture non-adherent cells, we quantified the dynamics of ERK signalling activity in human HSPCs upon cytokine stimulation. ERK signalling was chosen due to its known very fast dynamics,36,37 and activation in hHSPCs upon SCF stimulation.37–39 To observe ERK activity in real time, we delivered a translocation-based biosensor (ERKKTRmRUBY2)36 and a constitutive fluorescent nuclear membrane label (VENUSnucmem)40 for nuclear segmentation to human umbilical cord blood (UCB) derived HSPCs by lentiviral transduction.5 ERKKTRmRUBY2 translocation from the nucleus to the cytoplasm during ERK pathway activation enables visualization and quantification of ERK activity over time in living single cells (Fig. 3C). The resulting change in fluorescence distribution is very subtle and its quantification requires a more sensitive imaging and quantification approach than was possible by wide-field imaging in e.g. plastic bottom 384 well plates (data not shown). We imaged ERK activity every 3 minutes, beginning 30 minutes before cytokine stimulation to establish a baseline. Fresh media with or without SCF was then flowed into flow chambers every 30 min for a 120 min period. While no ERK activation was observed in HSPCs in response to control (no SCF) media (n = 46 cells, 2 experiments), rapid and transient ERK activity could be detected in 92% of HSPCs (n = 39, 2 experiments) stimulated with SCF within 3 minutes of exposure (Fig. 3D). Thus, the stimulation speeds in the developed chip are sufficient to enable detection of very rapid cellular responses, with low enough cell displacement for reliable single cell tracking, segmentation and quantification. We were unable to detect any difference in ERK activity, e.g. in activation time, between cells directly adjacent to capillary networks and cells on the outer edges of the culture chamber. This confirms that diffusion is fast enough for uniform stimulation of the whole culture chamber.Discussion
We describe the design and operation of a microfluidic chip capable of efficiently capturing rare cells in culture chambers while protecting them from fluid flow during rapid automated media exchange procedures. Several devices have previously been published demonstrating these individual capabilities separately.27,35,41 Here, we combine these features into a single device that is easy to assemble and operate by inexperienced users as established while training biologists with no prior experience in our group. Compared to more complex 3D devices, our two-layer design simplifies chip assembly and operation by eliminating microwell fabrication steps35 or complex bonding procedures.42 We placed the control channel PDMS layer between the glass slide and the flow layer channels containing cells since this can be produced more reliably. We found that when produced by inexperienced users, this reduced the frequency of imperfect bonding between PDMS layers which leads to undesired connections between control channels, allowing the transmission of pressure between valves intended to operate independently, thereby rendering chips inoperable. While we found no influence on the single-cell imaging used here (data not shown), having the control PDMS layer between the microscope objective and the cells to be imaged by reflected light may reduce the quality of images generated by higher-resolution microscopy. Several possible solutions to this problem exist, including placing the flow layer PDMS between the glass and the control layer PDMS, allowing cells to grow directly on the glass.The PDMS valves used in our design improve upon existing devices27,43 by allowing users to precisely control cell delivery into specific culture chambers and maintain multiple complex culture conditions automatically across a large number of chambers using our custom graphic user interface (https://bsse.ethz.ch/csd/Hardware/C4F4.html). This allows researchers to minimize time spent producing and operating these chips.
Our design is highly scalable as all chamber units are addressed using a binary multiplexing tree. The addition of 2n control channels allows the number of chambers to be increased by a factor of 2n. A standard 75 mm × 50 mm glass slide could accommodate 128 chambers based on our design. Additionally, increasing the culture chamber length would not compromise trapping efficiency or diffusion times but may prevent the chamber from being imaged in a single field of view. Increasing the culture chamber width would increase diffusion times as predicted by Fick's law of diffusion. We greatly reduce the risk of wasted valuable rare cells associated with seeding other chip designs. Our capillary networks trap more than 88% of arriving single cells, and up to 70% of human HSCs leaving the seeding tube were trapped in culture chambers. This eliminates the need for complex sequential seeding procedures29 and large input cell counts.27,33,41 The capillary network and chamber geometry enable rapid diffusion-based material exchange between flow chambers and culture chambers within 2–3 min without the use of 3D microwells,35 extensive channel infrastructure, or complex fabrication steps.28 Previous 2D chamber designs have achieved comparable media replacement times. However, these designs have no capacity to trap rare cell populations,24,41,44 or to automate media switching for multiple conditions.27,43 They also often suffer from long lasting cytokine gradients in their culture chambers. Indeed, we could not detect ERK signalling dynamics as quantified here when using a previously published device,24 due to long lasting cytokine gradients across the larger chamber space (data not shown). Computational modelling of diffusion times revealed predicted long-lasting cytokine gradients also in other published devices due to large distances between cytokine sources and cell populations. These would obfuscate ERK dynamics due to variation in cytokine stimulation intensity in individual cells.
The low flow rates enabled by our capillaries are comparable or better than in many other designs.24,27,35 Particle displacements over 60 s of continuous flow were under 1/3rd of very small cells, allowing easy tracking following a typical time-lapse imaging experiment. Finally, by optimizing our chamber dimensions to a typical microscopy field of view, we have optimized image acquisition efficiency, a feature that is frequently ignored when designing microfluidic chips.23,43 By combining these features into a single device, we were now able to observe ERK dynamics in human HSPCs with the required resolution that was previously impossible.36
Our chip is ideal for studying cells that are difficult to obtain in large quantities or are displaced by fluid flow like the murine and human HSPCs used here. Additional cytokine conditions and stimulation patterns can now be applied to many other cell types in future studies to comprehensively dissect the function of relevant signalling pathway components in their cell fate control.
Materials and methods
Chip design and fabrication
Using AutoCAD (Autodesk Inc.) we designed a two-layer chip and printed chrome masks at 40 kdpi resolution (Delta Mask B.V., Netherlands). Moulds for PDMS casting were produced using standard photolithography as previously described.21 Specifically, the 25 μm high flow layer and control layer channel networks were produced using SU-83025 (Microchem) spun at 3000 RPM while 4 μm high capillary networks were generated using SU-83005 (Microchem) spun at 6000 RPM on silicon wafers. 25 μm parabolic surfaces at valve positions were achieved by depositing and reflowing AZ-50XT (AZ Electronic Materials) using a previously reported protocol.21 After producing our silicon master moulds we mixed and degassed 66 g of PDMS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; polymer:catalyst) before coating the TMCS treated flow layer silicon wafer and curing for 45 min at 80 °C. Similarly, we poured 10 g of mixed and degassed PDMS (20:1; polymer:catalyst) over the TMCS treated control layer silicon wafer before spinning at 2000 rpm for 60 s and curing for 45 min at 80 °C. After punching inlet holes into the cured flow layer, we treated the flow layer with oxygen-plasma at 15 W for 20 s and aligned it to the control layer. After 2 h of thermal bonding at 80 °C, we punched control layer inlets and plasma treated the assembly at 15 W for 20 s before bonding the device to a plasma-treated coverslip. We then cured the completed chip for 16 h at 80 °C.
:1; polymer:catalyst) before coating the TMCS treated flow layer silicon wafer and curing for 45 min at 80 °C. Similarly, we poured 10 g of mixed and degassed PDMS (20:1; polymer:catalyst) over the TMCS treated control layer silicon wafer before spinning at 2000 rpm for 60 s and curing for 45 min at 80 °C. After punching inlet holes into the cured flow layer, we treated the flow layer with oxygen-plasma at 15 W for 20 s and aligned it to the control layer. After 2 h of thermal bonding at 80 °C, we punched control layer inlets and plasma treated the assembly at 15 W for 20 s before bonding the device to a plasma-treated coverslip. We then cured the completed chip for 16 h at 80 °C.
Particle capture calibration
All chips were controlled using a customized version of the solenoid-valve (Festo) microfluidic control manifolds designed by R. Gómez-Sjöberg as previously described.21 Control layer elements and flow layer channels were pressurized to 1.7 bar and 0.25 bar respectively. For bead capture experiments, polystyrene beads with a diameter of 10 μm (Spherotech) were flushed into the cell culture chambers treated with 0.1% Triton-X detergent in deionized water to prevent bead adhesion and clumping. Chambers were then imaged for 60 s with flow followed by 60 s without flow at an imaging interval of 1 s. An additional empty chamber was imaged in parallel in order to replicate the effect of stage motion on particle displacement during real experiments. Chips used in testing the effective capture rate of murine HSCs were first degassed with culture media before seeding the HSCs into chambers. In both cases flow through the culture chambers was applied for 5 min on a Nikon Ti-Eclipse inverted microscope that was equipped with an Orca Flash 4.0 camera (Hamamatsu Photonics K.K.), using a 10× objective (NA 0.45) (Nikon Instruments Europe B.V.) and particle capture was recorded using custom scripts written for Youscope (www.youscope.org).Diffusion-based cell culture and stimulation
Dynamic input calibration experiments were conducted using a 40 kDa FITC-conjugated dextran (c = 300 μM) (catalogue number: 60842-46-8, Sigma-Aldrich), which has a size comparable to low-molecular-weight proteins, such as cytokines. The chip was filled with PBS before a single pulse of 40 kDa FITC–dextran was flushed through the relevant flow chambers. Vessels containing all flow layer components were pressurized to 0.25 bar. Fluorescence in all chamber components was monitored by time-lapse imaging. Experiments assessing chip behaviour in dynamic stimulation modes were conducted using oscillating flow feeding phases on 15 min intervals and using 30 s of direct flow through each flow chamber. When assessing the maximum possible media exchange rate in our device, PBS containing FITC–dextran was pulsed through chambers on a 2 min interval, 2 min with flow followed by 2 min without flow.In order to provide fresh media to the culture chambers we opened a sequence of valves leading from the desired media input up to the target flow chamber for 10 s of unimpeded flow through a dedicated bypass channel. Following this washing step, we washed the flow chamber of the desired chamber unit for 30 s by opening another set of valves leading through the desired flow chamber. Diffusion occurred between culture chambers and flow chambers on 15 min intervals. Cytokine stimulations were produced by using this process to fill the desired flow chambers with control media or media containing the desired cytokines on 30 min intervals for 2.5 h.
Isolation of primary murine HSCs
For experiments with primary murine cells, 12–16 weeks old, male C57BL/6J mice expressing PU.1eYFP/GATA1mCherry45 were euthanized. Isolation of primary murine hematopoietic stem cells (HSC), phenotypically defined as Lineage−Sca1+cKIT+CD34−CD48−CD150+, was performed as previously described.45,46 mHSCs were cultured in phenolred free SFEM (Stem Cell Technologies) + penicillin/streptomycin (Gibco) + 10% FCS (PAA) + SCF (100 ng ml−1) + EPO (2 U ml−1) + IL3 (10 ng ml−1) + IL6 (10 ng ml−1) in either plastic-bottom 384 well plates (part number 788 161, Greiner Bio-One) or in our chip. All cytokines were purchased from Peprotech. All animal experiments were performed in accordance with the ordinance provided by Canton Basel-Stadt and approved by the veterinary office of Canton Basel-Stadt, Switzerland (approval no. 2655).UCB HSPC isolation
Anonymized human UCB samples were collected from healthy new-borns of both sexes at University Hospital Basel. Relevant ethical regulations were followed, according to the guidelines of the local Basel ethics committees (vote 13/2007V, S-112/2010, EKNZ2015/335). UCB cells were processed by density gradient centrifugation, and CD34+ cells were isolated using EasySep™ human cord blood CD34 positive selection kit II (cat# 17896, Stemcell Technologies, Vancouver, BC, Canada) and stored at −150 °C. Upon thawing, CD34+ cells were stained with the following antibodies: CD34 (APC conjugate, clone: 581, 1:100 dilution, BD Biosciences), CD38 (PEcy7, HB7, 1:200, BioLegend), CD45RA (FITC, HI100, 1:100, BioLegend), CD90 (PE, 5E10, 1:50, BD Biosciences) and CD49f (PEcy5, GoH3, 1:50, BD Biosciences) for 30 minutes at 4 °C. Cells were then washed and resuspended in PBS + 2% FCS with 1 μg ml−1 7-aminoactinomycin D (7-AAD, ThermoFisher Scientific). HSPCs, phenotypically defined as CD34+CD38−CD45RA−CD90+CD49f+, were sorted using a BD™ FACSAria III with 100 μm nozzle, purity mode and sorting purities ≥90%. Gates and thresholds were set according to fluorescence minus one (FMO) controls.
Lentivirus production and transduction
ERKKTR (#59138, Addgene)36 was fused to the fluorescent protein mRUBY2, linked with VENUSnucmem by the picornavirus 2A (P2A) sequence, and cloned into VSV-G pseudotyped lentivirus (third generation) constructs using the In-Fusion Cloning system (Takara Bio). Virus was produced and titrated as previously described.5,18 HSPCs were cultured overnight in 5% O2 5% CO2 at 37 °C and followed by 48 hours of infection in a serum-free medium as previously described38 supplemented with 100 ng ml−1 stem cell factor (SCF, R&D Systems, Minneapolis, MN, USA), 100 ng ml−1 FMS-like tyrosine kinase 3 ligand (FLT3L, R&D Systems), 50 ng ml−1 thrombopoietin (TPO, R&D Systems) and 100 nM UM171 (Stemcell Technologies).Environmental control for cell culture
All chip experiments were conducted on environmentally controlled microscopes maintained at 37 °C using a feedback-controlled heating unit (Life Imaging Services) installed in custom built aluminium-slot frame boxes lined with black, foamed insulation-boards. Humidity (>98%) and gas composition (5% CO2, 5% O2, 90% N2, PanGas) were maintained by constantly flowing 1 mL h−1 mixed gas into a custom made 3D printed stage-top enclosure (https://bsse.ethz.ch/csd/Hardware/3DPChipEnclosure.html) through a 3D printed temperature controlled air humidifier containing roughly 100 mL of water held at 37 °C (https://bsse.ethz.ch/csd/Hardware/3DPHumidifier.html). We monitored relative humidity using a data logger (Sensirion, part number SHT31-DIS-B).Generation and analysis of single-cell timeseries
For each tracked3,18,19,47,48 cell, the sum of nuclear fluorescence intensity of ERKKTR (mRUBY2) was divided by the sum of fluorescence intensity of VENUSnucmem at each timepoint, with the assumption that the sum of VENUSnucmem fluorescence intensity stays constant for the imaging period (150 min). Fold changes were then calculated by normalizing to the mean of baseline (0 to 27 minutes) values before growth factor stimulation. The resulting trajectories were mirrored along the horizontal axis to intuitively reflect ERK activity and a value of 2 was added to all timepoints in order to re-set the mean of baseline to 1 for each trajectory. log2 transformed values were plotted.Microscopy and image data analysis
All cell culture experiments were conducted on a Nikon Ti-Eclipse inverted microscope that was equipped with an Orca Flash 4.0 camera (Hamamatsu Photonics K.K.), using a 10× objective (NA 0.45) (Nikon Instruments Europe B.V.) for murine HSC experiments and a 20× CFI Plan Apochromat λ objective (NA 0.45)(Nikon Instruments Europe B.V.) for experiments involving human UCB HSPCs. A Spectra X light engine (Lumencor) was used as fluorescent light source. Pictures that were used for quantifications were saved in lossless PNG format and processing and data analysis was carried out using the “The Tracking Tool” and fastER software package.47,48Conclusions
We demonstrate the use of capillary networks for efficient rare cell capture, and for rapid diffusion-based media exchange without displacement of non-adherent cells. Optimized for microscopy, our device allows signalling dynamics quantification even in primary hematopoietic stem cells only available in limited quantities. Our design provides a basis for experiments exploring signal-processing mechanisms in rare non-adherent stem cell populations, which may lead to clinically relevant discoveries in the future.Author contributions
P. D. designed and implemented the chip, control hardware, control software, and environmental control apparatus. W. W., N. A., M. E. and Y. Z. provided feedback improving the functionality and user experience of the chip. W. W. and Y. Z. produced hHSPCs data, N. A. mHSC data. T. S. and D. L. developed continuous quantitative single-cell imaging and maintained it with T. K. C. L. contributed umbilical cord specimens. T. S. designed and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the D-BSSE single cell and clean room facilities for technical support. This work was supported by Swiss National Science Foundation grant 179490 to T. S. and a Swiss Initiative in Systems Biology Transition Postdoc fellowship to W. W.References
- M. Endele, M. Etzrodt and T. Schroeder, Exp. Cell Res., 2014, 329, 207–213 CrossRef CAS.
- N. Perrimon, C. Pitsouli and B.-Z. Shilo, Cold Spring Harbor Perspect. Biol., 2012, 4, a005975 Search PubMed.
- M. A. Rieger, P. S. Hoppe, B. M. Smejkal, A. C. Eitelhuber and T. Schroeder, Science, 2009, 325, 217–218 CrossRef CAS.
- T. Schroeder, Ann. N. Y. Acad. Sci., 2005, 1044, 201–209 CrossRef.
- M. Endele, D. Loeffler, K. D. Kokkaliaris, O. Hilsenbeck, S. Skylaki, P. S. Hoppe, A. Schambach, E. R. Stanley and T. Schroeder, Blood, 2017, 129, 1691–1701 CrossRef CAS.
- T. Schroeder, H. Kohlhof, N. Rieber and U. Just, J. Immunol., 2003, 170, 5538–5548 CrossRef CAS.
- R. A. Kellogg and S. Tay, Cell, 2015, 160, 381–392 CrossRef CAS.
- H. Ryu, M. Chung, M. Dobrzyński, D. Fey, Y. Blum, S. S. Lee, M. Peter, B. N. Kholodenko, N. L. Jeon and O. Pertz, Mol. Syst. Biol., 2015, 11, 838 CrossRef.
- M. Barberis, T. Helikar and P. Verbruggen, Front. Physiol., 2018, 9 DOI:10.3389/fphys.2018.00879.
- J. E. Purvis and G. Lahav, Cell, 2013, 152, 945–956 CrossRef CAS.
- J. X. Lin and W. J. Leonard, Annu. Rev. Immunol., 2019, 37, 295–324 CrossRef CAS.
- C. J. Marshall, Cell, 1995, 80(2), 179–185 CrossRef CAS.
- J. H. Levine, Y. Lin and M. B. Elowitz, Science, 2013, 342(6163), 1193–1200 CrossRef CAS.
- J. G. Albeck, G. B. Mills and J. S. Brugge, Mol. Cell, 2013, 49, 249–261 CrossRef CAS.
- P. S. Hoppe, D. L. Coutu and T. Schroeder, Nat. Cell Biol., 2014, 16, 919–927 CrossRef CAS.
- M. Etzrodt, M. Endele and T. Schroeder, Cell Stem Cell, 2014, 15, 546–558 CrossRef CAS.
- M. Endele and T. Schroeder, Ann. N. Y. Acad. Sci., 2012, 1266, 18–27 CrossRef CAS.
- D. Loeffler, A. Wehling, F. Schneiter, Y. Zhang, N. Müller-Bötticher, P. S. Hoppe, O. Hilsenbeck, K. D. Kokkaliaris, M. Endele and T. Schroeder, Nature, 2019, 573(7774), 426–429 CrossRef CAS.
- H. M. Eilken, S.-I. I. Nishikawa and T. Schroeder, Nature, 2009, 457, 896–900 CrossRef CAS.
- M. Mehling and S. Tay, Curr. Opin. Biotechnol., 2014, 25, 95–102 CrossRef CAS.
- R. A. Kellogg, R. Gómez-Sjöberg, A. A. Leyrat and S. Tay, Nat. Protoc., 2014, 9, 1713–1726 CrossRef CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS.
- R. Gómez-Sjöberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef.
- P. Dettinger, T. Frank, M. Etzrodt, N. Ahmed, A. Reimann, C. Trenzinger, D. Loeffler, K. D. Kokkaliaris, T. Schroeder and S. Tay, Anal. Chem., 2018, 90(18), 10695–10700 CrossRef CAS.
- S. A. Kobel, O. Burri, A. Griffa, M. Girotra, A. Seitz and M. P. Lutolf, Lab Chip, 2012, 12, 2843–2849 RSC.
- L. Weng, F. Ellett, J. Edd, K. H. K. Wong, K. Uygun, D. Irimia, S. L. Stott and M. Toner, Lab Chip, 2017, 17(23), 4077–4088 RSC.
- T. Cambier, T. Honegger, V. Vanneaux, J. Berthier, D. Peyrade, L. Blanchoin, J. Larghero and M. Théry, Lab Chip, 2015, 15, 77–85 RSC.
- L. Amato, Y. Gu, N. Bellini, S. M. Eaton, G. Cerullo and R. Osellame, Lab Chip, 2012, 12(6), 1135 RSC.
- V. Lecault, M. Vaninsberghe, S. Sekulovic, D. J. H. F. Knapp, S. Wohrer, W. Bowden, F. Viel, T. McLaughlin, A. Jarandehei, M. Miller, D. Falconnet, A. K. White, D. G. Kent, M. R. Copley, F. Taghipour, C. J. Eaves, R. K. Humphries, J. M. Piret and C. L. Hansen, Nat. Methods, 2011, 8, 581–586 CrossRef CAS.
- X. Han, Y. Ma, K. Zhang, P. Zhang, N. Shao and L. Qin, Proteomics, 2020, 20(13), 1900223 CrossRef CAS.
- S. Kobel, A. Valero, J. Latt, P. Renaud and M. Lutolf, Lab Chip, 2010, 10(7), 857–863 RSC.
- C. K. Fredrickson and Z. H. Fan, Lab Chip, 2004, 526–533 RSC.
- T. Frank and S. Tay, Lab Chip, 2013, 13, 1273–1281 RSC.
- A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef.
- C. Zhang, H.-L. Tu, G. Jia, T. Mukhtar, V. Taylor, A. Rzhetsky and S. Tay, Sci. Adv., 2019, 5(4), eaav7959 CrossRef CAS.
- S. Regot, J. J. Hughey, B. T. Bajar, S. Carrasco and M. W. Covert, Cell, 2014, 157, 1724–1734 CrossRef CAS.
- E. Wandzioch, C. E. Edling, R. H. Palmer, L. Carlsson and B. Hallberg, Blood, 2004, 104(1), 51–57 CrossRef CAS.
- E. Csaszar, D. C. Kirouac, M. Yu, W. Wang, W. Qiao, M. P. Cooke, A. E. Boitano, C. Ito and P. W. Zandstra, Cell Stem Cell, 2012, 10(2), 218–229 CrossRef CAS.
- D. J. H. F. Knapp, C. A. Hammond, N. Aghaeepour, P. H. Miller, D. Pellacani, P. A. Beer, K. Sachs, W. Qiao, W. J. Wang, R. K. Humphries, G. Sauvageau, P. W. Zandstra, S. C. Bendall, G. P. Nolan, C. Hansen and C. J. Eaves, Blood, 2017, 129(3), 307–318 CrossRef CAS.
- C. Okita, M. Sato and T. Schroeder, BioTechniques, 2004, 36(3), 418–424 CrossRef CAS.
- C. Frick, P. Dettinger, J. Renkawitz, A. Jauch, C. T. Berger, M. Recher, T. Schroeder and M. Mehling, PLoS One, 2018, 13(6) DOI:10.1371/journal.pone.0198330.
- Y.-S. Zeng, H. Fan, B. Xu, Z. Zhang, F.-F. Ren, C. Zhou, S.-Z. Wu, Y.-L. Hu, W.-L. Zhu, Y.-H. Su, J.-R. Chu, J.-W. Li, G.-Q. Li and D. Wu, Microfluid. Nanofluid., 2017, 21, 173 CrossRef.
- L. Wang, X. F. Ni, C. X. Luo, Z. L. Zhang, D. W. Pang and Y. Chen, Biomed. Microdevices, 2009, 11, 679–684 CrossRef CAS.
- P. M. Navarrete and J. Yuan, Micromachines, 2020, 11(4) DOI:10.3390/MI11040395.
- P. Hoppe, M. Schwarzfischer, D. Loeffler, K. Kokkaliaris, O. Hilsenbeck, N. Moritz, M. Endele, A. Filipczyk, A. Gambardella, N. Ahmed, M. Etzrodt, D. Coutu, M. Rieger, C. Marr, M. Strasser, B. Schauberger, I. Burtscher, O. Ermakova, A. Bürger, H. Lickert, C. Nerlov, F. Theis and T. Schroeder, Nature, 2016, 535, 299–302 CrossRef CAS.
- M. J. Kiel, Ö. H. Yilmaz, T. Iwashita, O. H. Yilmaz, C. Terhorst and S. J. Morrison, Cell, 2005, 121(7), 1109–1121 CrossRef CAS.
- O. Hilsenbeck, M. Schwarzfischer, S. Skylaki, B. Schauberger, P. S. Hoppe, D. Loeffler, K. D. Kokkaliaris, S. Hastreiter, E. Skylaki, A. Filipczyk, M. Strasser, F. Buggenthin, J. S. Feigelman, J. Krumsiek, A. J. J. van den Berg, M. Endele, M. Etzrodt, C. Marr, F. J. Theis and T. Schroeder, Nat. Biotechnol., 2016, 34, 703–706 CrossRef CAS.
- O. Hilsenbeck, M. Schwarzfischer, D. Loeffler, S. Dimopoulos, S. Hastreiter, C. Marr, F. J. Theis and T. Schroeder, Bioinformatics, 2017, 33(13), 2020–2028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00687d |
| This journal is © The Royal Society of Chemistry 2020 |