
Smartphone-based multiplex 30-minute nucleic acid test of live virus from nasal swab extract†

Abstract
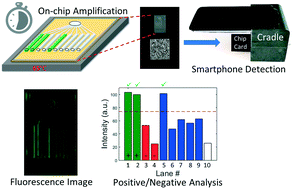
Rapid, sensitive and specific detection and reporting of infectious pathogens is important for patient management and epidemic surveillance. We demonstrated a point-of-care system integrated with a smartphone for detecting live virus from nasal swab media, using a panel of equine respiratory infectious diseases as a model system for corresponding human diseases such as COVID-19. Specific nucleic acid sequences of five pathogens were amplified by loop-mediated isothermal amplification on a microfluidic chip and detected at the end of reactions by the smartphone. Pathogen-spiked horse nasal swab samples were correctly diagnosed using our system, with a limit of detection comparable to that of the traditional lab-based test, polymerase chain reaction, with results achieved in ∼30 minutes.
- This article is part of the themed collections: Coronavirus articles - free to access collection and Coronavirus collection – Analytical
 Please wait while we load your content...
Please wait while we load your content...

