
Application of microphysiological systems in biopharmaceutical research and development
Norman C.
Peterson
 *a,
Prathap Kumar
Mahalingaiah
b,
Aaron
Fullerton
c and
Matteo
Di Piazza
d
*a,
Prathap Kumar
Mahalingaiah
b,
Aaron
Fullerton
c and
Matteo
Di Piazza
d
aClinical Pharmacology and Safety Sciences, AstraZeneca, One Medimmune Way, Gaithersburg, MD 20878, USA. E-mail: ncpdvmphd@gmail.com
bInvestigative Toxicology and Pathology, Abbvie, North Chicago, IL 60064, USA
cGenentech, Inc., 1 DNA Way, South San Francisco, CA, USA
dNonclinical Drug Safety, Boehringer Ingelheim Pharmaceuticals, Inc., 900 Ridgebury Rd, Ridgefield, CT 06877, USA
First published on 22nd January 2020
Abstract
Within the last 10 years, several tissue microphysiological systems (MPS) have been developed and characterized for retention of morphologic characteristics and specific gene/protein expression profiles from their natural in vivo state. Once developed, their utility is typically further tested by comparing responses to known toxic small-molecule pharmaceuticals in efforts to develop strategies for further toxicity testing of compounds under development. More recently, application of this technology in biopharmaceutical (large molecules) development is beginning to be more appreciated. In this review, we describe some of the advances made for tissue-specific MPS and outline the advantages and challenges of applying and further developing MPS technology in preclinical biopharmaceutical research.
Introduction
Complex in vitro experimental platforms such as tridimensional (3D) cultures and microphysiological systems have been largely developed and tested using small molecule pharmaceuticals with most applications in the field of toxicology. However, there is limited evidence corroborating applicability of these technologies to support the development of biopharmaceuticals. Yet, MPS present advanced features compared to classical monolayer cell-based assays that may enable more precise interpretation of preclinical efficacy testing, disease modeling, and safety assessment. Here we describe potentials, opportunities, and challenges of MPS technology for use in biopharmaceutical development.The terms MPS and biopharmaceuticals may be ambiguous, and for the purposes of this review will be defined as described below. In order for an in vitro system to qualify as an MPS, it must go beyond traditional 2D sandwich culture or monolayers and could include several of the following design aspects: having a multi-cellular environment within biopolymer or tissue-derived matrix, a 3D structure, mechanical factors such as stretch or perfusion (e.g. breathing, gut peristalsis, flow, etc.), incorporating primary or stem cell-derived cells, and/or inclusion of immune system components. Additionally, several MPS systems incorporate vascular components and microfluidic components that mimic natural tissue perfusion with dynamic provision and removal of nutrients, hormones, and cytokines. Biopharmaceuticals are non-chemically synthesized entities of biological origin or their synthesized replicas (e.g. peptides). Biopharmaceuticals can include a wide range of products such as recombinant therapeutic proteins and peptides, blood components, viruses and viral vectors, somatic cells, bacterial delivery systems, and to a certain extent liposomes and nanoparticles as non-viral carriers of therapeutic agents.1 From a safety perspective, biopharmaceuticals differ from small molecules in the sense that they are highly selective entities, have a much longer half-life, and may interact with the tissue microenvironment through multiple domains (complementarity determining regions, constant regions, and complex tertiary structures). The de-risking strategy is focused on identifying target distribution at sites different from the diseased organs or tissues, and to predict consequences of the adverse events and/or exaggerated pharmacology. Additionally, because of their biological origin, these products have the potential to be immunogenic, thus presenting the risk for the patient to display acute or delayed moderate to severe immune and autoimmune reactions.2
To gain the most comprehensive understanding of risks associated with the biopharmaceuticals under investigation, nonclinical safety studies are generally conducted in rodent and/or large animal species, the latter often being non-human primates due to the closer physiology to humans and/or lack of cross-reactivity to other species.3 Nonetheless, there are reported cases of severe clinical adverse effects related to exaggerated pharmacology4 or immunogenicity5 that were not observed in nonclinical investigations. Moreover, there is an increasing number of cases where the safety de-risking of a biopharmaceutical is mostly, if not solely, based on an in vitro assessment.6
Fig. 1 provides a theoretical perspective on the relative translational predictability of technologies currently used for biopharmaceuticals where the distance from each sphere to that of the individual patient (coordinates x = 1.0, y = 1.0, z = 1.0), reflects its relative predictive value. As the figure is hypothetical these distances should be interpreted as qualitative only, and not quantitative. Note that in the age of precision medicine, clinical studies may also error in predicting treatment response or toxicity of the individual patient. The graph is also built on the assumption that the best models retain cellular phenotypes (protein expression profile and physiology), tissue microenvironment, and tissues interact as if they would in the living patient. Animal models are invaluable and remain superior to MPS especially where whole-body assessments are required as in biodistribution studies and toxicity testing. For questions which are less affected by multi-tissue interactions (represented by z-scale), such as some mechanistic investigations within a tissue, the z-scale becomes less relevant and the relative predictability (closeness to the x = 1.0, y = 1.0, z = 1.0 coordinates) of MPS may even surpass that of the animal model.
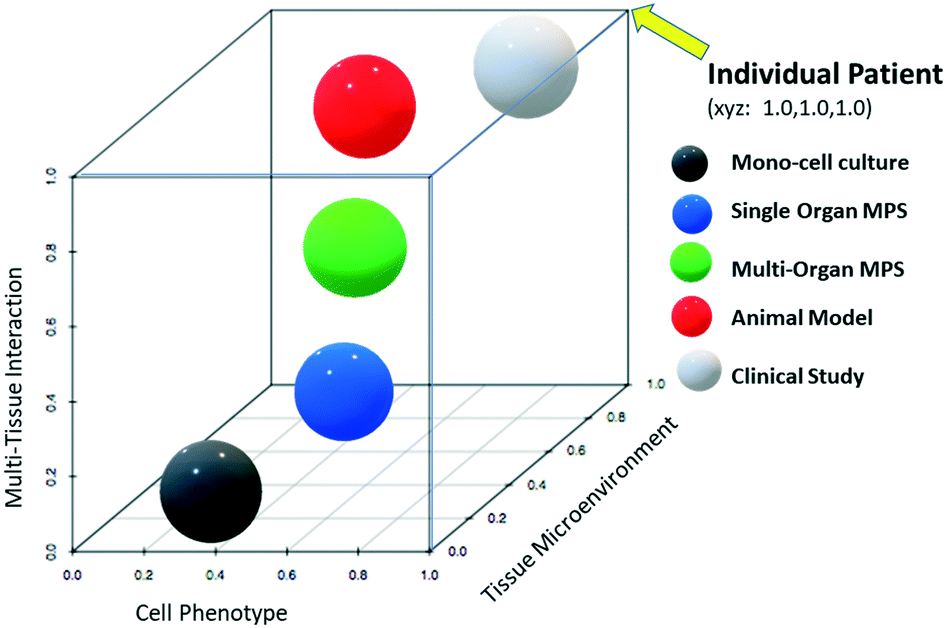 | ||
| Fig. 1 Theoretical relative translational predictability plot of technologies – categorical tools used to predict efficacy and safety of biopharmaceuticals are plotted on hypothetical scales to depict their relative strength in predicting outcomes. The closer they are to x = 1.0, y = 1.0, z = 1.0 the more predictable the technology. Testing the individual patient would plot to a perfect predictability. Human in vitro cell mono-cultures (black) maintain some molecular and gene expression characteristics (cell phenotype) of their natural environment. Human cell microphysiological systems (MPS, blue) provide a mixed cellular microenvironment that more closely resembles that found in the living human body which typically results in each cell type more closely retaining its natural phenotype. Although interconnected MPS platforms are being developed, they are far from being able to replicate natural systems. Animal models (red) are much better suited to replicate the tissue microenvironment of a cell and tissue interactions within the human body compared to the in vitro systems. However, differences in animal-human physiology and protein expression may cause disparities in translatability. Heterogeneity among human populations also causes clinical trial study (white) variations. | ||
MPS technology presents opportunities for biopharmaceutical development because of its ultimate goal to emulate the structure–function relationship of different cellular components within an organ, and the possibility to create a dynamic system where multiple organs are connected and properly reconstructed in scale.7
MPS is usually built by incorporating human primary cells isolated from adult organoids or derived from induced pluripotent stem cells (iPSC) which provide better predictability in testing species-specific targets when compared to traditional 2D cell cultures that exhibit an immortalized or transformed phenotype.8–10 Additionally, the presence of intact microenvironments and the integration of a functional vasculature allows testing of cell-to-cell or surface interactions, and replicates barriers inhibiting tissue and cell penetrance or exposure.11 Finally, recent efforts to incorporate tissue-associated immune components including draining lymph nodes, have promising potential for immunogenicity testing and efficacy and safety profiling of immune modulatory drugs.12
The status of the MPS technology is currently mostly suited to small molecule testing. A unique qualification for MPS in biopharmaceutical testing is the structure–function relationship of the system itself that effectively maintains normal gene expression profiles including targeted receptors, and enables formation of functional junctions ensuring realistic barrier properties. A main technical challenge is represented by the integration of imaging capabilities that would allow live monitoring of distribution patterns of labelled drug surrogates. While MPS applicability to biopharmaceuticals is a less explored territory, significant advancements have been achieved in the recent years. In the following sections, we will discuss specific examples of single and multi-organ MPS being developed for potential use in biopharmaceutical development.
Sensitivity differences between 2D and 3D co-culture systems
Before delving into MPS applications, it is important to understand the value and challenges of investing time and effort in these extra layers of complexity over more conventional methods. As mentioned above, 3D co-culture models/MPS platforms are expected to retain structural and functional characteristics including cell-to-cell interactions that mimic in vivo tissue/organ conditions. In addition, inclusion of mechanical stimuli such as media flow in 3D MPS models make them more physiologically relevant with enhanced potential for pre-clinical to clinical translatability compared to traditional 2D culture systems. However, for pharmaceutical applications, to the best of our knowledge, there are no such comprehensive comparative data available to evaluate sensitivity and translatability differences of 3D/MPS models compared to standard 2D cultures for different tissues/organ systems.The majority of studies using 3D models/MPS have focused on examining small molecules for toxicity or efficacy. Most of the available data from these studies suggest relatively simple standard 2D cultures are more sensitive to small molecule induced cytotoxicity endpoints compared to complex 3D co-culture models.13–18 Some of the potential reasons underlying this observation could be attributed to differences in exposure and/or diffusion of test item across cell types and extracellular material. Additionally, factors such as cell proliferation rate and expression of key receptors, transporters and/or signalling molecules involved in uptake and/or toxicity between 2D vs. 3D co-culture systems are also relevant. However, for functional injury readouts (biomarker endpoints), 3D models are expected to be more sensitive and predictive compared to 2D models with better correlation of organ-specific small molecule toxicants to in vivo results.13 It is important to note that sensitivity of complex in vitro models may also depend on long term stability of structural and functional characteristics over a period of time. For example, in contrast to acute studies, 3D models are reported to be more sensitive than 2D models in nephrotoxicity13,19 and hepatic toxicity20 studies under long term exposure conditions.
With regard to biopharmaceuticals, sensitivity of in vitro models primarily depends on multiple key functional characteristics affecting molecule distribution and exposure such as internalization kinetics (receptor-mediated or non-specific endocytosis), intracellular trafficking, recycling/transcytosis, and endosomal or lysosomal catabolism. To better understand the challenges and limitations of in vitro systems, comparison of the sensitivities of 2D cultures and 3D/MPS models for different biologic agents under acute and chronic exposures is needed. Comparison of key structural and functional characteristics (summarized in Table 1) across models would also better define appropriate applications of 3D/MPS models for pharmaceutical screenings.
| Structural | • Ratio of individual cell type in co-culture system |
| • Proper orientation and polarity of cells | |
| • Physicochemical properties of extracellular matrix material (including charge and chemical composition) | |
| • Presence of cell–cell interactions and immune component wherever necessary | |
| Functional | • Level of differentiation and functional maturation of cells with organ/tissue specific key receptors and signaling pathways |
| • Expression of key receptors involved in internalization, recycling and transcytosis (Eg., FcRn, FcgRs, megalin, cubilin etc.) | |
| • Cell proliferation rate and potential for regenerative/recovery response | |
| • Functional stability of cells over a study/assay period | |
| • Rate of receptor dependent and independent (non-specific) endocytosis/internalization, recycling and catabolism of proteins and IgG based therapeutics |
The blood–brain barrier and the brain
The importance of maintenance of 3D architecture described above is most evident in studies of the blood–brain barrier (BBB). The BBB is part of a larger structure, the neurovascular unit (NVU), consisting of endothelial cells, pericytes, astrocytes, microglia, neurons, and associated extracellular matrix proteins. It serves as a physical and functional barrier partitioning blood and neural tissues to maintain proper homeostasis in the brain. IgG antibodies are one such component that has restricted entry upon systemic administration21 and any breaches pose the risk of potential neurotoxicity. Different central nervous system toxicities ranging from transiently severe to lethal have been reported for complex biopharmaceuticals like T-cell engagers,22 antibody-drug conjugates,23 and CAR T-cells.24 Additionally, selection or engineering of therapeutic antibodies to enhance their ability to penetrate the BBB requires careful assessment of possible neurotoxicities.25,26 The complexity of the NVU renders standard in vitro assays inadequate to capture the dynamic interaction of multiple involved cell types, while in vivo experimentation is limited by species variations in expression profiles of transporter proteins,27 and by the limited reproducibility of the human pathophysiology by genetically modified animals.28,29 For these reasons the MPS technology represents a promising adjunct or alternative for biopharmaceutical testing.BBB MPS commonly use polydimethylsiloxane scaffolds separated by a porous polycarbonate membrane, with poly-D-lysine and fibronectin coating in the neural and vascular compartments, respectively. The model's functionality is tested by measuring transendothelial electrical resistance (TEER), permeability, and examining the presence of tight junctions.30 To date, pharmaceuticals have been used to help demonstrate correlation with, and predictability to, in vivo model findings and clinical observations.
In a vascular permeability study, Achyuta et al. cultured freshly isolated E-18 rat cortical cells and RBE4 rat brain endothelial cell line under static conditions, and used TNF-alpha treatment to assess leakage of dextran under inflammatory conditions.31 In modified design of the above, the brain compartment contained only media and ex vivo isolated human neutrophils were employed to study transmigration across the vascular compartment using IL-8 as a chemoattractant, TNF-alpha to induce inflammation, or oxygen and glucose depletion followed by reoxygenation to mimic ischemic conditions.32 An improvement towards a more translationally relevant physiological model was represented by the introduction of a human cerebral microvascular endothelial cell line (hCMEC/D3) with a fluidic component to model the hemodynamic shear stress effect.33
Further advancements in the BBB MPS concept focused on integrating additional cellular components and improving fluid dynamics. Brown et al. cultured primary human brain-derived microvascular endothelial cells (hBMVEC), human pericytes and astrocytes, and human iPSC-derived neurons under a constant shear flow, and used glutamate treatment to evaluate functionality of disrupted tight junctions.34 Herland et al. used hBMVEC cells and either primary human brain pericytes or astrocytes to measure G-CSF and IL-6 secretion profiles in response to TNF-alpha to identify distinct contributions of these supporting cells to the neuroinflammatory response.35 As advancements with BBB MPS models continue, strategies can be adapted to explore the utility of biopharmaceutical inflammatory-mediator antagonists as an effective way to reduce neuroinflammation.
The most advanced model of brain MPS is a modular structure consisting of three chips, where the brain chip is sandwiched between two BBB chips composed of hBMVECs. The brain compartment contains primary brain microvascular pericytes, astrocytes, glial cells, and glutamatergic, GABAergic, dopaminergic and serotonergic neurons. In this coupled NVU system shear stress is adjusted to be close-to-zero in the brain compartment to allow modeling influx from the first BBB chip to the brain parenchyma, and efflux into the second BBB chip. The ability to fine-tune this system closely resembles the in vivo brain model where diffusion-mediated molecular transport is dominant.36 These physiological and hydrodynamic forces will likely play an important role in the assessment of efficacy and toxicity of large (aberrant) molecules engineered to enter the brain parenchyma. The same group recently developed an advanced BBB model that shows reversible permeability in response to osmotic changes, and better recapitulates in vivo transcytosis of peptides and antibodies. Higher MPS functionality was achieved by generating differentiating hBMVECs from iPSC incubated under hypoxic condition, a protocol that mimics physiological conditions occurring during embryonic brain development.37
Intestine
Creating MPS platforms of the intestinal tract is particularly challenging owing to the diversity of cell types composing the organ, functional dependence upon structural orientation, regional functional differences, and exposure to dynamic forces of flow from digesta and peristalsis. With respect to potential use of MPS disease models for biotherapeutic development, inflammatory bowel disease (IBD) garners the greatest interest.38 The use of mixed enterocyte-macrophage 3D culture system is described below in the immune system section. In their work, Kim et al.39 developed a system that not only included enterocytes and peripheral blood mononuclear cells (PBMC), but was also adaptable to addition of probiotic or pathogenic bacteria and could be exposed to cyclic mechanical deformations mimicking peristalsis. Cell injury mirroring IBD condition was induced with a cocktail of cytokines containing IL-1beta, IL-6, TNF-alpha, and IL-8 were added to the system in the presence of PBMCs. Importantly, the villus disruption caused by exposure to the cytokine cocktail was prevented by the addition of a blocking monoclonal antibody (mAb) against IL-8.39As biopharmaceutical companies are also exploring the potential for manipulating the intestinal microbiome (i.e. fecal transplant) as novel treatment option, models such as those produced in Kim's laboratory offer the ability to readily manipulate the system in a more controlled environment. They showed that the addition of an eight-strain probiotic to their microfluidic system resulted in a gene expression profile that more closely represented that of the human ileum compared to the system without the probiotic bacteria. Additionally, the presence of pathogenic enteroinvasive E. coli (EIEC) in the lumen of the epithelial channel resulted in intestinal barrier dysfunction and villus architecture disruption compared to presence of nonpathogenic E. coli.39 Even if not tested with biopharmaceuticals, the same platform was adapted to monitor oxygen gradients in real-time, and to co-culture intestinal cells with aerobic and anaerobic human gut microbiota40 Further development and optimization of the intestinal MPS platforms which allow to readily alter the microflora and nutrients, and to monitor cytokine profiles in a controlled setting will likely result in novel insights for intestinal and inflammatory disease treatment.
Liver
The liver plays a major role in the clearance and metabolism of small molecule pharmaceuticals and biopharmaceuticals. Liver endothelial sinusoidal cells and Kupffer cells have an interactive role in antibody and lipid nanoparticle retention, respectively and metabolism, and are important components in any examined 3D/MPS model.41–43 The filter function of the liver is challenging to replicate using in vitro systems; however, attempts at approximating the natural state are commonly made by separating the parenchymal layer from a media stream/flow with an endothelial layer and membrane support.44 In addition to the biomarkers that are of interest to a specific study, cytochrome P450 expression, albumin, and urea are typically monitored to assess changes in the cell status of the liver MPS.45Liver cell co-cultures and 3D microfluidic systems have been used to elucidate the mechanisms of the accelerated clearance of simvastatin (cholesterol lowering drug) observed with concomitant treatment with anti-IL6 antibody (tocilizumab) in rheumatoid arthritis patients.46 Work by Long et al. demonstrated that cytochrome P450 3A4 isoform (CYP3A4) activity was suppressed when IL-6 was added to a microfluidic 3D hepatocyte-Kupffer cell co-culture system (LiverChip) at physiologic concentrations similar to that found in patients.47 However, as observed clinically,46 when an anti-IL6 antibody was added to the in vitro system, CYP3A4 activity was desuppressed. Additional experiments by the group revealed that the resulting increased CYP3A4 activity reduced the half-life ratio of simvastatin, a sensitive CYP3A4 substrate, when added to the system.47
The hepatic response to sepsis was mimicked in another microfluidic platform, “MOTiF” (multi organ tissue flow biochip). By combining hepatocytes and endothelial cells with and without primary monocytes in this system, Groger et al. analyzed the response of these cells to lipopolysaccharide and Toll-like receptor agonists. In the absence of monocytes, addition of the agonists resulted in decreased protein expression of ApoB, MRP2, and albumin in the hepatocytes and decreased expression of VE cadherin, zona occludens, ICAM, and VCAM on the endothelial cells. Addition of monocytes to the flowing media on the endothelial side of the platform abrogated these responses. These observations are similar to those seen in vivo where monocytes respond to sepsis in the liver and reduce tissue damaging inflammation.44
Advances in MPS models of disease to support efficacy and safety assessment is evolving and may have a pronounced impact on biopharmaceutical development. For example, hepatocytes have superior Hepatitis B Virus infectivity in a liver MPS when compared to spheroid models.48 In the non-alcoholic fatty liver disease arena, both liver-only49 and gut-liver50 MPS models were recently developed that successfully recapitulate clinical features of steatosis. Among other advantages these models will allow testing repeated exposure to drug.
These examples illustrate the capacity for hepatic MPS models to reflect in vivo observations and demonstrate the importance of co-cultured cells in more closely replicating physiologic responses to biopharmaceuticals. As these systems demonstrate a quantifiable, functioning, and interacting immune component, they offer the opportunity for exploring effects of immunomodulatory drugs and biopharmaceuticals on the liver.
Cardiovascular
One of the primary challenges for many large molecule therapeutics is that in order to be effective (or toxic), the biotherapeutic must cross the vascular barrier and penetrate into the heart tissue. If this paradigm is to be mimicked in vitro, the tissue should contain intact vessels with media/blood flow and have permeability properties similar to that of the organ in vivo. The latter is typically assessed by measurements of TEER or leakiness of a fluorescent-labeled blood pooling agent.51 As with other tissues, the molecular phenotype of the vessels should also reflect that of the tissues in situ. This may be challenging, as the molecular and physiologic characteristics of endothelial cells differ dependent upon which tissues they support and the endothelial tissue-specific molecular biomarkers may not be known.52 Exposure of the endothelial cells to flow of media may be important in maintaining the cell phenotype and to mimic natural physiology and sheer forces.53 This is especially pertinent when analyzing the ability of antibodies, nanoparticles, or cellular mediators to localize at, or cross the endothelial barrier. Studies may be designed to investigate vessel receptor targeting, toxicity, leakiness, biopharmaceuticals penetrance across the endothelial border or cellular interactions.In an example of translational application of MPS, Barrile et al. developed an in vitro endothelial vessel model that was perfused with human whole blood to mimic the clotting reaction observed in clinical findings of thromboembolic events that occurred in lupus patients being treated with huCD40L. Once the system was developed, it was used in a proof of concept study to demonstrate that a second generation huCD40L lacking the FcγRIIa did not cause clot formation.54 In order to model arterial stenosis in vitro, Korin et al. developed a microfluidic system in which the endothelial lined channel narrowed. Synthetically produced fibrin clots were added to the media and clotted in the vicinity of the constricted channel. The group then went on to demonstrate that when sheer-activated nanoparticles were added to the in vitro disease model, they adhered to the formed clot and were able to deliver tissue plasminogen activators that dissolved the clot.55 These two examples demonstrate how MPS models can be of value in candidate selection to minimize risk and in conducting proof of concept studies for biotherapeutic development.
As previously mentioned vasculature characteristics vary according to their supported tissue type and this holds true in tumors. Various 3D tumor tissue-endothelial vessel models have also been developed to investigate tumor endothelial specific targeting56 or to assess the potential of nanoparticles to traverse them.57 In a biometric microfluidic tumor microenvironment culture system, Tang et al. compared vessel leakiness in tumors. Using large liposomal drug carriers and fluorescence microscopy, they found that endothelial junctions were leakier in the presence of metastatic breast cancer cells or their conditioned media when compared to co-cultures with their non-metastatic counterparts.58 Understanding the characteristics of the vascular system, its function, and permeability within each of the tissues that it supports may be critical in accurately mimicking the ability, or inability, of a test biopharmaceutical to reach its targeted destination.
Lungs
Immunologically the lung is a major battlefield at the interface between allergens, infectious agents, toxicants and the immune system. As such, cytokines produced during these inflammatory wars can be both beneficial and harmful. Tissue damaging or disease intervention strategies focus largely on regulating the concentration of these cytokines in their microenvironment through antibody-binding of immune modulating proteins or cell receptors.59,60 In this respect, for a lung MPS to be effective in biotherapeutic development, it must emulate the immunologic milieu and responsiveness of its natural in vivo setting. One such example is in the utilization of a porous silicon-based fluidic microchip system designed by Punde et al. to mimic inflammatory conditions in human airways. Using this model, the migration of fibrocytes responding to bronchial cells stimulated with eosinophilic cationic protein was significantly decreased by the addition of anti-CXCR4 antibody.61Infectious disease modeling can also be performed in 3D cultures and MPS. When Staphylococcus aureus was added to human-derived tracheal/bronchial epithelial 3D cell culture, hematoxylin–eosin staining and assessment of lactate dehydrogenase levels revealed that the tissue was completely destroyed. However, this cytotoxic effect was inhibited by the addition of antibodies targeting the leukocidins produced by bacteria.62 The cytotoxic effect of these leukocidins on leukocytes was further demonstrated by transferring the bacteria-free expended media from the infected 3D cultures to human polymorphonuclear (PMN) cells. PMN cell death was inhibited when the media transfer was performed in the presence of the anti-leukocidin antibodies.63 More complex and dynamic lung MPS which can incorporate bacterial infection have been developed and may be adapted to similar studies.64
Kidneys
Proper in vitro reconstruction of renal architecture and functionality is challenging due to thecomplex compartmentalized microstructureof renal. Some systems have shown potential value for predicting renal toxicity of small molecules,65,66 although the number of reports documenting application for large molecules is very low. Of relevance, a 3D organoid proximal tubule culture model was used to assess nanoparticle toxicity. When exposed to hydroxylated generation-5 polyamidoamine dendrimers (G5-OH) the in vitro system compared similarly to published preclinical in vivo rodent nephrotoxicity data. G5-OH exposure resulted in induction of kidney injury molecule-1 and release of inflammatory cytokines, but was not severe enough to result in decreased cell viability. The latter was equated to the lack of changes in blood urea nitrogen and creatinine in mouse models as reported in the literature.67 Gold nanoparticles were also tested in the renal tubular system, but quickly abandoned due to their inability to diffuse through the hyaluronic acid hydrogel matrix.68 This failure exemplifies the need to test the support matrices used in MPS for interfering properties such as nonspecific binding and viscosity incompatibilities that may confound results.In parallel, significant efforts have been made to reproduce the most complex functional unit of the kidney, the glomerulus, which plays a crucial role in plasma ultrafiltration and homeostasis. Structural and functional interactions of three different cell types (fenestrated endothelial cells, mesangial cells and highly specialized podocytes) along with secreted glomerular basement membrane (GBM) are critical for an accurate size and charge selective glomerular filtration barrier.69 In addition, a FcRn-dependent active transport mechanism is also active in podocytes to remove immunoglobulins and possibly albumin that accumulate at the filtration barrier/GBM.70 Hence standard 2D culture systems poorly mimic complex structural and functional characteristics of glomerulus as well its sensitivity to drugs.71,72 Also, glomerular cells are normally subject to mechanical forces, such as stretching and relaxation from pulsing blood flow73 which is missing in 2D culture systems. Therefore, 3D co-culture systems/MPS incorporating all three cell types with physiologically relevant flow are required to recapitulate human glomerular function under in vitro conditions. This may be even more relevant for pharmaceutical-like recombinant therapeutic proteins and IgG-based therapeutics, which have a propensity to get deposited in the glomerulus and initiate complement activation with inflammatory cascade leading to pathological changes in the kidney.74,75
However, currently there is no published information on validated and functional 3D/MPS platforms incorporating all the three cell types to mimic complex physiological structure and functions of glomeruli in vivo. Work by Zhou et al. reported development of a mouse glomerulus on a chip microfluidic device using a glomerular endothelial cell and podocyte co-culture system to model hypertensive nephropathy in humans.76 For the development of chip models of human glomerulus, a lack of availability of functional human podocytes (key component of glomerulus) was considered as major limitation. However, recent success in differentiation of podocytes from human induced pluripotent stem cells is expected to overcome this challenge.77 Functional characterization of these human podocytes including GBM collagen secretion and differential clearance of proteins (albumin and inulin) when co-cultured with glomerular endothelial cells (separated by laminin-coated membrane) in an organ on a chip microfluidic device has shown promise for this model. Further, this human glomerulus on a chip model has also been shown to reproduce Adriamycin-induced podocyte injury under a clinically relevant concentration and exposure route (continuous infusion through the endothelium-lined vascular channel).77 However, there are no reports of testing any biopharmaceuticals using this model. Even though currently available 3D/MPS glomerular models are promising and have been shown to perform much better than 2D models, they are still comparatively simple and lack other key structural and functional components of glomerulus such as mesangial cells and GBM. Also, when glomerulus 3D/MPS models incorporating all the key cellular components are available they need to be fully characterized (e.g. FcRn-mediated active transport in podocytes, physicochemical complexity of GBM, phagocytic activity of mesangial cells, intercellular signalling etc.) to demonstrate physiological relevance to in vivo conditions in uptake and clearance of biotherapeutics.
The immune system
The necessity of a dynamic 3D in vitro system to properly emulate the functionality of the immune system is defined by its anatomical organization. Lymphoid organs, circulating immune cells, tissue resident cells and tissue embedded lymphoid bodies are all participating in the process of pathogen recognition, attack and clearance. Additionally, several lymphoid organs are involved in the differentiation, maturation, and expansion of the various immune cell populations. The importance of recapitulating all these features for biotherapeutic discovery and safety testing is related to the fact that virtually all biotherapeutics are immunogenic and can trigger immune responses with different degrees of severity.78 Furthermore, increasing numbers of biopharmaceuticals are in development for cancer immunotherapy and require careful analysis of immune activation and immune-mediated destruction of non-target organs.79 Although testing using laboratory animals remains necessary and provides valuable information, limitations in their predictive value for human patients80 highlights the need for novel platforms such as MPS technology.In this space, efforts have focused on the emulation of immunocompetent non-lymphoid 3D organ equivalents. A static transwell-based 3D inflamed intestinal mucosa was developed utilizing intestinal epithelial Caco-2 cells with primary, blood-derived macrophages and dendritic cells as components of the intestinal innate immune system.81 Treatment with IL-1beta was used to induce onset of chronic inflammation,81 and the system was tested with small molecules nanoformulations82 with the ultimate aim to study interaction of xenobiotics and nanoparticles with the inflamed intestinal epithelial barrier.
A more advanced system is available for the lung where human alveolar epithelial cells and endothelial cells are seeded on the apical and basolateral surface of a porous membrane, respectively. Application of a cyclic strain allowed the generation of an air-liquid interface in a dynamic 3D alveolar model where human neutrophils could be introduced in the basolateral component through the respective microfluidic channel. This functional alveolar barrier was tested for exposure to bacteria, inflammatory cytokines and nanoparticles.64
MPS are also employed to study the interplay between immunity and disease, and in particular the impact of the immune system in cancer therapeutics. In this field, a different approach is used where instead of layering cellular components, tumor fragments with or without embedded naïve tumor-infiltrating lymphocytes are loaded into microchannels that allow perfusion with oxygenated media. These complex systems recapitulate more closely features of both the tumor and its microenvironment, and have been used to test small molecules, checkpoint inhibitors, TCR engineered T cells, and autologous T cells (ref. 82–86).
The first human artificial lymph node was developed as a membrane-based perfusion bioreactor system consisting of a matrix-assisted culture area to allow dendritic-T-cell crosstalk, lymphoid follicle self-assembly and antigen-induced activation, and a peripheral fluidic space to mimic lymph drainage.83 Adaptive immune responsiveness was initially tested by in vitro immunization to hepatitis A and cytomegalovirus infections,84 and further improved by integrating rat mesenchymal stromal cells into the respective rat 3D lymph node to mimic the role of these cells in immune modulation.85 This model is being expanded for immunogenicity and immunotoxicity testing in the human system.12,84 As immunocyte localization and function is heavily dependent upon the microenvironment architecture within the lymphatic system, their progenitors also require contextual interaction within the bone marrow. Advanced reconstruction of a 3D bone marrow niche is based on the employment of porous hydroxyapatite scaffolds where human stromal and hematopoietic cells are embedded in perfusion channels organized in alternating directions. Validation of this model was achieved by assessing the effect of hematopoietic growth factors on differentiation and homeostasis of both mature hematopoietic cells and early multipotent progenitors.86
Very recently, MPS that make use of pharmaceuticals as diagnostic tool instead of therapeutic drugs were developed to detect minimal residual disease in cancer patients. These microfluidic devices utilize antibodies immobilized within affinity-selected subpopulations of circulating plasma cells.87 MPS modified with antibodies targeting CD33, CD34, and CD117 cell surface antigens were employed to identify aberrant markers for recurrence of acute myeloid leukemia while microfluidic devices modified with anti-CD138 antibodies were used for cytogenetic and molecular characterization of patients with multiple myeloma or monoclonal gammopathies.88 In both instances, MPS performance showed comparable accuracy and sensitivity to standard bone marrow biopsy analysis thus representing potential for a novel, less invasive clinical diagnostic method for routine monitoring.
Immunotoxicology
A discussion of the immune system in the context of MPS would not be complete without addressing its value as a technology platform for studying and screening pharmaceuticals to de-risk adverse events. As mentioned previously, biopharmaceuticals carry a potential to induce the release of proinflammatory cytokines from circulating immune cells. This represents a relatively unique safety hazard for biopharmaceuticals that has received increased scrutiny following the 2006 TGN1412 incident; where a mAb targeting CD28 induced life-threatening “cytokine storm” during the first human clinical administration.89 This event led to the general acknowledgement that better predictive in vitro models were needed to understand the potential risk of exaggerated target engagement. As a result, a large variety of cytokine release assays (CRAs) have been developed by various groups—with each spanning different formats and context of use.Incubation of mAbs with donor whole blood was initially expected to provide the most physiological response. However, the whole blood format was ultimately deemed insensitive and incapable of generating the magnitude of cytokine response observed clinically for mAbs associated with severe “cytokine storm” (i.e. TGN1412).90,91 Subsequent in vitro models have revealed the importance of effective mAb presentation in recapitulating this cytokine response. Utilizing an aqueous phase mAb presentation to donor PBMCs increased signal strength over the whole blood format and decreased donor-to-donor variability however, the cytokine profile was inconsistent with that of the TGN1412 clinical response.92 In contrast, solid phase mAb presentation by artificial immobilization on polypropylene microtiter plates followed by incubation with donor PBMCs elicited increased cytokine signals by exaggerating target cross-linking and providing high-density polarized stimulation of immune cells. While this solid phase assay format recapitulates the clinical TGN1412 cytokine profile and appears sufficient for use in hazard identification, it has come under scrutiny for its lack of a physiological corollary in regards to mAb presentation. Due to the requirements for test article immobilization in the model, there exists the potential for alteration of the biologic structural features and exposure of epitopes not present under normal physiological conditions. This phenomenon may explain the higher false positive rate observed in the immobilized assay format and the decreased ability to distinguish between those mAbs known to cause severe “cytokine storm” from those associated with much milder clinical responses.92
Adopting a more physiologically relevant approach, aqueous phase mAb incubation in a co-culture of PBMCs over endothelium-derived cells generates a model system that better mimics both the in vivo vascular microenvironment as well as elicits the appropriate cytokine response profile for TGN1412. Furthermore, this format minimizes artificial manipulation (i.e. immobilization) of the test article.4,92 Use of this PBMC/endothelial cell co-culture assay format also provides promise in the ability to differentiate between mAbs associated with severe “cytokine storm” compared to those with only mild cytokine release observed in the clinic.92 Such capabilities suggest this assay format could progress beyond simple hazard identification and better serve to translate in vitro dose-responses to clinical risk, allow elucidation of mechanisms of action and even inform selection of first-in-human dose levels. Further characterization of the endothelial-immune cell interface in these assays has revealed that maximizing predictive value will likely entail the continued optimization of the physiological relevance of the component factors. For example, assay performance is improved by the use of human AB serum over traditional fetal calf serum.93 Additionally, primary human umbilical vein endothelial cells have been shown to generate more responsive co-culture systems as compared to use of endothelial-derived cell lines.92 One report has shown that use of cell components (PBMCs and endothelial cells) from different donors results in a heterologous system that can elicit major histocompatibility complex reactions, whereas use of blood outgrowth endothelial cells and PBMCs from the same donor generates an autologous system with improved signal-to-noise detection of the cytokine release in response to mAbs.94
Furthermore, as these CRA models lend themselves to use in more mechanistic investigations it is necessary to characterize additional facets of the model system and incorporate endpoints adapted to the biopharmaceutical being evaluated. For example, screening of IgG1 mAbs may necessitate careful assessment in donor cells of variable factors such as FcγR polymorphisms, aspects which are less relevant for testing the immune response to an IgG4 mAb. Similarly, immune cell proliferation may be an important supplemental endpoint when evaluating biopharmaceuticals targeting leukocytes and less so with those targeting tissue-expressed or soluble antigens. These concerns specific to the use of biopharmaceuticals extend beyond the context of CRA models and should be carefully considered in any MPS tissue model that incorporates immune cell populations or is otherwise immunocompetent.
Additionally, MPS models currently in use present limitations in predicting risk of immune-related toxicities, in particular immunogenicity. Single-organ MPS can capture toxicities in the tissue implanted on the chip while anti-drug antibodies (ADA) often cause toxicities at distant sites. Moreover, severity of ADA reactions is dependent on the route of administration, and this variability can be only partially assessed with support of modelling and simulation exercises. Finally, low throughput MPS models do not allow for efficient generation of the numbers of replicates needed to account for inter-individual variability in immunogenicity. Still, a new European Commission Internal Market Information System95 initiative was recently started to develop more predictive multi-organ MPS for efficacy and safety assessment of immune-oncology drugs, indicating that effort is ongoing to improve the MPS technology in this direction.
Conclusion/future direction
It is likely that we have just touched the tip of the iceberg in the number of examples where MPS have been applied to biopharmaceutical development. Many such studies cannot be currently disclosed due to intellectual property concerns. Furthermore, the number of potential applications that can be generated through adaptation of small molecule test systems and creative strategies using newly developed and refined systems is limitless. For a more in-depth discussion on these systems a series of compressive manuscripts focusing on an in-depth discussion of neurology, gastrointestinal, hepatic, renal and immune system MPS technologies are currently in preparation.In summarizing accomplishments thus far, a few underlying requirements for MPS use in the biopharmaceutical space have become apparent. 1) The choice of matrix used to support cells is not only important for growth, orientation and the retention of the cellular phenotypes, but should also allow for normal diffusion of the test article without artificially interacting with it. To this effect, consideration must also be given to the materials used to construct the devices and their interface with the tissue components of the chip. Nonspecific binding of biopharmaceuticals to the devices and/or support matrices could result in reduction of available test article and possible underestimation of exaggerated pharmacology/toxicology.96 The issue of non-specific binding to PDMS, a widely used microfluidic device material is often highlighted for hydrophobic and high log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D compounds (lipophilic) small molecules.95 No published data is available investigating binding of biopharmaceuticals to PDMS or other chip materials. Similar to small molecules, surface charge or Isoelectric point or overall hydrophobicity of biopharmaceuticals may contribute to nonspecific binding and retention in chip material, hence it is critical to test tool biopharmaceuticals with varying surface modifications to understand extent of non-specific binding to microfluidic models. 2) The blood vessel wall is a major barrier for entry of large molecule therapeutics into the tissue parenchyma or tumors. Blood flow, shear stress, and pressure also influence exposure of the target to the biotherapeutics. MPS which incorporate blood vessel-like structures with a well-formed and functionally characterized endothelium and microfluidic components within the tissues being studied are more likely to be better models compared to systems lacking these features. Optimized matrix and integration of vasculature would additionally avoid diluting and washing away trophic factors, autocrine and paracrine signaling molecules as observed with current perfusion methods. 3) Numerous biopharmaceuticals being developed are immunomodulatory, and virtually all the others can be functionally influenced by the formation of anti-drug antibodies, receptor (Fc) mediated immune responses, cytokine release, and metabolism by resident and transient white blood cells. Major advances in the application of MPS platforms for biopharmaceutical development will depend upon incorporation of the functioning and responsive immune cells within the tissues being analyzed. 4) Large molecule biopharmaceuticals have the advantage that they can be relatively easily labeled with fluorescent tags and tracked. Optical imaging systems that are compatible with the MPS are likely to be very useful, if not essential, for these studies. 5) As complex 3D and MPS systems often involves co-culturing of multiple primary cells, donor matching and reliability of cell sourcing for each cell type will be a key factor to determine immune compatibility and overall predictability of the model. Use of primary cells from different donors may contribute for significant variation in level of intrinsic metabolic activity as well as key receptors associated with uptake or recycling of biopharmaceuticals. Therefore, it is recommended to use reliable donor matched cell source for all the primary cell types. However due to limited donor availability, it is practically very challenging to derive all cells types from a single donor. Use of well characterized fully differentiated iPSC derived cells from a single donor (autologous source) in complex MPS system may be helpful to overcome this challenge. 6) Lastly, MPS, and in particular multi-organ on a chip, devices are mainly low-throughput, non-standard disposables that typically include up to three organs without replicates. Development of throughput-scalable format with standard assay workflow and multiplexed reads is a necessary step to allow routine application of this technology. Currently, most advanced systems allow parallel testing in 40 independent modules (three-channels;97 or up to 1600 chambers (cell culture array; ref. 98) Estimating the scale ideally required for a high-throughput MPS to achieve tangible results is challenged by many variables such as the biopharmaceuticals mode of action (need for single- vs. multi-organ MPS) and the scope of testing (reduction or replacement of in vivo studies in one or two toxicology species). In this regard, recent innovations beyond efficacy and safety testing have been implemented that enable applicability of MPS to biopharmaceutical manufacturing, i.e. scalability and batch-to-batch variability of nanotherapeutics,99 or cell separation by acoustophoresis100 and nucleic acid delivery in a microfluidic three-layer device101 during CAR T-cells production.
D compounds (lipophilic) small molecules.95 No published data is available investigating binding of biopharmaceuticals to PDMS or other chip materials. Similar to small molecules, surface charge or Isoelectric point or overall hydrophobicity of biopharmaceuticals may contribute to nonspecific binding and retention in chip material, hence it is critical to test tool biopharmaceuticals with varying surface modifications to understand extent of non-specific binding to microfluidic models. 2) The blood vessel wall is a major barrier for entry of large molecule therapeutics into the tissue parenchyma or tumors. Blood flow, shear stress, and pressure also influence exposure of the target to the biotherapeutics. MPS which incorporate blood vessel-like structures with a well-formed and functionally characterized endothelium and microfluidic components within the tissues being studied are more likely to be better models compared to systems lacking these features. Optimized matrix and integration of vasculature would additionally avoid diluting and washing away trophic factors, autocrine and paracrine signaling molecules as observed with current perfusion methods. 3) Numerous biopharmaceuticals being developed are immunomodulatory, and virtually all the others can be functionally influenced by the formation of anti-drug antibodies, receptor (Fc) mediated immune responses, cytokine release, and metabolism by resident and transient white blood cells. Major advances in the application of MPS platforms for biopharmaceutical development will depend upon incorporation of the functioning and responsive immune cells within the tissues being analyzed. 4) Large molecule biopharmaceuticals have the advantage that they can be relatively easily labeled with fluorescent tags and tracked. Optical imaging systems that are compatible with the MPS are likely to be very useful, if not essential, for these studies. 5) As complex 3D and MPS systems often involves co-culturing of multiple primary cells, donor matching and reliability of cell sourcing for each cell type will be a key factor to determine immune compatibility and overall predictability of the model. Use of primary cells from different donors may contribute for significant variation in level of intrinsic metabolic activity as well as key receptors associated with uptake or recycling of biopharmaceuticals. Therefore, it is recommended to use reliable donor matched cell source for all the primary cell types. However due to limited donor availability, it is practically very challenging to derive all cells types from a single donor. Use of well characterized fully differentiated iPSC derived cells from a single donor (autologous source) in complex MPS system may be helpful to overcome this challenge. 6) Lastly, MPS, and in particular multi-organ on a chip, devices are mainly low-throughput, non-standard disposables that typically include up to three organs without replicates. Development of throughput-scalable format with standard assay workflow and multiplexed reads is a necessary step to allow routine application of this technology. Currently, most advanced systems allow parallel testing in 40 independent modules (three-channels;97 or up to 1600 chambers (cell culture array; ref. 98) Estimating the scale ideally required for a high-throughput MPS to achieve tangible results is challenged by many variables such as the biopharmaceuticals mode of action (need for single- vs. multi-organ MPS) and the scope of testing (reduction or replacement of in vivo studies in one or two toxicology species). In this regard, recent innovations beyond efficacy and safety testing have been implemented that enable applicability of MPS to biopharmaceutical manufacturing, i.e. scalability and batch-to-batch variability of nanotherapeutics,99 or cell separation by acoustophoresis100 and nucleic acid delivery in a microfluidic three-layer device101 during CAR T-cells production.
Since its inception, the biopharmaceutical industry has struggled with finding the best models for preclinical efficacy and toxicity studies. The use of nonhuman primates as primary animal models for toxicity testing is sometimes limited by differences in affinity/cross reactivity and mechanistic differences across species. Utilization of MPS which are developed to support human cells/tissues may be invaluable in filling in some of the knowledge gaps left from animal and conventional in vitro studies. Moreover, the development of MPS devices using cells from preclinical species (e.g. primates, rats or dogs) may allow better predictability of cross-species functionality of druggable targets, and better support the toxicology species selection process. As the technology, standardization, and acceptance of MPS improves, it is likely that these systems will become commonplace in the laboratory model arsenal for biopharmaceutical development.
This review focused on identifying areas where MPS models can offer upside potential in the efficacy and safety assessment of biotherapeutics. However, not all safety concerns can be addressed with the MPS technology and, although advanced in vitro approaches can supplement and, in some cases, possibly replace the in vivo studies, the latter are still necessary and relevant. On case-by-case basis the developer should carefully consider the use of MPS models to support, reduce or replace the use of animal studies.
Author contributions
Conception and design of the manuscript: NP, MDP. Drafting and revising of the manuscript: NP, PKM, AF, MDP. All authors have approved the final manuscript.Conflicts of interest
Authors are employed in the private pharmaceutical sector. There are no conflicts to declare.Acknowledgements
This manuscript was developed with the support of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ, http://www.iqconsortium.org). IQ is a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators and the broader research and development community. We thank the IQ MPS Affiliate reviewers for their constructive review of this manuscript.References
- R. A. Rader, Nat. Biotechnol., 2008, 26, 743–751 CrossRef CAS PubMed.
- N. Bhogal, Curr. Drug Saf., 2010, 5, 293–307 CrossRef CAS PubMed.
- J. L. Bussiere, P. Martin, M. Horner, J. Couch, M. Flaherty, L. Andrews, J. Beyer and C. Horvath, Internet J. Toxicol., 2009, 28, 230–253 CrossRef CAS.
- R. Stebbings, L. Findlay, C. Edwards, D. Eastwood, C. Bird, D. North, Y. Mistry, P. Dilger, E. Liefooghe, I. Cludts, B. Fox, G. Tarrant, J. Robinson, T. Meager, C. Dolman, S. J. Thorpe, A. Bristow, M. Wadhwa, R. Thorpe and S. Poole, J. Immunol., 2007, 179, 3325–3331 CrossRef CAS.
- E. Marshall, Science, 1999, 286, 2244–2245 CrossRef CAS.
- F. R. Brennan and A. Kiessling, Toxicol. In Vitro, 2017, 45, 296–308 CrossRef CAS PubMed.
- J. R. Coppeta, M. J. Mescher, B. C. Isenberg, A. J. Spencer, E. S. Kim, A. R. Lever, T. J. Mulhern, R. Prantil-Baun, J. C. Comolli and J. T. Borenstein, Lab Chip, 2016, 17, 134–144 RSC.
- F. Noor, J. Physiol., 2015, 593, 5043–5055 CrossRef CAS.
- D. Pamies, T. Hartung and H. T. Hogberg, Exp. Biol. Med., 2014, 239, 1096–1107 CrossRef.
- S. E. Park, A. Georgescu and D. Huh, Science, 2019, 364, 960–965 CrossRef CAS PubMed.
- A. Tourovskaia, M. Fauver, G. Kramer, S. Simonson and T. Neumann, Exp. Biol. Med., 2014, 239, 1264–1271 CrossRef PubMed.
- C. Giese and U. Marx, Adv. Drug Delivery Rev., 2014, 69-70, 103–122 CrossRef CAS PubMed.
- A. I. Astashkina, B. K. Mann, G. D. Prestwich and D. W. Grainger, Biomaterials, 2012, 33, 4712–4721 CrossRef CAS PubMed.
- E. J. Silva, N. K. Carvalho, C. T. Ronconi, G. De-Deus, M. L. Zuolo and A. A. Zaia, Braz. Dent. J., 2016, 27, 652–656 CrossRef.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS.
- R. G. Zhipan Wu, M. Tao, F. Lyu, G. Cao, M. Liua and J. Gaod, RSC Adv., 2017, 7, 12437–12445 RSC.
- V. Hongisto, S. Jernstrom, V. Fey, J. P. Mpindi, K. Kleivi Sahlberg, O. Kallioniemi and M. Perala, PLoS One, 2013, 8, e77232 CrossRef CAS.
- A. G. Souza, I. B. B. Silva, E. Campos-Fernandez, L. S. Barcelos, J. B. Souza, K. Marangoni, L. R. Goulart and V. Alonso-Goulart, Curr. Pharm. Des., 2018, 24, 1689–1694 CrossRef CAS PubMed.
- T. M. DesRochers, L. Suter, A. Roth and D. L. Kaplan, PLoS One, 2013, 8, e59219 CrossRef CAS PubMed.
- C. C. Bell, A. C. A. Dankers, V. M. Lauschke, R. Sison-Young, R. Jenkins, C. Rowe, C. E. Goldring, K. Park, S. L. Regan, T. Walker, C. Schofield, A. Baze, A. J. Foster, D. P. Williams, A. W. M. van de Ven, F. Jacobs, J. V. Houdt, T. Lahteenmaki, J. Snoeys, S. Juhila, L. Richert and M. Ingelman-Sundberg, Toxicol. Sci., 2018, 162, 655–666 CrossRef CAS PubMed.
- R. J. Boado, Q. H. Zhou, J. Z. Lu, E. K. Hui and W. M. Pardridge, Mol. Pharmaceutics, 2010, 7, 237–244 CrossRef CAS PubMed.
- A. C. Wilke and N. Gokbuget, Expert Opin. Drug Saf., 2017, 16, 1191–1202 CrossRef CAS.
- M. A. Fanale, A. Forero-Torres, J. D. Rosenblatt, R. H. Advani, A. R. Franklin, D. A. Kennedy, T. H. Han, E. L. Sievers and N. L. Bartlett, Clin. Cancer Res., 2012, 18, 248–255 CrossRef CAS.
- L. DeFrancesco, Nat. Biotechnol., 2017, 35, 6–7 CrossRef CAS PubMed.
- G. Thom, J. Hatcher, A. Hearn, J. Paterson, N. Rodrigo, A. Beljean, I. Gurrell and C. Webster, mAbs, 2018, 10, 304–314 CrossRef CAS PubMed.
- W. M. Pardridge, Expert Opin. Drug Delivery, 2015, 12, 207–222 CrossRef CAS PubMed.
- R. Shawahna, X. Decleves and J. M. Scherrmann, Curr. Drug Metab., 2013, 14, 120–136 CrossRef CAS PubMed.
- D. G. Hackam, BMJ, 2007, 334, 163–164 CrossRef.
- H. B. van der Worp, D. W. Howells, E. S. Sena, M. J. Porritt, S. Rewell, V. O'Collins and M. R. Macleod, PLoS Med., 2010, 7, e1000245 CrossRef PubMed.
- M. W. van der Helm, A. D. van der Meer, J. C. Eijkel, A. van den Berg and L. I. Segerink, Tissue Barriers, 2016, 4, e1142493 CrossRef PubMed.
- A. K. Achyuta, A. J. Conway, R. B. Crouse, E. C. Bannister, R. N. Lee, C. P. Katnik, A. A. Behensky, J. Cuevas and S. S. Sundaram, Lab Chip, 2013, 13, 542–553 RSC.
- H. Cho, J. H. Seo, K. H. Wong, Y. Terasaki, J. Park, K. Bong, K. Arai, E. H. Lo and D. Irimia, Sci. Rep., 2015, 5, 15222 CrossRef CAS PubMed.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS.
- J. A. Brown, V. Pensabene, D. A. Markov, V. Allwardt, M. D. Neely, M. Shi, C. M. Britt, O. S. Hoilett, Q. Yang, B. M. Brewer, P. C. Samson, L. J. McCawley, J. M. May, D. J. Webb, D. Li, A. B. Bowman, R. S. Reiserer and J. P. Wikswo, Biomicrofluidics, 2015, 9, 054124 CrossRef.
- A. Herland, A. D. van der Meer, E. A. FitzGerald, T. E. Park, J. J. Sleeboom and D. E. Ingber, PLoS One, 2016, 11, e0150360 CrossRef PubMed.
- B. M. Maoz, A. Herland, E. A. FitzGerald, T. Grevesse, C. Vidoudez, A. R. Pacheco, S. P. Sheehy, T. E. Park, S. Dauth, R. Mannix, N. Budnik, K. Shores, A. Cho, J. C. Nawroth, D. Segre, B. Budnik, D. E. Ingber and K. K. Parker, Nat. Biotechnol., 2018, 36, 865–874 CrossRef CAS.
- T. E. Park, N. Mustafaoglu, A. Herland, R. Hasselkus, R. Mannix, E. A. FitzGerald, R. Prantil-Baun, A. Watters, O. Henry, M. Benz, H. Sanchez, H. J. McCrea, L. C. Goumnerova, H. W. Song, S. P. Palecek, E. Shusta and D. E. Ingber, Nat. Commun., 2019, 10, 2621 CrossRef.
- A. Bein, W. Shin, S. Jalili-Firoozinezhad, M. H. Park, A. Sontheimer-Phelps, A. Tovaglieri, A. Chalkiadaki, H. J. Kim and D. E. Ingber, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 659–668 CrossRef.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7–E15 CrossRef CAS PubMed.
- S. Jalili-Firoozinezhad, F. S. Gazzaniga, E. L. Calamari, D. M. Camacho, C. W. Fadel, A. Bein, B. Swenor, B. Nestor, M. J. Cronce, A. Tovaglieri, O. Levy, K. E. Gregory, D. T. Breault, J. M. S. Cabral, D. L. Kasper, R. Novak and D. E. Ingber, Nat. Biomed. Eng., 2019, 3, 520–531 CrossRef CAS.
- A. Datta-Mannan, J. E. Croy, L. Schirtzinger, S. Torgerson, M. Breyer and V. J. Wroblewski, mAbs, 2016, 8, 969–982 CrossRef CAS.
- E. Endrerud, Size Matters: Liver Uptake, http://dliver.com/size-matters Search PubMed.
- B. Shi, E. Keough, A. Matter, K. Leander, S. Young, E. Carlini, A. B. Sachs, W. Tao, M. Abrams, B. Howell and L. Sepp-Lorenzino, J. Histochem. Cytochem., 2011, 59, 727–740 CrossRef CAS.
- M. Groger, K. Rennert, B. Giszas, E. Weiss, J. Dinger, H. Funke, M. Kiehntopf, F. T. Peters, A. Lupp, M. Bauer, R. A. Claus, O. Huber and A. S. Mosig, Sci. Rep., 2016, 6, 21868 CrossRef.
- M. Ortega-Ribera, A. Fernandez-Iglesias, X. Illa, A. Moya, V. Molina, R. Maeso-Diaz, C. Fondevila, C. Peralta, J. Bosch, R. Villa and J. Gracia-Sancho, Biotechnol. Bioeng., 2018, 115, 2585–2594 CrossRef CAS.
- C. Schmitt, B. Kuhn, X. Zhang, A. J. Kivitz and S. Grange, Clin. Pharmacol. Ther., 2011, 89, 735–740 CrossRef CAS.
- T. J. Long, P. A. Cosgrove, R. T. Dunn, 2nd, D. B. Stolz, H. Hamadeh, C. Afshari, H. McBride and L. G. Griffith, Drug Metab. Dispos., 2016, 44, 1940–1948 CrossRef CAS.
- A. M. Ortega-Prieto, J. K. Skelton, S. N. Wai, E. Large, M. Lussignol, G. Vizcay-Barrena, D. Hughes, R. A. Fleck, M. Thursz, M. T. Catanese and M. Dorner, Nat. Commun., 2018, 9, 682 CrossRef CAS.
- T. Kostrzewski, T. Cornforth, S. A. Snow, L. Ouro-Gnao, C. Rowe, E. M. Large and D. J. Hughes, World J. Gastroenterol., 2017, 23, 204–215 CrossRef.
- S. Y. Lee and J. H. Sung, Biotechnol. Bioeng., 2018, 115, 2817–2827 CrossRef CAS.
- C. M. Sakolish, M. B. Esch, J. J. Hickman, M. L. Shuler and G. J. Mahler, EBioMedicine, 2016, 5, 30–39 CrossRef PubMed.
- H. G. Augustin and G. Y. Koh, Science, 2017, 357, eaal2379 CrossRef PubMed.
- K. Hattori, Y. Munehira, H. Kobayashi, T. Satoh, S. Sugiura and T. Kanamori, J. Biosci. Bioeng., 2014, 118, 327–332 CrossRef CAS PubMed.
- R. Barrile, A. D. van der Meer, H. Park, J. P. Fraser, D. Simic, F. Teng, D. Conegliano, J. Nguyen, A. Jain, M. Zhou, K. Karalis, D. E. Ingber, G. A. Hamilton and M. A. Otieno, Clin. Pharmacol. Ther., 2018, 104, 1240–1248 CrossRef CAS.
- N. Korin, M. Kanapathipillai, B. D. Matthews, M. Crescente, A. Brill, T. Mammoto, K. Ghosh, S. Jurek, S. A. Bencherif, D. Bhatta, A. U. Coskun, C. L. Feldman, D. D. Wagner and D. E. Ingber, Science, 2012, 337, 738–742 CrossRef CAS.
- F. Jin, Z. Xie, C. J. Kuo, L. W. Chung and C. L. Hsieh, Cancer Gene Ther., 2005, 12, 257–267 CrossRef CAS.
- J. H. Choi, J. Lee, W. Shin, J. W. Choi and H. J. Kim, Nano Convergence, 2016, 3, 24 CrossRef.
- Y. Tang, F. Soroush, J. B. Sheffield, B. Wang, B. Prabhakarpandian and M. F. Kiani, Sci. Rep., 2017, 7, 9359 CrossRef.
- J. Nixon, P. Newbold, T. Mustelin, G. P. Anderson and R. Kolbeck, Pharmacol. Ther., 2017, 169, 57–77 CrossRef CAS.
- J. Kearley, J. S. Erjefalt, C. Andersson, E. Benjamin, C. P. Jones, A. Robichaud, S. Pegorier, Y. Brewah, T. J. Burwell, L. Bjermer, P. A. Kiener, R. Kolbeck, C. M. Lloyd, A. J. Coyle and A. A. Humbles, Am. J. Respir. Crit. Care Med., 2011, 183, 865–875 CrossRef CAS PubMed.
- T. H. Punde, W. H. Wu, P. C. Lien, Y. L. Chang, P. H. Kuo, M. D. Chang, K. Y. Lee, C. D. Huang, H. P. Kuo, Y. F. Chan, P. C. Shih and C. H. Liu, Integr. Biol., 2015, 7, 162–169 CrossRef CAS.
- H. Rouha, S. Weber, P. Janesch, B. Maierhofer, K. Gross, I. Dolezilkova, I. Mirkina, Z. C. Visram, S. Malafa, L. Stulik, A. Badarau and E. Nagy, Virulence, 2018, 9, 231–247 CrossRef CAS.
- S. Mairpady Shambat, P. Chen, A. T. Nguyen Hoang, H. Bergsten, F. Vandenesch, N. Siemens, G. Lina, I. R. Monk, T. J. Foster, G. Arakere, M. Svensson and A. Norrby-Teglund, Dis. Models Mech., 2015, 8, 1413–1425 CrossRef PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- S. Kim, S. C. LesherPerez, B. C. Kim, C. Yamanishi, J. M. Labuz, B. Leung and S. Takayama, Biofabrication, 2016, 8, 015021 CrossRef.
- M. K. Vormann, L. Gijzen, S. Hutter, L. Boot, A. Nicolas, A. van den Heuvel, J. Vriend, C. P. Ng, T. T. G. Nieskens, V. van Duinen, B. de Wagenaar, R. Masereeuw, L. Suter-Dick, S. J. Trietsch, M. Wilmer, J. Joore, P. Vulto and H. L. Lanz, AAPS J., 2018, 20, 90 CrossRef.
- S. Matsuura, H. Katsumi, H. Suzuki, N. Hirai, H. Hayashi, K. Koshino, T. Higuchi, Y. Yagi, H. Kimura, T. Sakane and A. Yamamoto, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10511–10516 CrossRef CAS.
- A. I. Astashkina, C. F. Jones, G. Thiagarajan, K. Kurtzeborn, H. Ghandehari, B. D. Brooks and D. W. Grainger, Biomaterials, 2014, 35, 6323–6331 CrossRef CAS PubMed.
- K. Tryggvason and J. Wartiovaara, Physiology, 2005, 20, 96–101 CrossRef PubMed.
- S. Akilesh, T. B. Huber, H. Wu, G. Wang, B. Hartleben, J. B. Kopp, J. H. Miner, D. C. Roopenian, E. R. Unanue and A. S. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 967–972 CrossRef CAS.
- R. Hausmann, C. Kuppe, H. Egger, F. Schweda, V. Knecht, M. Elger, S. Menzel, D. Somers, G. Braun, A. Fuss, S. Uhlig, W. Kriz, G. Tanner, J. Floege and M. J. Moeller, J. Am. Soc. Nephrol., 2010, 21, 2053–2058 CrossRef CAS PubMed.
- S. J. Shankland, J. W. Pippin, J. Reiser and P. Mundel, Kidney Int., 2007, 72, 26–36 CrossRef CAS PubMed.
- L. Ewart, E. M. Dehne, K. Fabre, S. Gibbs, J. Hickman, E. Hornberg, M. Ingelman-Sundberg, K. J. Jang, D. R. Jones, V. M. Lauschke, U. Marx, J. T. Mettetal, A. Pointon, D. Williams, W. H. Zimmermann and P. Newham, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 65–82 CrossRef CAS PubMed.
- J. L. Robertson, Toxicol. Pathol., 1998, 26, 64–72 CrossRef CAS PubMed.
- C. Gomez-Guerrero, N. Duque, M. T. Casado, C. Pastor, J. Blanco, F. Mampaso, F. Vivanco and J. Egido, J. Immunol., 2000, 164, 2092–2101 CrossRef CAS PubMed.
- M. Zhou, X. Zhang, X. Wen, T. Wu, W. Wang, M. Yang, J. Wang, M. Fang, B. Lin and H. Lin, Sci. Rep., 2016, 6, 31771 CrossRef CAS.
- S. Musah, A. Mammoto, T. C. Ferrante, S. S. F. Jeanty, M. Hirano-Kobayashi, T. Mammoto, K. Roberts, S. Chung, R. Novak, M. Ingram, T. Fatanat-Didar, S. Koshy, J. C. Weaver, G. M. Church and D. E. Ingber, Nat. Biomed. Eng., 2017, 1, 0069 CrossRef CAS.
- Z. E. Sauna, D. Lagasse, J. Pedras-Vasconcelos, B. Golding and A. S. Rosenberg, Trends Biotechnol., 2018, 10, 1068–1084 CrossRef PubMed.
- A. Shimabukuro-Vornhagen, P. Godel, M. Subklewe, H. J. Stemmler, H. A. Schlosser, M. Schlaak, M. Kochanek, B. Boll and M. S. von Bergwelt-Baildon, J. Immunother. Cancer, 2018, 6, 56 CrossRef.
- M. Leist and T. Hartung, Arch. Toxicol., 2013, 87, 563–567 CrossRef CAS PubMed.
- F. Leonard, E. M. Collnot and C. M. Lehr, Mol. Pharmaceutics, 2010, 7, 2103–2119 CrossRef CAS PubMed.
- F. Leonard, H. Ali, E. M. Collnot, B. J. Crielaard, T. Lammers, G. Storm and C. M. Lehr, ALTEX, 2012, 29, 275–285 CrossRef.
- C. Giese, C. D. Demmler, R. Ammer, S. Hartmann, A. Lubitz, L. Miller, R. Muller and U. Marx, Artif. Organs, 2006, 30, 803–808 CrossRef CAS PubMed.
- C. Giese, A. Lubitz, C. D. Demmler, J. Reuschel, K. Bergner and U. Marx, J. Biotechnol., 2010, 148, 38–45 CrossRef CAS PubMed.
- M. Seifert, A. Lubitz, J. Trommer, D. Konnig, G. Korus, U. Marx, H. D. Volk, G. Duda, G. Kasper, K. Lehmann, M. Stolk and C. Giese, Int. J. Artif. Organs, 2012, 35, 986–995 CrossRef CAS PubMed.
- N. Di Maggio, E. Piccinini, M. Jaworski, A. Trumpp, D. J. Wendt and I. Martin, Biomaterials, 2011, 32, 321–329 CrossRef CAS PubMed.
- J. M. Jackson, J. B. Taylor, M. A. Witek, S. A. Hunsucker, J. P. Waugh, Y. Fedoriw, T. C. Shea, S. A. Soper and P. M. Armistead, Analyst, 2016, 141, 640–651 RSC.
- J. W. Kamande, M. A. M. Lindell, M. A. Witek, P. M. Voorhees and S. A. Soper, Integr. Biol., 2018, 10, 82–91 CrossRef CAS PubMed.
- G. Suntharalingam, M. R. Perry, S. Ward, S. J. Brett, A. Castello-Cortes, M. D. Brunner and N. Panoskaltsis, N. Engl. J. Med., 2006, 355, 1018–1028 CrossRef CAS PubMed.
- L. Findlay, D. Eastwood, R. Stebbings, G. Sharp, Y. Mistry, C. Ball, J. Hood, R. Thorpe and S. Poole, J. Immunol. Methods, 2010, 352, 1–12 CrossRef CAS PubMed.
- B. Wolf, H. Morgan, J. Krieg, Z. Gani, A. Milicov, M. Warncke, F. Brennan, S. Jones, J. Sims and A. Kiessling, Cytokine+, 2012, 60, 828–837 CrossRef CAS PubMed.
- L. Findlay, G. Sharp, B. Fox, C. Ball, C. J. Robinson, C. Bird, R. Stebbings, D. Eastwood, M. Wadhwa, S. Poole, R. Thorpe and S. J. Thorpe, Cytokine+, 2011, 55, 141–151 CrossRef CAS PubMed.
- C. Grimaldi, D. Finco, M. M. Fort, D. Gliddon, K. Harper, W. S. Helms, J. A. Mitchell, R. O'Lone, S. T. Parish, M. S. Piche, D. M. Reed, G. Reichmann, P. C. Ryan, R. Stebbings and M. Walker, Cytokine+, 2016, 85, 101–108 CrossRef CAS PubMed.
- D. M. Reed, K. E. Paschalaki, R. D. Starke, N. A. Mohamed, G. Sharp, B. Fox, D. Eastwood, A. Bristow, C. Ball, S. Vessillier, T. T. Hansel, S. J. Horpe, A. M. Randi, R. Stebbings and J. A. Mitchell, FASEB J., 2015, 29, 2595–2602 CrossRef CAS PubMed.
- Emerging translational safety technologies and tools for interrogating human immuno-biology, https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/imi2-2018-15-04).
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- N. R. Wevers, R. van Vught, K. J. Wilschut, A. Nicolas, C. Chiang, H. L. Lanz, S. J. Trietsch, J. Joore and P. Vulto, Sci. Rep., 2016, 6, 38856 CrossRef CAS PubMed.
- V. Lecault, M. Vaninsberghe, S. Sekulovic, D. J. Knapp, S. Wohrer, W. Bowden, F. Viel, T. McLaughlin, A. Jarandehei, M. Miller, D. Falconnet, A. K. White, D. G. Kent, M. R. Copley, F. Taghipour, C. J. Eaves, R. K. Humphries, J. M. Piret and C. L. Hansen, Nat. Methods, 2011, 8, 581–586 CrossRef CAS PubMed.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS PubMed.
- B. L. Levine, Cancer Gene Ther., 2015, 22, 79–84 CrossRef CAS PubMed.
- C. Lissandrello, R. Dubay, K. T. Kotz and J. Fiering, SLAS Technol., 2018, 23, 352–363 CAS.
| This journal is © The Royal Society of Chemistry 2020 |