
Tuning the coordination number of Fe single atoms for the efficient reduction of CO2†
Huihuang
Chen‡
 a,
Xu
Guo‡
a,
Xiangdong
Kong
a,
Yulin
Xing
a,
Yan
Liu
a,
Bolong
Yu
a,
Qun-Xiang
Li
a,
Zhigang
Geng
*a,
Rui
Si
*b and
Jie
Zeng
*a
a,
Xu
Guo‡
a,
Xiangdong
Kong
a,
Yulin
Xing
a,
Yan
Liu
a,
Bolong
Yu
a,
Qun-Xiang
Li
a,
Zhigang
Geng
*a,
Rui
Si
*b and
Jie
Zeng
*a
aHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Strongly-Coupled Quantum Matter Physics, Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gengzg@ustc.edu.cn; zengj@ustc.edu.cn
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, P. R. China. E-mail: sirui@sinap.ac.cn
First published on 29th September 2020
Abstract
CO2 electroreduction into valuable products holds great promise for energy supply and environmental remediation but remains a challenge due to the lack of high-performance electrocatalysts. Herein, we developed an efficient strategy to prepare highly active Fe single-atom catalysts (Fe–N5/Fe–N6) by tuning the coordination number of Fe with N towards CO2 electroreduction. The faradaic efficiency of CO for Fe–N5 exceeded 90% ranging from −0.35 to −0.65 V versus the reversible hydrogen electrode (vs. RHE) towards CO2 electroreduction. The turnover frequency of CO for Fe–N5 reached 5006 h−1 at −1.05 V vs. RHE, which was 3.8 times that (1324 h−1) of Fe–N6. Besides, CO was generated at an overpotential as low as 50 mV over Fe–N5, smaller than that (90 mV) over Fe–N6. Density functional theory calculations demonstrated that Fe–N5 facilitated the adsorption and activation of CO2 to form the *COOH intermediate, thus leading to superior selectivity and activity for CO2 electroreduction.
1. Introduction
The ever-increasing combustion of depletable fossil fuels has resulted in a serious energy crisis and environmental pollution worldwide due to the massive release of atmospheric carbon dioxide (CO2).1,2 CO2 electroreduction into value-added carbon products is considered as an ideal approach to mitigate the energy and environmental issues.3,4 To achieve high energy efficiency and feasible scalability, the reaction must occur rapidly and selectively at low overpotentials. However, this technology is still hindered by the high overpotential, low activity, and limited product selectivity of electrocatalysts nowadays, arising from the chemical inertness of linear CO2 and the thermodynamically favorable competing hydrogen evolution reaction.5–12 To overcome these drawbacks, it is crucial to develop efficient and robust catalysts towards CO2 electroreduction.Carbon monoxide (CO) has emerged as one of the most practical products of CO2 electroreduction owing to its indispensable role in the preparation of synthetic liquid fuels via the industrial Fischer–Tropsch process.13 The benchmark electrocatalysts for the CO2-to-CO conversion are Ag- and Au-based materials, which demonstrated over 90% faradaic efficiency for CO formation with an overpotential of 240 mV,14–16 but their high price, scarcity and inadequate activity warrant further research endeavors to explore low-cost and Earth-abundant nonprecious-metal catalysts to replace those noble-metal catalysts. Recently, single-atom catalysts (SACs) have been identified as promising candidates in the field of electrochemical conversion processes.17–21 This is because the unique features of SACs such as tailorable electronic structure, maximized atomic utilization, and unsaturated coordination environment render them intriguing properties for catalysis dramatically different from their bulk counterparts.22 To this end, various strategies (e.g. defect engineering, structural manipulation, and interfacial regulation) have been proposed to develop nonprecious-metal (Fe, Co, Ni, and Sb) SACs for the CO2-to-CO conversion.23–31 Nevertheless, their electrocatalytic performance still suffers from either the large overpotential or the tradeoff between high faradaic efficiency and appreciable current density.32–34 Typically, the faradaic efficiency of CO2 electroreduction drops rapidly with increasing potential in order to deliver reasonable current density. In such a context, it remains a challenge to develop robust nonprecious-metal SACs to address the aforementioned two limitations for the practical implementation of CO2 electroreduction.
Herein, we developed an efficient strategy to prepare highly active Fe SACs (Fe–N5/Fe–N6) by tuning the coordination number of Fe with N towards CO2 electroreduction. The mass loading of Fe in Fe–N5 and Fe–N6 was determined to be 0.57% and 0.58%, respectively, by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The faradaic efficiency (FE) of CO (FECO) for Fe–N5 exceeded 90% within a wide potential window from −0.35 to −0.65 V versus the reversible hydrogen electrode (vs. RHE). Meanwhile, the turnover frequency (TOF) of CO (TOFCO) for Fe–N5 reached 5006 h−1 at −1.05 V vs. RHE, which was 3.8 times as high as that (1324 h−1) for Fe–N6. Besides, CO was generated at an overpotential as low as 50 mV over Fe–N5, smaller than that (90 mV) over Fe–N6. Experimental results and density functional theory (DFT) calculations demonstrated that Fe–N5 facilitated the adsorption and activation of CO2 to form the key *COOH intermediate, thus leading to the superior selectivity and activity for CO2 electroreduction. Our findings highlight the significance of regulating the coordination number of active centers in SACs to simultaneously enhance the catalytic activity and selectivity towards CO2 electroreduction.
2. Experimental methods
2.1. Materials and chemicals
Anhydrous ferric chloride (FeCl3, 99%, Sigma), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%, Aladdin), 2-methylimidazole (2-mIm, 99%, Sigma), potassium hydroxide (KOH, 99%, Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China)), potassium bicarbonate (KHCO3, 99%, Sigma), iron nanoparticles (Fe NPs, 99%, Sigma), ferric oxide (Fe2O3, 99%, Sigma), ferriferrous oxide (Fe3O4, 99%, Sigma), methanol (99.8%, Fisher), and ethanol (99.8%, Fisher) were used as received without further purification. Carbon paper, Nafion 115 membrane, and Nafion solution (5% wt%) were supplied by Sigma-Aldrich. Ultrapure Millipore water (18.2 MΩ cm, Milli-Q, Millipore Co.) was used in all experiments.2.2. Preparation of catalysts
In a typical synthetic process of Fe-doped ZIF-8, 0.09 g of FeCl3 and 5.03 g of Zn(NO3)2·6H2O were dissolved in 480 mL of methanol containing 11.1 g of 2-mIm. Then the mixture was kept at 25 °C for 12 h. The resultant solid products were separated by centrifugation and thoroughly washed with methanol five times, followed by drying at 60 °C under vacuum overnight. The synthetic procedure of ZIF-8 was the same as the above except no FeCl3 was introduced. The as-obtained Fe-doped ZIF-8 was placed in a tube furnace and pyrolyzed (900 °C for Fe–N5, 800 °C for Fe–N6) for 6 hours with a heating rate of 5 °C min−1 under an Ar flow (100 mL min−1). After naturally cooling to room temperature, Fe–N5 and Fe–N6 were obtained as black powder and directly used without any further treatment. The synthetic process of pyrolyzed ZIF-8 was the same with Fe–N5/Fe–N6 except that the precursor was pure ZIF-8 instead of Fe-doped ZIF-8. Pyrolyzed ZIF-8 at 800 and 900 °C is denoted as ZIF-8-800 and ZIF-8-900, respectively.2.3. Electrochemical measurements
All electrochemical experiments were carried out on a CHI760E electrochemical workstation (CHI660E, Shanghai CHI Instruments Company) at atmospheric pressure and ambient temperature. In a typical preparation procedure of the working electrode, 3 mg of the catalyst and 37 μL of Nafion solution was dispersed in 2.0 ml of water–ethanol solution with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, followed by sonication for 1 hour to obtain the homogeneous catalyst ink. The ink was then uniformly loaded onto one side of carbon paper with an area of 2 × 2 cm2, which was directly used for the electrochemical measurements after drying. A flow-cell was used as the electrolyzer for CO2 electrochemical reduction in 1.0 M KOH. The cathodic chamber and anodic chamber were separated by a proton exchange membrane (Nafion 115). A mass flow controller was used to set the CO2 flow gas at 10 sccm. The deposited carbon paper, Pt wire, and Ag/AgCl served as the working electrode, counter electrode, and reference electrode, respectively. All potentials reported in this study were vs. RHE via the equation E(vs. RHE) = E(vs. Ag/AgCl) + 0.197 V + 0.0592 × pH. The pH value of the electrolyte was determined with a Mettler Toledo FE 28 Desktop pH meter (Mettler Toledo International Inc., Shanghai, China). Controlled potential electrolysis was performed at each potential for 40 min. The current density was normalized with the geometric surface area of the carbon paper. Cyclic voltammetry measurements of Fe–N5 and Fe–N6 were carried out from 1.08 to 1.18 V vs. RHE and 1.06 to 1.16 V vs. RHE at various scan rates under a CO2 atmosphere to obtain the double layer capacitance (Cdl). The Cdl was determined by plotting the current density difference Δj (ja − jc) at 1.13 and 1.11 V vs. RHE against the scan rates, where ja and jc represent the anodic and cathodic current density, respectively. The Cdl is half of the linear slope and positively correlated with the electrochemical active surface area.
:3, followed by sonication for 1 hour to obtain the homogeneous catalyst ink. The ink was then uniformly loaded onto one side of carbon paper with an area of 2 × 2 cm2, which was directly used for the electrochemical measurements after drying. A flow-cell was used as the electrolyzer for CO2 electrochemical reduction in 1.0 M KOH. The cathodic chamber and anodic chamber were separated by a proton exchange membrane (Nafion 115). A mass flow controller was used to set the CO2 flow gas at 10 sccm. The deposited carbon paper, Pt wire, and Ag/AgCl served as the working electrode, counter electrode, and reference electrode, respectively. All potentials reported in this study were vs. RHE via the equation E(vs. RHE) = E(vs. Ag/AgCl) + 0.197 V + 0.0592 × pH. The pH value of the electrolyte was determined with a Mettler Toledo FE 28 Desktop pH meter (Mettler Toledo International Inc., Shanghai, China). Controlled potential electrolysis was performed at each potential for 40 min. The current density was normalized with the geometric surface area of the carbon paper. Cyclic voltammetry measurements of Fe–N5 and Fe–N6 were carried out from 1.08 to 1.18 V vs. RHE and 1.06 to 1.16 V vs. RHE at various scan rates under a CO2 atmosphere to obtain the double layer capacitance (Cdl). The Cdl was determined by plotting the current density difference Δj (ja − jc) at 1.13 and 1.11 V vs. RHE against the scan rates, where ja and jc represent the anodic and cathodic current density, respectively. The Cdl is half of the linear slope and positively correlated with the electrochemical active surface area.
2.4. Determination of reduction products
The products of CO2 electrochemical reduction were determined by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy. Particularly, the gas products were monitored by an online gas chromatgraph (Shimadzu 2010 plus GC) equipped with a thermal conductivity detector (TCD) and Molsieve 5 A column every five minutes. The liquid products were analyzed using BRUKER AVANCE III HD 400 NMR by mixing 0.5 mL of electrolyte after electrolysis, 0.1 mL of deuterium oxide (D2O) and 0.1 mL of 6 mM1-propanesulfonic acid 3-(trimethylsilyl) sodium salt (DSS) solution. CO and H2 are the only two gas products and no liquid products are detected after electrolysis for both Fe–N5 and Fe–N6.3. Results and discussion
3.1. Structural characterization
Fe–N5 and Fe–N6 were obtained via pyrolyzing an Fe-containing derivative of zeolitic imidazolate framework-8 (ZIF-8) at 900 and 800 °C, respectively. The synthetic scheme of Fe–N5 and Fe–N6 is illustrated in Fig. 1a. The Fe-containing derivative of ZIF-8 was synthesized through the standard synthetic route of ZIF-8 except for partially substituting Zn(NO3)2 with FeCl3. As shown by transmission electron microscopy (TEM) images, the Fe-containing derivative of ZIF-8 exhibited a homogeneous rhombododecahedral morphology with an average size of 220 nm (Fig. S1†). Scanning electron microscopy (SEM), TEM, and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of Fe–N5 suggested that the original structure of the Fe-containing derivative of ZIF-8 shrank into regular microporous rhombododecahedra with an average size of 200 nm after the pyrolysis process (Fig. 1b–d). Magnified HAADF-STEM images of Fe–N5 clearly demonstrated that individual Fe atoms were uniformly dispersed in the microporous frame (Fig. 1e and f). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping of Fe–N5 displayed the homogeneous distribution of C, N, and Fe over the entire architecture (Fig. 1g). Compared with Fe–N5, Fe–N6 displayed a similar morphology and elemental composition (Fig. S2†).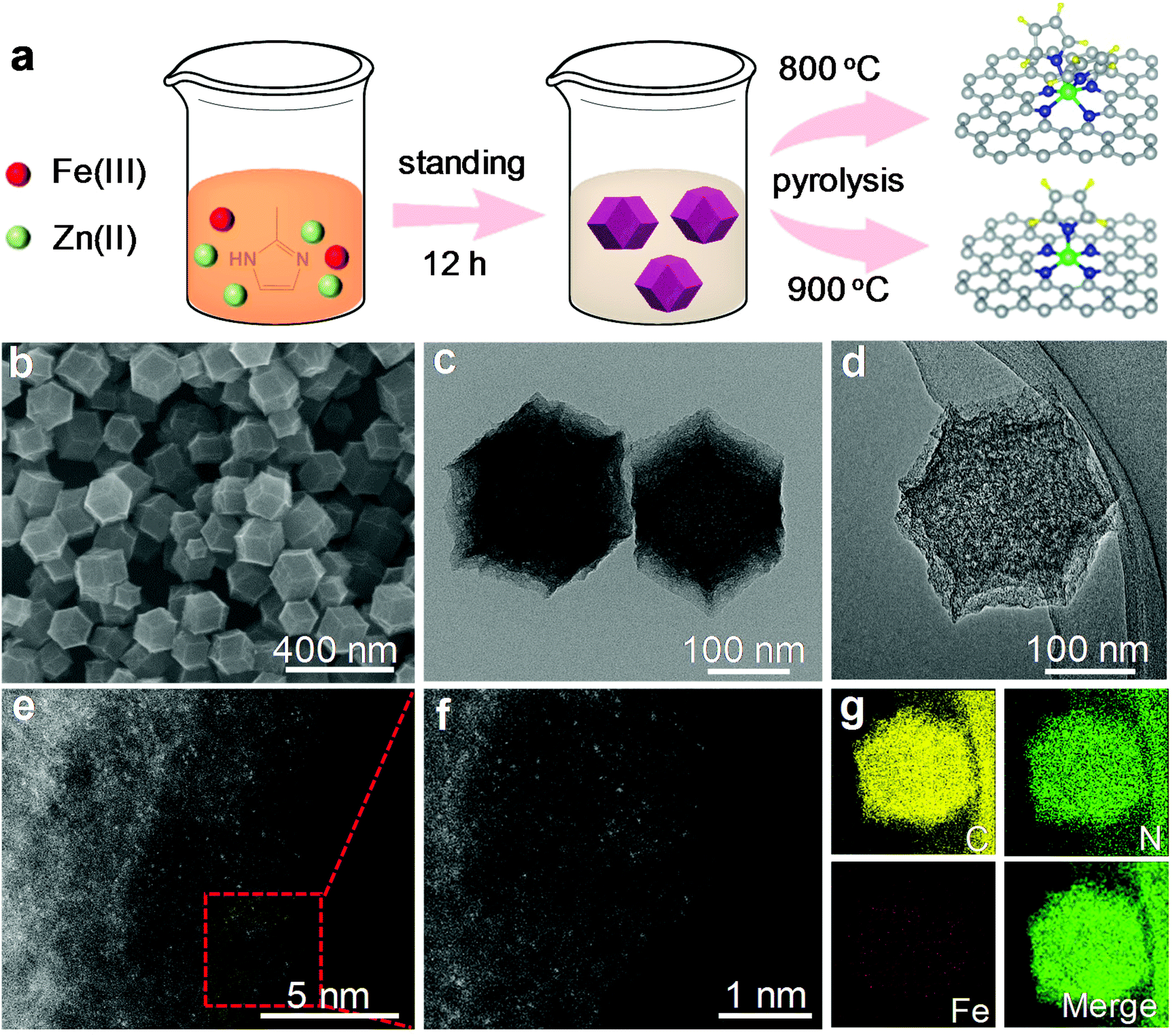 | ||
| Fig. 1 (a) Schematic of the formation process of Fe–N6 and Fe–N5. (b) SEM, (c) TEM, and (d) HAADF-STEM images of Fe–N5. (e) and (f) Magnified HAADF-STEM images of Fe–N5 demonstrate the atomic dispersion of Fe atoms. (g) EDS elemental mapping reveals the homogeneous distribution of Fe, N, and C in Fe–N5. | ||
To gain insights into the structure and composition information of Fe–N5 and Fe–N6, we conducted a series of characterization techniques. The X-ray diffraction (XRD) patterns of Fe–N5 and Fe–N6 exhibited two broad peaks at 24.1° and 43.3°, which can be assigned to the (002) and (101) facets of graphite carbon, respectively (Fig. 2a).8 No characteristic peaks of Fe or FeOx crystals appeared in the XRD patterns of Fe–N5 and Fe–N6, which further demonstrated that Fe species were atomic Fe in Fe–N5 and Fe–N6. As displayed in Fig. 2b, the N 1s spectra of Fe–N5 and Fe–N6 were deconvoluted into four distinct peaks at 398.6, 399.7, 400.9, and 402.3 eV, which could be assigned to pyridinic N, Fe–N, pyrrolic N, and graphitic N, respectively.35,36 Notably, the fraction of Fe–N decreased from 15.16% for Fe–N6 to 8.44% for Fe–N5, indicating that the elevated temperature reduced the coordination number of Fe with N (Table S1†). We further carried out Raman measurements to study the structure difference between Fe–N5 and Fe–N6. As shown in Fig. S3,† two peaks at 1350 and 1585 cm−1 are assigned to the D band and G band of graphite carbon, respectively.37,38 The intensity ratio of D band to G band (ID/IG) for Fe–N5 is 0.99, which is lower than that (1.02) for Fe–N6. This implies that a higher temperature enhanced the graphitization of carbon in Fe–N5. To further investigate the electronic and coordination structure of Fe–N5 and Fe–N6, we conducted X-ray absorption fine structure (XAFS) spectroscopy measurements. Fig. 2c shows the Fe K-edge X-ray absorption near-edge structure (XANES) profiles for Fe–N5, Fe–N6, FeCl2, FeCl3, and Fe foil. Fe–N5 and Fe–N6 exhibited similar energy absorption edge profiles with FeCl3 in the energy range between 7120 and 7124 eV, indicating that the valence state of Fe species in both Fe–N5 and Fe–N6 was ca. +3. As shown in Fig. 2d, the extended X-ray absorption fine structure (EXAFS) spectra of both Fe–N5 and Fe–N6 exhibited only one prominent peak at approximately 2.0 Å, which could be attributed to Fe–N bonds. No significant contribution of the Fe–Fe bond was observed at 2.49 Å, further revealing the atomic dispersion of Fe atoms throughout the whole Fe–N5 and Fe–N6 structure. We also conducted a least-squares EXAFS fitting. When the EXAFS fitting parameter of bond length was set to 2.00 Å with the coordination number of ca. 5.4 and 6.0 for Fe–N5 and Fe–N6, respectively, the fitting curves of Fe–N5 and Fe–N6 perfectly matched with the EXAFS result (Table S2 and Fig. S4†). The coordination number of Fe with N decreased when increasing the pyrolysis temperature from 800 to 900 °C, which is due to the fact that more FeIII-imidazolate-FeIII linkages would decompose to release CN fragments at elevated temperatures, thus resulting in the decay of Fe–N bonds.
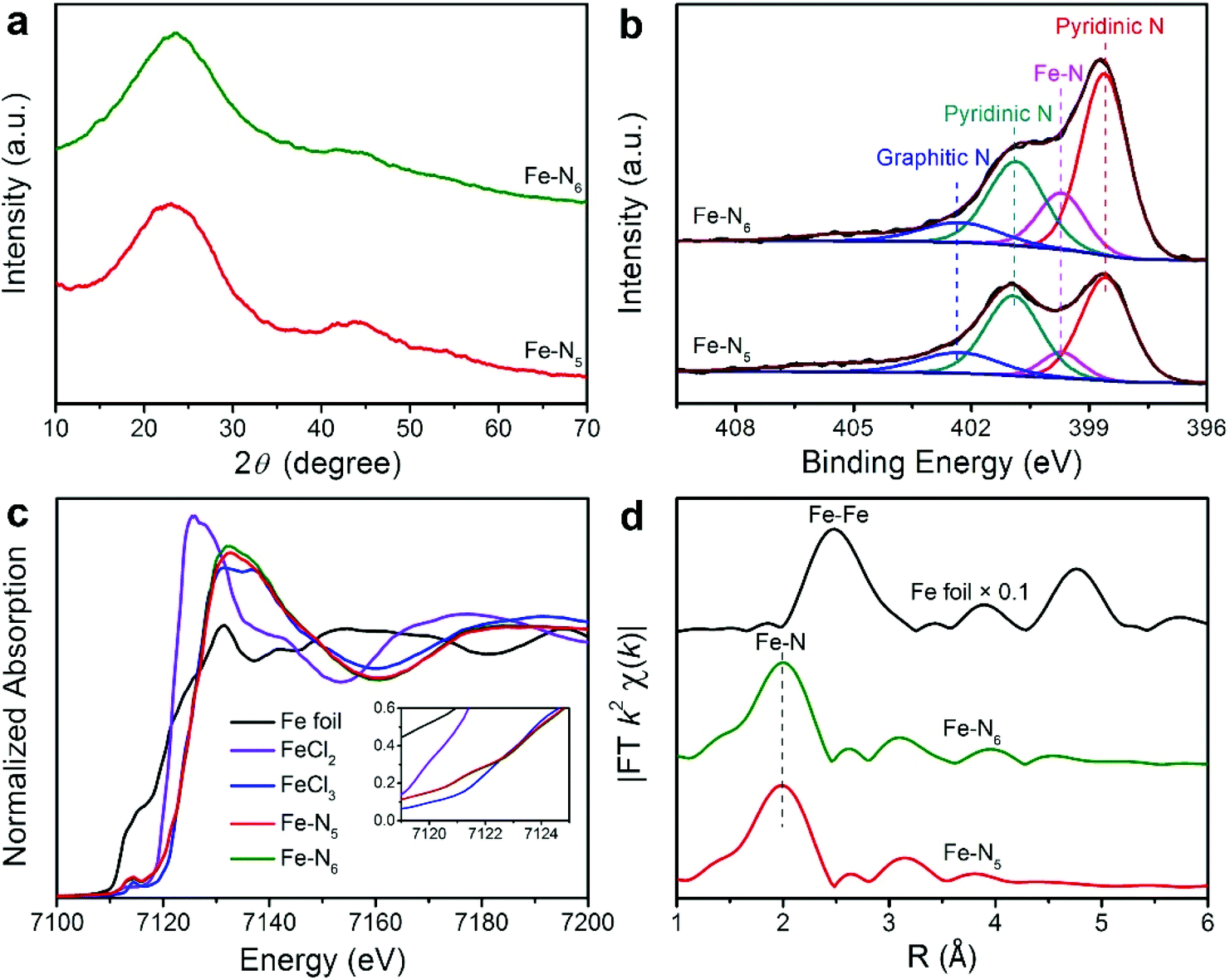 | ||
| Fig. 2 (a) XRD patterns of Fe–N5 and Fe–N6. (b) High-resolution N 1s XPS spectra of Fe–N5 and Fe–N6. (c) Fe K-edge XANES spectra of Fe–N5, Fe–N6 and Fe foil/FeCl2/FeCl3 reference materials. (d) Fe K-edge k2-weighted FT-EXAFS spectra of Fe–N5, Fe–N6, and Fe foil. | ||
3.2. Electrocatalytic performance towards CO2 electroreduction
We used a flow-cell system with 1.0 M KOH as the electrolyte to evaluate the electrocatalytic performance of Fe–N5 and Fe–N6 towards CO2 electroreduction (Fig. S5†). During the process of chronoamperometry electrolysis, the gaseous products of H2 and CO were quantified via online gas chromatograph (GC) (Fig. S6a†). Meanwhile, no liquid products were detected in the cathodic electrolyte via1H nuclear magnetic resonance (1H NMR) (Fig. S6b†). Fe–N5 demonstrated a significantly higher reduction current density than Fe–N6 throughout the potential range based on the cyclic voltammograms (CVs), indicating the superior electrocatalytic performance of Fe–N5 compared to Fe–N6 (Fig. S7†). Fig. 3a shows the FECO of Fe–N5 and Fe–N6 at various applied potentials. Fe–N5 exhibited a higher FECO than Fe–N6 at each applied potential. In particular, the FECO of Fe–N5 was higher than 90% within a wide electrochemical window from −0.35 to −0.65 V vs. RHE. Note that the FECO of Fe–N5 reached nearly 100% (99%) at −0.35 V vs. RHE. More importantly, CO was already detected at a low potential of −0.16 and −0.20 V vs. RHE for Fe–N5 and Fe–N6, respectively (Fig. S8†). Considering the CO2/CO equilibrium potential is −0.11 V vs. RHE, the overpotential to produce CO was merely 50 and 90 mV for Fe–N5 and Fe–N6, respectively, even outperforming that for noble-metal catalysts such as Ag and Au.39–43Fig. 3b shows the partial current density of CO (jCO) of Fe–N5 and Fe–N6. Fe–N5 exhibited higher jCO at all applied potentials. At −1.05 V vs. RHE, the jCO of Fe–N5 was up to −20.8 mA cm−2, which was 3.8 times as high as that for Fe–N6. The Cdl (2.58 mF cm−2) of Fe–N5 was close to that (2.25 mF cm−2) of Fe–N6 (Fig. S9†), implying that Fe–N5 and Fe–N6 possessed comparable electrochemical active surface area. Given the significantly higher FECO and jCO of Fe–N5 relative to Fe–N6, it is safe to conclude that Fe–N5 had a higher intrinsic activity towards CO2 electroreduction. To probe the active center and eliminate the interference of the substrate (N-doped porous carbon), we further pyrolyzed pure ZIF-8 at 800 and 900 °C, which was denoted as ZIF-8-800 and ZIF-8-900, respectively (Fig. S10†). It is worth noting that the FECO of ZIF-8-800 and ZIF-8-900 was lower than 25% (Fig. S11†), demonstrating that Fe atoms in Fe–N5 and Fe–N6 are largely responsible for the electroreduction of CO2 to CO. Moreover, the FECO of commercial Fe nanoparticles, Fe3O4, and Fe2O3 were negligible at −0.55 V vs. RHE, demonstrating that atomically distributed Fe sites are the active center rather than bulk Fe species (Fig. S12†). As shown in Fig. 3c, the TOF of CO (TOFCO) was normalized by the mass loading of Fe at different applied potentials. It can be seen that the TOFCO of Fe–N5 significantly exceeded that of Fe–N6 throughout the applied potential range. Particularly, for Fe–N5, the TOFCO reached 5006 h−1 at −1.05 V vs. RHE, which was 3.8 times that for Fe–N6. All these results strongly corroborate the intrinsically higher CO2 electroreduction performance of Fe–N5 relative to that of Fe–N6, which is comparable to the best reported SACs for CO2 electroreduction (Table S3†). In view of the excellent selectivity and activity of Fe–N5 for CO2 electroreduction, we performed potentiostatic measurements to assess its durability in 1.0 M KOH aqueous solution at −0.4 V vs. RHE. As shown in Fig. 3d, the current density and the FECO of Fe–N5 remained basically unchanged during the 24 h continuous electrolysis. TEM and HAADF-STEM images of Fe–N5 after the stability test show that the Fe single atoms were still atomically dispersed on the N-doped carbon support (Fig. S13†). The N1s XPS spectra of spent Fe–N5 resembled that of pristine Fe–N5, indicating the superior stability after the reaction (Fig. S14 and Table S4†).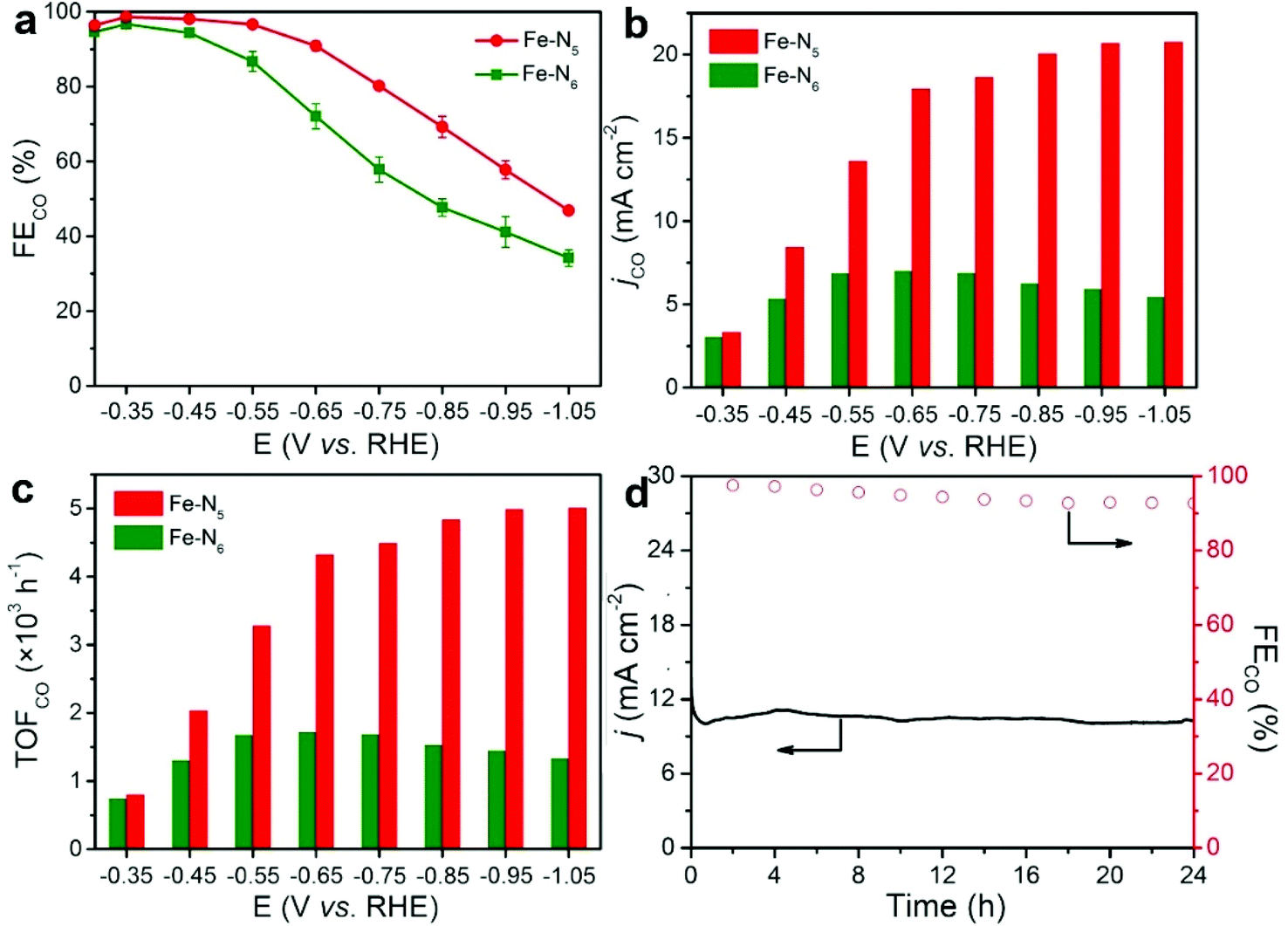 | ||
| Fig. 3 CO2 electroreduction catalytic performance of Fe–N5 and Fe–N6. (a) Faradaic efficiency of CO at different potentials. (b) jCO in 1.0 M KOH electrolyte. (c) TOFCO at various applied potentials. (d) 24 h stability test of Fe–N5 at −0.4 V vs. RHE and the corresponding FECO. | ||
3.3. Mechanistic studies on CO2 electroreduction
To further investigate the origin of the intrinsically high electrocatalytic performance of Fe–N5, we conducted CO2 temperature-programmed desorption (CO2-TPD). As shown in Fig. 4a, the CO2 desorption peak of Fe–N5 was located at 141 °C, which was higher than that (118 °C) of Fe–N6. As such, the binding strength of CO2 on Fe–N5 was stronger than that on Fe–N6. The strong binding of CO2 on Fe–N5 indicated the facilitated adsorption of CO2, thus resulting in the high activity of Fe–N5.44,45 This is in line with the enhanced CO2 adsorption on Fe–N5 compared to that on Fe–N6 (Fig. S15†). To provide a theoretical insight into the relationship between the coordination number and the reaction pathway, we calculated the Gibbs free energy for each step involved in CO2 electroreduction (Table S5†). The structural models of Fe–N5 and Fe–N6 were established via the obtained XPS and EXAFS fitting results (Table S2 and Fig. S16†). The structural model of Fe–N6 with one pyrrole ring on each side of carbon matrix was excluded after structure optimization (Fig. S17†). As shown in Fig. 4b, the formation of *COOH required the highest energy barrier among all the steps of CO2 electroreduction on both Fe–N5 and Fe–N6. Therefore, the formation of *COOH serves as the potential-limiting step over Fe–N5 and Fe–N6 towards CO2 electroreduction. Particularly, the Gibbs free energy for the formation of *COOH on Fe–N5 was 1.34 eV, which was significantly lower than that on Fe–N6 (2.35 eV). As shown in Fig. 4c and d, the C–Fe bond length of adsorbed *COOH for Fe–N5 and Fe–N6 increased from 2.09 to 2.26 Å, respectively, further revealing that Fe–N5 was more beneficial for *COOH adsorption relative to Fe–N6. Apart from that, the C atom of adsorbed *COOH on Fe–N5 displays higher electron density compared to that on Fe–N6, confirming the stronger stabilization of *COOH on Fe–N5 (Fig. 4e and f).46 As such, Fe–N5 promoted the activation of CO2, leading to the enhanced electrocatalytic activity for CO2 electroreduction relative to Fe–N6.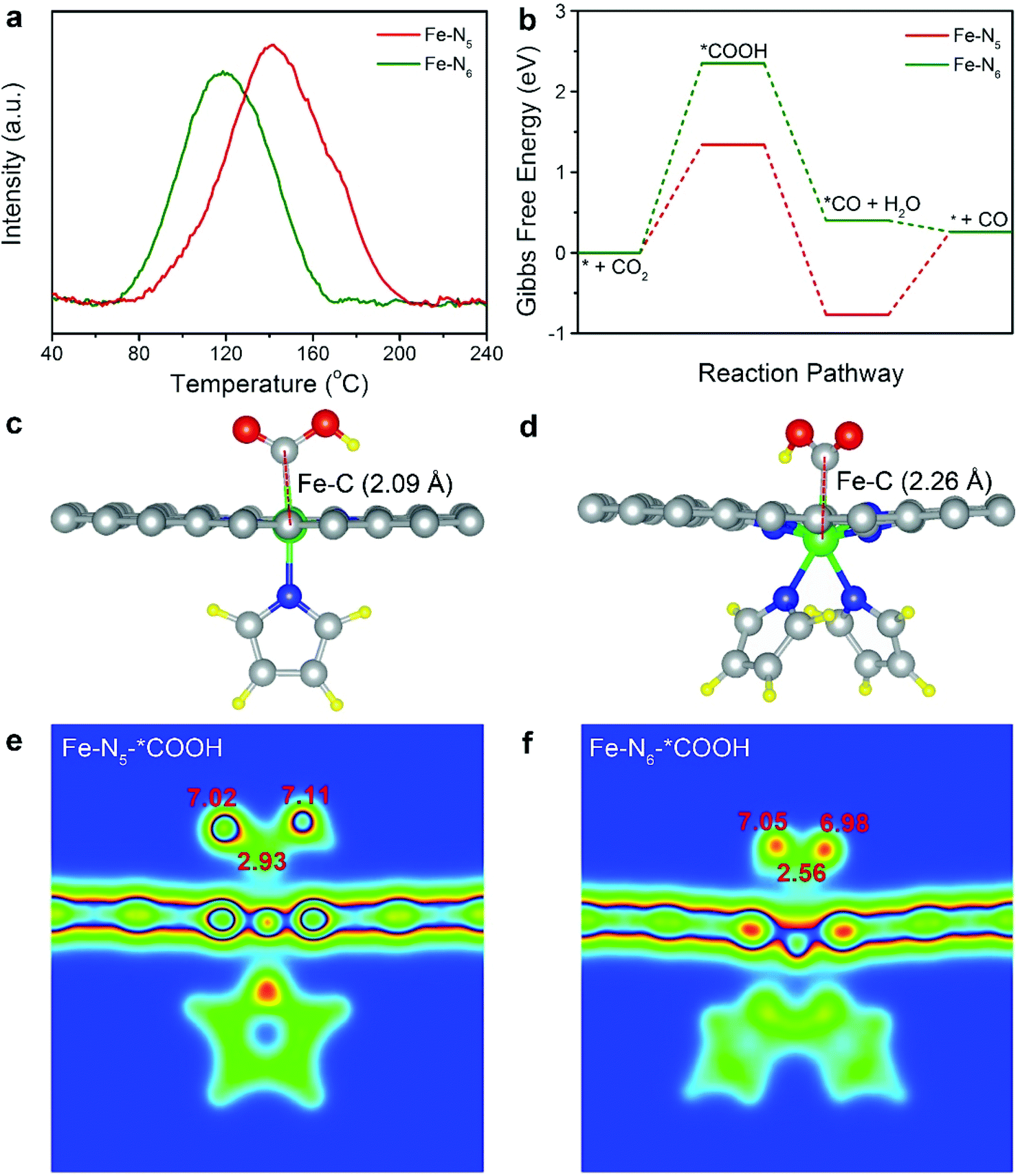 | ||
| Fig. 4 (a) CO2-TPD profiles for Fe–N5 and Fe–N6. (b) Calculated Gibbs free energy diagrams for CO2 electroreduction to CO over Fe–N5 and Fe–N6. (c) and (d) Optimized structures of Fe–N5 and Fe–N6 with adsorbed *COOH, * denotes the adsorption site. The gray, green, blue, red and yellow spheres represent C, Fe, N, O, and H atoms, respectively. (e) and (f) Slice of calculated charge densities of Fe–N5 and Fe–N6 with adsorbed *COOH. Bader partial atomic charges are labeled by the red numbers. | ||
4. Conclusions
In summary, we have synthesized two Fe SACs for CO2 electroreduction. Compared with Fe–N6, Fe–N5 demonstrated a lower onset potential (−0.16 V vs. RHE), higher turnover frequency (5006 h−1), and remarkable faradaic efficiency (99%) for CO production. This was attributed to the facile adsorption and activation of CO2 arising from the favourable coordination structure. This work should pave a promising avenue to rationally design novel single-atom electrocatalysts with a tunable coordination environment in energy and environment related fields.Conflicts of interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (21925204), China Postdoctoral Science Foundation (2019M662192), NSFC (U1932146 and U19A2015), National Key Research and Development Program of China (2019YFA0405600), Key Research Program of Frontier Sciences of the CAS (QYZDB-SSW-SLH017), Fundamental Research Funds for the Central Universities, and USTC Research Funds of the Double First-Class Initiative (YD2340002002).References
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- A. Goeppert, M. Czaun, G. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- R. Daiyan, X. Tan, R. Chen, W. H. Saputera, H. A. Tahini, E. Lovell, Y. H. Ng, S. C. Smith, L. Dai, X. Lu and R. Amal, ACS Energy Lett., 2018, 3, 2292–2298 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. D. Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS.
- Y. Xing, J. Ku, W. Fu, L. Wang and H. Chen, Chem. Eng. J., 2020, 395, 125149 CrossRef CAS.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., 2020, 59, 2705–2709 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- M. Jia, S. Hong, T.-S. Wu, X. Li, Y.-L. Soo and Z. Sun, Chem. Commun., 2019, 55, 12024–12027 RSC.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji and X. Zheng, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie and A. S. Jalilov, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, 1903668 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2018, 52, 656–664 CrossRef.
- X. Rong, H.-J. Wang, X.-L. Lu, R. Si and T.-B. Lu, Angew. Chem., 2020, 59, 1961–1965 CrossRef CAS.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- N. Leonard, W. Ju, I. Sinev, J. Steinberg, F. Luo, A. S. Varela, B. R. Cuenya and P. Strasser, Chem. Sci., 2018, 9, 5064–5073 RSC.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang and D. Su, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS.
- V. Strauss, K. Marsh, M. D. Kowal, M. El-Kady and R. B. Kaner, Adv. Mater., 2018, 30, 1704449 CrossRef.
- Z. Ma, C. Tsounis, P. V. Kumar, Z. Han, R. J. Wong, C. Y. Toe, S. Zhou, N. M. Bedford, L. Thomsen and Y. H. Ng, Adv. Funct. Mater., 2020, 30, 1910118 CrossRef CAS.
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301–1306 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- S. Zhao, R. Jin and R. Jin, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- Y. S. Ham, S. Choe, M. J. Kim, T. Lim, S. K. Kim and J. J. Kim, Appl. Catal., B, 2017, 208, 35–43 CrossRef CAS.
- S. M. Lee, H. Lee, J. Kim, S. H. Ahn and S. T. Chang, Appl. Catal., B, 2019, 259, 118045 CrossRef CAS.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 CrossRef CAS.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS.
- F. Y. Gao, S. J. Hu, X. L. Zhang, Y. R. Zheng, H. J. Wang, Z. Z. Niu, P. P. Yang, R. C. Bao, T. Ma and Z. Dang, Angew. Chem., 2020, 132, 8784–8790 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures, tables and methods related to this article. See DOI: 10.1039/d0gc02689a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |