
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated Raman based cell sorting with 3D microfluidics†
Yingkai
Lyu
 ab,
Xiaofei
Yuan
ac,
Andrew
Glidle
a,
Yuchen
Fu
a,
Hitoshi
Furusho
d,
Tianxin
Yang
*b and
Huabing
Yin
*a
ab,
Xiaofei
Yuan
ac,
Andrew
Glidle
a,
Yuchen
Fu
a,
Hitoshi
Furusho
d,
Tianxin
Yang
*b and
Huabing
Yin
*a
aDivision of Biomedical Engineering, School of Engineering, University of Glasgow, Glasgow G12 8LT, UK. E-mail: huabing.yin@glasgow.ac.uk
bKey Laboratory of the Ministry of Education on Optoelectronic Information Technology, School of Precision Instrument and Optoelectronics Engineering, Tianjin University, Tianjin 300072, China. E-mail: tyang@tju.edu.cn
cSchool of Environmental Science and Engineering, Southern University of Science and Technology, Shenzhen, 518055, China
dNissan Chemical Ltd., 5-1, Nihonbashi 2-Chome, Chuo-ku, Tokyo 103-6119, Japan
First published on 8th October 2020
Abstract
Raman activated cell sorting has emerged as a label-free technology that can link phenotypic function with genotypic properties of cells. However, its broad implementation is limited by challenges associated with throughput and the complexity of biological systems. Here, we describe a three-dimensional hydrodynamic focusing microfluidic system for a fully automated, continuous Raman activated cell sorting (3D-RACS). The system consists of a 3D printed detection chamber (1 mm3) that is integrated with a PDMS based sorting unit, optical sensors and an in-line collection module. It has the ability to precisely position cells in the detection chamber for Raman measurements, effectively eliminating spectroscopic interference from the device materials. This enables the sorting of a range of cell sizes (from 1 μm bacteria to 10's μm mammalian cells) with stable operation over >8 hours and high throughput. As a proof-of-concept demonstration, Raman-activated sorting of mixtures of Chlorella vulgaris and E. coli has demonstrated a purity level of 92.0% at a throughput of 310 cells per min. The platform employed in this demonstration features a simple “Raman window” detection system, enabling it to be built on a standard, inverted microscope. Together with its facile and robust operation, it provides a versatile tool for function-based flow cytometry and sorting applications in the fields of microbiology, biotechnology, life science and diagnostics.
Introduction
Cell sorting is a key requirement for many applications in life science and clinical diagnosis; however, high throughput isolation of functional cells from a complex community still represents a critical challenge.1 In recent years, Raman-activated cell sorting (RACS) based on single cell Raman spectra (SCRS) has been explored as a label-free tool for functional-based isolation of cells.2–4 The Raman spectrum of a single cell provides information about its intrinsic biochemical profile5 which, in turn, can be used to characterise its phenotype,6 metabolic activity and function.7,8 Notably, the capability of sorting a microbial community based on SCRS has offered great potential for the discovery of new functional species and genes from the vast array of untapped natural resources.4,9,10RACS in flow can be both non-invasive and have higher throughput than methods that rely on measuring spectra of cells that are (momentarily or permanently) stationary, followed by cell picking. Since spontaneous Raman spectra are often inherently weak, to date, most of the methods that have used cells in microfluidic systems have used a “trap-and-release” approach, employing methods such as optical tweezing4,11–15 or dielectrophoresis.16,17 Although “cell trapping” can meet the need for long signal acquisition, ultimately the trapping mechanism limits throughput, and the efficiency of the trap is often dependent on cell size, medium conductivity, refractive index and/or flow rate.18,19 To overcome this, recently, we developed a trapping-free RACS system capable of both continuous and automated sorting of individual cells based on SCRS20 and 2D hydrodynamic focusing. Significantly, hydrodynamic focussing is independent of the physical properties of cells and medium, and thus universally applicable to a wide variety of biological systems.
As with the majority of microfluidic cell sorting platforms,21,22 microfluidic devices used for most of the RACS systems to date have employed polymer materials (e.g. PDMS, SU-8) to define the microchannels (in general of 10's μm height and 100's μm width). A consequence of this is, that the Raman signals from the device materials often overlap with those of the cells, making on-the-fly spectral classification of weak signals difficult.4,16,20,23 Furthermore, problems associated with cell adhesion to channel walls and channel blockages regularly occur with prolonged usage. This can be a significant inhibitor in the uptake of these technologies. Although enlarging the channel dimensions could alleviate these problems, this approach would require the capability to accurately control the position of cells in 3D, at the focus point.
Here, we present the development of a novel three-dimensional (3D) hydrodynamic-focusing Raman activated cell sorting platform that can precisely position cells in 3D, has long term stable operation and achieves significantly higher throughput than before through the inclusion of additional sensors within the platform. Although 3D hydrodynamic flow focusing has been widely used in flow cytometry for optical detection of cells,24 reports on 3D hydrodynamic flow focusing in microfluidic systems with active sorting mechanisms are very few and limited to traditional wholly soft lithography fabricated devices.25,26 However, as the development reported below shows recent advances in 3D printing technology have allowed us to realise a modular high-fidelity 3D microfluidic system that interface a 3D printed, 3D hydrodynamic focusing chamber for sample detection with a soft lithography-based pressure-driven sorting module.
The development and characterisation of this system were pursued by first using fluorescent samples (various sized beads, fluorescently labelled mammalian cells) and then the more challenging Raman sorting of untagged mixtures of different types of microbes (Chlorella vulgaris and E. coli). It is worth noting the significant achievement of tight 3D flow focusing of the small E. coli (∼1 μm) in the large 1 mm3 detection chamber in order to address some of the issues above, since focusing such small cells in microfluidic channels of 100's μm width and height remains a significant challenge.10,25,26
Compared to existing RACS systems, the 3D-RACS system has achieved 1) Raman measurements without background signals of the device materials; 2) the ability to sort a large range of cell sizes and mixtures of them without blockage, and 3) long-term stable operation (>8 h) with high throughputs (e.g. >310 cells per min for sorting of cells based on their intrinsic Resonance Raman signal).
Finally, as part of the platform development, a simple “single-band” Raman acquisition system was used for the Raman-activated sorting of the microbes to demonstrate the deployment of a relatively simple detection method. However, it should be noted that the system could also be readily interfaced with an optical fibre based spectrometer, or implemented on commercial Raman spectrometer when more complex Raman identification is required. Thus, together with its ability of fully automated operation and in-line collection, this system could become a universally applicable, high-throughput flow cytometry and sorting platform for cells that can be distinguished by their Raman spectra or other weak optical signals.
Experimental section
3D hydrodynamic focusing and device design
The 3D-RACS device comprises two juxtaposed subunits: a 3D hydrodynamic focusing detection chamber and a pressure-driven cell sorting module (Fig. 1A). The device is a hybrid construction of the 3D printed detection chamber with a PDMS-based sorting unit (ESI† S1, device design, fabrication and assembly, ESI† Fig. S1 and S2).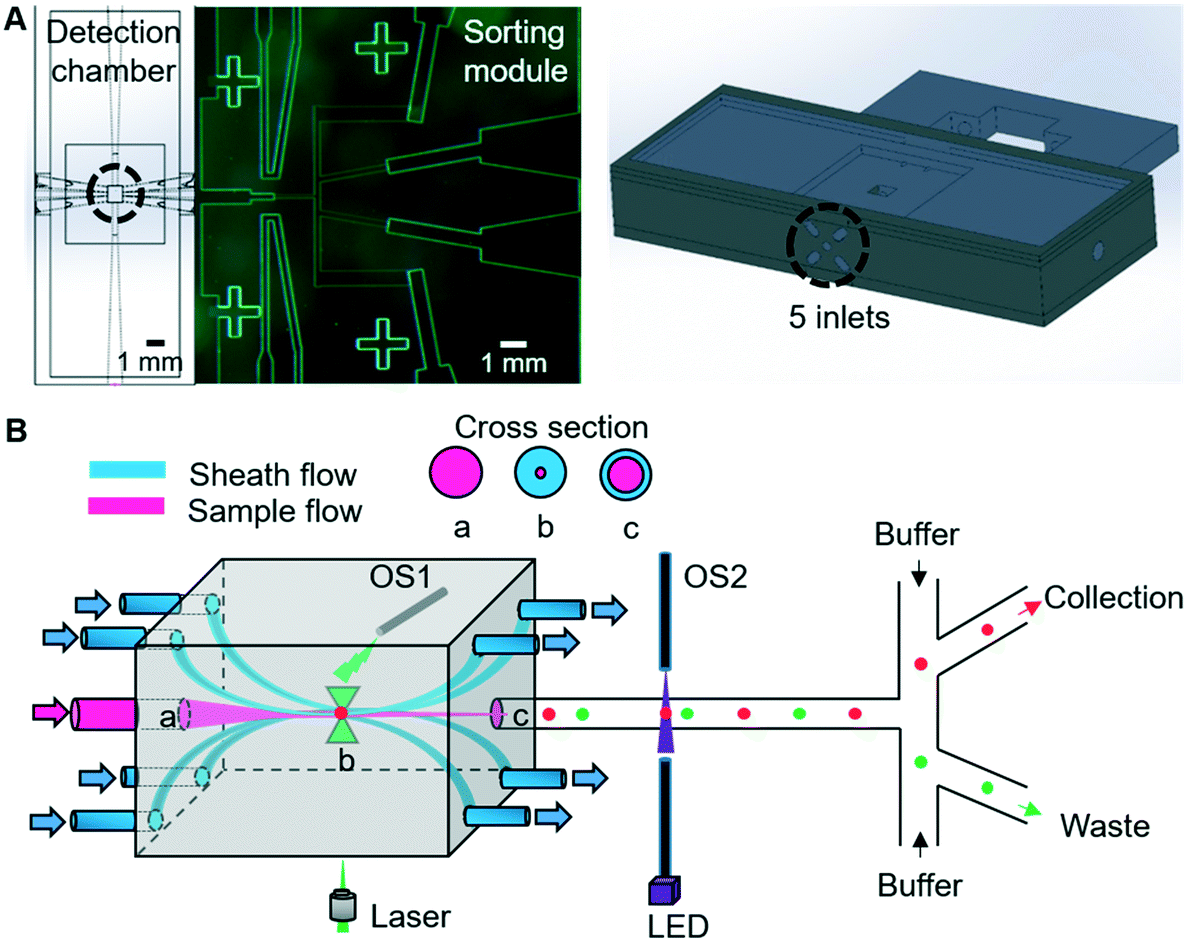 | ||
| Fig. 1 (A) Left: 2D view of the microfluidic device. Right: 3D view of the 3D printed unit. (B) Illustration of 3D hydrodynamic focusing. The sheath flows confine the sample flow in the Centre of the detection chamber and leave from the opposite side of the chamber. The sample flow (a) is hydrodynamically focussed so as to pass through the laser focus (b), before being directed into the sorting unit (c). OS1 and OS2 correspond to the optical fibre based sensors to detect when cells pass through particular points in the detection chamber or sorting module. | ||
The 3D flowing focusing is realised by surrounding the sample stream with multiple sheath streams, as shown in Fig. 1B. By tuning the velocity of the sample and sheath streams, we can control the tightness and shape of the sample stream in 3D. In comparison to the previous 3D hydrodynamic focusing devices,24,27 a unique feature of our design is to remove the sheath flows via the mirrored outlets on the opposite side of the detection chamber (Fig. 1B). This not only allows flexible steering of the focused sample stream but also the separation of sheath flow and sample flow after detection. Consequently, significant dilution of the sorted cells and fluid disturbance at the optical interrogation area resulting from the instantaneous pressure changes during sorting is avoided.
Additional important elements of the device include two sets of optical fibre sensors (105 μm core diameter, multimode, Thorlabs) that are used to synchronise a detection event with subsequent pressure-driven sorting actuation (Fig. 1B). The first fibre is inserted into a hole in the detection chamber, with the end of the fibre pointing to the position of the focused laser spot. When a cell passes through the focused laser spot, it generates a large Rayleigh scattering signal. This scattered light is collected by the fibre and converted to an electronic timing signal using a photodiode.
The second pair of fibres are located in the most upstream point of the sorting module (Fig. 1B) and configured in the form of a simple beam-break sensor that provides a timing mark related to when a cell enters the sorting module. Here, 405 nm light from a laser diode (Thorlabs) traverses the channel and is collected by an opposing fibre, with the signal being converted into a second electronic signal using a photodiode. The transient fluctuations in this signal when a cell passes through the pencil beam crossing the channel are detected and amplified using an offsetting amplifier (SRS instruments).
Automated, continuous sorting process
The 3D-RACS sorting process is illustrated in Fig. 2, including 1) 3D flow focusing to deliver cells precisely to the detection point, 2) detection (fluorescence or single window Raman and scattered light); 3) on-the-fly classification; 4–5) synchronisation between the time when the cell is detected and when it enters the sorting module; 6) sorting actuation to divert a target cell into the collection channel and in-line collection.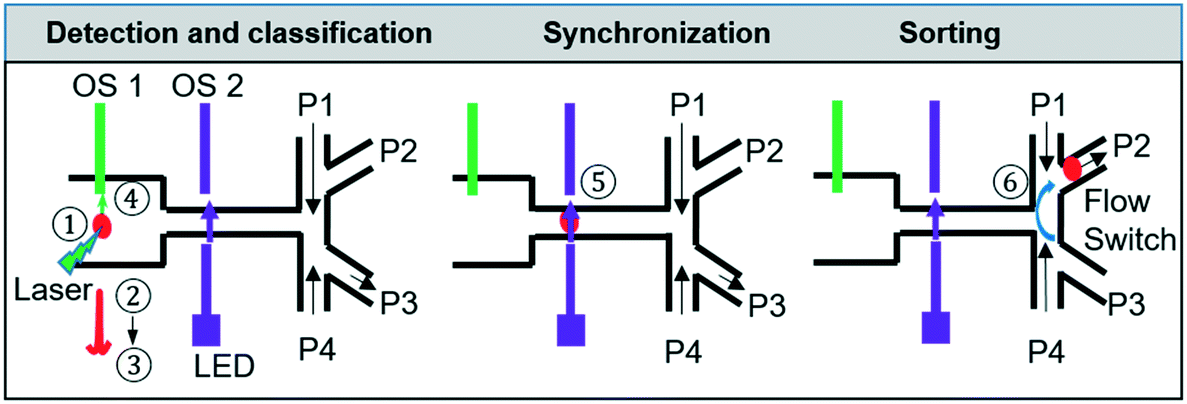 | ||
| Fig. 2 Flow chart of the steps of the sorting process. ① 3D flow focusing to deliver cells to the detection point; ② fluorescence or single-band Raman detection, ③ on-the-fly classification; ④ & ⑤synchronization (the 1st optical sensor indicates the arrival of a cell at the detection point, and the 2nd optical sensor indicates the arrival of a cell at the sorting channel junction) ⑥ sorting actuation. Red sphere indicates a cell. OS1 and OS2: optical sensors. P1–P4: different outlet pressures. | ||
To maximise the throughput, one computer controlled the acquisition and processing of the Raman (or fluorescence) signal while another computer controlled the optical sensor interrogation and the sorting actuation. Timestamped trigger signals to synchronise the sorting actuation were sent from the first computer to the 2nd one via an RS232 connection. This parallel operation greatly improves the speed of the process flow and therefore throughput. The whole process was controlled and automated using a customised LabVIEW program (ESI† Fig. S3).
Device fabrication and assembly of the platform
The device was modelled using COMSOL 3.5 (ESI† Fig. S1) and designed using Solidworks or L-Edit (ESI† Fig. S2). The detection chamber is 3D printed using (Object30 Prime printer) whereas the cell sorting module is fabricated using standard lithography (Fig. 3A and B and ESI† S1). The fluid connection between the two units was made by a short length (∼1 cm) of 50 μm internal diameter fused silica tubing (ESI† Fig. S2). The two modules were then assembled in a customised holder suitable for placing on a standard x–y stage of an inverted microscope (Fig. 3C).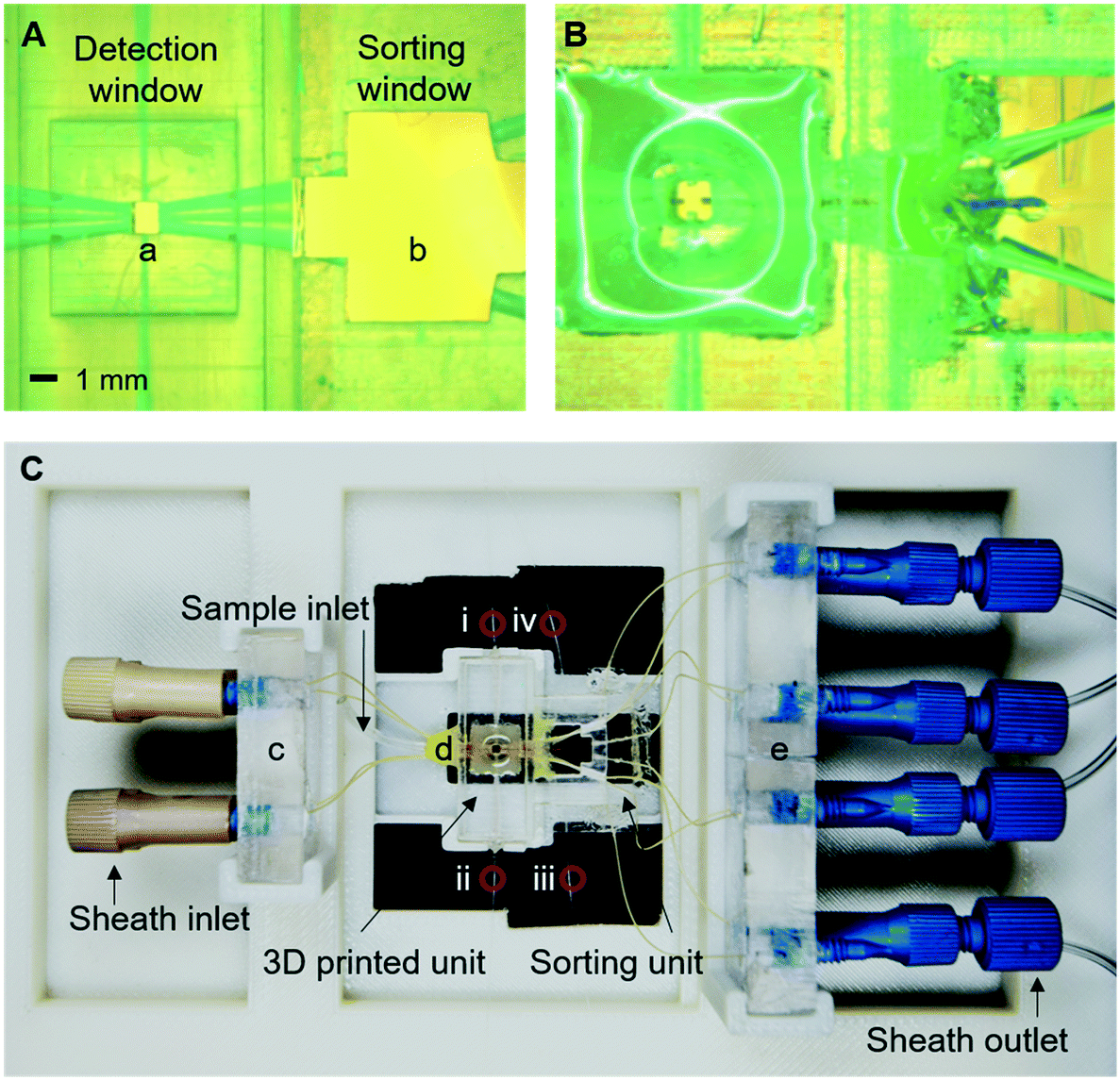 | ||
| Fig. 3 Optical images of (A) 3D-printed detection chamber and sort module holder, (B) combined detection and sorting subunits, and (C) the device and holder. Key components are denoted as (a) detection window, (b) sorting observation window, (c) sheath flow holders, (d) the integrated device, and (e) outlet sheath flow and sorting flow holders. i: Receiving fibre for the detection of the scattered signal at the detection point; ii: balancing fibre in the detection chamber (not used); iii: emitting fibre of beam-break sensor; iv: receiving fibre of beam-break sensor. | ||
The system setup
The overall 3D-RACS system operates on an inverted fluorescence microscope platform (Zeiss AxioObserver D1) (ESI† Fig. S4A). The Raman (or fluorescence) stimulation is provided by the laser source focused through an objective lens. Detection of the Raman (or fluorescence) signal also occurs via this objective, using a standard epi-configuration. In order to characterise the accuracy of the cell sorting actuation process in situ, an overhead microscope is positioned above the switching junction of the sorting module. This too worked in an epi-configuration, equipped with high speed camera for imaging fluorescently labelled cells or beads as they pass through the sorting module. However, to evaluate the Raman sorting performance when using non-labelled cells (such as the microbes here), an in-line collection module and post collection analyses were used instead.The detailed setup for sorting (excluding the overhead microscope) is shown in ESI† Fig. S4B. The Raman (or fluorescence) signals were collected using a 20×/0.4 NA objective lens and an Andor Neo sCMOS camera. Illumination was provided by either a fibre coupled 150 mW Oxxius 532 nm laser or a 100 W HBO lamp connected to the rear illumination port of the microscope. Rhodamine and FITC filters (Omega Optical) were used for fluorescence measurements. For Raman measurements, a 532 nm notch filter was placed in front of the camera (to remove Rayleigh scattered 532 nm excitation light) and narrow-band filters selected so as to detect a single Raman band of interest on the sCMOS camera (see later for details). It should be noted that simple replacement of the camera with a fibre coupled spectrometer would enable acquisition of full Raman spectra.
The inlet and outlet sheath flow pressured were controlled by Fluigent pumps (MFCS-EZ), with sorting actuation being controlled via pressure switching using an Elveflow pump (OB1 MK3+). The optical images and videos from the overhead microscope were acquired using an Andor DU 885 CCD and Andor Solis software. Image J was used to process optical images.
Cell culture and preparation
Cells and culture medium were bought from ATCC unless otherwise indicated. K562 cells from human bone marrow (∼10–15 μm) were cultured at 37 °C, 5% CO2, with passaging every 2 to 3 days and using IMDM (Iscove's Modified Dulbecco's Medium), supplemented with 10% (v/v) FBS (Fetal Bovine Serum) and 5% (v/v) penicillin–streptomycin. For characterisation of the sorting system, living cells were stained with either Calcein AM (green fluorescence) or CellTrace Yellow (red fluorescence, ThermoFisher) and mixed in different ratios in PBS buffer.As a proof-of-concept, Raman activated cell sorting of the system was characterised using samples containing both Chlorella vulgaris (4–6 μm) and E. coli (∼1 μm). Chlorella vulgaris 211/110 (Culture Collection of Algae and Protozoa, CCAP, UK) was cultured under continuous halogen light illumination (60–80 μmol photons m−2 s−1, 120 W) at 25 °C on a shaking table and in 3N-BBM+V medium (CCAP, UK). GFP expressing E. coli (MC1061 pBAD HheC WT) was a gift from Professor Lixia Tang and cultured in LB broth. Mixed population samples were prepared by combining these types of cells at different ratios in PBS buffer.
Raman spectra acquisition
Raman spectra of Chlorella vulgaris and E. coli were acquired using a LabRAM HR800 (Horiba Ltd) equipped with a 532 nm laser, a 600 grating and a Synapse charge-coupled-device (CCD) camera. Cells were washed with deionised water three times and deposited onto an Al-coated slide and air-dried prior to Raman analysis. A 63×/0.7 NA objective lens was used for spectra collection from randomly selected individual cells. The laser power at the sample was adjusted to 2 mW using an optical density filter. The acquisition time of Chlorella vulgaris and E. coli are 1 second and 50 seconds respectively.Raman activated cell sorting and recovery
Chlorella vulgaris contains carotenoids which have a characteristic Resonance Raman spectra.28 Its peak at 1516 cm−1 is widely used to characterise carotenoids containing cells or tissues.29,30 To detect Raman signals using an ordinary epi-configured microscope, we adopted a simple method of creating a narrow band spectral ‘Raman Window’ using bandpass filters.32 The Raman band with the central peak of ∼1521 cm−1 (90 cm−1 width, from 1496 cm−1 to 1585 cm−1) was used to detect Chlorella vulgaris. When a 532 nm excitation laser is used, this corresponded to the wavelength range from 578 nm to 581 nm, based on eqn (1).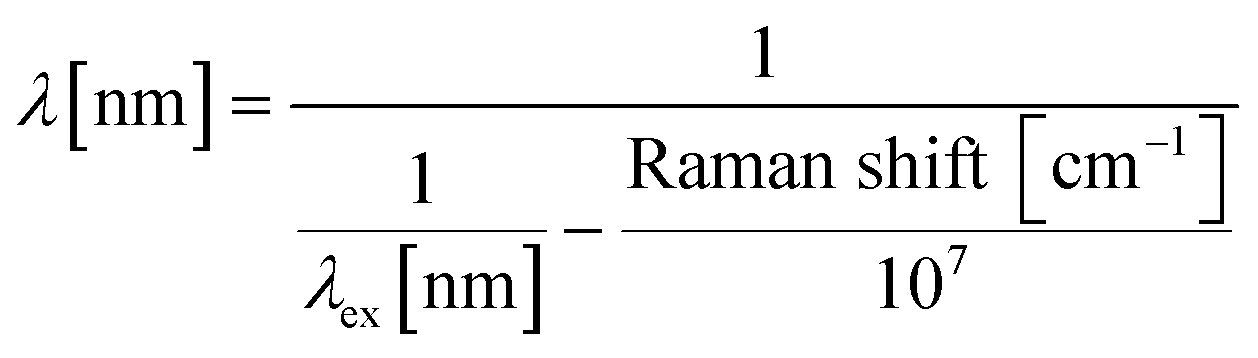 | (1) |
A simple ‘Raman window’ for this peak can be created by overlapping two narrow band filters (571–581 nm from Semrock and 578–640 nm from Omega) to give a band pass of 578–581 nm (note: if desired, a narrower bandpass filter can be created by tilting one or other the filter pair to move their cut-on/cut-off wavelengths). The sorted cells were collected using a ∼0.45 μm nanopore membrane integrated in-line with the collection and waste channels. Cells captured on the membrane were then identified and counted using standard microscopy.
Results and discussion
Characterisation of 3D hydrodynamic focusing and sorting
To facilitate the observation of the sample flow and 3D flow focusing, the sample stream was loaded with 5 μm diameter green fluorescent polystyrene beads, with or without the addition of ∼1 μM of fluorescein or rhodamine in DI water. As shown in Fig. 1B, the sample flow is connected to the central inlet in the leftmost face of the detection chamber, with the inlet sheath flows being connected to the four surrounding inlets. Thus, by adjusting the flow rate in the inlet sheath flows, the sample flow can be confined in 3D so that the region occupied by the cells at the flow focus is sufficiently tight that all of the cells pass through the optical focus of the Raman stimulating laser (Fig. 4A). The flow focus can be easily adjusted to be as small as 3 μm in this 1 mm3 detection chamber (ESI† Fig. S5 and Video S1), enabling reliable and consistent detection of each cell. Independent control of the outlet sheath-flow channel flow-rates provides additional flexibility and control of this focusing method. As illustrated in Fig. 4B, the inlet sample stream can be directed to any of the sheath flow outlets or the designated central sample outlet and onwards to the sorting module, as desired. This feature can be used to avoid contamination of the sample collection stream in the sorting unit when the system is being initially set-up or being cleaned part way through a long-term sorting process.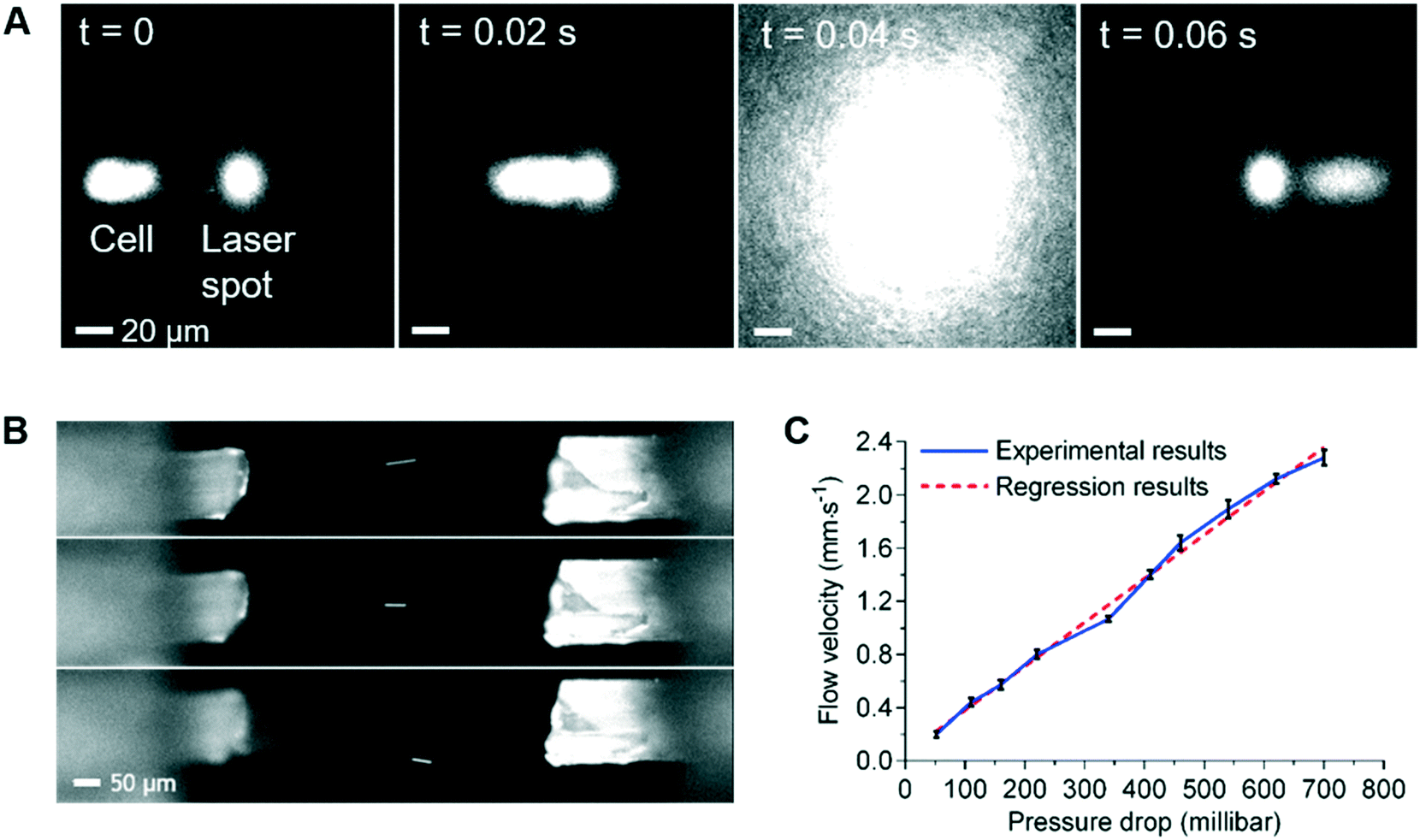 | ||
| Fig. 4 (A) Time-lapse images showing precise delivery of a K562 cell to the laser point. Cells are stained with CellTrace yellow and generate the fluorescence signal at T = 0.04 s. (B) Flexible tuned 3D hydrodynamic focusing using sheath flows. The bright-line is an image of a 5 μm diameter red fluorescent bead in flow. (C) Relationship between flow velocity at the optical interrogation area and pressure drop between inlets and outlets of the 3D hydrodynamic focusing chamber. | ||
By varying the pressure difference between the inlet and outlet sheath flow channels, cell velocity at the 3D focus position can be easily controlled. Examination of velocities that range from 0.2 mm s−1 to 2.2 mm s−1 at the flow focus, showed excellent stability and a close to a linear relationship between the velocity and the pressure difference between the inlet and outlet sheath flows (Fig. 4C). This ability to control the velocity allows us to extend the acquisition time for cells with weak Raman signals if necessary. For example, if the laser spot was defocused so as to be 20 μm in diameter, these flow rates would correspond to effective acquisition times of ∼10 ms to ∼100 ms, which are sufficient to collect signals from the weakest of the test samples used here. If, however, slower flow rates are required (e.g. to achieve longer Raman signal acquisition times), 3D printed detection chambers which are both longer and wider can be used. These changes in the dimension of the detection chamber enable the cell velocity to be reduced, whilst maintaining the degree of flow focusing control required to consistently position every cell in the focus of the laser.
As shown in Fig. 2, once cells entered the sorting module (location 5), they are diverted to either the collection or waste channels by adjusting the pressures applied to these channels. To collect or discard the cells, several different pre-programmed pressure settings are used. These pressures are set in such a way that the pressures of P1 and P4 are always slightly greater than those of P2 and P3 respectively to prevent backflow of cells. The short sequence of video frames in ESI† Fig. S6 (Video S2†) illustrates the operation of this switching protocol to send a non-target cell into the waste stream, and then a target cell into the collection channel, before switching the flow back to the waste stream. A series of switching times were tested and 75 ms was used for the sorting experiments below in which the channel segment from the T-junction of the sorting network to the collection or waste stream branch was 400 μm long. This distance was chosen to facilitate the characterisation of the high speed sorting process using video acquisition, rather than to optimise the throughput of cells. It should be noted that a faster switching could be achieved by using a shorter T-junction to collection branch distance, or by using other methods, such as piezoelectric actuator-driven pumps.31
Synchronising and automation
To achieve automated and reliable operation for extended operation, hardware/software integration was developed to allow reliable synchronisation of Raman acquisition, real-time signal processing and sorting. To maximise the throughput, the software that detected signals from the target cells on the sCMOS camera ran independently of that which controlled the pressure driven switching in the sorting unit, as shown in ESI† Fig. S3.Thus, to reliably synchronise the two operations, it is necessary to predict the transit time from the detection point to the beam-break sensor in the sorting channel network so as not to miss the arrival of the target cell. This is readily achieved with the two optical sensors (Fig. 1c, OS1&OS2), as shown in Fig. 5A. Optical sensor 1 collects the Rayleigh scattered light from a cell as it passes through the focussed laser spot and generates an electrical signal (red trace, Fig. 5A). Optical sensor 2 generates a second electrical signal when that cell passes the beam-break sensor (black trace, Fig. 5A). The time interval between the two signals corresponds to the transit time between the two modules. The average and standard deviation of these transit times were then calculated using a signal processing routine implemented in the LabVIEW program. As shown in Fig. 5B, these times form a close to a normal distribution with a standard deviation that is close to the square root of the mean. These parameters are used to control the time that the sorting computer waits after having received a trigger from the Raman detection computer, before starting to monitor optical sensor 2 (see ESI† Fig. S3). When the target cell has reached the beam-break sensor, the pressure actuation sequence in the sorting channel network is started. The accuracy of sorting actuation as a function of the transit time and its standard deviation has been simulated based on Poisson statistics for the passage and temporal distribution of target and non-target cells through the detection chamber (ESI† Fig. S7A) and shows that the accuracy of sorting could be improved effectively by improving the prediction of the transit time or reducing the spread in transit times.
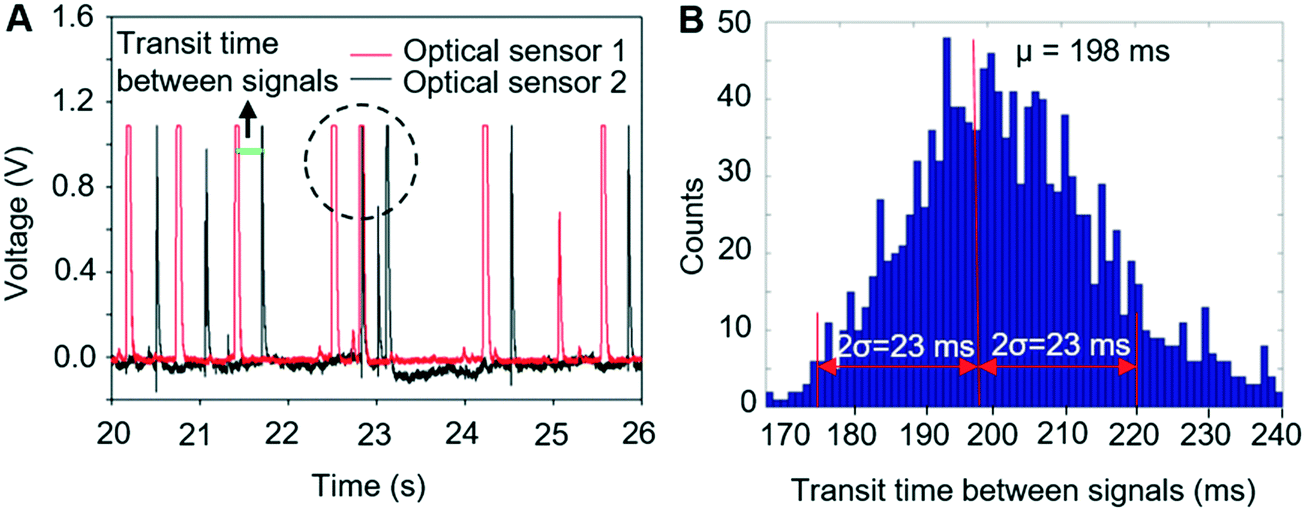 | ||
| Fig. 5 (A) Electrical signals from two optical sensors. The transit time is shown as the gap between signals generated by one cell from both optical sensors. The dotted circle represents the signals generated by two cells close to each other. These situations occur randomly. (B) Histogram distribution of the transit time between two optical sensors. The mean value (198 ms) and standard deviation (11.5 ms) were automatically calculated and transmitted to the part of the program that controls the pressure driven switching. | ||
Characterising sorting performance
Three characteristic parameters of the sorting system were evaluated, namely 1) accuracy of sorting – the percentage of detected target cells that are directed into the collection channel; 2) sorting throughput – the number of cells analysed per second; 3) purity – the percentage of cells that are diverted into the collection channel that are actually target cells.After initial characterisation experiments involving fluorescent polymer beads, in the first instance to facilitate real-time imaging of the sorting performance when using cells, the platform was evaluated using mixtures of living K562 cells stained with either Calcein AM (green fluorescence) or Cell Trace Yellow (red fluorescence). A variety of target cell percentages (10.8%, 21.2%, and 43.5%) and different total cell concentrations (2 × 105, 5 × 105, and 1 × 106 cells per ml) were tested. A selection of these results is shown in Fig. 6, where it can be seen that for all cell concentrations, the accuracy of sorting is ∼90% (i.e. ∼10% of the target cells that are detected are not collected) (Fig. 6A). This loss is a consequence of the slight skewness in the distribution of transit times (Fig. 5B), and the known probability that the beam-break sensor triggers on a non-target cell that enters the sorting module in front of the target cell. Nevertheless, this is close to the theoretical prediction based on Poisson statistics (ESI† Fig. S7B) and further optimisation of the parameters can improve the sorting accuracy to ∼98% as detailed in ESI† S2.
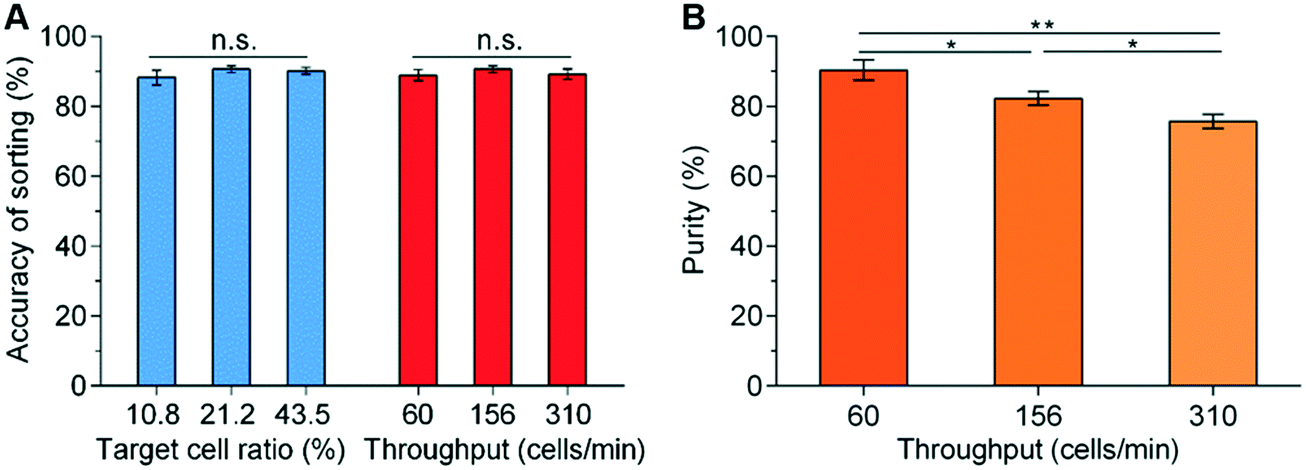 | ||
| Fig. 6 Result of sorting performance using K562 cells. (A) Sorting efficiency of K562 cells at different target ratios with a throughput of ∼156 cells per min, and (B) purity of collected cells at different cell concentration in the samples at a fixed target ratio of 21.2% and throughput rates of 60, 156 and 310 cells per min. The throughput was achieved by varying total cell concentration to 0.2, 0.5 and 1 × 106 cells per ml respectively. Three independent experiments were conducted for each condition. ns: not significant; *: p < 0.05; **: p < 0.01 (n = 3). The sorting actuation time was fixed at 75 ms. | ||
As with many in flow cell sorting systems, the interrelation of sample throughput and purity of the sorted cells is determined in large part by time taken to perform the switching, detecting when a target cell arrives in the sorting network, factors associated with the acquisition of signals, and Poisson statistics. Fig. 6 shows the influence of total cell concentration and throughput on purity as a consequence of the time taken to perform the sorting actuation. At a fixed initial target percentage of 21.2%, the increase of the total cell concentrations leads to an increase in throughput (i.e. 0.2, 0.5 and 1 × 106 cells per ml corresponding to 60, 156 and 310 cells per min respectively).
Similarly, as the throughput increases, the purity falls since it is more likely that there will be a non-target cell close to the target one during the switching actuation (Fig. 6B). Statistically, the purity could be predicted based on the throughput (ESI† 2 and Fig. S7B). Under the conditions of Fig. 6B (i.e. the switching time is 75 ms) and the throughput is 310 cells per min, the predicted purity based on Poisson statistics is 80.6% (ESI† Fig. S7B), which is not far from the experimentally determined value of 75.5% (Fig. 6B). Similarly, when increasing the throughput to ∼540 cells per min, the sorting accuracy reduces slightly to 79.0% and the purity to 70.5%. However, these experimental data are close to the predicted theoretical values (i.e. accuracy of 79.5% and purity of 70.9%) (ESI† Fig. S7B), illustrating excellent performance of the system.
The robustness of this automated system was further demonstrated in long-period sorting experiments (>8 h) by an hourly sampling of the accuracy of sorting and purity (ESI† Table S1). When using a sample that contains 10% of target beads, 90% non-target beads and throughput rate of ∼260 beads per min, the purity was ∼76.6%, i.e. ∼7.5 fold refinement, and the standard deviation of the hourly measurements was only 1.6% of its average value. Similarly, the accuracy was 84.4% with a variation of ∼4.3%. It is worth noting that in this long duration test demonstration, the switching parameters were selected to allow the observation of the movement of individual beads in the sorting module, and each of the purity and accuracy metrics can be improved on by decreasing the switching time and using shorter channel lengths for segments of the sorting module (ESI† S2).
Sorting microbes based on their Raman signals
An immediate benefit of using a 1 mm3 sized detection chamber is the elimination of background Raman scattering from the chamber walls and windows. As shown in Fig. 7, the Raman spectrum of water acquired at a central height, the middle of the detection chamber is relatively featureless in the fingerprint and C–H region (although the bands due to water can be seen, depending on the NA of the objective used). In contrast, that acquired using a typical 2D PDMS channel (of 25 μm height) show strong Raman peaks corresponding to PDMS and the glass substrate. Such a low background is key to being able to make the single “Raman window” measurements as well as being able to improve sample signal/background measurements when using a spectrometer-based system (Fig. 7).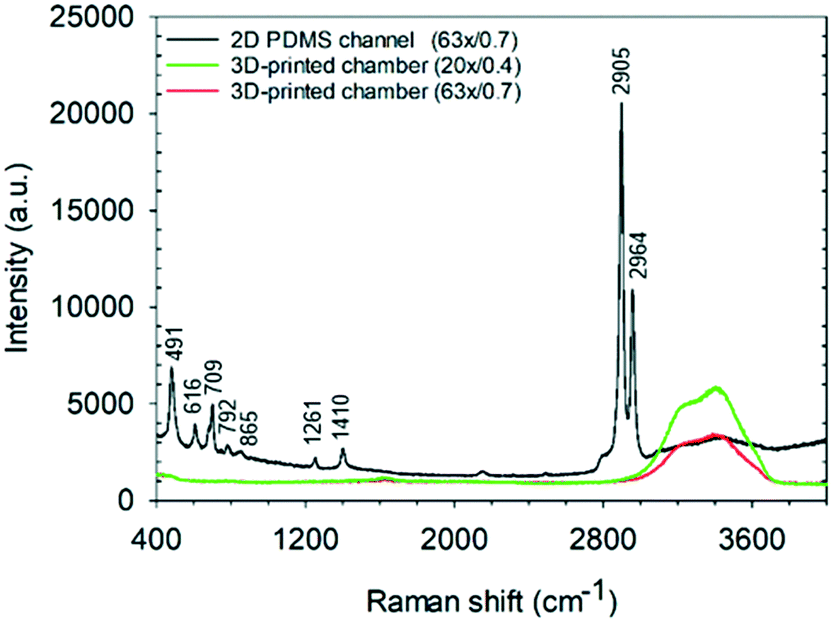 | ||
| Fig. 7 Raman spectra of water taken in the middle of the 3D-printed detection chamber (1 mm3) and a 2D PDMS microchannel (of 25 μm height) with two objective lenses (63×/0.7 NA and 20×/0.4 NA). | ||
A further advantage of the large detection chamber is that it is not prone to blockage in the way that 2D microfluidic systems can be. However, in the absence of 3D flow focusing, such a large chamber could encounter difficulties when detecting small cells (such as 1 μm bacteria). Therefore, to illustrate how this design can be successfully used to perform Raman activated cell sorting, this 3D-RACS system was evaluated using mixtures of Chlorella vulgaris algae and E. coli. (∼1 μm) as described below.
Chlorella vulgaris contains carotenoids, which have strong characteristic Resonance Raman bands around (1002 cm−1, 1152 cm−1 and ∼1521 cm−1),9,30 whilst Raman signals of E. coli are very weak under the same acquisition condition using a conventional Raman spectrometer (Fig. 8A). During the 3D-RACS sorting, when Chlorella passes through the laser spot, a bright Raman image (∼1521 cm−1) is obtained through the single “Raman window”, whereas that corresponding to E. coli is very weak (Fig. 8B). The signal-to-background ratio of Chlorella (102.6 ± 78.5) is ∼10 times higher than that of E. coli (9.9 ± 6.2) (Fig. 8C), leading to a high degree of confidence in classifying the target cells.
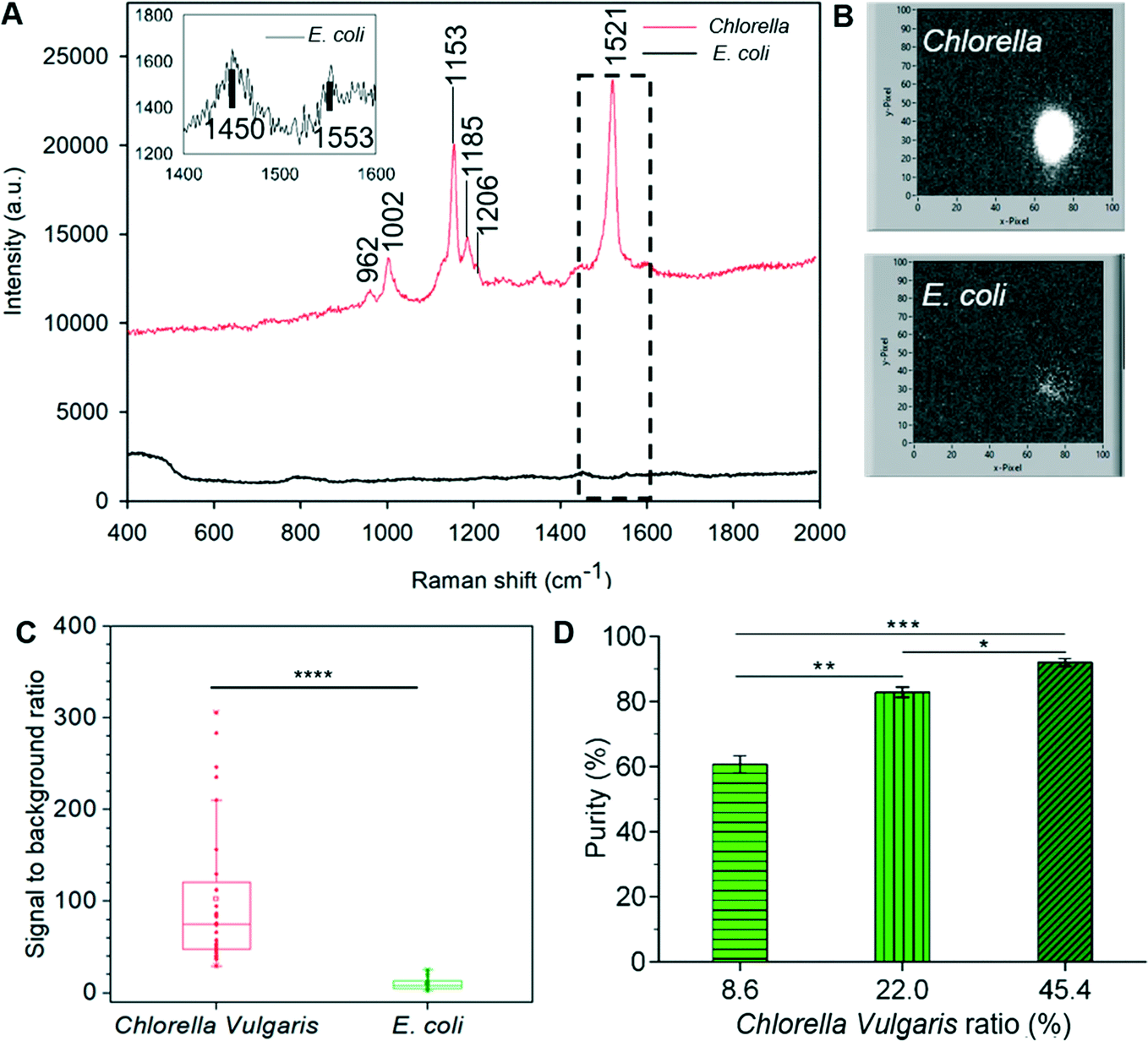 | ||
| Fig. 8 (A) Single-cell Raman spectra of Chlorella vulgaris and E. coli using 532 nm laser. The enlarged view of E. coli spectrum is shown at the top left. (B) LabVIEW interfaces that show the Raman signal of Chlorella vulgaris and E. coli from the “Raman window” with 10 ms acquisition. (C) Signal-to-background ratio of Chlorella vulgaris and E. coli detected by single Raman channel filter. n = 30. ****: p < 0.0001. (D). Purity of Chlorella vulgaris cells in the sorted collection versus different percentages of Chlorella vulgaris in the initial samples at throughput of 310 cells per min (three independent measurements). *: p < 0.05; **: p < 0.01; ***: p < 0.001. | ||
To collect the cells for off-line processes, we placed a membrane filter in line with the target collection and waste channels (ESI† Fig. S8A). Here, the advantage of the removal of the sheath flows after the detection chamber can be seen by minimising the dilution of the sorted target bacteria. A second advantage is the low volumetric flow rate passing into the collection channel. This means that the pressure drop across the membrane is small; and so reduces any potential damage to the cells as well as enabling them to be collected within a small volume, facilitating harvesting for subsequent processing.
The sorting performance of the device based on the Raman signals was evaluated with a series of experiments were performed with different E. coli and Chlorella vulgaris ratios. The operations were performed in the same manner as with the mammalian cells above. To evaluate its potential for sorting a large heterogeneous population, the throughput of 310 cells per min was used for all the conditions.
Here, for identification purposes, advantage was taken of Chlorella vulgaris and E. coli exhibiting distinct red and green fluorescence intensity respectively when illuminated by 488 nm light (ESI† Fig. S8B and C). Hence the bacteria collected on the filter paper could be readily identified and quantified (using Image J). Quantitative assessment of the purity of the collected target cells is shown in Fig. 8D and has achieved a purity level of 92.0% at this throughput (i.e. 310 cells per min). Although this drops to ∼60% (which is nevertheless a 7-fold refinement) when the initial target cell concentration is still relatively low (8.6%), this can be compensated for either by repeated sorting or further optimising the sorting conditions as discussed above (ESI† 2).
Finally, it is noted that several benefits could be gained from the use of this narrow band “Raman window” configuration, including an imaging mode (due to the use of an sCMOS or CCD detector) and potentially enhanced signal collection through the elimination of losses that arise due to the entrance slit and grating present in most Raman spectrometers. These in conjunction with the simplicity of constructing the narrow “Raman window” system on an ordinary optical microscope, could facilitate wider adoption of Raman signals for label-free cell analysis and sorting. The demonstration here of sorted cells is based on the presence of carotenoids, which widely exist in algae, plants and nearly all photosynthetic cells.33,34 Sorting carotenoid-containing cells from those without carotenoids could help discover novel unculturable phototrophic bacteria in untapped natural resources,9 as well as in screening culture conditions that can promote carotenoid-production in engineered microorganisms.35 Other narrow-band windows for Raman bands such as 750 cm−1 for Cytochrome c to detect apoptosis,8 2850–2930 cm−1 for C–H to detect lipids or carbohydrate,36 or 2040–2300 cm−1 for C–D band in the Raman silent region to detect generic metabolic activity,13 can be easily implemented by appropriate selection of filter sets.
However, it is also worth highlighting the trade-off between simplicity/cost and the rich information of full spectra. Traditional Raman spectroscopy and various enhanced Raman techniques (e.g. coherent anti-Stokes Raman scattering,37 stimulated Raman Scattering38) are still needed for the applications requiring multiplexing detection and high Raman resolution39 and these can be accessed here by use of a fibre coupled spectrometer in place of the filter and sCMOS camera.
Conclusions
The important roles of individual heterogeneity in disease development40 and the function of microbial communities41 have become widely recognised, which has stimulated rapid developments single-cell technologies.42 Raman activated cell sorting has demonstrated its unique strength in function-based isolation, label-free approaches of individual cells, and the ability to establish a direct link between the phenotypic functions and the genomics of members in a community.4,9 These promises could provide a solution to many long-standing challenges, for example, in the search for rare but key functional subset from complex environmental microbial communities. However, the speed and robust operation required to sort large, complex populations are yet to be met by the current RACS technologies.3,4In this context, the fully automated 3D-RACS system presented here provides a label-free, quantitative, and highly effective method to sort a heterogeneous population based on specific function at the single-cell level. With its novel 3D hydrodynamic focusing method and large detection chamber (1 mm3), it has successfully overcome blockage problems that is commonplace when using 2D microfluidic sorting devices over extended periods.43 This robustness is also accompanied by long-period of stable sorting performance (<5% variation in purity and accuracy over 8 hours).
As a trapping free system, it holds great potential for being high throughput. Without extensive optimisation, it has achieved 540 cells per min with a sorting accuracy of 79%, close to that theoretically predicted for this throughput. The proof-of-concept demonstration of Raman activated sorting of Chlorella vulgaris and E. coli illustrated a high throughput of 310 cells per min with 92% purity which is higher than the existing continuous platforms.3,17,20 Most importantly, the universal applicability of hydrodynamic focusing through its independence on the physical properties of cells (size and shape, etc.) and media, provides a unique advantage in dealing with real-world complex samples.
Finally, the use of a simple “Raman window” microscope allows easy integration Raman with other common optical imaging techniques (e.g. fluorescence, phase-contrast), which will in the future expand the scope of cell functions that can be probed. This, coupled with the rapid development in 3D printing technologies promises fully 3D printed 3D-RACS devices in the near future. As it stands, however, this platform could be adapted to be used as a robust and flexible platform for flow cytometry and activated cell sorting based on optical signals that could be either Raman, fluorescence, or bioluminescence. Consequently, this will expand the list of high throughput cell and particle sorting tools readily available to researchers in a broad range of fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank support from both Nissan Chemical Ltd and the University of Glasgow for funding YL's studentship, Industrial Biotechnology Innovation Centre (IBioIC, Project-2016-152), and Natural Environment Research Council (NE/P003826/1). We also thank the technical team of the James Watt Nanofabrication Centre (JWNC) at the University of Glasgow for the support in fabricating the devices.References
- H. R. Hulett, W. A. Bonner, J. Barrett and H. La, Science, 1969, 166, 747–749 CrossRef CAS.
- M. Q. Li, J. Xu, M. Romero-Gonzalez, S. A. Banwart and W. E. Huang, Curr. Opin. Biotechnol., 2012, 23, 56–63 CrossRef CAS.
- Y. Z. Song, H. B. Yin and W. E. Huang, Curr. Opin. Chem. Biol., 2016, 33, 1–8 CrossRef CAS.
- K. S. Lee, M. Palatinszky, F. C. Pereira, J. Nguyen, V. I. Fernandez, A. J. Mueller, F. Menolascina, H. Daims, D. Berry, M. Wagner and R. Stocker, Nat. Microbiol., 2019, 4, 1035–1048 CrossRef CAS.
- W. E. Huang, M. Q. Li, R. M. Jarvis, R. Goodacre and S. A. Banwart, in Advances in Applied Microbiology, ed. A. I. Laskin, S. Sariaslani and G. M. Gadd, Elsevier Academic Press Inc, San Diego, 2010, vol. 70, pp. 153–186 Search PubMed.
- S. Pahlow, S. Meisel, D. Cialla-May, K. Weber, P. Rosch and J. Popp, Adv. Drug Delivery Rev., 2015, 89, 105–120 CrossRef CAS.
- H. J. van Manen, Y. M. Kraan, D. Roos and C. Otto, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10159–10164 CrossRef CAS.
- M. Okada, N. I. Smith, A. F. Palonpon, H. Endo, S. Kawata, M. Sodeoka and K. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 28–32 CrossRef CAS.
- Y. Z. Song, A. K. Kaster, J. Vollmers, Y. Q. Song, P. A. Davison, M. Frentrup, G. M. Preston, I. P. Thompson, J. C. Murrell, H. B. Yin, C. N. Hunter and W. E. Huang, Microb. Biotechnol., 2017, 10, 125–137 CrossRef CAS.
- K. S. Lee, M. Wagner and R. Stocker, Microb. Cell, 2020, 7, 62–65 CrossRef CAS.
- S. Dochow, C. Krafft, U. Neugebauer, T. Bocklitz, T. Henkel, G. Mayer, J. Albert and J. Popp, Lab Chip, 2011, 11, 1484–1490 RSC.
- A. Y. Lau, L. P. Lee and J. W. Chan, Lab Chip, 2008, 8, 1116–1120 RSC.
- D. Berry, E. Mader, T. K. Lee, D. Woebken, Y. Wang, D. Zhu, M. Palatinszky, A. Schintimeister, M. C. Schmid, B. T. Hanson, N. Shterzer, I. Mizrahi, I. Rauch, T. Decker, T. Bocklitz, J. Popp, C. M. Gibson, P. W. Fowler, W. E. Huang and M. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E194–E203 CrossRef CAS.
- W. E. Huang, A. D. Ward and A. S. Whiteley, Environ. Microbiol. Rep., 2009, 1, 44–49 CrossRef CAS.
- L. Collard, F. Sinjab and I. Notingher, Biophys. J., 2019, 117, 1589–1598 CrossRef CAS.
- P. R. Zhang, L. H. Ren, X. Zhang, Y. F. Shan, Y. Wang, Y. T. Ji, H. B. Yin, W. E. Huang, J. Xu and B. Ma, Anal. Chem., 2015, 87, 2282–2289 CrossRef CAS.
- X. X. Wang, L. H. Ren, Y. T. Su, Y. T. Ji, Y. P. Liu, C. Y. Li, X. R. Li, Y. Zhang, W. Wang, Q. Hu, D. X. Han, J. Xu and B. Ma, Anal. Chem., 2017, 89, 12569–12577 CrossRef CAS.
- J. Voldman, Annu. Rev. Biomed. Eng., 2006, 8, 425–454 CrossRef CAS.
- A. Ashkin, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4853–4860 CrossRef CAS.
- D. McIlvenna, W. E. Huang, P. Davison, A. Glidle, J. Cooper and H. B. Yin, Lab Chip, 2016, 16, 1420–1429 RSC.
- C. W. Shields, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249 RSC.
- A. Wolff, I. R. Perch-Nielsen, U. D. Larsen, P. Friis, G. Goranovic, C. R. Poulsen, J. P. Kutter and P. Telleman, Lab Chip, 2003, 3, 22–27 RSC.
- S. Lee, J. Choi, L. Chen, B. Park, J. B. Kyong, G. H. Seong, J. Choo, Y. Lee, K. H. Shin, E. K. Lee, S. W. Joo and K. H. Lee, Anal. Chim. Acta, 2007, 590, 139–144 CrossRef CAS.
- M. A. Daniele, D. A. Boyd, D. R. Mott and F. S. Ligler, Biosens. Bioelectron., 2015, 67, 25–34 CrossRef CAS.
- N. Nitta, T. Sugimura, A. Isozaki, H. Mikami, K. Hiraki, S. Sakuma, T. Iino, F. Arai, T. Endo, Y. Fujiwaki, H. Fukuzawa, M. Hase, T. Hayakawa, K. Hiramatsu, Y. Hoshino, M. Inaba, T. Ito, H. Karakawa, Y. Kasai, K. Koizumi, S. Lee, C. Lei, M. Li, T. Maeno, S. Matsusaka, D. Murakami, A. Nakagawa, Y. Oguchi, M. Oikawa, T. Ota, K. Shiba, H. Shintaku, Y. Shirasaki, K. Suga, Y. Suzuki, N. Suzuki, Y. Tanaka, H. Tezuka, C. Toyokawa, Y. Yalikun, M. Yamada, M. Yamagishi, T. Yamano, A. Yasumoto, Y. Yatomi, M. Yazawa, D. Carlo, Y. Hosokawa, S. Uemura, Y. Ozeki and K. Goda, Cell, 2018, 175, 266–276 CrossRef CAS.
- Y. Suzuki, K. Kobayashi, Y. Wakisaka, D. Deng, S. Tanaka, C. J. Huang, C. Lei, C. W. Sun, H. Q. Liu, Y. Fujiwaki, S. Lee, A. Isozaki, Y. Kasai, T. Hayakawa, S. Sakuma, F. Arai, K. Koizumi, H. Tezuka, M. Inaba, K. Hiraki, T. Ito, M. Hase, S. Matsusaka, K. Shiba, K. Suga, M. Nishikawa, M. Jona, Y. Yatomi, Y. Yalikun, Y. Tanaka, T. Sugimura, N. Nitta, K. Goda and Y. Ozeki, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15842–15848 CrossRef CAS.
- Y. J. Chiu, S. H. Cho, Z. Mei, V. Lien, T. F. Wu and Y. H. Lo, Lab Chip, 2013, 13, 1803–1809 RSC.
- T. H. Lee, J. S. Chang and H. Y. Wang, Anal. Chem., 2013, 85, 2155–2160 CrossRef CAS.
- T. R. Hata, T. A. Scholz, I. V. Ermakov, R. W. McClane, F. Khachik, W. Gellermann and L. K. Pershing, J. Invest. Dermatol., 2000, 115, 441–448 CrossRef CAS.
- Y. Y. Huang, C. M. Beal, W. W. Cai, R. S. Ruoff and E. M. Terentjev, Biotechnol. Bioeng., 2010, 105, 889–898 CAS.
- S. Sakuma, Y. Kasai, T. Hayakawa and F. Arai, Lab Chip, 2017, 17, 2760–2767 RSC.
- I. V. Ermakov, M. R. Ermakova and W. Gellermann, Appl. Spectrosc., 2005, 59, 861–867 CrossRef CAS.
- M. Griffiths, W. R. Sistrom, G. Cohenbazire and R. Y. Stanier, Nature, 1955, 176, 1211–1214 CrossRef CAS.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS.
- P. C. Lee and C. Schmidt-Dannert, Appl. Microbiol. Biotechnol., 2002, 60, 1–11 CrossRef CAS.
- H. W. Wu, J. V. Volponi, A. E. Oliver, A. N. Parikh, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3809–3814 CrossRef CAS.
- K. Hiramatsu, T. Ideguchi, Y. Yonamine, S. Lee, Y. Z. Luo, K. Hashimoto, T. Ito, M. Hase, J. W. Park, Y. Kasai, S. Sakuma, T. Hayakawa, F. Arai, Y. Hoshino and K. Goda, Sci. Adv., 2019, 5, 8 Search PubMed.
- C. Zhang, K. C. Huang, B. Rajwa, J. J. Li, S. Q. Yang, H. N. Lin, C. S. Liao, G. Eakins, S. H. Kuang, V. Patsekin, J. P. Robinson and J. X. Cheng, Optica, 2017, 4, 103–109 CrossRef CAS.
- Y. Wang, W. E. Huang, L. Cui and M. Wagner, Curr. Opin. Biotechnol., 2016, 41, 34–42 CrossRef CAS.
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS.
- S. V. Avery, Nat. Rev. Microbiol., 2006, 4, 577–587 CrossRef CAS.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS.
- J. W. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00679c |
| This journal is © The Royal Society of Chemistry 2020 |