
Low-energy catalytic methanolysis of poly(ethyleneterephthalate)†
Duong Dinh
Pham
 ab and
Joungmo
Cho
*ab
ab and
Joungmo
Cho
*ab
aResearch Centre for Green Carbon Catalysis, Korea Research Institute of Chemical Technology (KRICT), Daejeon, 34114, Republic of Korea. E-mail: jmcho@krict.re.kr; Fax: +82-42-860-7388; Tel: +82-42-860-7699
bDepartment of Advanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon, 34113, Republic of Korea
First published on 8th December 2020
Abstract
Methanolysis is a chemical pathway for depolymerizing post-consumer PET plastic waste into monomeric feedstock, which can be utilized as a starting component to produce polymer materials with the same quality as the original polymer or other valuable products. In general, conventional methanolysis is carried out at a high reaction temperature under high pressure, demanding high capital and operating costs and in turn leading to an adverse effect on the environment from CO2 emissions. In this study, we developed a low-energy catalytic route for methanolysis to convert PET resin to dimethyl terephthalate (DMT). Potassium carbonate (K2CO3), which is an inexpensive and nontoxic salt, was used as a catalyst, and the effects of cosolvents on the catalytic performance were investigated to develop a new decomposition pathway towards DMT at ambient temperature. Compared to existing methanolysis processes, the overall reaction rate of the proposed system was relatively slow and steady, but the PET resins were completely decomposed into monomers within 24 hours. Intriguingly, a high selectivity of DMT was obtained at a mild temperature range of 20–35 °C. The yield of DMT, obtained by methanolysis at 25 °C, was 93.1% as the molar ratios of methanol, dichloromethane and K2CO3 to PET repeating units were 50, 50, and 0.2, respectively. In this experimental setup, the initial molar ratio of moisture to PET repeating units was adjusted to be 0.4. 2-Hydroxyethyl methyl terephthalate and monomethyl terephthalate were the major by-products created during the process. It was demonstrated that the ideal conversion of PET into DMT could be achieved by controlling the moisture level. In addition to several analytical methods for product characterization, we performed a parametric study to probe the most likely reaction steps and to observe the reaction behaviour of PET methanolysis. Based on the proposed mechanism, a kinetic model was developed and compared with the experimental data to estimate kinetic parameters. In the reaction system, PET depolymerization apparently proceeded through two series of reaction steps, and the degradation of PET had a relatively low activation energy of 66.5 kJ mol−1, which was accountable for catalytic methanolysis of PET at ambient conditions.
1. Introduction
Global plastic waste reached 260 million tons per year in 2016 and is projected to continuously rise to 460 million tons per year by 2030.1 The disposal of nonbiodegradable plastics has accelerated serious environmental problems and an irreversible waste of resources.2,3 Despite recent global awareness and regulations to reduce plastics, it is expected that a complete ban or elimination of plastic waste is unlikely.4,5 Recycling technologies, which convert waste plastics into original feedstocks or valuable products, can be an essential strategy to mitigate plastic waste issues.6In the past, plastic recycling technologies were not actively developed because a large price gap between recycled and petroleum-derived feedstocks. Post-consumer plastics are usually discharged in a mixed form and should be recycled after separation. The recycling of plastics can be performed in a physical, thermal, chemical or combined manner.7 However, physical and thermal methods are not substantial strategies for achieving a circular economy in recycled plastics due to the deterioration or alteration of their properties,8 while a chemical conversion process allows for a facile method to produce monomers that are equivalent to the original petroleum-derived feedstocks.9 As technologies in the chemical industry are driven by economic merits, an efficient and low-cost technology to convert plastics to valuable monomers could aid in securing an ideal circular economy while alleviating current global environmental issues.10
Polyester is a polymeric material that contains ester functional groups. Owing to its transparent, mechanically stable, lightweight, chemically stable and nontoxic properties, polyethylene terephthalate (PET) is the most commonly and widely used thermoplastic polyester, with applications in soft drink bottles, textiles, plastic films, food packaging and insulation.11 Several chemical recycling technologies for PET have been well developed, and some are commercially available.12
Solvolysis is a chemical recycling pathway that depolymerizes PET resin in the presence of a counterreacting solvent.13 The most widely practised approaches include glycolysis with ethylene glycol (EG),14,15 methanolysis with methanol and hydrolysis under acidic, basic or neutral conditions.16–18 Recently, aminolysis and amonolysis have also been suggested as feasible options for converting PET into value-added products.19,20 The glycolysis of PET is an energy-intensive depolymerization process because it requires an elevated reaction temperature (above the boiling point of ethylene glycol) and post-treatment steps to purify the products.12 Nevertheless, glycolysis is the most commonly applied chemical recycling method, mainly due to the direct applicability of recycled products (i.e., bis(2-hydroxyethyl) terephthalate) as a single raw feedstock for the reproduction of PET. During the hydrolysis process, PET undergoes decomposition to form terephthalic acid (TPA) and EG under strongly acidic, basic or neutral conditions. However, the corrosion of process units and operating costs associated with wastewater treatment makes this option less attractive.
Methanolysis is a catalytic process that employs high-temperature and high-pressure methanol to decompose PET into dimethyl terephthalate (DMT) and EG.21 Owing to its relatively low solubility in water, DMT can be easily purified to a polymer grade applicable to an alternative feedstock for the production of PET.22 Another commercially important application of DMT is hydrogenation into 1,4-cyclohexanedimethanol (CHDM), which is a diol monomer for the modification of the glycol portion of PET resins.23,24
The commercial methanolysis processing of PET flakes is performed at 180–280 °C and 2–4 MPa.25 In the early 1960s, Hans et al. reported a two-stage methanolysis process that obtained high-purity DMT initiated by melted PET being in contact with methanol, followed by the completion of methanolysis in an excess amount of heated methanol.26 In the mid-1990s, Eastman Kodak developed an integrated process to produce high-purity DMT.27 As the complete decomposition of PET via methanolysis is preferentially achieved at a high temperature and pressure, there have been few studies to advance the performance of methanolysis of PET in a supercritical fluid (SCF) of methanol. Depolymerization under a supercritical state of methanol allows a high conversion of PET and an improved DMT yield (up to 95%) within 1 h under optimal reaction conditions (260–270 °C, 9–11 MPa).28,29 In this approach, the reaction must be conducted at a high pressure, and thus, high capital and operating costs are the primary disadvantages. Another approach to achieve a high yield of DMT in a short reaction time involves the utilization of microwaves. It was reported that more than 80% of DMT yield can be obtained within only 30 min at 160 °C by utilizing a zinc acetate catalyst and microwave irradiation.30
Methanolysis is essentially a transesterification reaction. Various metal salt forms, including metal acetates (e.g., zinc acetate, lead acetate), metal oxides (e.g., aluminium isopropoxide, sodium silicate) and metal hydroxides, are known as common catalysts for this reaction.31–35 Although these metal salt catalysts exhibit a high catalytic performance for conventional methanolysis, a low reaction temperature far lower than the melting point of PET (Tm = 260 °C) is not appropriate to favourably proceed with PET decomposition in a selective and rapid manner. Recently, Loop Industries has filed a patent for a low-temperature methanolysis of PET. In this technology, stoichiometric (or sub-stoichiometric) amounts of alkali methoxides and cosolvents are used as active ingredients to decompose PET at a lower temperature than the boiling point of methanol.36 To facilitate the reactivity of PET solvolysis, several studies employed a cosolvent-assisted reaction system, although the role of cosolvents was not clearly described; for instance, aromatic solvents assisted in the dissolution of PET due to intermolecular interactions,37p-xylene adjusted the thermodynamic compatibilities of the reactant,38 dimethylsulfoxide was used for PET dissolution in glycolysis,39 ethereal solvents aided in the percolation and chelation of hydroxyl ions,40 and halogenated solvents were used for swelling in the alkali decomposition of PET.41
In this study, we demonstrated a new catalytic route of methanolysis that efficiently converts PET into DMT with low-energy consumption. It was found that a highly active and selective catalytic system for the complete decomposition of PET can be achieved with a very specific combination of catalyst and cosolvent. Among several metallic salt catalysts, a prominent catalytic performance appeared only when potassium carbonate (K2CO3) was used. When combined with the catalyst, an appropriate aprotic polar solvent was also determined to boost the catalytic activity and selectivity of DMT under ambient conditions.
2. Experimental procedure
2.1. Materials
Postconsumer PET bottles and coloured PET label wastes were obtained from the Lotte Chemical Co. (Daejeon, South Korea) in the form of prewashed flakes. The plastic resins were washed with an excessive amount of ethanol three times and then rinsed with acetone, followed by drying in an oven at 60 °C for 3 h. PET granules containing 30% glass reinforcer were purchased from Sigma-Aldrich. A sample of bis(2-hydroxyethyl) terephthalate (BHET) was obtained from Sigma-Aldrich (in a mixture of monomer, dimer and oligomer). The individual BHET components were collected by an extraction method reported elsewhere,42 and HPLC was used to confirm their purities (purities of extracted BHET and dimer were measured to be 99.1% and 98.5%, respectively). Several catalysts, including alkali and alkaline earth metal salts, are applied to the PET methanolysis. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD), potassium carbonate (K2CO3) and potassium methoxide (CH3OK) were purchased from Sigma-Aldrich, and potassium bicarbonate (KHCO3), potassium acetate (KOAc), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), sodium acetate (NaOAc), calcium carbonate (CaCO3), magnesium carbonate (MgCO3), and calcium oxide (CaO) were purchased from Samchun Chemical. Anhydrous solvents, including methanol, dichloromethane, chlorobenzene, acetone, tetrahydrofuran, and acetonitrile, were purchased from Sigma-Aldrich and used as organic solvents in PET methanolysis as received. HPLC-grade methanol and DI water (Samchun), without any purification, were used to prepare a mobile phase for HPLC analysis.2.2. General procedure for PET methanolysis
The methanolysis reaction was carried out in a 50 mL round-bottomed flask. Typically, 8.31 g of methanol (260 mmol), 22.10 g of dichloromethane (260 mmol) and 143.52 mg of K2CO3 were added to the flask. Thereafter, the initial molar ratio of water-to-PET repeating units in the reaction solution was adjusted to 0.4 by adding deionized water. After thorough mixing, the volumetric Karl Fisher titration method (Metrohm 852 Titrando) was used to confirm the water concentration of the resulting solution. The flask was immersed in a water bath in which the reaction temperature was accurately maintained (within ±0.1 °C) by a PID-controlled hot plate and circulating chilled water through coils. PET methanolysis was initiated by adding 1 g of PET chips (5.2 mmol). The reaction solution was continuously stirred using a magnetic stirrer (at a speed of 500 rpm) for 24 h. The reaction was completed by a separation of the catalyst (for solid catalyst) or by the addition of a pre-measured amount of cold water (for liquid catalyst) when PET methanolysis was accomplished for a desired reaction time. The reaction mixture was then filtered and separated into two parts: liquid filtrate and insoluble solids. The above reaction procedures and conditions, unless otherwise specified, were applied to formulate the reaction mixture for methanolysis in this report.2.3. Analysis of methanolysis products
The liquid filtrate contained several decomposed products. To determine the compositions in the solution, several known concentrations of standard solutions made of DMT (Aldrich, >99%), mono-methyl terephthalate (MMT; TCI chemicals, >98%), 1-(2-hydroxyethyl) 4-methyl terephthalate (HEMT, TCI chemicals, >97%) and TPA (Aldrich, >99%) were prepared and applied to the HPLC calibration. In the catalysis using alkali metals, the calibration for MMT and TPA was carried out using the standard solutions of corresponding metal salts (TCI chemicals). In each quantitative analysis, 0.2 ml of the liquid filtrate was removed and diluted 1000 times in a methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water solution (70:30 (v/v)). The rest of the reaction solution was used to collect DMT by consecutive purification steps of solvent removal and recrystallization in water. First, liquid solvents and EG were separated using a rotary evaporator. The nondistillable products, mostly containing terephthalate components, were mixed with 25 g of distilled water and stirred for 6 h. The resulting solution was stored at 4 °C overnight. It was then filtered and completely dried in a vacuum drying oven kept at 60 °C overnight. A white needle-like crystal powder was obtained as the final product. It was confirmed by HPLC analysis that the product was almost pure DMT. The sample was kept for further structural analysis by 1H-NMR and EI-MS.
:water solution (70:30 (v/v)). The rest of the reaction solution was used to collect DMT by consecutive purification steps of solvent removal and recrystallization in water. First, liquid solvents and EG were separated using a rotary evaporator. The nondistillable products, mostly containing terephthalate components, were mixed with 25 g of distilled water and stirred for 6 h. The resulting solution was stored at 4 °C overnight. It was then filtered and completely dried in a vacuum drying oven kept at 60 °C overnight. A white needle-like crystal powder was obtained as the final product. It was confirmed by HPLC analysis that the product was almost pure DMT. The sample was kept for further structural analysis by 1H-NMR and EI-MS.
The insoluble solid product consists of unreacted PET, solid catalyst, and cation-exchanged forms of MMT. TPA salts were also found in the mixture only when a high dosage of alkali metal catalysts (≥0.75 mole per mole of PET repeating units) was applied. The insoluble product was completely dried in a vacuum oven prior to any further analysis. A small fraction of solid powder, which did not contain unreacted PET, was carefully removed for X-ray diffraction measurements to identify the nature of the solid sample. The rest of the solid product was used to determine the concentration of decomposed products. The sample was mixed in a premeasured amount of water. Unreacted PET pieces were then separated from the solution by filtration. The concentration of decomposed products in the filtrate was measured by HPLC analysis. Finally, the filtrate solution was acidified by 1 M hydrochloric acid (HCl) solution to convert metal salt products into an acidic form of TPA and MMT. The filtration and complete drying steps were repeated to obtain a powder sample of TPA and MMT mixture. The sample was kept for structural analysis by 1H-NMR and EI-MS, and the overall experimental procedures are summarized in Fig. S1.†
PET conversion and product yields were calculated from the mass balance and the concentration of each component in liquid and solid products:
| XPET = (m0 − m)/m0 × 100 | (1) |
| YDMT = NDMT/N0 × 100 | (2) |
| YHEMT = NHEMT/N0 × 100 | (3) |
| YMMT = NMMT/N0 × 100 | (4) |
| YTPA = NTPA/N0 × 100 | (5) |
2.4. Characterization of products
NMR spectra of the main product and by-products were collected by a Bruker Avance III HD NMR spectrometer operating at 400 MHz in chloroform-d (CDCl3) and dimethyl sulfoxide (DMSO)-d6 solvents. An MS (JEOL JMS-700) instrument equipped with electron ionization (EI) was used to analyse the molecular weights of the products. FT-IR analysis of the products was carried out using a Bruker ALPHA-P infrared spectrometer. X-ray diffraction (XRD) patterns were collected on an X-ray diffractometer operated at 40 kV and 40 mA with Cu Kα radiation (λ = 1.54060 A). The quantification of methanolysis products was carried out by HPLC (YL-3000 HPLC instrument) equipped with a UV detector set at 254 nm and a reverse-phase C18 column kept at 40 °C. A mixture of methanol and water (70:30 in a volume ratio) was used as a mobile phase at a flow rate of 0.7 mL min−1.
3. Results and discussion
The methanolysis of PET involves transesterification of the PET polymer with methanol to produce DMT and ethylene glycol. Owing to its high potential to obtain pure monomeric compounds, flexibility in combining with other processes (e.g., a hybrid process by combining with glycolysis43), and direct applicability to the polymerization process, methanolysis has been one of the most widely practised processes to commercially recycle postconsumer PET waste for several decades.26,35,36,44–46 However, current commercial technologies still favour high reaction temperatures, which lead to high pressure due to the low boiling point of methanol. Therefore, the methanolysis of PET has been generally regarded as a process that requires not only a large consumption of energy but also a prohibitive cost for plant construction, which makes this option less attractive when selecting a recycling process for post-consumer PET waste.The utilization of cosolvents is an alternative strategy for lowering the energy barrier in PET decomposition, although their roles remain unclear. Orbay et al.38 claimed that xylene helped to improve the reactivity in PET glycolysis due to preferential mass transfer of decomposed products to the inorganic phase. Liu et al.37 noted that the addition of polar solvent, e.g., DMSO, aided in PET solvation, which led to rapid depolymerization when exposed to glycolysis conditions. Most recently, Loop Industries successfully proved that PET can be depolymerized into TPA or DMT at a sufficiently low temperature to avoid highly pressurized reactions by adding a secondary polar solvent.36,41
3.1. Solvent-aided catalytic methanolysis
Fig. 1 shows the product distributions for the catalytic methanolysis of PET at 25 °C for 24 h in the presence of various cosolvents. In catalytic methanolysis, DMT was obtained as a major product. HEMT and MMT were obtained as by-products in methanolysis. It has been reported that conventional PET methanolysis, which is performed at a high temperature (typically ≥200 °C), undergoes slow decomposition steps of the polymer into fragments with lower molecular weights, as these high concentrations of oligomers and dimers are observed at an initial stage of methanolysis.28,29,47 It is notable that any partially decomposed products, such as oligomers and dimers, were not detected during the low-temperature catalytic methanolysis reactions performed in this study. All products were verified by several characterization tools, including NMR, EI-MS, FT-IR, and HPLC, and calibrated by standard materials (detailed in Fig. S2 and S3†). The characterization of all liquid and solid products revealed that transesterification occurs at the terminal group and leads to the direct conversion of PET into DMT in the presence of catalyst. The reaction steps for thermally-driven and cosolvent-driven PET methanolysis are schematically depicted in Scheme 1.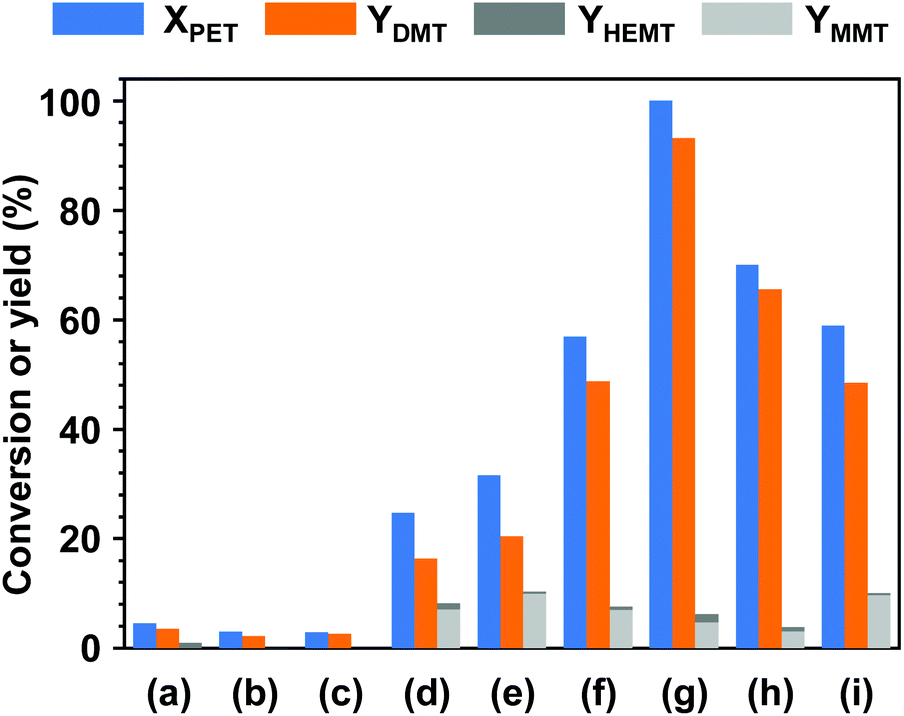 | ||
| Fig. 1 Effect of cosolvent on PET methanolysis for 24 h at an ambient temperature (T = 25 °C) in the presence of (a) only methanol and (b) heptane, (c) cyclohexane, (d) acetone, (e) acetonitrile, (f) tetrahydrofuran, (g) dichloromethane, (h) chloroform and (i) chlorobenzene cosolvents in the same molar amount of methanol. The molar ratios of catalyst (K2CO3), methanol, and cosolvent (if any) to PET repeating unit were adjusted to be 0.2, 50, and 50, respectively. | ||
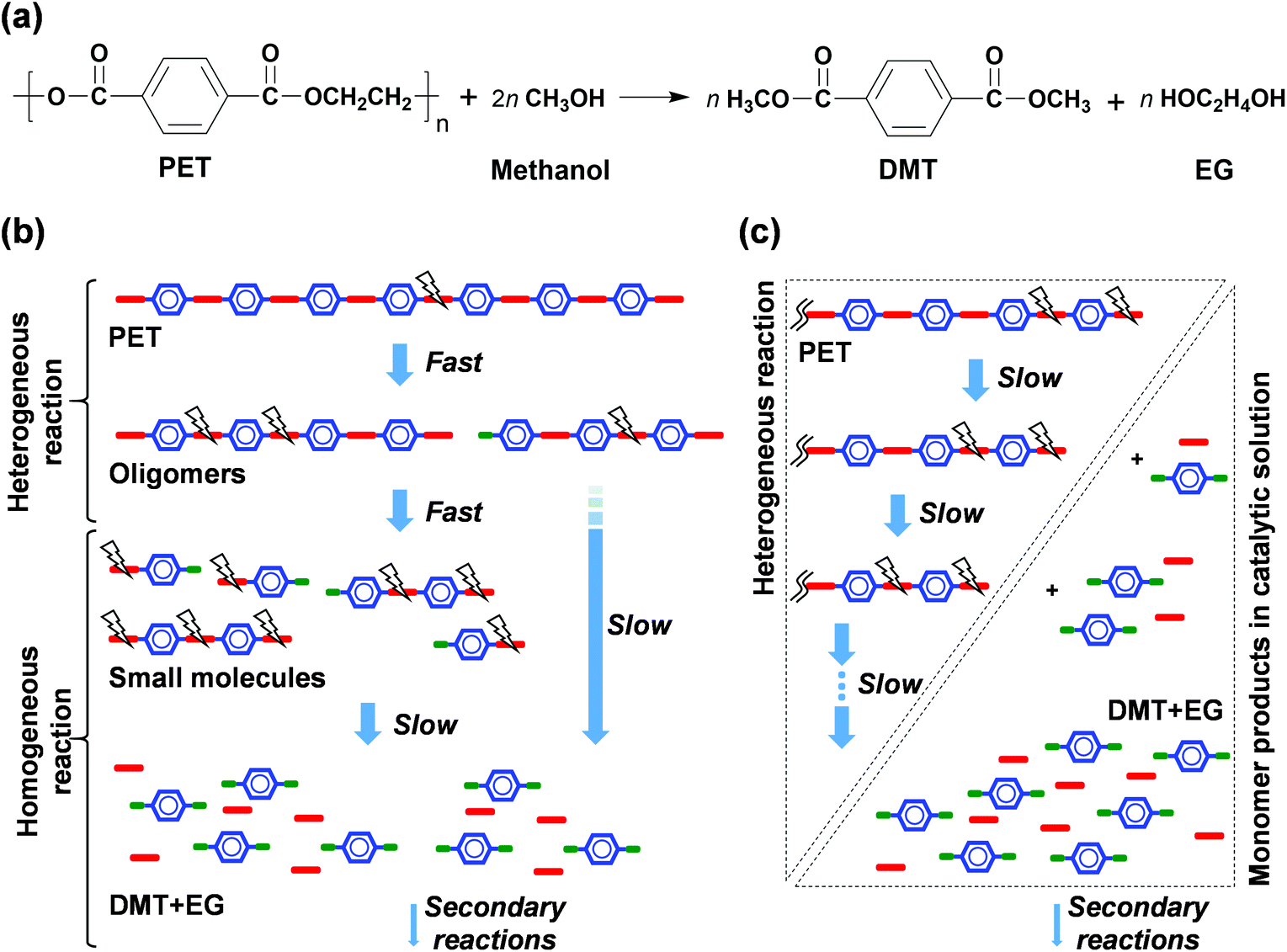 | ||
| Scheme 1 Reaction models of PET depolymerisation via methanolysis; (a) major reaction step, (b) random scission model for conventional methanolysis,28,29,47 and (c) end-group scission model for solvent-aided catalytic methanolysis (this work). Blue, red and green symbols indicates bonded functional groups of terephthalic ring, ethylene glycol, and methanol, respectively. | ||
To measure the effect of each cosolvent on the rate of decomposition, the catalytic reactions were performed under identical reaction conditions. In the experimental setup, the added amount of methanol was adjusted to have a weight ratio of 8.3 (g/g PET) corresponding to a molar ratio of 50 per repeating unit of PET. For the addition of cosolvents, the same molar amount to methanol was applied to the reaction system. With only methanol, it was obvious that PET rarely undergoes methanolysis. Only 4.5% of PET was degraded into products, and the obtained DMT yield was approximately 3.6% in this case. Without a cosolvent, HEMT was obtained only as a by-product (0.9%) whose one side group was replaced by a functional group of ethylene glycol. To investigate the effect of solvent addition on methanolysis, two nonpolar solvents (1-heptane and cyclohexane) were first tested. In both cases, no significant improvements in catalytic performance could be found. In contrast, the reaction resulted in a slight decrease in reactivity, while it exerted slightly better selectivity for DMT. Although the addition of nonpolar solvents did not affect the catalytic performance over PET decomposition, the dilution effect by additional solvents presumably introduced slight differences in reactivity for the whole system. Several types of polar aprotic solvents were further applied to the catalytic methanolysis of PET. As noted in Fig. 1(d) through (i), a significant increase in catalytic performance appeared in the reaction systems with polar cosolvents. Among the cosolvents, the addition of DCM allowed the best performance in the methanolysis of PET. Within a given condition (methanolysis at 25 °C for 24 h), all PET particles were degraded, and the yield of DMT reached 93.1%. It is noteworthy that the reaction system did not result in similar DMT selectivity. Chlorinated methane solvents, herein referring to DCM and chloroform, showed relatively good selectivity to DMT, and in turn, a relatively small fraction of by-products was formed. The results indicated that the overall methanolysis was characterized by different reaction steps, and their reaction rates were unevenly facilitated by polar aprotic cosolvents, although they were not directly involved in the reaction. Although the role of cosolvents has yet to be clearly clarified, the improved activity in PET decomposition with polar solvent could be explained by the facilitated catalytic action associated with the dielectric secondary β relaxation of amorphous PET at a low temperature. The relaxation process, particularly in the presence of chlorinated methane compounds, significantly limited the intramolecular motions of short-range segments in the amorphous phase of PET with diffuse solvent molecules.48 As a consequence, the relaxation process restricted the mobility of ester bonds,49 where noncooperative motions of carbonyl groups were effectively catalysed by potassium carbonate.
3.2. Effective catalysts on solvent-aided methanolysis
Fig. 2 presents the results of PET methanolysis with various catalysts using DCM as a cosolvent. First, several alkali and alkaline earth metal salts, which are barely soluble in the reaction mixture of methanol and DCM, were applied to evaluate catalytic capability in the solvent-aided methanolysis of PET. Interestingly, only potassium carbonate (K2CO3) showed an excellent catalytic performance (Fig. 2(a)), while the addition of equivalent amounts of other insoluble metal oxides or insoluble metal salts resulted in very low or no activity in PET methanolysis. Within 24 h, PET was completely decomposed into monomer products, and nearly 93% of DMT was obtained by catalytic methanolysis with K2CO3. The relatively high activity of K2CO3 is ascribed to the higher rate of surface-controlled dissolution into polar aprotic solvent at a reaction temperature of 100 °C or above.50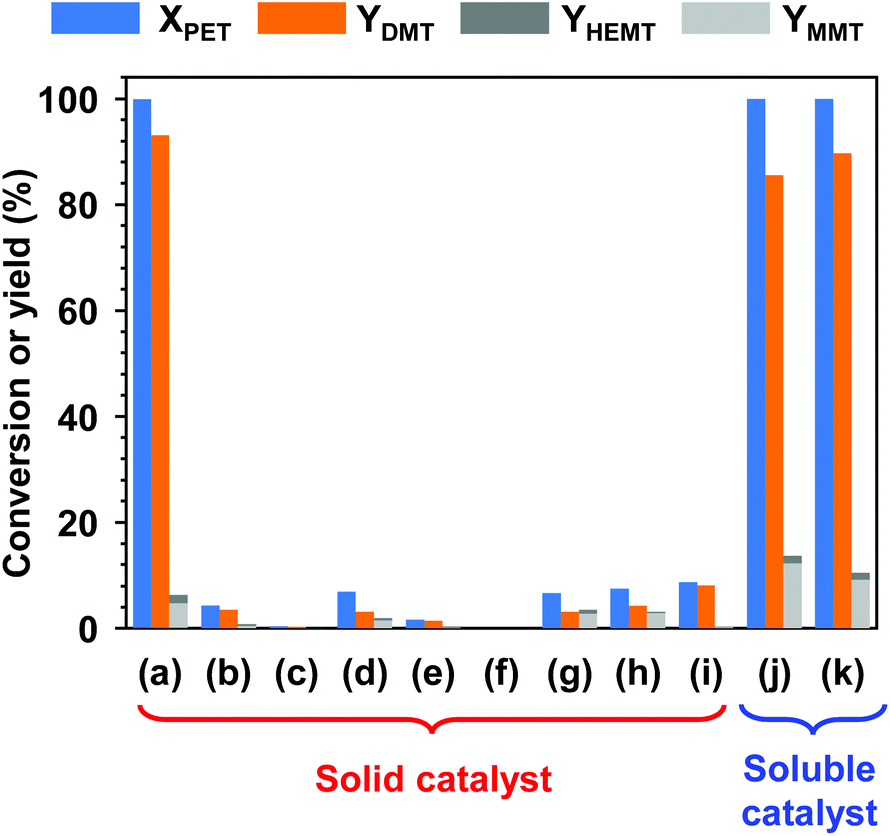 | ||
| Fig. 2 Catalytic methanolysis of PET at 25 °C for 24 h; applied catalysts are (a) K2CO3, (b) KHCO3, (c) KOAc, (d) Na2CO3, (e) NaHCO3, (f) NaOAc, (g) MgCO3, (h) CaCO3, (i) CaO, (j) CH3OK, and (k) TBD. The molar ratios of catalyst, methanol, and DCM to PET repeating units were adjusted to be 0.2, 50, and 50, respectively. | ||
The catalytic screening tests were extended to methanolysis by soluble catalysts. Methanolysis was basically carried out under the same conditions, but a homogeneous catalytic solution was prepared by completely dissolving the catalyst in a mixture of methanol and cosolvent prior to the addition of PET. For comparison, equimolar concentrations of CH3OK and TBD, whose performances were validated by the Loop Industries36 and IBM,43 were applied to the PET methanolysis. CH3OK is a highly reactive inorganic material, and TBD is a basic organocatalyst. Both are soluble in methanol. It is expected that the catalytic performance of solid catalysts is much less effective than that of soluble catalysts because it is highly limited by mass transfer at the boundary between the catalytic phase and polymer surface. As expected, both catalysts exerted a high reactivity over PET decomposition. PET decomposition was complete within 12 h. However, the final yields of DMT were 85.5% and 89.3%, as denoted in Fig. 2(j) and (k).
Although catalytic particles were not in homogeneous contact with the PET surface, the catalytic methanolysis system with K2CO3 produced a higher yield of DMT. This was ascribed to the selective catalytic performance of K2CO3 at the phase boundary between liquid and solid, where surface-controlled dissolution process plays a critical role.50 K2CO3 promoted the methanolysis of PET more effectively than hydrolysis, while the other catalysts accelerated both reactions significantly. It is meaningful and motivating to overcome the economic and energy barriers that hinder most chemical recycling technologies. In the catalytic methanolysis system, K2CO3, which is considered a stable, inexpensive, abundant and less harmful material, has the potential to be reused and to enable easy purification of monomer products mainly because K2CO3 and the monomer show opposite solubility behaviours in both reaction solution and water.
3.3. Effects of solvent and catalyst amount on PET methanolysis
Fig. 3 depicts the effect of solvent on the catalytic performance over methanolysis. The effect of methanol on methanolysis is shown in Fig. 3(a). When the molar amount of methanol per PET repeating unit (NMeOH) was limited to 10, a very low reactivity was observed, as only approximately 40% of PET decomposed. With an increase in the added methanol amount (NMeOH ≥ 20), a sharp increase in the decomposition rate was observed. After PET decomposition for 24 h, the conversion reached approximately 95%, but a low DMT yield (only 53.1%) was observed when the molar amount of added methanol was approximately 20. The major by-product was a partially hydrolysed monomer MMT (potassium exchanged form), which accounted for the low selectivity. The result indicated that the reaction rate of hydrolysis was comparable to that of PET methanolysis. A further increase in methanol led to a gradual increase in DMT yield, while PET particles were completely decomposed into monomeric products. From the observation of PET methanolysis behaviour by limiting an initial amount of methanol, one can conclude that an excessive amount of methanol (NMeOH > 50) is required not only to obtain a high yield of DMT but also to suppress the formation of by-products.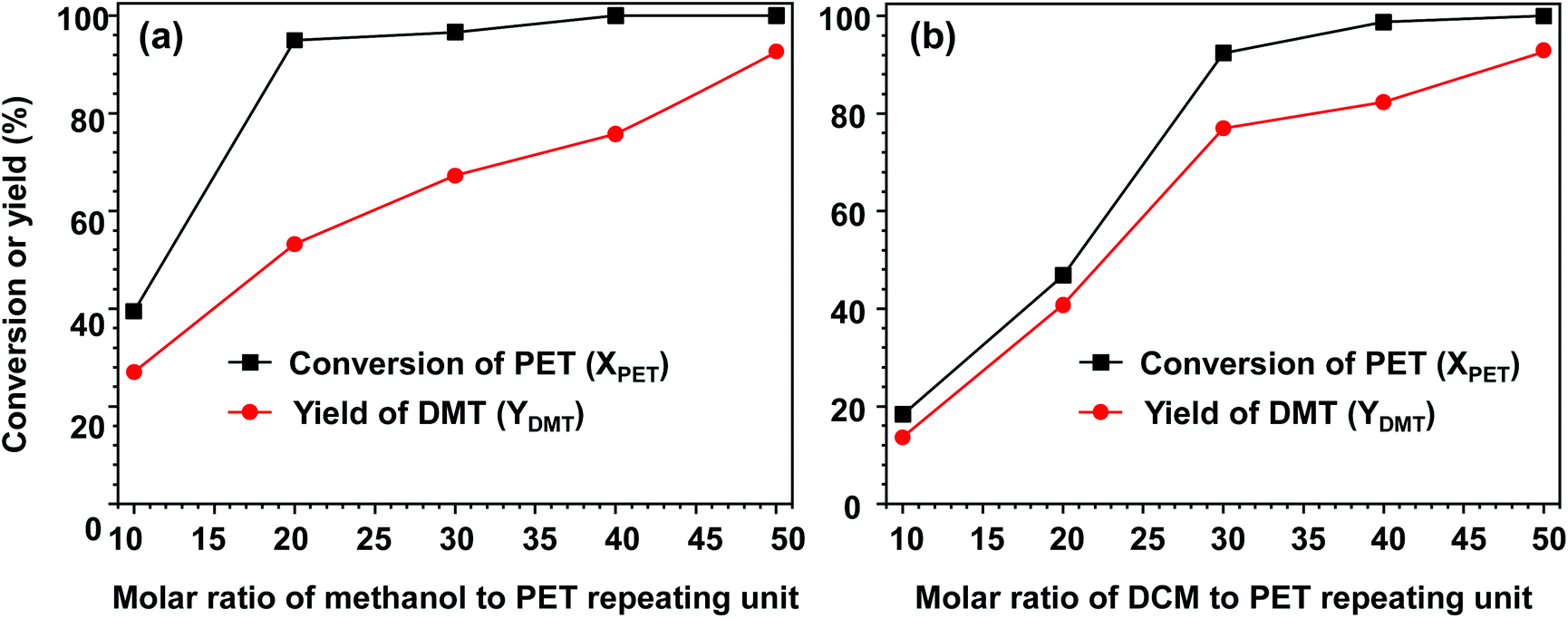 | ||
| Fig. 3 Effect of (a) methanol and (b) DCM amount on the PET methanolysis reaction at 25 °C for 24 h. The molar ratio of catalyst to PET repeating unit was adjusted to be 0.2. The added amount of methanol was varied for the case of (a) and DCM for the case of (b). In each case, the molar ratio of the other solvent to PET repeating unit was 50. | ||
Fig. 3(b) shows the effect of polar cosolvent (DCM) on PET methanolysis. In methanolysis, methanol participates in transesterification, but the cosolvent remains inert through PET decomposition. Therefore, the total amount applied to the reaction can be fully recoverable and recyclable. Although the consumption is nearly zero in methanolysis, it was found that the concentration of cosolvents could significantly influence the overall methanolysis even at a fixed temperature of 25 °C, as exemplified in Fig. 3(b). In the absence of polar cosolvent, no significant catalytic activity of methanolysis was observed at 25 °C, even though a sufficiently high amount of methanol was provided. When the molar ratio of DCM to PET repeating unit (NDCM) was adjusted to 10, the reaction mixture gave rise to prominent PET decomposition. As a result of 24 h PET methanolysis, 18.3% of PET was decomposed and converted into DMT in a relatively high yield (up to 13.6%). It is notable that an increase in the DCM amount in methanolysis resulted in a parallel increase in PET conversion and DMT yield. This explains why the catalytic performance of methanolysis can be significantly improved by adding DCM. At a high concentration (NDCM > 30), the PET conversion exceeded 90%. As Liu et al. discussed in their PET glycolysis study,37 polar solvents may play a coupled role of dissolution-degradation in facilitating PET decomposition at the phase boundary between the solid surface of PET and solvents. DCM is a polar and hydrophobic compound with good miscibility with methanol. In addition to the elevated decomposition rate, it is believed that a relatively high diffusive flux of methanol into the phase boundary of the PET surface allows the high selectivity of DMT in the presence of DCM.
Fig. 4 presents the effect of catalyst amount on PET methanolysis at 25 °C. To establish reaction conditions critically promoted by the catalyst, the molar doses of K2CO3 to the PET repeating unit (Ncat) were varied from 0.1 to 1.0 while all other compositions were fixed. In addition, a series of experiments were repeatedly carried out for different reaction times (6, 12, and 24 hours) to investigate the evolution of product distribution and kinetic behaviours in the vicinity of equilibrium. Conversional curves along the molar amount of catalysts, drawn as solid lines in Fig. 4(a), indicated that a large amount of catalyst and a lengthened exposure to the catalytic solution favourably completed PET decomposition. For a short reaction time of 6 h, the amount of catalysts strongly affected the reactivity of methanolysis. The highest conversion (approximately 78%) could be found when the molar amount of added catalyst was almost equal to that of PET repeating units. Regardless of catalytic amounts, DMT was initially produced in a very selective manner, although the conversional rate was kinetically limited. This was attributed to the relatively slow and subsequent hydrolysis of DMT. When the reaction time was prolonged to 12 h, an increase in the catalytic amount resulted in the appearance of by-products to a significant degree. Depolymerized molecules in continuous contact with moisture could increase the chance of partial hydrolysis, which should be responsible for MMT production. HEMT was also detected as a by-product, but its concentration was not amplified by the amount of catalyst. Noticeably, HEMT formation was accompanied by PET decomposition, and its equilibrium concentration was kept low (approximately 1.5% upon complete decomposition of PET). Thus, HEMT could be considered a by-product coming from the glycolysis of DMT with EG rather than the reactive intermediate derived directly from the decomposition of PET. After 24 h of reaction, all fractions of PET were decomposed into monomer products with the addition of a smaller catalytic amount (Ncat ≥ 0.2). Although complete decomposition of PET is attainable, the reaction conditions for the high yield of DMT can be found in a narrow range of catalyst concentrations. The highest yield of DMT (93.1%) was obtained at a catalytic molar ratio to PET (Ncat) of 0.2. Above this critical value, the DMT yield significantly decreased with a large amount of catalyst. This indicated that K2CO3 catalysed not only PET decomposition by methanolysis but also DMT decomposition by hydrolysis. After 24 h of reaction with a high dosage of catalysts (Ncat ≥ 0.7), a small fraction of TPA, which is fully hydrolysed terephthalate, was detected in our analysis. After 24 h of reaction with an addition of 0.7 moles of catalyst per PET, approximately 0.7% of TPA was collected in a solid form (recovered as dipotassium terephthalate).
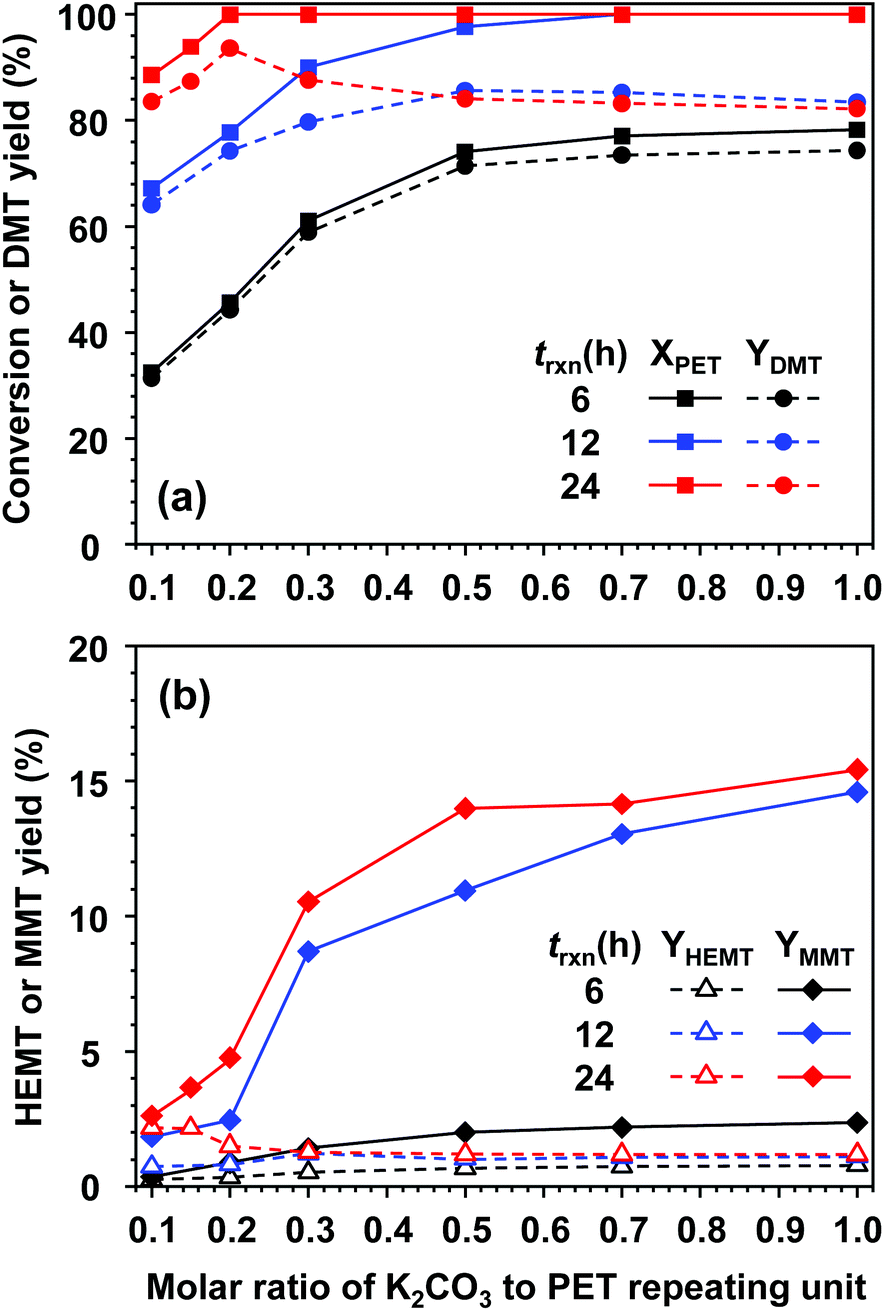 | ||
| Fig. 4 Effect of catalytic amounts on the PET methanolysis reaction at 25 °C for 6, 12 and 24 h. (a) PET conversion and DMT yield, (b) by-product (HEMT and MMT) yields. The molar ratios of both methanol and DCM to PET repeating units were set to 50, and the molar catalytic amount of [K2CO3] was varied from 0.1 to 1.0. | ||
To confirm the nature of the solid parts in the reaction medium, XRD measurements were further carried out. Two different solid product samples, starting with molar catalytic amounts (Ncat) equal to 0.2 and 0.7, were taken after 24 h of reaction (plotted as red symbols in Fig. 4). In each XRD measurement, 30 mg was used to collect XRD patterns for solid samples and compared with those of pure components, as depicted in Fig. 5. Although PET dominantly underwent methanolysis, characteristic peaks accounting for DMT (XRD peaks for DMT are presented in Fig. S4†) were not found in either solid sample. This explains why nearly all fractions of DMT remained dissolved in the reaction solution with a sufficiently high solubility. The XRD pattern for the solid part from PET decomposition at a low dosage of K2CO3 (Ncat = 0.2) included characteristic peaks of potassium monomethyl terephthalate and relatively weak peaks attributed to insoluble K2CO3. In contrast, strong characteristic peaks for dipotassium terephthalate (K-TPA) predominantly appeared in the solid sample derived from PET decomposition at a high dosage of K2CO3 (Ncat = 0.7). It was evident that a larger amount of K2CO3 catalysed hydrolysis of DMT in a more rapid and frequent manner, leading to the occurrence of consecutive hydrolysis of two ester bonds in a DMT molecule. Apart from the abovementioned analysis for the by-products, XRD patterns revealed that the active species catalysing methanolysis is K2CO3 presented in a solid phase. The balance amount of K2CO3 was also confirmed by gas chromatographic analysis of carbon dioxide released after HCl addition. Meanwhile, the existence of potassium methoxide as an active catalytic species in soluble and insoluble parts of the reaction mixture was also checked. In a comparison with the XRD analysis for mixed reaction solutions with pure substances of alkali metal or its methoxide form, it was confirmed that potassium methoxide, characterized by XRD patterns in Fig. 5(b), did not appear as an active catalytic intermediate during PET decomposition.
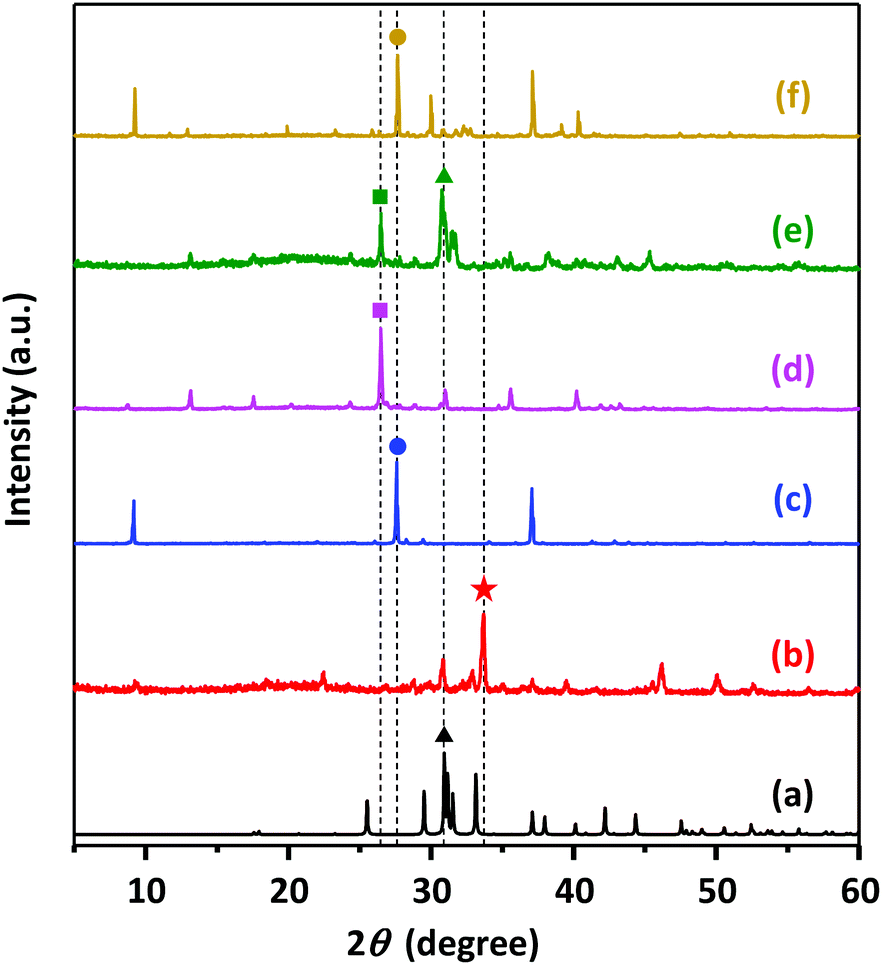 | ||
| Fig. 5 XRD patterns of pure materials of (a) K2CO3 (b) CH3OK, (c) dipotassium terephthalate (K-TPA), (d) potassium monomethyl terephthalate (K-MMT) and solid residue samples obtained after 24 h methanolysis at 25 °C starting with molar catalytic amounts ([K2CO3]/[PET repeating units]) of (e) 0.2 and (f) 0.7. | ||
3.4. Effect of initial moisture on PET methanolysis
Metal salt catalysts are commonly used for PET decomposition. In particular, zinc acetate is the most widely used transesterification catalyst for methanolysis and glycolysis. In the presence of water in the reaction solution, transesterification catalysts significantly promote the hydrolysis of the methanolysis product.51 The hydrolysis of DMT involves two decomposition steps of DMT conversion into MMT and MMT into TPA. In each reaction step, one mole of methanol is discarded and consumes one mole of water to form a hydrolysed by-product. If the concentration of water is not sufficiently high, hydrolysis induces the precipitation of hydrolysed products, which is mainly caused by cation exchange with metal salts. Consequently, the reaction system consumes the metal salt and loses its catalytic function to a significant degree. Through a catalytic activity test that replaced K2CO3 with dipotassium terephthalate or potassium mono-methyl terephthalate, it was confirmed that the metal ion-exchanged terephthalates exerted almost zero catalytic reactivity under the typical conditions of methanolysis applied herein.Fig. 6 shows the effect of the water content in the reaction solution on the catalytic methanolysis. The high reactivity of K2CO3 leading to a complete decomposition of PET was observed in a molar ratio range of water to PET repeating units from 0 to 0.9 (Nwater ≤ 0.9). In this reaction regime, the initial amount of moisture significantly affected the product distribution. The maximum conversion of PET to DMT could be achieved when the concentration of moisture approached zero. In the projection to the moisture-free condition, the highest DMT yield was expected to be approximately 98.5%. This small gap from ideal DMT production was mainly attributed to an equilibrium concentration of HEMT, which will be discussed in section 3.7. As the initial moisture content increased in the reaction solution, the decomposition of PET resulted in a decreased DMT yield that accounted for the appearance of MMT as a by-product. Although the high reactivity, leading to a complete decomposition of PET, was mainly driven by methanolysis in this regime, the hydrolysis, promoted by the solid K2CO3 catalyst, induced further decomposition of DMT into MMT so that it became a determinative reaction step for the final product distribution. Similar reaction behaviours were observed in the case of the soluble metal methoxide catalyst, which was more sensitive to the initial moisture (the moisture sensitivity to each catalytic system is compared in Fig. S5†).
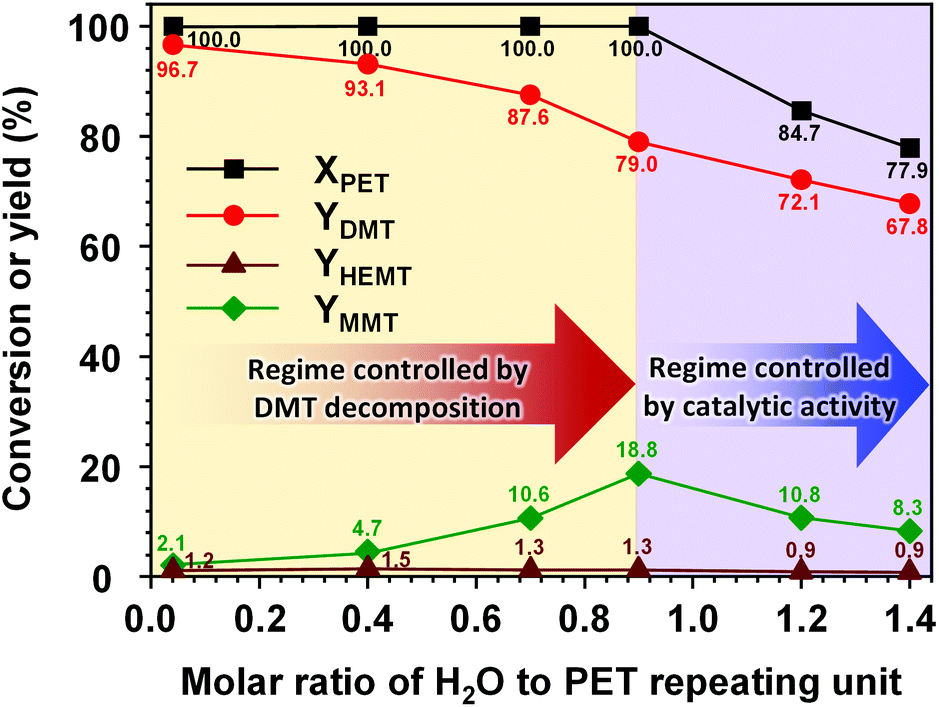 | ||
| Fig. 6 Effect of initial moisture contents on the product distribution of PET methanolysis at 25 °C for 24 h. The molar ratios of catalyst, methanol, and DCM to PET repeating unit were adjusted to be 0.2, 50, and 50, respectively. The molar ratio of water to PET repeating units varied from 0.1 to 1.4. | ||
A continuous increase in the molar ratio value of water to PET repeating units exceeding 0.9 (Nwater > 0.9) gave rise to a completely different reaction behaviour. Above this critical point, the reaction rate of PET decomposition was significantly lowered, and the overall product yield decreased. In this regime, the increase in the water molar amount decreased not only the production rate of MMT but also that of DMT. The results revealed that an excessive amount of water could dissolve a large fraction of K2CO3 and limit its catalytic activity in the reaction solution. The loss in catalytic activity limited both methanolysis and hydrolysis reactions. The experimental results revealed that the presence of water in the reaction solution significantly affected not only the selectivity of the desired product but also the catalytic activity. In addition, even a small moiety of water could take part in the further decomposition of DMT into hydrolysed products. This suggests that the pre-treatment of moisture removal from the reaction solution could be a crucial strategy for achieving an ideal conversion of PET into DMT via methanolysis and lowering the formation of by-products, which in turn could enable the efficient purification of the products, otherwise requiring several energy-intensive purification steps.
3.5. Mass-transfer limitation to methanolysis
During PET decomposition via solvolysis, the size, shape and degree of polymerization significantly affect the degradation behaviour and reaction rate.52 Catalytic methanolysis involves both heterogeneous and homogeneous reactions at the surface boundary of PET. The ideal control of manipulated variables, e.g., high stirring rate, may not help to overcome the mass transfer limitation caused by geometrical factors of the plastic wastes. Table 1 shows the methanolysis results for various feedstock materials possessing ester bonds connecting terephthalate and EG. Methanolysis was carried out under identical conditions described in the Experimental section (see section 2.2). Their reaction behaviours significantly varied, although all transesterifications occurred at the same ester bonds connecting terephthalate and EG. BHET was the smallest molecular unit of terephthalate connected to two moles of EG by two ester bonds, while its dimer possessed four ester bonds with two terephthalate and three EG molecules. BHET and its dimer completely decomposed into monomer products within a half hour, while most polymers required more than 18 h for complete decomposition. In contrast, only 63% of granular PET particles were degraded into monomer compounds within 24 h. In this case, complete PET decomposition was significantly delayed mainly due to several extrinsic factors, including a complex transport mechanism restricted by large particle sizes and a refractory PET structure characterized by high crystallinity (XRD patterns are shown in Fig. S6†). The methanolysis of finely ground PET particles was also tested to investigate the effect of particle size on the decomposition rate. Among the tested samples, the decompositions starting with single-layered PET bottle flakes did not show much discernible deviation from the reaction behaviour associated with the mass transfer limitation. The methanolysis of waste PET label samples basically showed a fairly good agreement with the decomposition behaviours of single-layered PET bottle flakes, but its decomposition resulted in a slightly smaller amount of DMT (3% less) attributable to the impurity of non-PET material. From the above hypothetical approach to the limitation, single-layered PET bottle flakes were practically adopted as a feedstock to study the further kinetics of PET methanolysis.| Feedstock | Dimension in average (mm) |
t
decomp
(h) |
Densitye (g cm−3) | X PET (%) | Y DMT (%) |
|---|---|---|---|---|---|
| a The sample contains 30% glass reinforcer. b BHET and its dimer are obtained from the hot water filtration of mixed BHET sample (Sigma-Aldrich BHET > 80%). c (t), (ϕ) and (h) indicates thickness, diameter and height, respectively. The dimension was determined by SEM analysis. d t decomp denotes a reaction time required for full conversion of PET. e A 25 ml pycnometer was used to determine polymer densities. | |||||
| PET bottle (single-layer) | 8 × 8 cut (t)c 0.18 | 14 | 1.34 | 100.0 | 93.1 |
| PET bottle (double-layer) | 8 × 8 cut (t) 0.39 | >24 | 1.34 | 91.3 | 85.5 |
| Coloured PET labels | 5 × 5 cut (t) 0.04 | 14 | 1.33 | 100.0 | 90.2 |
| PET bottle (powder) | 0.001–0.01 mm | 12 | 1.34 | 100.0 | 92.8 |
| PET granulesa | (ϕ)c 1.8 × (h)c 3.0 | >24 | 1.69 | 63.0 | 58.8 |
| BHET monomerb | Powder | 0.5 | 1.38 | 100.0 | 95.7 |
| BHET dimerc | Powder | 0.4 | 1.37 | 100.0 | 95.3 |
SEM analysis was used to further investigate detailed structural changes in PET during degradation. Fig. 7 presents the SEM snapshots representing the morphology changes during PET degradation by catalytic methanolysis. A similar decomposition process has been described by Zhou et al.53 during other solvent-aided PET transesterification processes. As depicted in Fig. 7(a), fresh PET (double-layered waste PET bottle flakes in Table 1) has a relatively flat and smooth surface, and its lateral size was approximately 0.39 mm. After 12 h (Fig. 7(b)), several voids appeared on the surface, and the thickness of PET flakes decreased to 0.246 mm. With methanolysis for 24 h (Fig. 7(c)), 91.3% of PET was degraded into monomer products, and a similar ratio of size reduction was observed in the direction perpendicular to the flat PET surface (0.035 mm thick). The one-dimensional size reduction of PET samples indicated that decomposition dominantly occurred at the surface of the polymer substrate and continued to form a number of pores through the internal polymer matrix. The development of internal pores increased the surface area in contact with the reaction solution. As a consequence, the rate of polymer decomposition was drastically accelerated.
 | ||
| Fig. 7 SEM images of the polymer surface (left) and cross-section (right) after methanolysis of double-layered waste PET for (a) 0 hours, (b) 12 hours and (c) 24 hours at 25 °C. | ||
3.6. Catalytic methanolysis reactions with soluble and insoluble metal salts
Fig. 8 illustrates the reaction behaviours of methanolysis catalysed by homogeneous and heterogeneous catalytic solutions. López-Fonseca et al. claimed that the cation plays a catalytic role in the transesterification of PET in their kinetic study of PET glycolysis with metal carbonates.11 Although the decomposition behaviours were expected to vary with the thermodynamic properties of catalysts in the reaction solution, the catalytic activity difference between K2CO3 and CH3OK was measured by applying equivalent amounts, based on the molar numbers of cations and anions, to the same reaction solutions. CH3OK, which is completely soluble in a mixture of methanol and DCM, showed a high initial activity. DMT yields reached 93.0% after 9 h for the addition of 0.2 mole CH3OK per PET repeating unit and 92.6% after 5 h for 0.4 mole of CH3OK. However, DMT started to rapidly decompose into MMT by virtue of hydrolysis.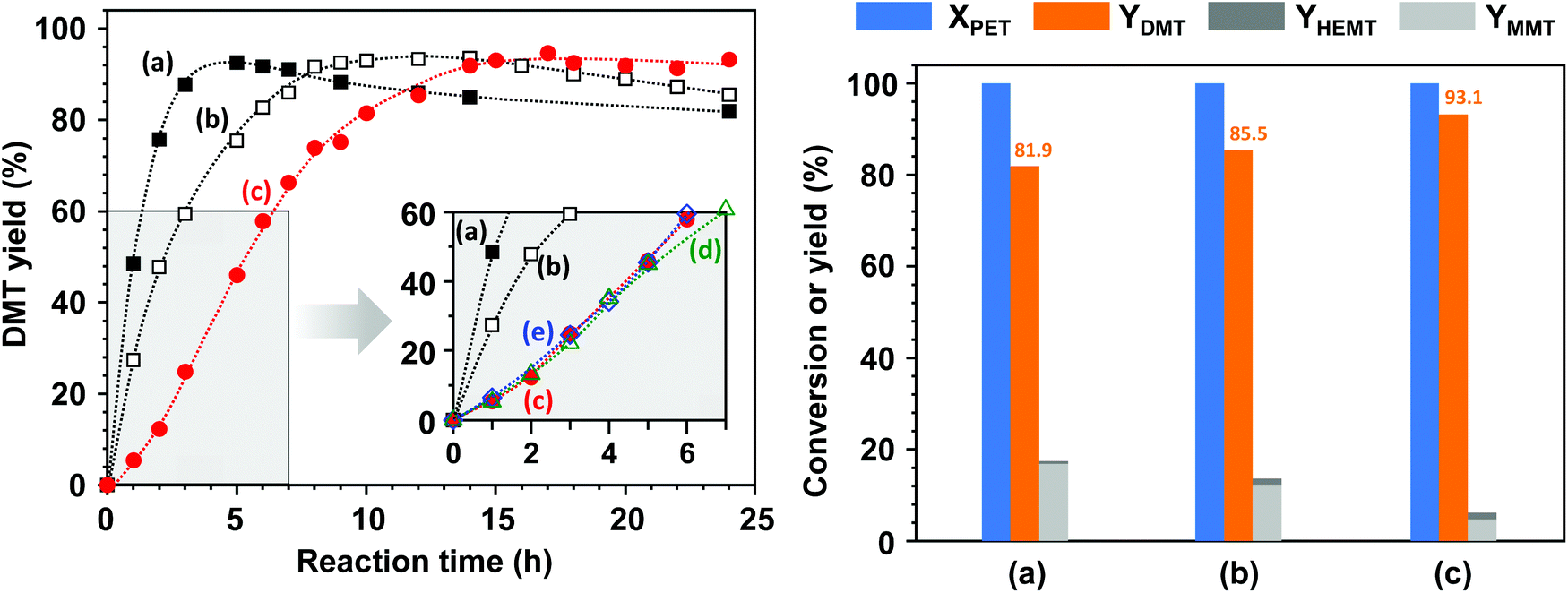 | ||
| Fig. 8 Production rate of DMT (left) and the final product distributions (right) after 24 h methanolysis catalysed by (a) 0.4 mol CH3OK, (b) 0.2 mol CH3OK, (c) 0.2 mol K2CO3 without any pre-treatment, (d) 0.2 mol K2CO3 after PET soaking in a mixture of solvents for 12 h, and (e) 0.2 mol K2CO3 premixed in a mixture of solvents for 12 h prior to the initiation of reaction by adding PET. The molar ratios of catalyst, methanol, and DCM to PET repeating units were adjusted to be 0.2, 50, and 50, respectively. | ||
In contrast to methanolysis by CH3OK, solid K2CO3 showed a slightly delayed reactivity initially, and decomposition proceeded slowly. To examine a possible cause of the peculiar induction period at an early stage, the initial reaction rate was repeatedly monitored by including two pre-treatment steps: (1) sufficiently long soaking in a mixture of solvents to inspect the effect of polymer swelling or solvation and (2) premixing of K2CO3 with solvents to probe in situ catalytic activation, e.g., the formation of reactive methoxide (CH3OK) by deprotonation of methanol. The corresponding results are denoted by (d) and (e) in the inset of Fig. 8 (left); additional pre-treatment steps did not noticeably alter the reaction behaviours. In addition, the decomposition process into lower molecular weight fragments was unlikely to contribute to the process because any smaller fractions, including dimers and oligomers, were not detected during catalytic methanolysis. As progressive surface erosions were found (depicted in Fig. 7), the delayed reactivity was attributed to the multiple processes by which polar solvent induced the dielectric relaxation of polymer substrate and then facilitated the mass transfer of solvent molecules to the polymer matrix, and the metal salt formed a molecular complex on the PET surface, leading to a gradual decomposition. Nevertheless, a DMT yield higher than 92% was achieved after 14 h, but thereafter, no significant degradation into hydrolysed products was observed even after prolonged exposure to the catalytic solution containing moisture for 24 h. The final product yields are compared in the right bar chart of Fig. 8; the DMT yield was approximately 93.1% as a result of catalytic methanolysis by K2CO3, while only 85.5% and 81.9% were attainable for the reaction in the presence of 0.2 mole and 0.4 mole of CH3OK, respectively. The results indicate that a catalytic system with solid K2CO3 has a high potential to provide an efficient strategy to convert PET to DMT in a stable and selective manner.
3.7. Development of a kinetic model for solvent-aided catalytic methanolysis
In general, the depolymerization of PET is a complex process that involves multiple chemical reactions and the continuous transport of heat and mass through a multiphase reaction medium. Nevertheless, several efforts have been made to develop a simplified kinetic model describing the reaction behaviour during PET decomposition. The development of the lumped kinetic model is the simplest approach to estimate apparent rate parameters for PET decomposition.54 However, the model cannot be used as a useful tool for the design or development of new processes, mainly because of its failure to predict overall reaction behaviours. To resolve these errors, mathematical correlations allowing the best fit to the empirical data are often used as a predictive model. Typical examples of the correlation include the solid-state model52 and the shrinking core model.55 In the correlation, a descriptive mathematical function can be chosen to correlate experimental data, although the predicted parameters lack physical meaning or are impractical for process description in detail. Although generalized kinetic models have not yet been established, there have been several efforts to mechanistically describe the decomposition behaviours of PET. Genta et al.28 developed a kinetic model describing the multiple steps of PET depolymerization in supercritical methanol. More recently, they developed a continuous kinetic model incorporating the distribution functions of molecular weight changes in polymers during the supercritical methanolysis of PET.47 Although these efforts provide a plausible prediction for decomposed product distribution, their application has been limited to a few particular systems in which the depolymerization steps into smaller fragments are relatively slow.Based on the observed reaction behaviours and the proposed reaction mechanism (presented in Scheme 1), a realistic kinetic model, mechanistically describing the multiple reaction steps in catalytic methanolysis, was developed. Polymeric compounds were decomposed into monomer products by two consecutive reaction steps.
At an initial stage of the catalytic methanolysis of PET, a delayed decomposition behaviour was observed. Based on this observation, it was assumed that the transformation into readily degradable polymer matrices, denoted by ‘PET*’, is presumably initiated by intramolecular rearrangement.
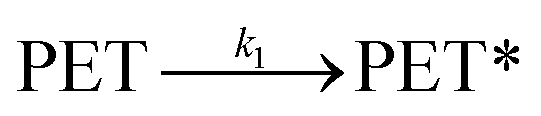 | (6) |
After the induction period, the direct decomposition of PET into the monomer product starts to proceed. In a conventional methanolysis process at a high reaction temperature (>150 °C), significant concentrations of oligomers and dimers were measured as reactive intermediates.28,47 In contrast, DMT was obtained as a major dominant product, and interestingly, no partially decomposed products were detected during the catalytic methanolysis systems in this study. Thus, it is assumed that the decomposition of activated polymer proceeded in a single step.
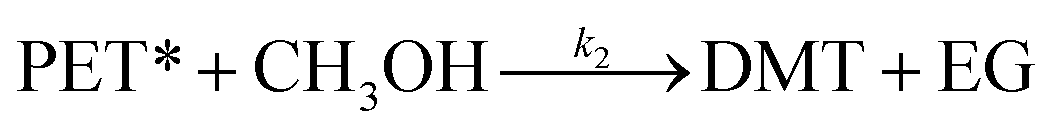 | (7) |
The reversibility of the above reaction was also explored by formulating equivalent amounts of DMT and EG in identical catalytic solutions. During the observation, no polymeric nuclei or repolymerized compounds were detected. Instead, a much lower molar amount of HEMT than DMT began to appear in the solution. Thus, a reverse reaction of eqn (7) was unlikely to occur. In the presence of the catalyst, the glycolysis of DMT produced HEMT until it reached equilibrium.
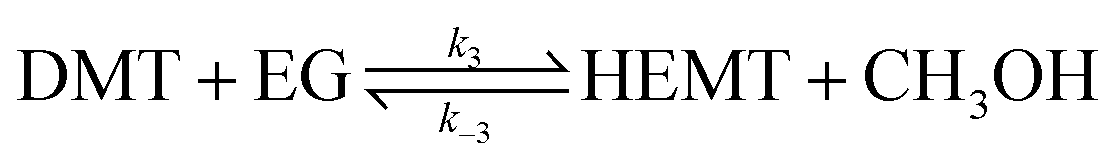 | (8) |
The decomposition of DMT is not a desired reaction step in methanolysis. In the presence of moisture, monomer products are further decomposed into hydrolysed compounds.
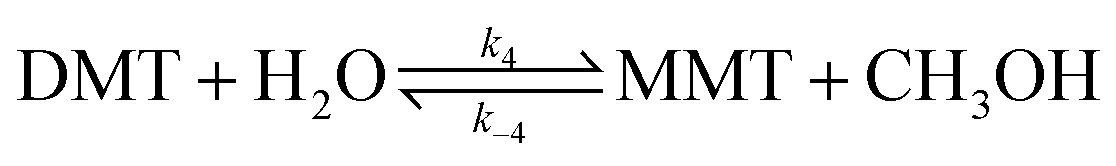 | (9) |
With a large amount of metallic catalyst or water, MMT can be further decomposed into dipotassium terephthalate, which can be converted into terephthalic acid by acid titrations substituting metal cations with protons.
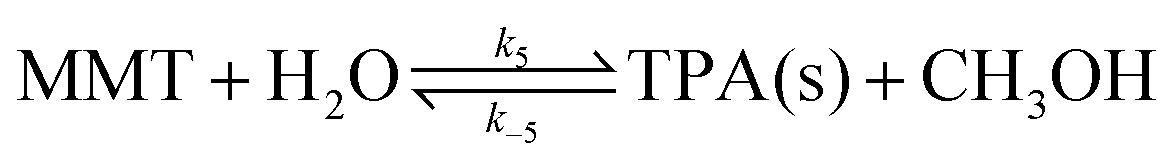 | (10) |
Dipotassium terephthalate, produced from the successive hydrolysis of DMT, was completely insoluble in the reaction solution. The second hydrolysis proceeded at an extremely slow rate only when an equivalent amount of catalyst to PET was present in the reaction solution. Thus, the reaction steps, denoted by k5 and k−5, were ignored in the reaction model.
The kinetic model was compared with all experimental data measured during PET decomposition. The overall reaction mainly consisted of PET decomposition, equilibrium with HEMT and hydrolysis steps with DMT. It was assumed that all reaction steps were first order with respect to each involved chemical species and independent of one another. Arrhenius equations were used to calculate rate coefficients and combined with the kinetic model to correlate a series of experimental data measured at different reaction temperatures.
| ki = ki,0exp[−Ei/(RT)] | (11) |
Based on this hypothesis, a kinetic model for DMT decomposition was first investigated to acquire rate parameters for the formation of by-products. Two series of experiments were repeatedly carried out to interpret the decomposition pathways in a reaction temperate range of 10–35 °C. In the first experimental set, only DMT was initially added to the hydrous catalytic solution of methanol and DCM (the molar ratios of catalyst, methanol, and DCM to DMT were adjusted to be 0.2, 50, and 50, respectively). The initial molar amount of water was 0.4. As expected, slow hydrolysis of DMT was observed in the observed temperature range. As a result of hydrolysis, only partially hydrolysed product (K-MMT) was detected as a decomposed product, which was distributed in both solution and precipitation (the detailed decomposition profiles are described in Fig. S7†). In the next experimental set, an equimolar amount of EG to DMT was added to the catalytic solution, and the appearance of the products was continuously monitored. This case is more practical for building a kinetic model for the overall reaction since nearly equimolar amounts of DMT and EG are released from PET methanolysis. In contrast to the previous observation, mixed reaction behaviours of glycolysis and hydrolysis, which are described by eqn (8) and (9), respectively, were observed in a temperature range of 10–35 °C. The observed data and the kinetic model fittings are presented in Fig. 9. Compared to the previous case (no initial concentration of EG), a slightly lower hydrolysis rate was detected, while a very small amount of HEMT appeared and rapidly reached a constant value. The equilibrium concentration of HEMT to DMT was approximately 0.0152 (K3 = k3/k−3 ≈ [HEMT]eq/[DMT]eq). It is notable that the equilibrium concentration of HEMT was found to be weakly dependent on the reaction temperature. Apart from the correlation, the reactions starting with uneven molar amounts of EG and DMT (or limiting one of reactants) were also tested to validate the current assumption of the first-order reaction with respect to each reactant (the results are shown in Fig. S8†).
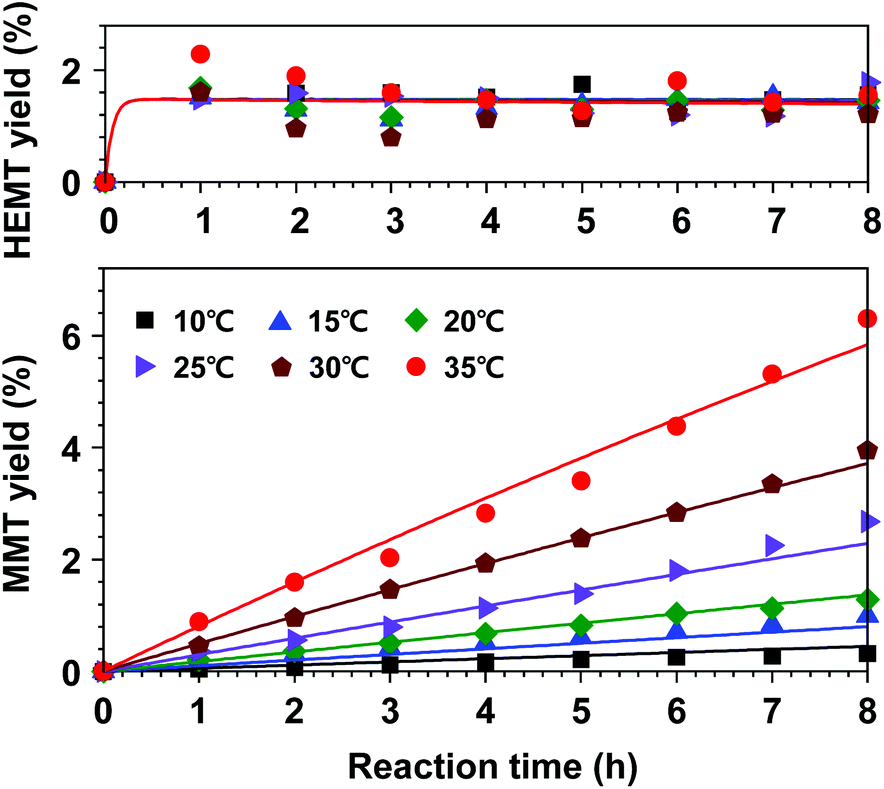 | ||
| Fig. 9 Kinetic model fit (lines) for DMT decomposition to MMT and equilibrium with HEMT. The molar ratios of catalyst, EG, methanol, and DCM to DMT were set to be 0.2, 1.0, 50, and 50, respectively. | ||
Next, we built a kinetic model for the overall reaction. The kinetic model for DMT decomposition and the estimated parameters (k3, k−3, and k4) were combined with the kinetic model of PET decomposition to fully describe the overall reaction. The experimental data for product yields in the PET decomposition are plotted in Fig. 10 along with the kinetic model fit. The model shows an excellent correlation for the complete experimental set of all reaction temperatures. The estimated kinetic parameters are summarized in Table 2.
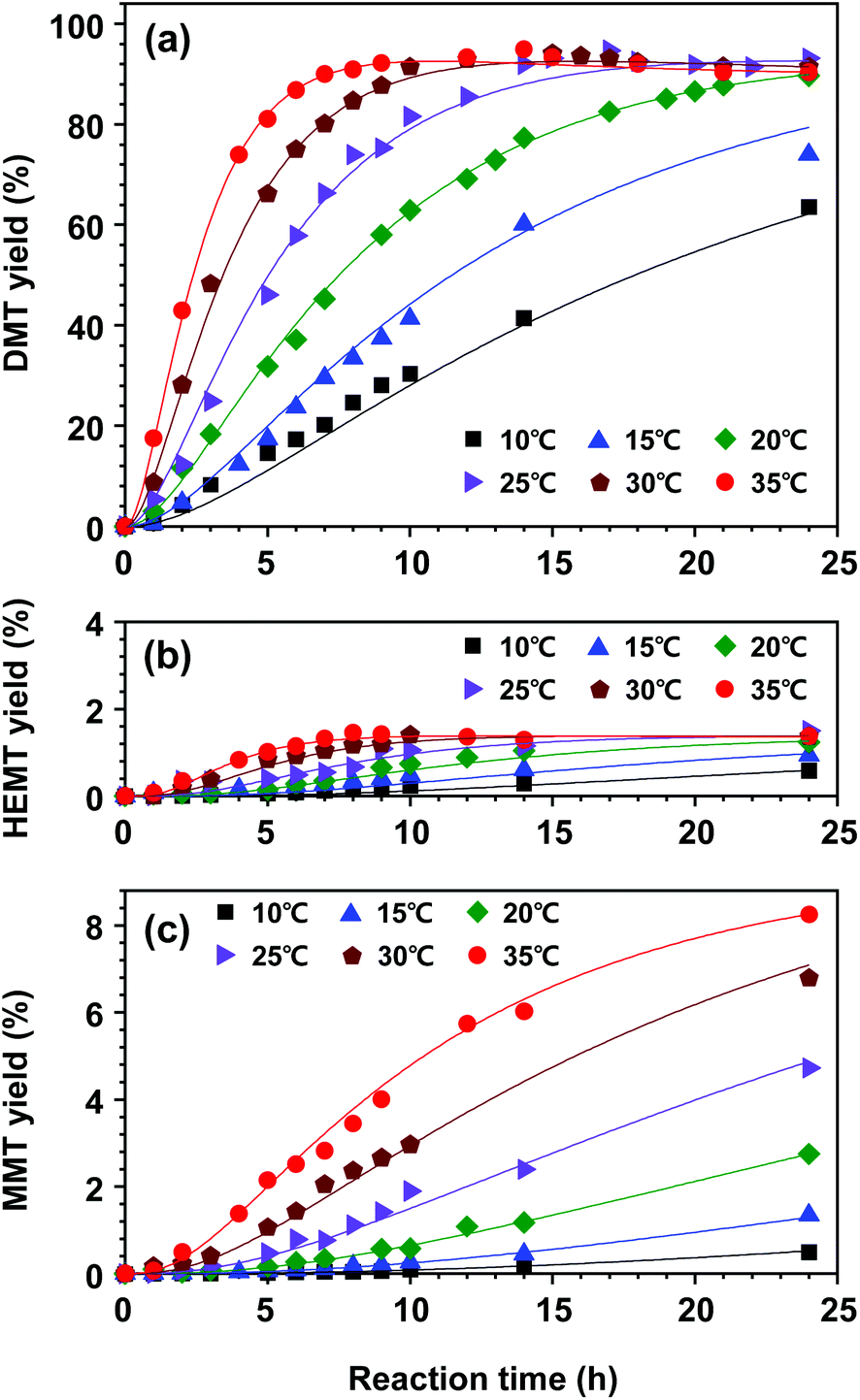 | ||
| Fig. 10 Evolution of monomer products, (a) DMT, (b) HEMT, and (c) MMT, from the catalytic methanolysis of PET at different reaction temperatures (10–35 °C). Solid lines correspond to the kinetic model for the overall reaction. | ||
| Reaction step, i | Arrhenius parameters | |
|---|---|---|
| log10ki,0 (h−1) | E i (kJ mol−1) | |
| a The rate coefficients are lumped with the concentration of solvent reactant because the concentration remains almost constant; ki = k′iC0. b The measured equilibrium constant was used to describe relative rates of association and dissociation between DMT and EG. | ||
| 1 | 6.5 | 38.1 |
| 2 | 11.0 | 66.5 |
| 4a | 11.5 | 77.2 |
| −4a | 18.3 | 114.9 |
| 3 & −3a | K 3 = 1.52 × 102 | |
The initial reaction rate of PET decomposition increased with an increased reaction rate. Obviously, a higher temperature helped shorten the reaction time to fully decompose applied PET flasks. Under the reaction conditions of 20–35 °C, the final DMT yield after 24 h of reaction was found to be almost independent of the reaction temperature. The results indicated that the high selectivity of DMT could be conserved such that methanolysis proceeded dominantly, but further decomposition of DMT was not significantly promoted by the catalytic system. However, DMT significantly decomposed into hydrolysis products if the reaction system was exposed to a high reaction temperature for a long period, which led to an irreversible consumption of the metal salt catalyst. Although the hydrolysis of DMT was relatively slow and the equilibrium concentration of HEMT was very low, the reaction temperature could significantly reduce the amount of DMT. This tendency could be found more clearly in PET methanolysis at a higher temperature of 40–60 °C (the effect of temperature on product yields is presented in Fig. S9†). As seen in Fig. 10(c), the final MMT yield varied from 0.5% at 10 °C to 8.3% at 35 °C. The result revealed that PET methanolysis at a lower temperature was beneficial for attaining a higher yield of DMT, but an optimal reaction time should be considered. To further investigate the catalytic reaction system providing a high selectivity of DMT, several catalytic methanolysis systems, in which the co-solvent was replaced by other polar solvents, were tested at 60 °C, and the observed results are presented in Fig. S10.†
3.8. Proposed reaction mechanism for solvent-aided catalytic methanolysis
Based on the aforementioned experimental results, the possible reaction mechanism for PET solvent-aided catalytic methanolysis in the presence of K2CO3 catalyst is delineated in Scheme 2. It is generally accepted that the transesterification of PET is attributed to the Lewis acid–base synergistic effect. Similarly, potassium cations, which basically work as Lewis acids, protonate the carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in both ester groups, functionalize the carbonyl group to be more electrophilic and vulnerable to nucleophilic attack by methanol. Meanwhile, carbonate anion CO32− pulls the hydrogens of the hydroxyl group in methanol towards its side, and accordingly, the oxygens in methanol possess high electron density and are prone to attack carbonyl groups in ester bonds. Thereafter, both ester and hydroxyl bonds are broken, leading to the molecular abstract from the polymer substrate by making a new C–O bond between the carbonyl in PET and the oxygen in methanol. It is believed that the above catalytic transesterification proceeds on the ester groups located on both sides of the terephthalate. As a consequence, DMT is selectively formed as a major product of methanolysis. The formation of DMT also accompanies the release of the same molar amount of free EG in the reaction solution.
O) in both ester groups, functionalize the carbonyl group to be more electrophilic and vulnerable to nucleophilic attack by methanol. Meanwhile, carbonate anion CO32− pulls the hydrogens of the hydroxyl group in methanol towards its side, and accordingly, the oxygens in methanol possess high electron density and are prone to attack carbonyl groups in ester bonds. Thereafter, both ester and hydroxyl bonds are broken, leading to the molecular abstract from the polymer substrate by making a new C–O bond between the carbonyl in PET and the oxygen in methanol. It is believed that the above catalytic transesterification proceeds on the ester groups located on both sides of the terephthalate. As a consequence, DMT is selectively formed as a major product of methanolysis. The formation of DMT also accompanies the release of the same molar amount of free EG in the reaction solution.
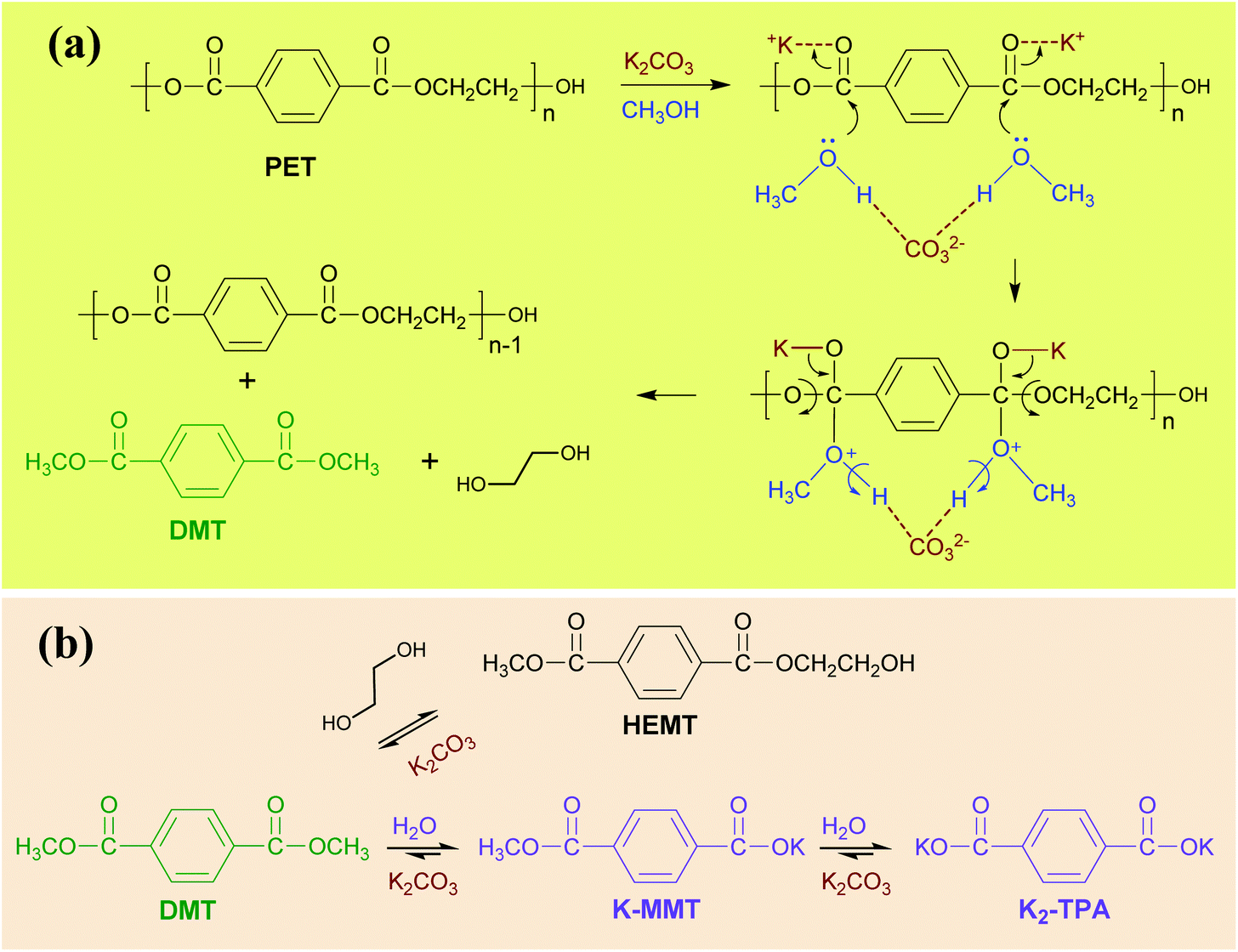 | ||
| Scheme 2 Proposed mechanism for PET decomposition. The decomposition comprises two phases of (a) PET methanolysis and (b) DMT decomposition. | ||
DMT and EG molecules, released from PET methanolysis, undergo recombination to generate HEMT. HEMT hardly develops into a high molecular weight due to successive glycolysis. The production of HEMT is totally reversible, and the corresponding equilibrium constant is a weak function of reaction temperature. The equilibrium process of HEMT in the reaction solution can be evidently monitored by kinetic NMR measurement of the reaction system (presented in Fig. S11†), which characterizes the evolution of soluble products in the reaction solution. In the combined presence of the transesterification catalyst and moisture, the hydrolysis of DMT can be catalysed to form hydrolysed products such as MMT and TPA. Since K2CO3 particles are present in the reaction solution, metal cation exchanged forms are more preferentially obtained as hydrolysed by-products. At a low concentration of catalyst, K-MMT is produced as a major by-product. DMT hydrolysis can continue until a majority of the initial moisture is consumed. If the total amount of K-MMT goes beyond the solubility limit of the solution, it starts to precipitate and accumulates in the solid part. The addition of a large amount of catalyst, which is comparable to the molar amount of PET repeating unit, can result in the appearance of K2-TPA, which is mostly insoluble in the catalytic reaction solution.
4. Conclusion
In this work, we developed a new catalytic reaction system of methanolysis that selectively converts PET to DMT. In the reaction system, potassium carbonate, which is insoluble in a reaction mixture, was used as a catalyst, and a mixture of methanol and polar aprotic solvent was used to achieve the complete decomposition of PET within 24 h under ambient conditions. In a typical condition (the reaction temperature was 25 °C and the molar ratios of catalyst, moisture, methanol, and DCM to PET repeating unit were 0.2, 0.4, 50, and 50), a single layer of waste PET bottle flakes were fully decomposed into monomers within 14 h, and the DMT yield reached 93.1% during methanolysis for 24 h. In a parallel comparison with other recent technologies, our catalytic system exhibited a superior catalytic performance, which could be characterized by a high DMT yield, a low moisture sensitivity, a long-term stability against hydrolysis, and an easy material recyclability. Several parametric studies were carried out to unveil the detailed reaction mechanism and to investigate the effects of temperature, catalytic amount, initial moisture level, molar composition of solvent and feedstock geometry. Based on the experimental observations, a kinetic model mechanistically describing PET decomposition was developed. This model included a description of the initial catalytic methanolysis and DMT decomposition into by-products. The model was compared with experimental data measured at a temperature range of 10–35 °C. The PET decomposition proceeded with two reaction steps in series. The first step was featured by intermediate polymer matrices, which exhibited delayed decomposition behaviour. The second step provided a depolymerization step with a higher activation energy of 66.5 kJ mol−1, which was estimated by the model. It was found that the hydrolysis of DMT could also be promoted by the transesterification catalyst, and the reaction rate was increased by elevating the reaction temperature. In our catalytic reaction system, DMT degradation consumed catalyst to produce by-products in metallic salt forms via two consecutive hydrolysis steps. PET decomposition at a low temperature could benefit the high selectivity of DMT, but a decrease in activity significantly delayed the decomposition of PET. Alternatively, an ideal DMT yield (up to 98.5%) could be achieved by controlling the initial moisture level.This study demonstrates a new catalytic methanolysis route to convert PET into recyclable monomer products in an efficient and economical manner. Owing to the utilization of inexpensive green catalysts, easy scale-up, low-energy consumption, and a low separation cost, the proposed reaction system may provide a feasible option to achieve a circular plastic economy and in turn mitigate ongoing threats to the global environment from waste plastics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ministry of Trade, Industry & Energy (MOTIE), Korea Institute for Advancement of Technology (KIAT) through the Develoment Program for Industrial Innovative Technology and Infrastructure (Grant Number: P0014836) and by the Institutional Research Program of Korea Research Institute of Chemical Technology (KRICT, Project Number: KK2011-00).References
- McKinsey Online Article, https://www.mckinsey.com/industries/chemicals/our-insights/how-plastics-waste-recycling-could-transform-the-chemical-industry, (accessed October, 2020).
- J. Gigault, A. Halle, M. Baudrimont, P.-Y. Pascal, F. Gauffre, T.-L. Phi, H. El Hadri, B. Grassl and S. Reynaud, Environ. Pollut., 2018, 235, 1030–1034 CrossRef CAS PubMed.
- Trends in global CO2 and total greenhouse gas emissions, https://www.pbl.nl/sites/default/files/downloads/pbl-2017-trends-in-global-co2-and-total-greenhouse-gas-emissons-2017-report_2674.pdf, (accessed October, 2020).
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef.
- F. Awaja and D. Pavel, Eur. Polym. J., 2005, 41, 1453–1477 CrossRef CAS.
- H. Zhang and Z.-G. Wen, Waste Manage., 2014, 34, 987–998 CrossRef CAS.
- I. Vollmer, M. J. Jenks, M. C. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- J. Chen, C. Ou, Y. Hu and C. Lin, J. Appl. Polym. Sci., 1991, 42, 1501–1507 CrossRef CAS.
- U. Vaidya and V. Nadkarni, J. Appl. Polym. Sci., 1989, 38, 1179–1190 CrossRef CAS.
- J. R. Campanelli, M. Kamal and D. Cooper, J. Appl. Polym. Sci., 1993, 48, 443–451 CrossRef CAS.
- M. Collins and S. Zeronian, J. Appl. Polym. Sci., 1992, 45, 797–804 CrossRef CAS.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil and J. E. Rice, Polym. Chem., 2013, 4, 1610–1616 RSC.
- A. Jain and R. Soni, J. Polym. Res., 2007, 14, 475–481 CrossRef CAS.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- C. Pudack, M. Stepanski and P. Fässler, Chem. Ing. Tech., 2020, 92, 452–458 CrossRef CAS.
- A. B. Hungria, R. Raja, R. D. Adams, B. Captain, J. M. Thomas, P. A. Midgley, V. Golovko and B. F. Johnson, Angew. Chem., Int. Ed., 2006, 45, 4782–4785 CrossRef CAS PubMed.
- H. Jia, Z. Yang, X. Yun, X. Lei, X. Kong and F. Zhang, Ind. Eng. Chem. Res., 2019, 58, 22702–22708 CrossRef CAS.
- J. Scheirs, Polymer recycling: science, technology and applications, John Wiley & Sons Ltd, 1st edn, 1998 Search PubMed.
- G. Hans, H. Walter and M. Heinz, US Pat, 3403115A, 1968 Search PubMed.
- W. E. Toot Jr. and B. R. DeBruin, US Pat, 5414022A, 1995 Search PubMed.
- M. Genta, T. Iwaya, M. Sasaki, M. Goto and T. Hirose, Ind. Eng. Chem. Res., 2005, 44, 3894–3900 CrossRef CAS.
- Y. Yang, Y. Lu, H. Xiang, Y. Xu and Y. Li, Polym. Degrad. Stab., 2002, 75, 185–191 CrossRef CAS.
- M. N. Siddiqui, H. H. Redhwi and D. S. Achilias, J. Anal. Appl. Pyrolysis, 2012, 98, 214–220 CrossRef CAS.
- J.-T. Du, Q. Sun, X.-F. Zeng, D. Wang, J.-X. Wang and J.-F. Chen, Chem. Eng. Sci., 2020, 115642 CrossRef CAS.
- H. Kurokawa, M.-A. Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- S. Liu, Z. Wang, L. Li, S. Yu, C. Xie and F. Liu, J. Appl. Polym. Sci., 2013, 130, 1840–1844 CrossRef CAS.
- S. Mishra and A. S. Goje, Polym. Int., 2003, 52, 337–342 CrossRef CAS.
- J. L. Hedrick, R. C. Pratt and R. M. Waymouth, US Pat, 8309618B2, 2012 Search PubMed.
- A. Essaddam and F. Essaddam, US Pat, 10252976B1, 2019 Search PubMed.
- B. Liu, X. Lu, Z. Ju, P. Sun, J. Xin, X. Yao, Q. Zhou and S. Zhang, Ind. Eng. Chem. Res., 2018, 57, 16239–16245 CrossRef CAS.
- G. Güçlü, A. Kaşgöz, S. Özbudak, S. Özgümüs and M. Orbay, J. Appl. Polym. Sci., 1998, 69, 2311–2319 CrossRef.
- V. Sharma, P. Parashar, P. Srivastava, S. Kumar, D. Agarwal and N. Richharia, J. Appl. Polym. Sci., 2013, 129, 1513–1519 CrossRef CAS.
- L.-C. Hu, A. Oku, E. Yamada and K. Tomari, Polym. J., 1997, 29, 708–712 CrossRef CAS.
- H. Essaddam, US Pat, 9550713B1, 2017 Search PubMed.
- M. Imran, B.-K. Kim, M. Han, B. G. Cho and D. H. Kim, Polym. Degrad. Stab., 2010, 95, 1686–1693 CrossRef CAS.
- K. Mukai and M. Nakashima, US Pat, 7959807B2, 2011 Search PubMed.
- L. Rudolf, W. Gerhard and N. Clemens, US Pat, 3321510A, 1967 Search PubMed.
- A. A. Naujokas and K. M. Ryan, US Pat, 5501528A, 1991 Search PubMed.
- B. R. DeBruin, A. A. Naujokas and W. J. Gamble, US Pat, 5414022A, 1995 Search PubMed.
- M. Genta, M. Goto and M. Sasaki, J. Supercrit. Fluids, 2010, 52, 266–275 CrossRef CAS.
- T. Corrales, C. Peinado, P. Bosch and F. Catalina, Polymer, 2004, 45, 1545–1554 CrossRef CAS.
- J. Menegotto, P. Demont, A. Bernes and C. Lacabanne, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3494–3503 CrossRef CAS.
- C. L. Forryan, R. G. Compton, O. V. Klymenko, C. M. Brennan, C. L. Taylor and M. Lennon, Phys. Chem. Chem. Phys., 2006, 8, 633–641 RSC.
- M. Sim and M. Han, J. Chem. Eng. Jpn., 2006, 39, 327–333 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376–5394 RSC.
- L. Zhou, X. Lu, Z. Ju, B. Liu, H. Yao, J. Xu, Q. Zhou, Y. Hu and S. Zhang, Green Chem., 2019, 21, 897–906 RSC.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2018, 7, 3292–3300 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03536j |
| This journal is © The Royal Society of Chemistry 2021 |