
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolic engineering of Escherichia coli for production of non-natural acetins from glycerol†
Bakht
Zada‡
 a,
Seongjoon
Joo‡
b,
Chonglong
Wang‡
c,
Tenzin
Tseten
a,
Seong-Hee
Jeong
a,
Hogyun
Seo
b,
Jung-Hoon
Sohn
d,
Kyung-Jin
Kim
*b and
Seon-Won
Kim
*a
a,
Seongjoon
Joo‡
b,
Chonglong
Wang‡
c,
Tenzin
Tseten
a,
Seong-Hee
Jeong
a,
Hogyun
Seo
b,
Jung-Hoon
Sohn
d,
Kyung-Jin
Kim
*b and
Seon-Won
Kim
*a
aDivision of Applied Life Science (BK21 Plus), PMBBRC, Gyeongsang National University, Jinju 52828, Republic of Korea. E-mail: swkim@gnu.ac.kr; bakhtqau@gmail.com
bSchool of Life Sciences, KNU Creative BioResearch Group, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: kkim@knu.ac.kr; wntjdwnswkd@naver.com
cSchool of Biology and Basic Medical Sciences, Soochow University, Suzhou, People's Republic of China. E-mail: clwang@suda.edu.cn
dSynthetic Biology and Bioengineering Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 34141, Republic of Korea. E-mail: sohn4090@kribb.re.kr
First published on 23rd September 2020
Abstract
Mono-, di- and triacetin are three glycerol esters which are usually synthesized via costly and environmentally unfriendly chemical synthesis methods. Here, Escherichia coli is metabolically engineered for the production of mono-, di- and triacetin using glycerol as a substrate. First, a novel biosynthetic route of mono- and diacetin is established by overexpression of a native enzyme, maltose O-acetyltransferase (MAA). Next, the biosynthetic pathway is extended to produce a mixture of mono-, di- and triacetin by overexpression of chloramphenicol-O-acetyltransferase (CAT). By successive strategies, including heterologous gene expression, metabolic engineering, and culture optimization, a recombinant E. coli is enabled to produce more than 27 g L−1 of a mixture of mono-, di- and triacetin in shake flask cultures, which is a >650-fold increase over the initial production of 0.04 g L−1. In vitro studies confirm the acetylation of glycerol to mono- and diacetin by MAA, and the additional acetylation to triacetin by CAT. When crude glycerol is used as a substrate, the engineered strain produced a total of 25.9 g L−1 of the acetin mixture, about the same as that achieved from pure glycerol. To our knowledge, this is the first successful report of microbial production of the artificial chemicals, acetins.
Introduction
Glycerol is a by-product accompanying the production of biodiesel. It is reported that 10 lb of crude glycerol is generated for every 100 lb of biodiesel produced by trans-esterification of vegetable oils or animal fats.1 Thus, the worldwide production of glycerol in 2020 is estimated to be six times more than its demand,2 resulting in waste of glycerol. In recent years there has been growing interest in biorefinery industries towards the utilization of surplus glycerol. Bioconversions of surplus glycerol into 1,3-propandiol, propionic acid, citric acid, succinic acid, butanol, single cell oil and hydrogen gas have been developed,3–5 which can be called Zero Waste Biorefinery. This biotechnology conserves and recovers all resources to eliminate the volume of waste materials produced by industry that would be otherwise be burned or buried. The goal of Zero Waste Biorefinery is thus to process and convert waste streams produced at various industrial sites into valuable chemical compounds, thereby closing the production loop with no net waste. However, various products obtained from glycerol such as polymers (e.g. Sorona®, a co-polymer of 1,3-propanediol and terephthalates) and Susterra® propanediol are not associated with biodiesel and must be transported to their application sites for further utilization.Triacetin, also called glycerin triacetate, could be a potential alternative derived from waste glycerol. As a tri-ester of glycerol, it can be used as a fuel additive to improve the freezing and viscosity properties of biodiesel as well as an antiknock agent which can reduce engine knocking in gasoline.6,7 Engine performance can be significantly improved by blending 10% triacetin with biodiesel.8 The production of triacetin along with biodiesel production could serve as a complete on-site Zero Waste Biorefinery (Fig. 1A). Triacetin also has applications in cosmetics, food additives, plasticizers and precursors for isoamyl acetate synthesis.9–11 It is considered to be a possible source of food energy in the artificial food regeneration system for long space missions.12 Of the same importance, mono- and diacetin have received a great deal of attention in recent years as high-value specialty chemicals. They are currently being used in pharmaceuticals, food industries and cosmetics, as well as in the chemical industry as plasticizers, softening and tanning agents, explosives and raw materials for biodegradable polyesters.13,14 Like triacetin, diacetin could also be used as a biofuel additive.15
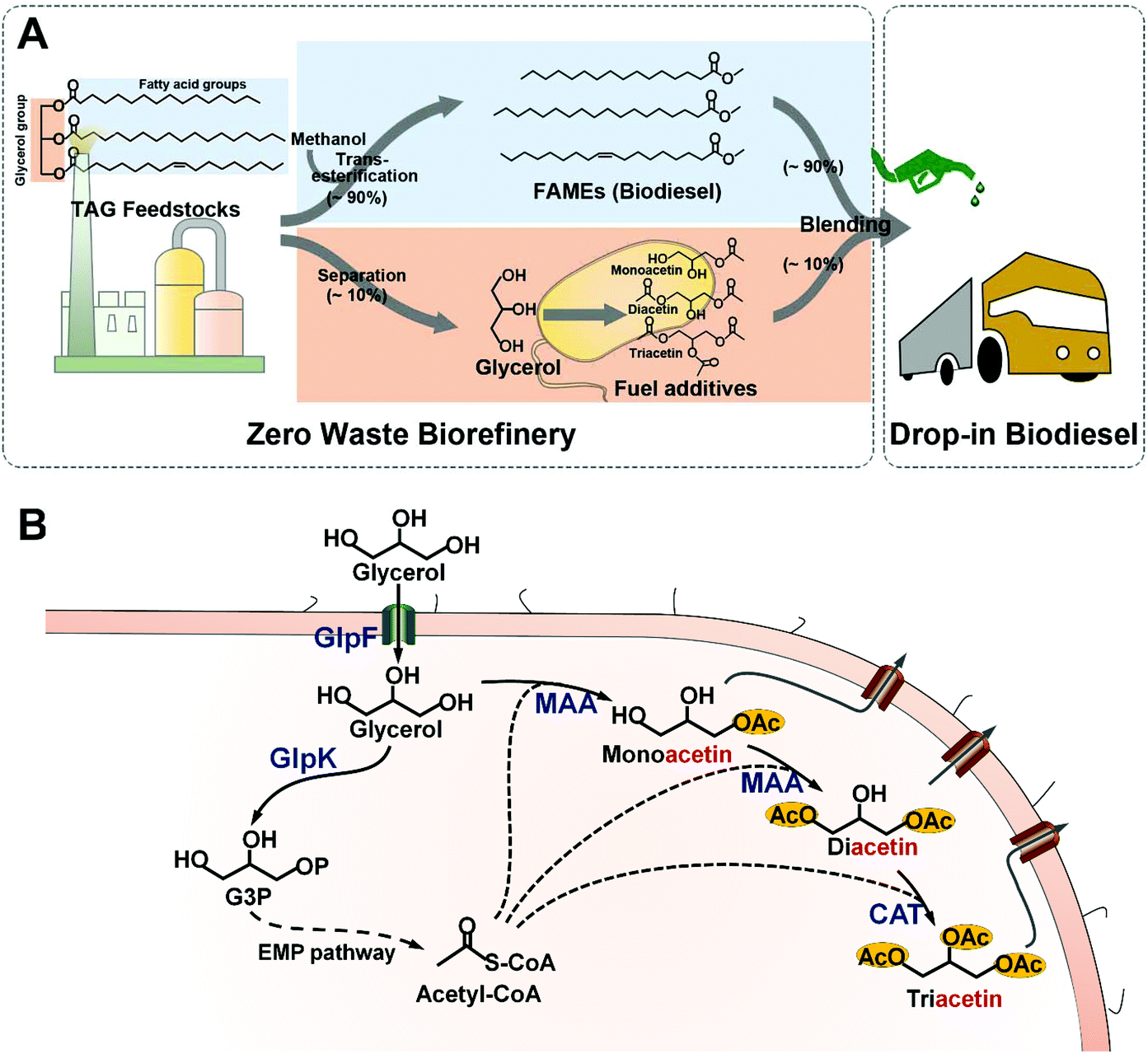 | ||
| Fig. 1 Schematic overview of Zero Waste Biorefinery and acetin biosynthesis in recombinant E. coli. (A) Approx. 10% surplus glycerol is produced by the trans-esterification of triacylglycerol (TAG) and methanol during biodiesel synthesis. A biological pathway is devised to convert surplus glycerol to the biofuel additive acetins that are used for blending with biodiesel to improve freezing and viscosity properties. The on-site application of surplus glycerol suggests a Zero Waste Biorefinery with no net waste. (B) Monoacetin is synthesized via co-condensation of glycerol and acetyl-CoA catalyzed by E. coli native enzyme, MAA. The monoacetin is further acetylated to diacetin by the action of the same enzyme, MAA. The CAT enzyme further transforms diacetin into triacetin by involving a third molecule of acetyl-CoA. The acetyl-CoA as a co-substrate is generated via the EMP pathway. The abbreviations of enzymes are as follows: GlpF, glycerol facilitator; GlpK, glycerol kinase; MAA, maltose O-acetyltransferase; CAT, chloramphenicol O-acetyltransferase. | ||
An acetin mixture of mono-, di-, and triacetin has been produced by the chemical esterification of glycerol with acetic acid or acetic anhydride using inorganic catalysts (Fig. S1A†).16–18 However, the chemical method suffers from the requirements for expensive catalysts, a high reaction temperature and pressure, the use of hazardous materials, and the release of toxic intermediates causing environmental pollution. Besides, the chemically synthesized acetins contain toxic byproducts and radicals, which limits their applications in the pharmaceutical, food, and cosmetic industries. The biological production of acetins using a commercial lipase, Novozyme 435 (CalB), has been adopted to catalyze the trans-esterification reaction of glycerol with alkyl acetate to acetins (Fig. S1B†).19,20 However, the CalB enzyme is too costly to support the feasible and economical production of acetins. Moreover, the trans-esterification reaction catalyzed by commercial lipase is reversible, and the complex of the final reaction mixture, comprising reactants (glycerol and alkyl acetate) and alcohol byproducts beyond the target acetins, increases the difficulty and cost of further processing to get pure acetins. The co-substrate, alkyl acetate, for acetin synthesis would incur an extra cost in the lipase-based approach. Perhaps this approach might also be restricted to pure glycerol as a substrate because most enzymes are highly sensitive to impurities and crude glycerol contains various impurities. Therefore, the microbial production of acetins using glycerol as a substrate remains the method of choice due to its economic feasibility as well as environmental friendliness.
In the last few decades, paramount interest is being paid to the use of microbes for the production of high-value specialty chemicals. Recent advances in metabolic engineering, protein engineering and synthetic biology have revolutionized the ability to design and engineer biosynthetic pathways in microbial hosts for the production of a wide variety of commodity products, such as silk proteins,21 pharmaceuticals,22 bio-plastics,23 platform chemicals24 and biofuels.25,26 Therefore, it will be an immense benefit to the industrial world to design and establish a novel biological route in host microorganisms for the production of non-natural acetins. Among the limited number of microorganisms harnessed as cell factories, E. coli is the most popular platform cell factory because of its high growth rate, well-known physiology, and easy culture and genetic manipulation.27E. coli has been metabolically engineered for the production of many useful chemicals and biofuels from glycerol as a carbon source.28–32 As acetins are glycerol-based compounds, E. coli could be an ideal host for the production of acetins, if a biosynthetic pathway for non-natural acetins can be successfully created and engineered. Only a few microorganisms such as Klebsiella oxytoca, Enterobacter aerogenes and some other Enterobacter sp. have been reported to produce monoacetin naturally in a trace amount.33 However, a synthetic pathway for monoacetin (or di- and triacetin) has not been elucidated in any living organisms. In this study, for the first time, we have designed and established a novel biosynthetic pathway for mono-, di- and triacetin from glycerol in E. coli.
Results and discussion
Formation of mono- and diacetin in E. coli overexpressing O-acetyltransferase
So far, no report has been available on the production of mono- and diacetin in living organisms, and annotated metabolic pathways do not exist for a reference. As acetins are acetyl-esterified compounds of glycerol, it was hypothesized that some O-acetyltransferases could catalyze the acetylation of glycerol to acetins. In E. coli, there are several O-acetyltransferases catalyzing the acetyl-CoA dependent transformation of a diverse set of substrates, including carbohydrates and amino acids. However, the functional activities of most of these endogenous acetyltransferases have not been fully explored. Five candidate genes of lacA, maa, cysE, wecH, and nhoA encoding galactoside O-acetyltransferase, maltose O-acetyltransferase, serine acetyltransferase, O-acetyltransferase and arylamine N-acetyltransferase, respectively, as well as three putative acetyltransferase genes of yjgM, yjaB and yiiD are screened by function-mediated mining and proposed to test acetin synthesis from glycerol in E. coli. These candidate genes of Ec-lacA, Ec-cysE, Ec-yjgM, Ec-yjaB, Ec-yiiD, Ec-wecH, Ec-nhoA and Ec-maa were cloned into a medium copy vector pTrc99A with a strong Ptrc promoter and transformed into E. coli DH5α to create recombinant strains DH-MD1 to DH-MD8 (Fig. 2A & Table S1†). E. coli DH5α harboring the empty plasmid pTrc99A (DH-MD0) was used as a control. All the strains were cultivated at 37 °C in a shake flask with 50 mL of M9 medium containing 5 g L−1 yeast extract (M9YE), 2% (v/v) glycerol, and 0.5 mM IPTG. Compared to strains DH-MD0 to DH-MD7 that failed to produce either mono- or diacetin, strain DH-MD8, overexpressing Ec-maa produced mono- and diacetin, as detected by GC-FID (Fig. S2†) and consequently confirmed by GC-MS (Fig. S3†). After 48 h of culture, strain DH-MD8 produced 31.8 mg L−1 and 8.9 mg L−1 of mono- and diacetin, respectively (Fig. 2B). The broad substrate specific Ec-MAA acetylates various sugars, including maltose and glucose, and the most effective substrate is glucose which acetylates at the C6 position.34–36 Strain DH-MD8 did not produce any acetin when glucose was substituted instead of glycerol as the substrate (data not shown). Thus, this study newly reveals that E. coli maltose O-acetyltransferase (Ec-MAA) can acetylate even glycerol (C3) to monoacetin (C5) and diacetin (C7) besides its native substrates of maltose and glucose. For the first time, we have successfully created a biosynthetic pathway through the overexpression of MAA to produce mono- and diacetin in E. coli using glycerol as a substrate (Fig. 1B), whereas a fully acetylated product of glycerol, triacetin, which was among our target products but not produced by this engineered strain.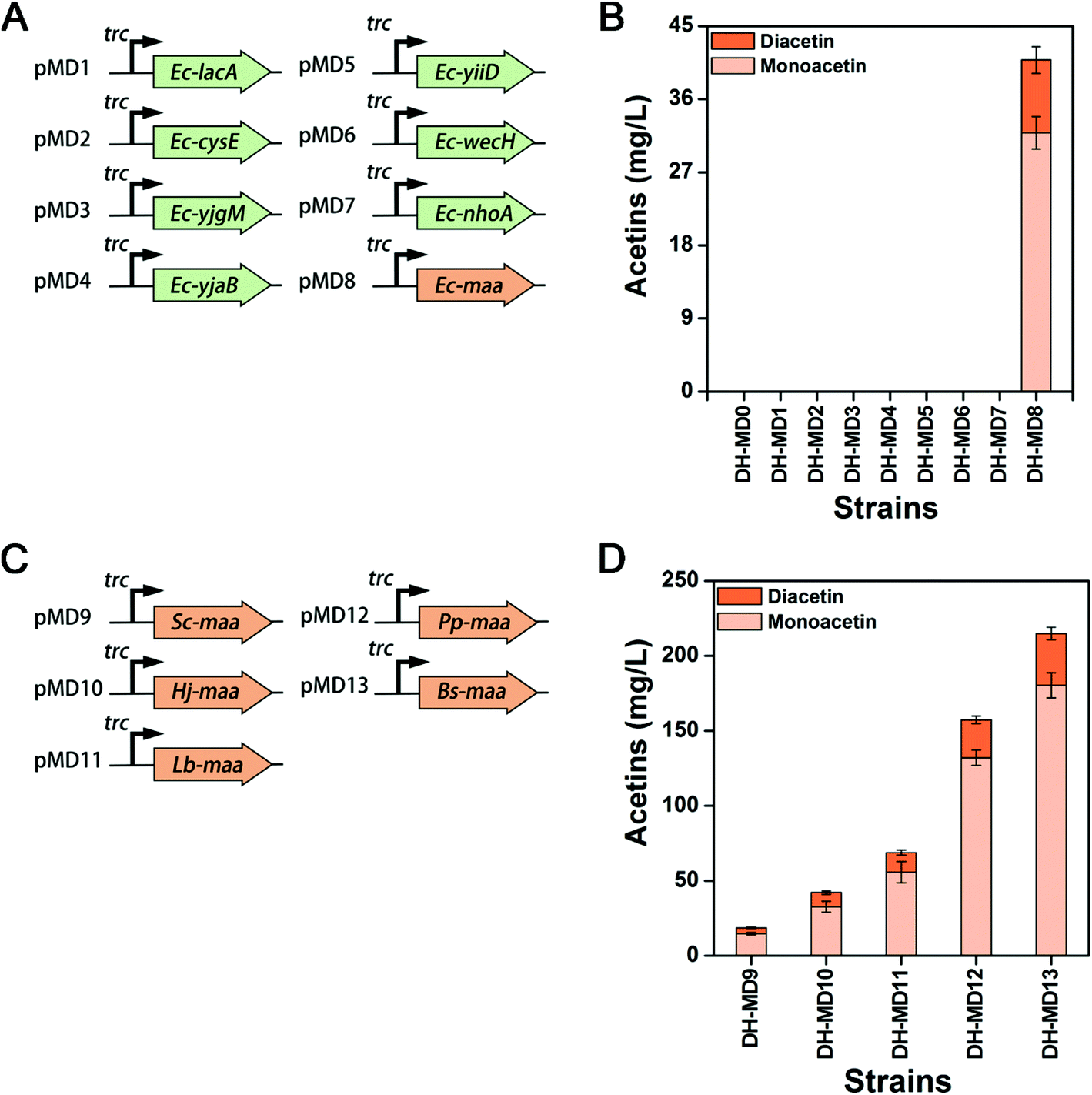 | ||
| Fig. 2 Production of mono- and diacetin from recombinant E. coli. (A) The construction scheme of E. coli endogenous O-acetyltransferases. Eight O-acetyltransferases genes lacA, cysE, yjgm, yjaB, yiiD, wecH, nhoA, and maa are constructed under a strong promoter trc. (B) Production of mono- and diacetin from the recombinant E. coli overexpressing endogenous O-acetyltransferases. The strains DH-MD1 to DH-MD8 are E. coli DH5α transformed with the plasmids pMD1 to pMD8, respectively. The control strain DH-MD0 is E. coli DH5α harboring the empty vector pTrc99A. (C) The construction scheme of heterologous maltose O-acetyltransferase (MAA) from different organisms. The genes Sc-maa, Hj-maa, Lb-maa, Pp-maa, and Bs-maa are derived from S. carnosus, H. jeotgali, L. brevis, P. putida and B. subtilis, respectively. (D) Production of mono- and diacetin from strains DH-MD9 to DH-MD13 transformed with plasmids pMD9 to pMD13, respectively. The means and standard divisions are from two biological replicates. | ||
Structural basis for acetins synthesis by MAA
To provide the structural basis for mono- and diacetin biosynthesis, we analyzed the crystal structures of maltose O-acetyltransferase from E. coli (Ec-MAA). First, we attempted to determine the crystal structure of Ec-MAA in complex with glycerol (Fig. S4 & Table S2†). However, neither co-crystallization nor a soaking experiment with glycerol was successful because the tris(hydroxymethyl)aminomethane (Tris) molecule was bound at the substrate binding site of Ec-MAA (Fig. 3A). Fortunately, the Tris-complexed structure provides a more explicit explanation for the acetylation of glycerol. In fact, Tris and glycerol have quite similar structures; Tris contains an amine-group head and three hydroxymethyl-group arms, and glycerol is composed of a hydroxyl-group head and two hydroxymethyl-group arms. In our structure, the Tris molecule is mainly stabilized with residues of Asp69, Asn83, His113 and Glu125 through hydrogen bonds (Fig. 3B). The first hydroxymethyl-group arm (position I) is stabilized with the catalytic His113 residue by a hydrogen, suggesting that the hydroxymethyl-group might be acetylated by the enzyme (Fig. 3B). Asp69 forms hydrogen bonds with the second (position II) and third hydroxymethyl-group (position III) arms, and Asn83 and Glu125 stabilize the second hydroxymethyl-group arm and the amine-group head, respectively (Fig. 3B). Based on these observations, we suggest that glycerol might bind in two different orientations so both 1′- and 3′-aceylated monoacetins can be synthesized. 1′-Acetylated monoacetin can be synthesized when glycerol binds in a mode with two hydroxymethyl-groups at position I and position II (Fig. 3C), while 3′-acetylated monoacetin can be synthesized by binding in a mode with two hydroxymethyl-groups at position I and position III (Fig. 3D). Like the fashion for glycerol binding, the binding of monoacetin isomers would show two different orientations in the diacetin synthesis, where 1′- or 3′-acetyl-hydroxymethyl-groups of monoacetins are located at position III and position II, respectively. In both cases, the free-hydroxymethyl-groups are located to position I to be acetylated. It should be noted that binding of the 3′-acetyl-hydroxymethyl-group to position III might be much weaker than that of the 1′-acetyl-hydroxymethyl-group to position II, because position II does not seem to have enough space to accommodate the 3′-acetyl-group of 3′-acetylated monoacetin (Fig. 3A). We suspect that this phenomenon can cause incomplete conversion of 3′-acetylated monoacetin to diacetin, which might explain the accumulation of a large portion of monoacetin in this in vivo acetin biosynthesis and serve as the rational basis for protein engineering to produce specific acetins.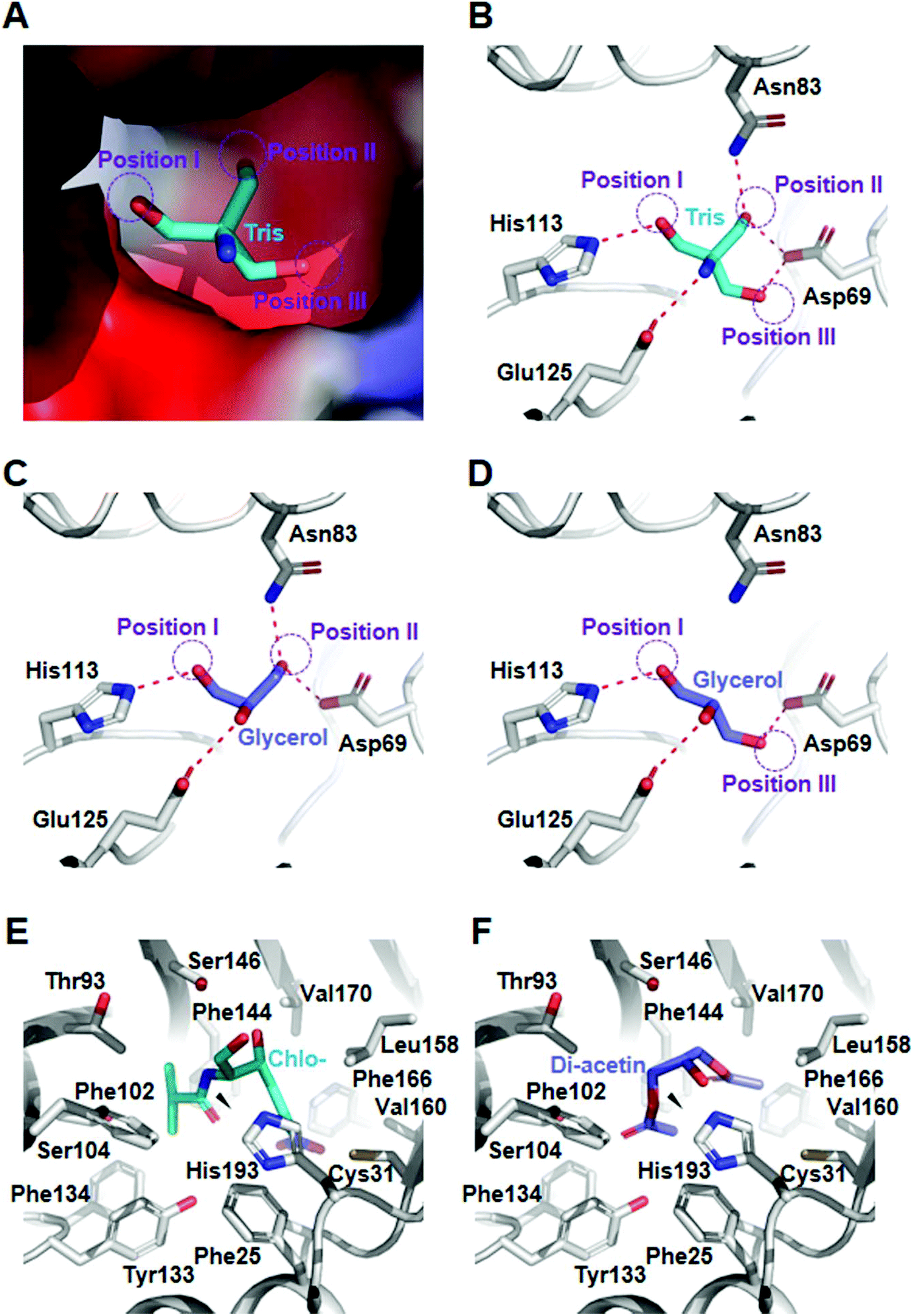 | ||
| Fig. 3 Structural basis for acetin biosynthesis by Ec-MAA and CAT. (A) Substrate binding pocket of Ec-MAA. The Ec-MAA structure is presented as an electrostatic potential surface model. The Tris molecule bound in Ec-MAA is shown as a stick model with a cyan color. The purple circles show the positions of the three hydroxymethyl groups of Tris. (B–D) Binding mode of the Tris and glycerol molecules. The Ec-MAA structure is presented as a cartoon diagram with a gray color. The Tris and glycerol molecules are shown as sticks with cyan and light-blue colors, respectively. Residues involved in the stabilization of the Tris and glycerol molecules in Ec-MAA are shown as sticks. The purple circles show the position of the hydroxymethyl group of Tris. (E) The binding mode of chloramphenicol in CAT and docked diacetin. The CAT structure is presented as a cartoon diagram with a gray color. The chloramphenicol molecule is shown as sticks with a cyan color. Residues forming the chloramphenicol binding pocket are shown as gray-colored sticks. (F) The binding mode of simulated diacetin. The simulated diacetin molecules are shown as sticks with a light-blue color. | ||
Screening of efficient heterologous maltose O-acetyltransferase
To screen an efficient heterologous MAAs for acetin production, MAAs from other microbial species were retrieved by similarity searches of amino acid sequences using Ec-maa. Five MAA candidates from Staphylococcus carnosus, Halalkalicoccus jeotgali, Lactobacillus brevis, Bacillus subtilis and Pseudomonas putida were selected by a BLAST search (Fig. S5 & S6†). To gain the optimal expression of the heterologous genes in E. coli, the ORF sequences of all these genes were optimized by using the GenScript algorithm (https://www.genscript.com/codon_opt_pr.html) with a codon adaptation index (CAI) of more than 0.8 (Table S3†). The optimized genes were cloned into pTrc99A (pMD9 to pMD13) and transformed in E. coli DH5α to create recombinant strains DH-MD9 to DH-MD13 (Fig. 2C & Table S1†). Mono- and diacetin were consequently evaluated from all these newly constructed strains (Fig. 2D). Strains DH-MD11, DH-MD12 and DH-MD13 show higher titers of mono- and diacetin when compared to strain DH-MD8. The highest titer of 214.9 mg L−1, equivalent to a 5.2-fold increase over strain DH-MD8, was achieved from strain DH-MD13, which overexpressed an efficient catalyzer, Bs-MAA, for mono- and diacetin production.Extension of mono- and diacetin biosynthetic pathway to triacetin in E. coli
Various metabolic engineering strategies have been developed and applied to increase the production of chemicals of interest. A canonical strategy is to balance the expression level of protein constituents of biosynthetic pathways, which can be easily addressed by harnessing promoters of various strengths. Thus, a stronger PT7 promoter and a weaker Plac promoter than Ptrc were applied to optimize the expression level of Bs-MAA. The gene Bs-maa was thus cloned into pET-28a(+) and pSTV28, respectively, resulting in pMD14 and pMDT1 plasmids (Fig. 4A). E. coli DH5α (DE3) and DH5α harboring pMD14 and pMDT1 are accordingly called DH-MD14 and DH-MDT1. Strain DH-MD14 produced 125.5 mg L−1 of mixed mono- and diacetin after 48 h of culture, which is 42% lower than that of strain DH-MD13 (Fig. 4B). The decrease in acetin production from the PT7 promoter is likely to be due to a metabolic burden on the cells, since the cell growth of DH-MD14 was much lower than that of DH-MD13 (Fig. 4C). An unexpected peak was observed in the GC chromatogram (Fig. S2†) of strain DH-MDT1 which was consequently identified as triacetin by GC-MS (Fig. S3†). After 48 h of culture, strain DH-MDT1 produced 581.1 mg L−1 of total acetins, consisting of 300.8 mg L−1 of monoacetin, 75.5 mg L−1 of diacetin and 204.8 mg L−1 of triacetin (Fig. 4B). The total acetin production of DH-MDT1 was 2.7-fold higher than that of DH-MD13 (214.9 mg L−1), accompanied by better cell growth of DH-MDT1 than DH-MD13. The remaining key task of screening O-acetyltransferase for triacetin formation was fortunately attained from strain DH-MDT1. The triacetin generation from strain DH-MDT1 was suspected to be due to the cat gene of pMDT1, encoding chloramphenicol O-acetyltransferase (CAT). It generally functions as an antibiotic marker to detoxify chloramphenicol to either chloramphenicol 3-acetate or chloramphenicol 1-acetate, by covalently attaching an acetyl group to hydroxyl groups of the antibiotic.37 Nevertheless, CAT is known to have a broad substrate specificity and catalyzes the non-specific esterification of various alcohols to ester derivatives such as 3-methyl-1-butyl acetate, isobutyl isobutyrate, and 2-phenylethyl acetate.38,39 CAT has also been reported for the esterification of isoprenoid alcohols, such as isopentenol, perillyl alcohol, geraniol and retinol, to corresponding acetylated products.40–43 The esterification of diacetin to triacetin by CAT can be speculated from the broad substrate specificity of this enzyme.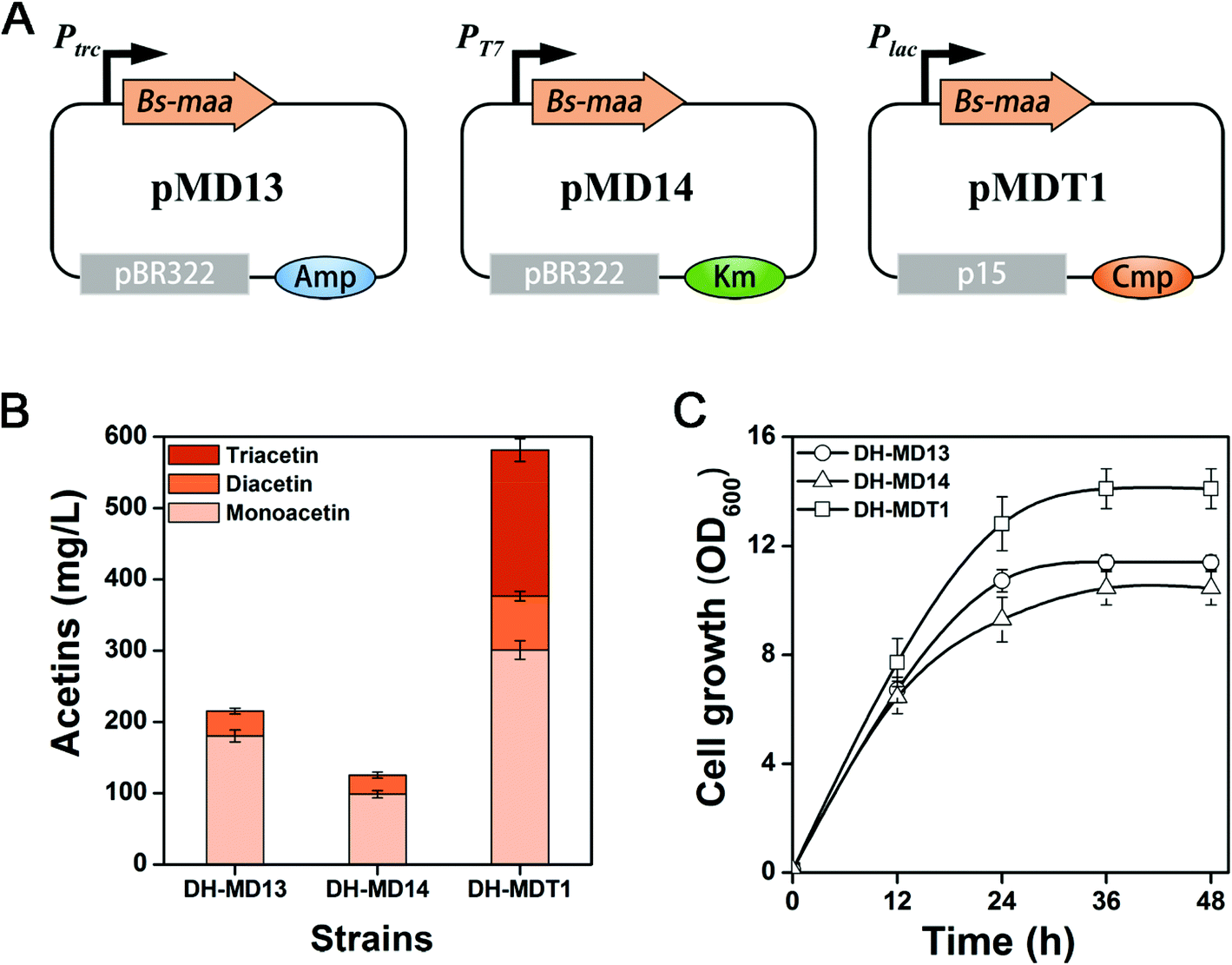 | ||
| Fig. 4 Production of mono-, di-, and triacetin from recombinant E. coli. (A) The construction scheme of pMD13, pMD14, and pMDT1. (B) Production of acetins and (C) cell growth of recombinant E. coli strains DH-MD13, DH-MD14, and DH-MDT1. The means and standard divisions are from two biological replicates. | ||
In order to elucidate whether CAT does three consecutive acetylations of free glycerol to triacetin or recruits the mono- or diacetin generated by MAA, the DH-CAT strain harboring pT-CAT (pTrc99A containing cat under Ptrc, Table S1†) was cultured in M9YE medium supplemented with 2% (v/v) glycerol. No acetin was observed from the culture of strain DH-CAT (data not shown). Strain DH-CAT was also cultured in M9YE medium containing 2% (v/v) of glycerol and 5000 mg L−1 of the acetin mixture, which is composed of 3800 mg L−1 of monoacetin and 1200 mg L−1 of diacetin (Fig. S7†). Strain DH-MD0 harboring an empty plasmid pTrc99A was used as a control. After 36 h of incubation, diacetin was completely transformed to triacetin by strain DH-CAT while there was no conversion of diacetin to triacetin by strain DH-MD0. There was no significant variation in monoacetin in cultures of either strain. Thus, these results indicate that CAT contributes to the acetylation of diacetin to triacetin, and a schematic diagram of mono-, di- and triacetin biosynthesis is presented in Fig. 1B.
Structural basis for acetin synthesis by CAT
To understand the molecular mechanism of acetylation of diacetin to triacetin by CAT, we performed a molecular docking simulation of diacetin into the CAT structure and compared it with the structure of CAT in complex with chloramphenicol (PDB: 3U9F) (Fig. S8†). In the chloramphenicol-complexed CAT structure, the V-shaped chloramphenicol molecule is mainly stabilized by hydrophobic residues, and the hydroxymethyl-group to be acetylated is located in the vicinity of the catalytic site (Fig. 3E). Our docking simulation result showed that diacetin binds to the substrate binding site in a mode similar to that of chloramphenicol (Fig. S8†). The simulated diacetin molecule also exhibits a V-shape with an acetylated arm at either side (Fig. 3F). The hydroxyl-group to be acetylated is also located near the hydroxymethyl-group of chloramphenicol to be acetylated (Fig. 3F), indicating that acetylation of diacetin can occur at the 2′-hydroxyl-group in this binding mode. We suspect that the two acetylated arms play a crucial role in the binding of diacetin into CAT, explaining why the enzyme cannot utilize glycerol or monoacetin as a substrate.Overexpression of CAT to improve triacetin production
We succeed in the production of triacetin catalyzed by CAT that acts as an antibiotic marker in the pMDT1 plasmid. Originally the cat gene in the pMDT1 plasmid (pSTV28 based vector) is under a weak CAT promoter, which expresses CAT just for the detoxification of the antibiotic, chloramphenicol. Moreover, pMDT1 is a p15A Ori based vector (Fig. 4A) with a low copy number (10–15 copies). Therefore, we hypothesized that the high-level expression of CAT via using a strong promoter and a high copy vector could increase triacetin production. Thus, the cat gene was cloned into the upstream of Bs-maa under a strong trc promoter in a high copy vector pMD13 (pTrc99A based vector with 20–30 copies), resulting in the pMDT2 plasmid (Fig. 5A). The resulting strain DH-MDT2 was cultured on M9YE medium with 2% (v/v) glycerol. After 48 h of culture, DH-MDT2 produced 2.62 g L−1 of total acetins, which represented an increase of 4.5-fold over the production of DH-MDT1 (Fig. 5B). Among the total acetins produced from strain DH-MDT2, 56.1% is the triacetin constituent which is a 62.7% improvement over strain DH-MDT1 (Fig. 5C). This result shows that the expression of CAT from the pMDT2 plasmid not only increases the production titer of total acetins but also enhances the proportion of triacetin among the total acetins. Thus, the increased production of triacetin in DH-MDT2 is ascribed to the improved expression of CAT as it is under a strong promoter and in a high copy vector.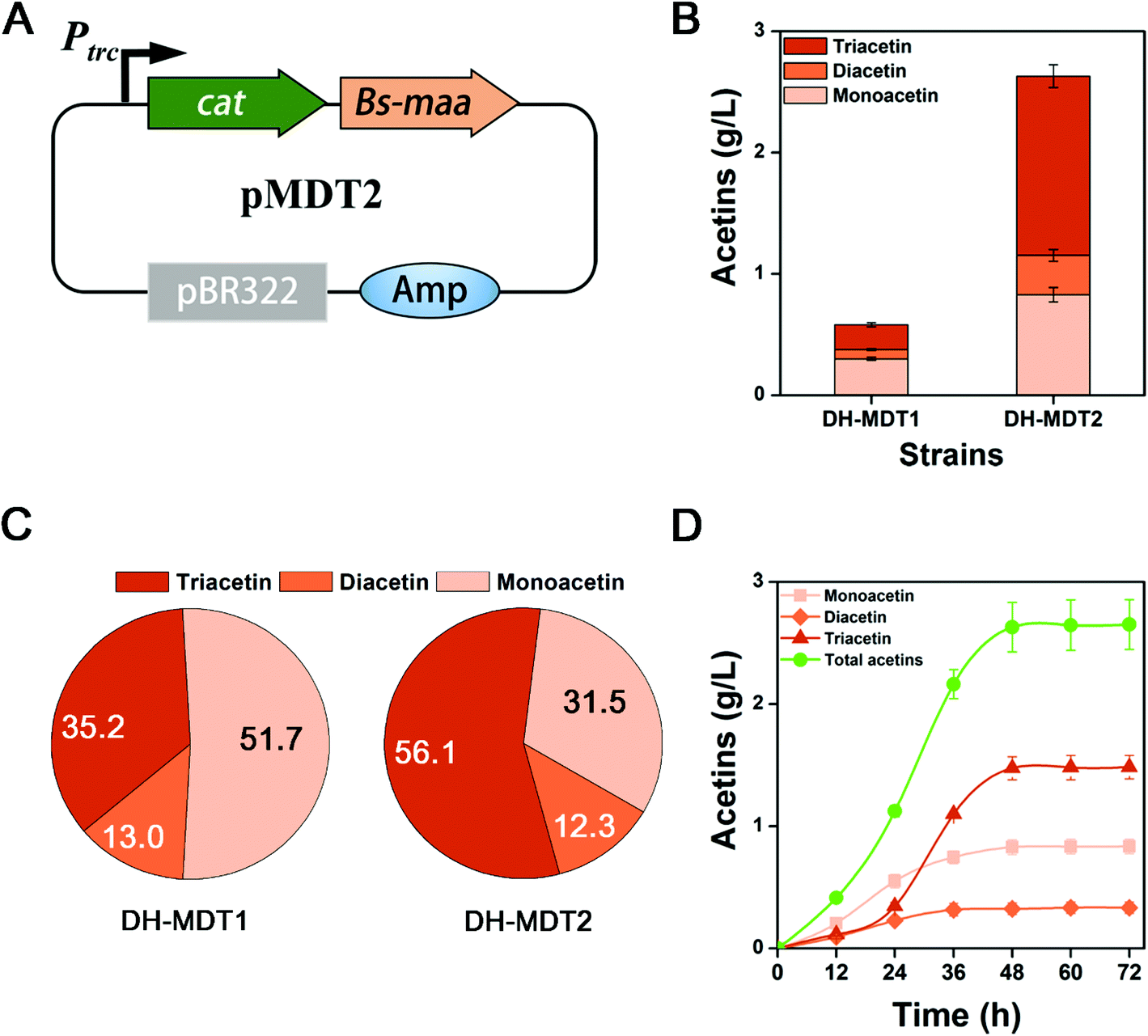 | ||
| Fig. 5 Strong expression of CAT increasing acetin productivity. (A) The construction scheme of pMDT2 plasmid expressing cat and Bs-maa. (B) Production of acetins and (C) percentage composition between strains DH-MDT1 and DH-MDT2. (D) Time course production of acetins from strain DH-MDT2. The means and standard divisions are from two biological replicates. | ||
The production of acetins was evaluated over culture time in strain DH-MDT2 in order to find out the culture time for maximum production. The production reached its maximum at 48 h of culture and there was no further increase after that (Fig. 5D). To find out the reason for no increase in production after 48 h, the cells were collected at 12 h intervals from the culture of strain DH-MDT2, resuspended to an OD600 of 1 in fresh M9 medium with 2% (v/v) glycerol, and incubated for 12 h at 37 °C with 250 rpm shaking. The production of acetins by the harvested cells was compared at different culture times (Fig. S9†). The maximum acetin production was achieved from the harvesting at 36 h while a trace amount of acetins was observed in the harvest at 48 h (Fig. S9†). It is speculated that the aged E. coli cells at 48 h could not generate acetyl-CoA, resulting in no increase in acetin production after 48 h of culture.
Screening of CAT-like O-acetyltransferases
In order to find a better O-acetyltransferase than CAT that could efficiently catalyze the acetylation of diacetin to triacetin, a BLAST search was performed with the amino acid sequences of CAT, by which five O-acetyltransferases from Bacillus cereus (Bc-Oat), Pseudomonas aeruginosa (Pa-Oat), Clostridium acetobutylicum (Ca-Oat), Mycobacterium abscessus (Ma-Oat), and Lactobacillus brevis (Lb-Oat) were retrieved from the GenBank database (Fig. S10 & S11†). For optimal expression of the heterologous genes in E. coli, all these genes were codon-optimized to achieve a CAI of more than 0.8 (Table S3†). The optimized genes were cloned in front of Bs-maa in pMD13, resulting in plasmids of pMDT3 to pMDT7, and transformed into E. coli DH5α to create recombinant strains DH-MDT3 to DH-MDT7 (Fig. 6A & Table S1†). Triacetin was indeed produced from strains DH-MDT3, DH-MDT4 and DH-MDT5 overexpressing Bc-Oat, Pa-Oat, and Ca-Oat, respectively, while there was no production from strains DH-MDT6 and DH-MDT7 with Ma-Oat and Lb-Oat (Fig. 6B). This suggests that Ma-Oat and Lb-Oat are unable to convert diacetin to triacetin. Disappointingly, the newly recruited O-acetyltransferases did not outperform CAT in triacetin biosynthesis and produced less triacetin than CAT, although no significant difference in cell growth was observed among the recombinant strains (Fig. 6C). Moreover, no colonies were observed, when strains DH-MDT3 to DH-MDT7 were plated on an LB solid medium containing 50 mg L−1 chloramphenicol (data not shown). This result indicates that the newly recruited O-acetyltransferases lack the chloramphenicol (antibiotic) resistance activities.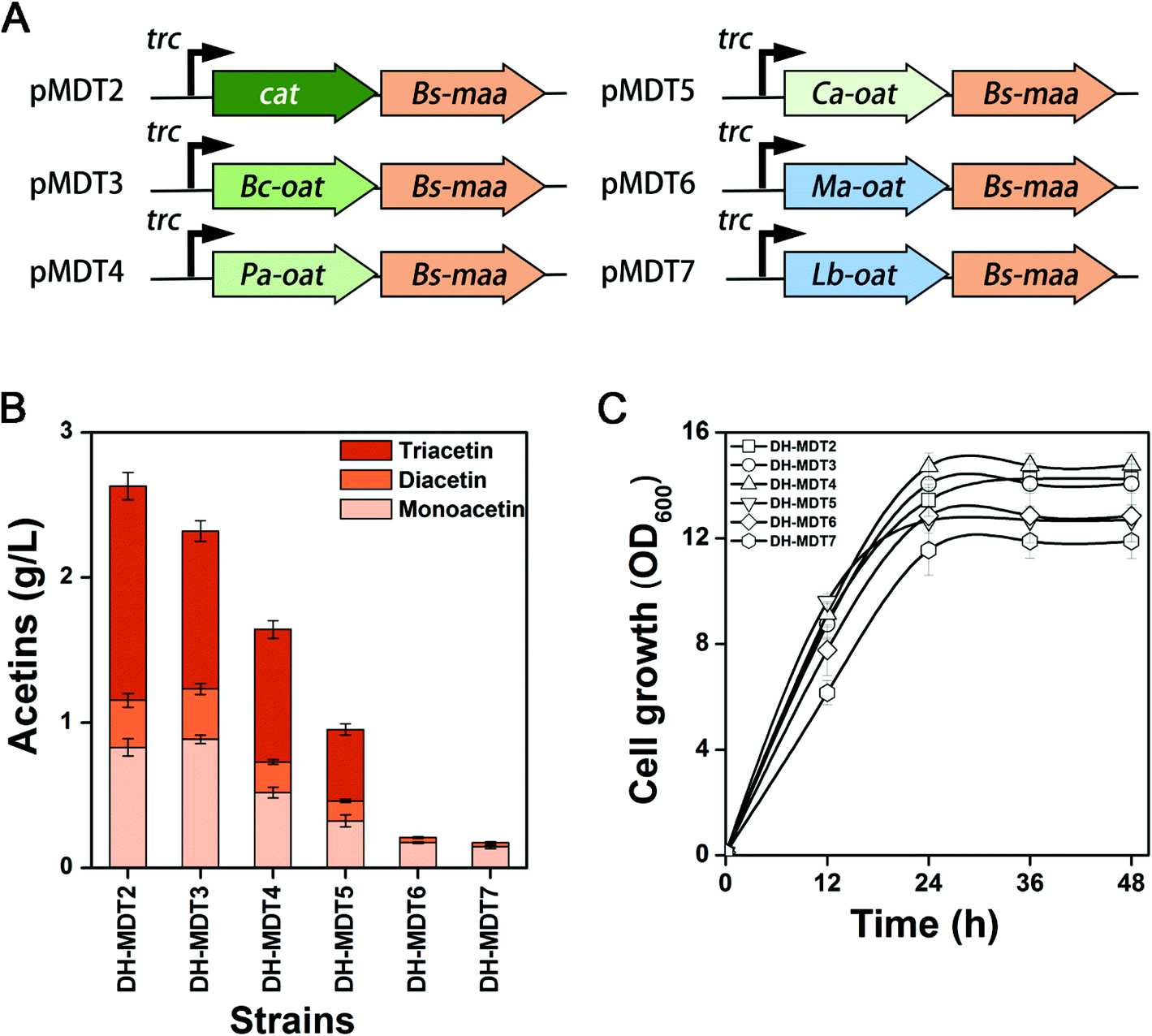 | ||
| Fig. 6 Screening of CAT-like O-acetyltransferases. (A) The construct scheme of CAT-like O-acetyltransferases from different organisms. Genes of Bc-Oat, Pa-Oat, Ca-Oat, Ma-Oat, and Lb-Oat are derived from B. cereus, P. aeruginosa, C. acetobutylicum, M. abscessus and L. brevis, respectively. (B) Production of acetins and (C) cell growth from strains DH-MDT2 to DH-MDT7 harboring plasmids pMDT2 to pMDT7, respectively. The means and standard divisions are from two biological replicates. | ||
Optimization of host strain and culture condition for acetin production
To achieve a high titer of acetins, the screening of an appropriate host strain and culture condition other than the incessant pathway optimization was performed. Production of acetins was firstly assessed from the genetic background of E. coli BW25113, W3110, MG1655 and AceCo (a talented strain in acetyl-CoA generation44) in comparison with the control strain E. coli DH5α (Fig. 7A). The highest production of 3.92 g L−1 of total acetins is obtained from strain MG-MDT2, which shows a 1.4-fold increase over strain DH-MDT2 (Fig. 7A), and no significant difference in cell growth was observed for any of the recombinant strains (Fig. 7B). As the acetins are biosynthesized from glycerol and acetyl-CoA, we expected a higher titer of acetins from AceCo than from the other strains. However, the obtained results are contrary to expectations, suggesting that the availability of acetyl-CoA is not the limiting factor of acetin biosynthesis in E. coli.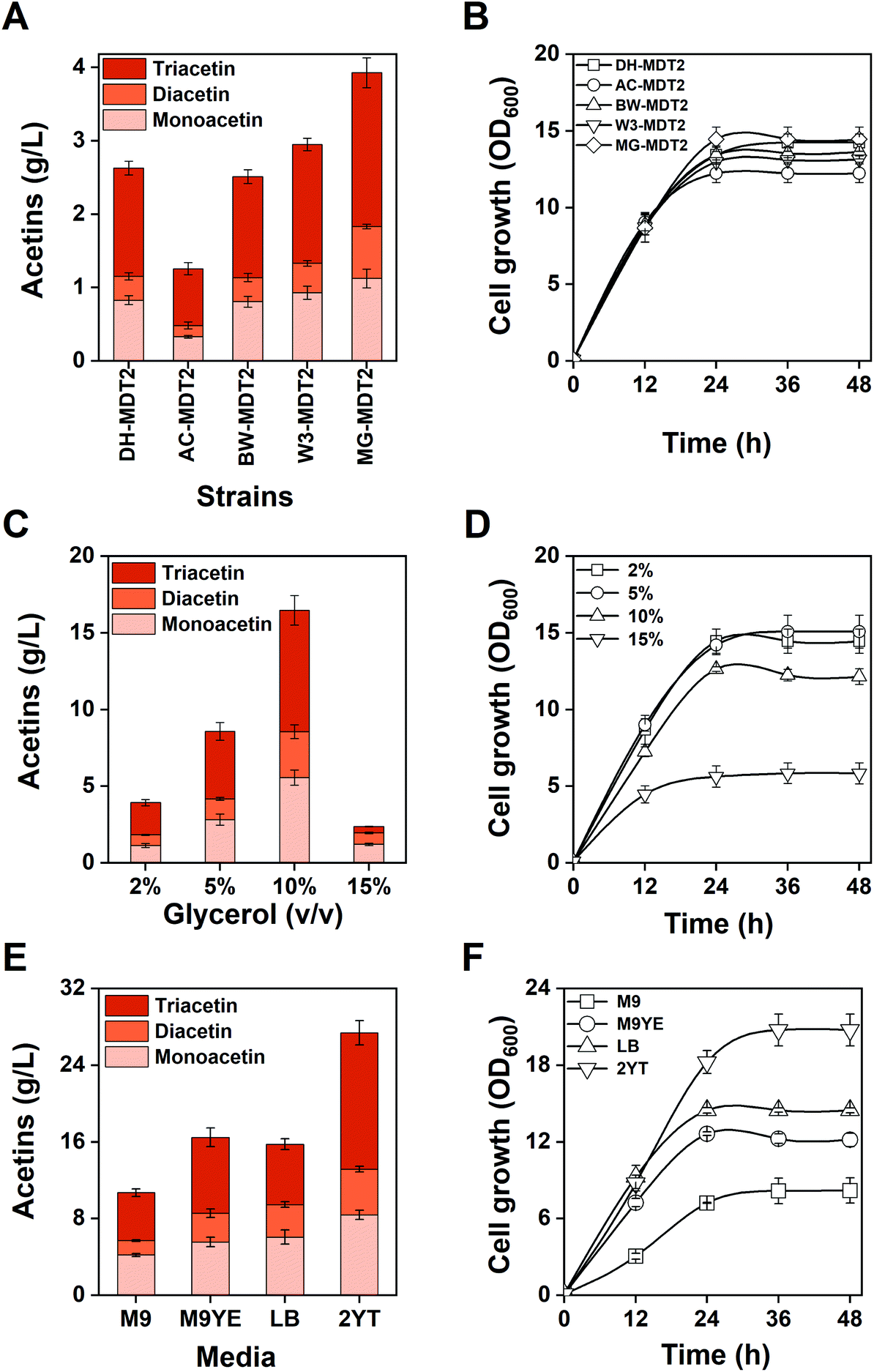 | ||
| Fig. 7 Effect of different host strains, glycerol concentrations, and culture media on acetin production and cell growth. (A) Production of acetins and (B) cell growth from E. coli strains with different genetic backgrounds. Strains DH-MDT2, AC-MDT2, BW-MDT2, W3-MDT2 and MG-MDT2 are created by transformation of pMDT2 to E. coli DH5α, AceCo, BW25113, W3110, and MG1655, respectively. (C) Production of acetins and (D) cell growth of strain MG-MDT2 fed on varying glycerol concentrations of 2% to 15% (v/v). (E) Production of acetins and (F) cell growth of strain MG-MDT2 cultivated in different culture media. The means and standard divisions are from two biological replicates. | ||
The supplied glycerol serves both as a precursor of acetins as well as a carbon and energy source of cell mass. The effect of glycerol concentration was investigated on the production of acetins using the best strain MG-MDT2 to find an optimal glycerol supply for the production (Fig. 7C). The highest production of 16.5 g L−1 of total acetins is obtained with the addition of 10% (v/v) glycerol, which is an increase of 4.2 fold over the addition of 2% (v/v) glycerol. Production of acetins can be enhanced with an increase in glycerol supply below 15% (v/v) glycerol. A significant retardation of cell growth appeared at 15% (v/v) glycerol, which was probably owing to severe osmotic stress generated from the extraordinarily high glycerol concentration (Fig. 7D). Thus, glycerol should not be supplied beyond the level causing osmotic stress.
Strain MG-MDT2 was cultivated in four different media of M9, M9 with 5 g L−1 of yeast extract (M9YE), LB and 2YT with the addition of 10% (v/v) glycerol (Fig. 7E). The highest production of 27.4 g L−1 of total acetins was obtained from the 2YT medium supporting the best cell growth (Fig. 7F). The 2YT medium enables E. coli MG-MDT2 to produce 2.5 fold more acetins than the M9YE medium. After the analysis of residual glycerol in 2YT medium culture, it was found that E. coli MG-MDT2 consumed 4.1% (v/v) (52.7 g L−1) of glycerol for the production of 27.4 g L−1 of total acetins. Based on the stoichiometric calculation (Fig. S12†), strain MG-MDT2 theoretically requires 43.0 g L−1 of glycerol to produce 27.4 g L−1 of total acetins. This means that 81.6% of consumed glycerol is converted to acetins, which is a significant conversion rate. However, the conversion rate is lowered to 34.1% due to the 5.9% (v/v) of residual glycerol, provoking us to continuously develop a more efficient strategy for an improved total conversion rate.
Acetin production using crude glycerol
Biodiesel along with bioethanol constitutes more than 90% of commercial biofuels. During biodiesel production, a huge amount of waste glycerol is generated. It is reported that one portion of crude glycerol is generated for every 10 portions of biodiesel.1 Accordingly, more than 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of glycerol are generated from a 30-million-gallon plant per year. Such a fact has a negative influence on the biodiesel prices as well as on the environment. To increase the value of the biodiesel industry, one solution is to employ the microbial conversion of crude glycerol into essential chemicals, such as 1,3-propanediol, hydrogen, butanol, citric acid, polyhydroxyalkanoates, docosahexaenoic acid and succinic acid. However, the crude glycerol needs to be transported from the biodiesel production site to various industrial sites in order to produce other chemicals. Since di- and triacetin are used as biodiesel additives, it would be more convenient and economical to use crude glycerol obtained from the biodiesel refinery for the production of acetins, as shown in Fig. 1A.
000 tons of glycerol are generated from a 30-million-gallon plant per year. Such a fact has a negative influence on the biodiesel prices as well as on the environment. To increase the value of the biodiesel industry, one solution is to employ the microbial conversion of crude glycerol into essential chemicals, such as 1,3-propanediol, hydrogen, butanol, citric acid, polyhydroxyalkanoates, docosahexaenoic acid and succinic acid. However, the crude glycerol needs to be transported from the biodiesel production site to various industrial sites in order to produce other chemicals. Since di- and triacetin are used as biodiesel additives, it would be more convenient and economical to use crude glycerol obtained from the biodiesel refinery for the production of acetins, as shown in Fig. 1A.
E. coli MG-MDT2 was fed with crude glycerol composed of 88.9% glycerol, 3.4% water, 4.7% methanol, and 3% other contents (salts, fatty acids, and ash). After 48 h of culture, MG-MDT2 produced 25.9 g L−1 of total acetins from 10% (v/v) of crude glycerol (Fig. 8A). This result shows that the engineered E. coli strain is capable of utilizing crude glycerol directly derived from the biodiesel side stream for acetin production. There was no difference in acetin production or in cell growth of strain MG-MDT2 while feeding on crude glycerol or pure glycerol (Fig. 8). These results indicate that the crude glycerol containing impurities is not toxic to strain MG-MDT2 and can be applied for the efficient production of acetins. Generally, the production of chemicals from crude glycerol is lower than that of pure glycerol. For example, the production of 1,3-propanediol using pure glycerol is 93.7 g L−1 while it is 76.2 g L−1 from crude glycerol.45 Similarly, the production of citric acid from pure glycerol is 98 g L−1 when the titer declined to 71 g L−1 with a 27.4% reduction from crude glycerol.46 The lowered production from crude glycerol is generally considered to be due to the presence of toxic impurities, especially a high concentration of salts which cause cell growth inhibition. Therefore, the crude glycerol used herein contains a trace amount of salts and does not inhibit cell growth and acetin production.
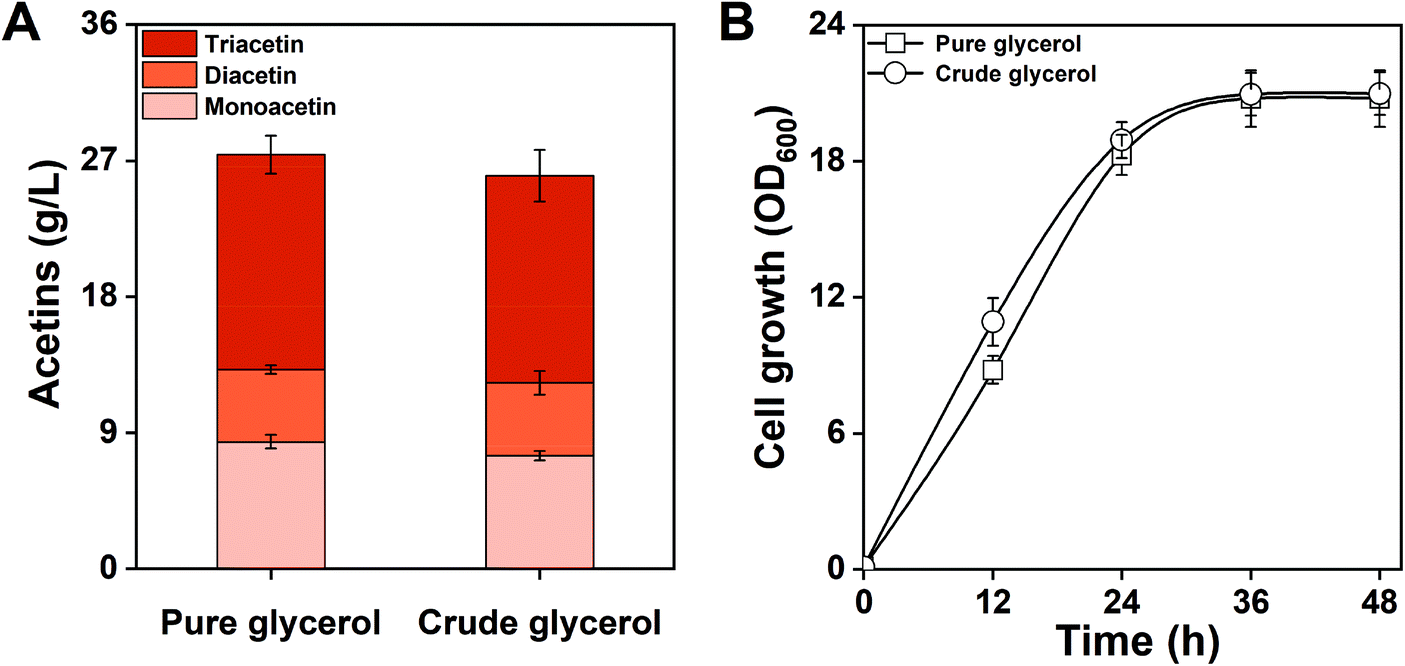 | ||
| Fig. 8 Comparable production of acetins using pure and crude glycerol. (A) Acetin production and (B) cell growth of E. coli MG-MDT2 strain fed on pure and crude glycerol. The means and standard divisions are from two biological replicates. | ||
Conclusions
To our knowledge, this work provides for the first time an efficient biosynthetic route in a living cell for the large-scale production of acetins, hitherto inaccessible molecules from a renewable resource, glycerol. The use of microbial hosts for the production of acetins offers several advantages over chemical synthesis methods because it provides safe products in a sustainable and environmentally friendly way. Our strategy of Zero Waste Biorefinery would benefit the biodiesel industry by utilizing waste glycerol that is generated during biodiesel production. By a myriad of approaches, including gene mining, homolog substitution, and optimization of the gene expression and culture media, the titer, yields, and conversion rate were improved in a stepwise fashion (Table 1). A lucrative titer of 27.4 g L−1 is achieved from the engineered strain MG-MDT2, which is promising for commercialization to rival the bioprocess producing 1,2-propanediol or 1,3-propanediol. We expect that further efforts to improve the host strain along with fermentation process optimization will guarantee a higher production rate and titer of acetins.| Strains | Media | Acetin titer (g L−1) | Yield (mg g−1 glycerol) | Conversion rate (%) |
|---|---|---|---|---|
| DH-MD8 | M9YE medium, 2% (v/v) glycerol | 0.039 | 1.5 | 0.2 |
| DH-MD13 | M9YE medium, 2% (v/v) glycerol | 0.214 | 8.5 | 1.2 |
| DH-MDT1 | M9YE medium, 2% (v/v) glycerol | 0.582 | 23.1 | 3.5 |
| DH-MDT2 | M9YE medium, 2% (v/v) glycerol | 2.629 | 104.3 | 16.4 |
| MG-MDT2 | M9ME medium, 2% (v/v) glycerol | 3.927 | 155.8 | 24.6 |
| MG-MDT2 | M9ME medium, 10% (v/v) glycerol | 16.518 | 131.1 | 20.5 |
| MG-MDT2 | 2YT medium, 10% (v/v) glycerol | 27.385 | 217.3 | 34.1 |
| MG-MDT2 | 2YT medium, 10% (v/v) crude glycerol | 25.975 | 206.2 | 32.5 |
Materials and methods
Strains, culture media and culture conditions
Escherichia coli DH5α was used for cloning purposes. For the construction of plasmids, cells were grown at 37 °C overnight in LB (Luria Bertani) medium (10 g of tryptone, 5 g of yeast extract and 10 g of sodium chloride per 1 L). E. coli DH5α, DH5α (DE3), AceCo, BW25113, W3110, and MG1655 were used as host strains for the production of acetins (Table S1†). E. coli DH5α (DE3) strain was constructed using the λDE3 lysogenization method (Novagen, Darmstadt, Germany). Briefly, E. coli DH5α was grown in LB medium supplemented with 0.2% maltose and 10 mM MgSO4 at 37 °C to an OD600 of 0.5. A 5 μL of culture were mixed with 108 pfu λDE3, 108 pfu helper phage and 108 pfu selection phage, and then incubated at 37 °C for 20 min before plating on LB agar. After overnight cultivation, the λDE3 lysogen was verified using T7 Tester phage.The seed culture preparation was performed in LB medium. For the production of acetins, strains are grown either in M9 minimal salt medium (Na2HPO4·7H2O 12.8 g L−1, KH2PO4 3 g L−1, NaCl 0.5 g L−1, NH4Cl 1 g L−1, MgSO4 0.12 g L−1, CaCl2 0.01 g L−1), M9YE (M9 medium containing 5 g L−1 yeast extract), LB (peptone 10 g L−1, yeast extract 5 g L−1, NaCl 10 g L−1) or 2YT (peptone 16 g L−1, yeast extract 10 g L−1, NaCl 5 g L−1) containing 2% (v/v) of glycerol as a carbon source, unless otherwise specified. Antibiotics were added to culture media at a final concentration of 100 mg L−1 ampicillin, 50 mg L−1 kanamycin and 50 mg L−1 chloramphenicol as required. To make the seed culture, colonies were picked for each construct and inoculated in 5 mL of LB medium with appropriate antibiotics and grown overnight at 37 °C with 250 rpm shaking. The production culture was made by inoculating the seed culture into a 300 mL flask with 50 mL of media to an OD600 of 0.1. The production culture was initially induced with 0.5 mM IPTG and incubated for 48 h with 200 rpm shaking in a rotary shaker at 37 °C, unless otherwise specified. Cellular growth of culture was measured in terms of OD600 by UV spectrophotometer. A complete list of strains used in this study is provided in Table S1.†
Plasmid construction
The polymerase chain reaction (PCR) primers used in this study are listed in Table S4.† PCR was performed using Pfu DNA polymerase (SolGent, Daejeon, Korea) according to the manufacturer's instructions for amplification of DNA fragments. The genes encoding O-acetyltransferases, Ec-lacA [GenBank: NC_000913.3 (361249..361860)], Ec-cysE [GenBank: NC_000913.3 (3781741..3782562)], Ec-yjgM [GenBank: NC_000913.3 (4479034..4479537)], Ec-yjaB [GenBank: NC_000913.3 (4213680..4214123)], Ec-yiiD [GenBank: NC_000913.3 (4077449..40784380], Ec-wecH [GenBank: NC_000913.3 (3725887..3726882)], Ec-nhoA [GenBank: NC_000913.3 (1534024..1534869)], and Ec-maa [GenBank: NC_000913.3 (479367..479918)] were PCR amplified from genomic DNA of E. coli MG1655 with primer sets of Ec-lacA-F/Ec-lacA-R, Ec-cysE-F/Ec-cysE-R, Ec-yjgm-F/Ec-yjgM-R, Ec-yjaB-F/Ec-yjaB-R, Ec-yiiD-F/Ec-yiiD-R Ec-wecH-F/Ec-wecH-R, Ec-nhoA-F/Ec-nhoA-R, and Ec-MAA-F/Ec-MAA-R, respectively. The PCR products were digested with BamHI and XbaI restriction enzymes, and ligated between the corresponding restriction sites of pTrc99A to create pMD1, pMD2, pMD3, pTMD4, pMD5, pMD6, pMD7, and pMD8 plasmids, respectively. Genes of Sc-maa [GenBank: AM295250.1 (284780..285337)], Hj-maa [GenBank: NC_014297.1 (2655659..2656213)], Lb-maa [GenBank: NC_008497.1 (1691113..1691676)], Pp-maa [GenBank: NC_002947.4 (5887693..5888259)], and Bs-maa [GenBank: NC_000964.3 (4195778..4196332)] were optimized according to codon usage of E. coli and synthesized at GenScript Corporation (860 Piscataway, NJ). For the construction of pMD9, pMD10, pMD11, pMD12, and pMD13 plasmids, the genes were PCR amplified with primer sets of Sc.maa-F/Sc.maa-R, Hj.maa-F/Hj.maa-R, Lb.maa-F/Lb.maa-R, Pp.maa-F/Pp.maa-R and Bs.maa-F/Bs.maa-R, respectively, digested with BamHI and XbaI restriction enzymes, and ligated between the corresponding restriction sites of pTrc99A. For the construction of pMD14 and pMDT1, Bs-maa fragment was PCR amplified with primers of Bs-maa-F and Bs-maa-R1, digested with BamHI and SalI restriction enzymes, and inserted between the corresponding restriction sites of pET28a (+) and pSTV28, respectively. The DNA fragment of cat [GenBank: NC_023277.2 (187866..188525)] encoding chloramphenicol O-acetyltransferase (CAT) was PCR amplified from pSTV28 with primers of Cat-F and Cat-R, digested with SacI and BamHI restriction enzymes, and inserted between the corresponding restriction sites of pMD13 to create pMDT2. Plasmids containing CAT-like O-acetyltransferases and Bs-maa were constructed like as follows. The Bs-maa along with pTrc99A backbone was first amplified from pMD13 with primers of Trc-Bs.maa-F and Trc-Bs.maa-R. The genes encoding CAT-like O-acetyltransferases, Bc-Oat [GenBank: CP001407.1 (2311034..2311684)], Pa-Oat [GenBank: CP000744.1 (347344..347778)], Ca-Oat [GenBank: NC_001988.2 (61507..62169)], Ma-Oat [GenBank: NZ_NQUY01000003.1 (1..690)], and Lb-Oat [GenBank: NC_008497.1 (1786705..1787373)] were also codon-optimized and synthesized by GenScript Corporation, and PCR amplified with primer sets of Bc.Oat-F/Bc.Oat-R, Pa.Oat-F/Pa.Oat-R, Ca.Oat-F/Ca.Oat-R, Ma.Oat-F/Ma.Oat-R and Lb.Oat-F/Lb.Oat-R, respectively. The DNA fragment of Bs-maa-containing pTrc99A was assembled with each of Bc-Oat, Pa-Oat, Ca-Oat, Ma-Oat, and Lb-Oat DNA fragments via HiFi DNA assembly method (NEBuilder HiFi DNA Assembly Master Mix, New England BioLab, catalog no. E2621S) to produce pMDT3, pMDT4, pMDT5, pMDT6 and pMDT7 plasmids, respectively. A complete list of plasmids used in this study is provided in Table S1.†Protein expression and crystallization
The gene coding for Ec-MAA was amplified by PCR using genomic DNA from E. coli strain MG1655 as a template. The PCR product was then subcloned into pET30a, and the resulting expression vector pET30a-Ec-maa was transformed into E. coli strain BL21 (DE3)-T1R, which was grown in 5 L of Lysogeny broth medium containing kanamycin at 37 °C. After induction by the addition of 0.5 mM IPTG, the culture was maintained for a further 16 h at 18 °C. The culture was then harvested by centrifugation at 4000g for 12 min at 20 °C. The cell pellet was re-suspended in buffer A (50 mM Na2HPO4, pH 7.0) and then disrupted by ultra-sonication. The cell debris was removed by centrifugation at 13500g for 30 min and the lysate was applied to a Ni-NTA agarose column (Qiagen). After washing with buffer A containing 36 mM imidazole, the bound proteins were eluted with 300 mM imidazole in buffer A. Finally, trace amounts of contaminants were removed by size-exclusion chromatography using a Superdex 200 prep-grade column (320 ml, GE Healthcare) equilibrated with buffer A. All purification experiments were performed at 4 °C. The degree of protein purification was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The purified protein was concentrated to 50 mg mL−1 in Buffer A. Crystallization of the purified protein was initially performed with the following crystal screening kits: Index, PEG/Ion (Hampton Research) and Wizard I and II (Rigaku) using the hanging-drop vapor-diffusion technique at 18 °C. Each experiment comprised mixing 1.0 μL protein solution with 1.0 μL reservoir solution and then equilibrating against 50 μL of the reservoir solution. The best quality Ec-MAA crystals appeared in 20% polyethylene glycol 3350, 0.1 M Tris (pH 8.0) and 0.2 M NaCl. The crystals were transferred to a cryoprotectant solution containing 20% (v/v) polyethylene glycol 3350, 0.1 M Tris (pH 8.0), 0.2 M NaCl and 30% (v/v) glycerol, extracted with a loop larger than the crystals and flash-frozen by immersion in liquid nitrogen.
X-ray diffraction data collection and structure determination
Data were collected at the 100 K beamline 7A in the Pohang Accelerator Laboratory (Pohang, Korea) with a Quantum 270 CCD detector (ADSC, USA). The data were then indexed, integrated, and scaled using the HKL2000 software suite.47 The Ec-MAA crystals belonged to the space group P3, with unit cell parameters of a = 62.104 Å, b = 62.104 Å and c = 81.128 Å. With two molecules of Ec-MAA per asymmetric unit, the Matthews coefficient was 2.26 Å3 Da−1, which corresponds to a solvent content of approximately 45.57%.48 The structure of Ec-MAA was determined by molecular replacement with the CCP4 version of MOLREP49 using the structure of maltose transacetylase from Geobacillus kaustophilus (GkMAA, PDB: 2IC7, 67% sequence identity) as a search model. The model building was performed using the WinCoot program50 and the refinement was performed with REFMAC5.51 The data statistics are summarized in Table S2.† The refined model of Ec-MAA has been deposited in the Protein Data Bank with PDB code 6AG8.Molecular docking simulation
Molecular docking of diacetin to CAT structure was carried out by using AutoDock Vina.52 The ligand of CAT was prepared with WinCoot50 and PRODRG53 and nonpolar H atoms were merged onto both the ligands and the targets using AutoDock Tools prior to performing these docking simulations. The grid box was designated with (x: 20.681, y: 15.693, z: 50.01) and the size of the grid was 24.0 × 18.0 × 18 Å. The model structure of diacetin was calculated and the best models of each compound were selected by the theoretical affinity of the binding (ΔGbind). The ΔGbind of the docked molecule was −4.6 kcal mol−1. The final conformation produced in this simulation was checked using PyMOL software.Phylogenetic tree analysis
Iterative searching for maltose O-acetyltransferases and chloramphenicol O-acetyltransferases was performed with the Basic Local Alignment Search Tool (BLAST) in the National Center for Biotechnology Information (NCBI) server using the position-specific iterated BLAST (PSI-BLAST) method. Multiple alignments were performed with Clustal Omega. The evolutionary history was inferred by using the maximum likelihood method and Le_Gascuel_2008 model.54 The bootstrap consensus tree inferred from 100 replicates is taken to represent the evolutionary history of the taxa analyzed.55 Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with a superior log-likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.6051)) and (5 categories (+G, parameter = 1.6680)), respectively. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 1.91% sites) in the phylogenetic tree of chloramphenicol O-acetyltransferases. These analyses involved 61 and 56 amino acid sequences, respectively. There were a total of 289 and 236 positions in the final dataset, respectively. Evolutionary analyses were conducted in MEGA X.56Analysis of acetins by GC-FID and GC-MS
Acetins were extracted from the culture broth with ethyl acetate, and subjected to instrumental analysis. Mono-, di-, and triacetin produced from engineered strains were identified by GC-MS (GC-MS-QP2010, Shimadzu, Japan). All products were quantitatively analyzed with GC, equipped with a flame ionization detector (FID) and an auto-sampler (GC-FID, Agilent Technologies 7890A, California 95051, USA). Each sample of 1 μL was injected into an HP-INNOWax column (19091N-133, 30 m in length, 0.250 mm in internal diameter and 0.25 μm film thicknesses). Nitrogen was used as the carrier gas at an inlet pressure of 39 psi. The GC oven temperature started at 50 °C and was raised with a gradient of 20 °C min−1 until 90 °C. It was then raised at a rate of 15 °C min−1 until 150 °C, at rate of 20 °C min−1 until 190 °C, and at rate of 15 °C min−1 until 230 °C, where it was held for 2 min. The detector temperature was maintained at 280 °C. Standard curves of mono-, di-, and triacetin were prepared for quantitative calculation of the production (Fig. S13†).Quantification of residual glycerol
Concentration of residual glycerol was determined by high performance liquid chromatography (HPLC) (Shimadzu, Kyoto, Japan) with a refractive index detector and Kromasil KR100-5NH2 column (250 mm × 4.6 mm, Eka Chemicals, Bohus, Sweden). The culture broth was first centrifuged at 5000g for 10 minutes. The supernatant obtained was filtered through a 0.22 μm membrane filter (Minisart) and then injected into the HPLC for the quantification of residual glycerol. The mobile phase was 75% acetonitrile in water at 35 °C and was run at a flow rate of 1.5 mL min−1. External standards of pure glycerol were applied for the identification and quantification of peak areas.Calculation of yields and conversion rates
The yield is defined as the total production of acetins (mg) per g of glycerol. The theoretical yields of acetins are based on the stoichiometric calculation (Table S5 & Fig. S12†). The total conversion rate is the percentage of the glycerol converted to acetins, which is calculated from the total production of acetins according to each theoretical yield. The equations are as follows:and
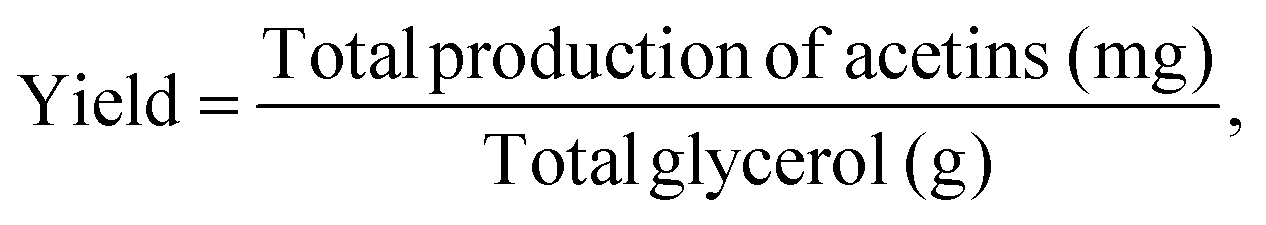
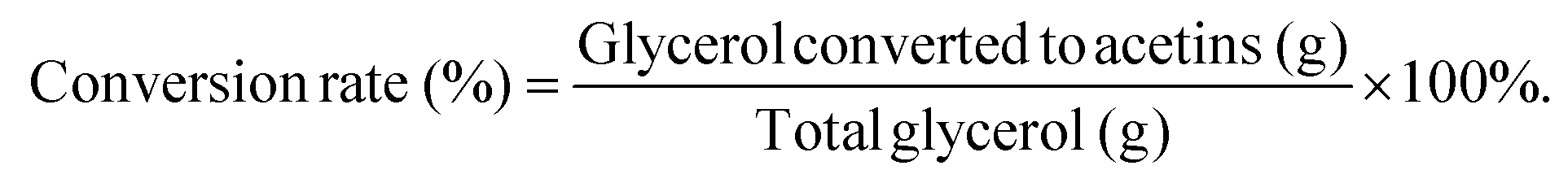
Feeding mono- and diacetin to culture
A mixture consisting of 3.8 g L−1 of monoacetin and 1.2 g L−1 of diacetin, which was extracted from the culture of strain DH-MD13, was added into 5 mL of M9YE medium containing 2% (v/v) of glycerol as a carbon source, which was inoculated with strains DH-MD0 and DH-CAT. The culture was initially induced with 0.5 mM IPTG and incubated at 37 °C with 250 rpm shaking in a rotary shaker for 48 h. Conversion of monoacetin or diacetin to triacetin was analyzed during the culture by using GC-FID.Source of crude glycerol
The crude glycerol was obtained from a 300 L pilot-scale of enzymatic biodiesel production unit using an immobilized recombinant CalB (Candida antarctica lipase B) produced in Saccharomyces cerevisiae. Soybean oil was used as a substrate in the process. The crude glycerol was simply separated by a gravity separation method without any prior purification process. The composition of the crude glycerol was analyzed as 88.9% glycerol, 3.4% water, 4.7% methanol, and 3% other contents (salts, fatty acids and ashes).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant#: PJ01326501), RDA, Korea.References
- S. S. Yazdani and R. Gonzalez, Curr. Opin. Biotechnol, 2007, 18, 213–219 CrossRef CAS.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Ullmann’s Encyclopedia of Industrial Chemistry, 2006 Search PubMed.
- S. Papanikolaou, S. Fakas, M. Fick, I. Chevalot, M. Galiotou-Panayotou, M. Komaitis, I. Marc and G. Aggelis, Biomass Bioenergy, 2008, 32, 60–71 CrossRef CAS.
- G. P. da Silva, M. Mack and J. Contiero, Biotechnol. Adv., 2009, 27, 30–39 CrossRef.
- A. Vlysidis, M. Binns, C. Webb and C. Theodoropoulos, Biochem. Eng. J., 2011, 58–59, 1–11 CrossRef CAS.
- N. K. Mayank Gupta, Renewable Sustainable Energy Rev., 2012, 16, 4551–4556 CrossRef.
- J. R. Dodson, T. Leite, N. S. Pontes, B. Peres Pinto and C. J. Mota, ChemSusChem, 2014, 7, 2728–2734 CrossRef CAS.
- P. V. Rao and B. V. A. Rao, Int. J. Therm. Technol., 2011, 1, 100–106 Search PubMed.
- P. H. Silva, V. L. Goncalves and C. J. Mota, Bioresour. Technol., 2010, 101, 6225–6229 CrossRef CAS.
- M. Z. Fiume and P. Cosmetic Ingredients Review Expert, Int. J. Toxicol., 2003, 22(Suppl 2), 1–10 Search PubMed.
- A. Wolfson, A. Atyya, C. Dlugy and D. Tavor, Bioprocess Biosyst. Eng., 2010, 33, 363–366 CrossRef CAS.
- J. Shapira, A. D. Mandel, P. D. Quattrone and N. L. Bell, Life Sci. Space Res., 1969, 7, 123–129 CAS.
- M. I. Galan, J. Bonet, R. Sire, J. M. Reneaume and A. E. Plesu, Bioresour. Technol., 2009, 100, 3775–3778 CrossRef CAS.
- F. Russo, A. d'Angelo, G. Vesce, L. Avallone, F. Roperto, C. Migliaresi, C. Carfagna, L. Nicodemo and L. Nicolais, Boll. - Soc. Ital. Biol. Sper., 1983, 59, 560–564 CAS.
- V. L. C. Gonçalves, B. P. Pinto, J. C. Silva and C. J. A. Mota, Catal. Today, 2008, 133–135, 673–677 CrossRef.
- Z. Mufrodi, R. Rochmadi, S. Sutijan and A. Budiman, Eng. J., 2013, 18, 29–39 CrossRef.
- R. Marzieh and H. S. Ghaziaskar, Green Chem., 2009, 11, 710–715 RSC.
- S. B. Stefanie Van Dammea and F. Contino, Energy Procedia, 2014, 61, 1852–1859 CrossRef.
- I. Costa, I. Junior, M. C. Flores, A. Clara Lourenço, S. Leite, L. S. Miranda, I. Leal and R. de Souza, Biocatalyzed Acetins Production under Continuous-Flow Conditions: Valorization of Glycerol Derived from Biodiesel Industry, 2013 Search PubMed.
- S. Oh and C. Park, Enzyme Microb. Technol., 2015, 69, 19–23 CrossRef CAS.
- X. X. Xia, Z. G. Qian, C. S. Ki, Y. H. Park, D. L. Kaplan and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14059–14063 CrossRef CAS.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 CrossRef CAS.
- Y. K. Jung, T. Y. Kim, S. J. Park and S. Y. Lee, Biotechnol. Bioeng., 2010, 105, 161–171 CrossRef CAS.
- S. J. Park, E. Y. Kim, W. Noh, H. M. Park, Y. H. Oh, S. H. Lee, B. K. Song, J. Jegal and S. Y. Lee, Metab. Eng., 2013, 16, 42–47 CrossRef CAS.
- J. Lee, Y. S. Jang, S. J. Choi, J. A. Im, H. Song, J. H. Cho, Y. Seung do, E. T. Papoutsakis, G. N. Bennett and S. Y. Lee, Appl. Environ. Microbiol., 2012, 78, 1416–1423 CrossRef CAS.
- Y. S. Jang, A. Malaviya, J. Lee, J. A. Im, S. Y. Lee, J. Lee, M. H. Eom, J. H. Cho and Y. Seung do, Biotechnol. Prog., 2013, 29, 1083–1088 CrossRef CAS.
- J. Nielsen and J. D. Keasling, Nat. Biotechnol., 2011, 29, 693–695 CrossRef CAS.
- S. Shams Yazdani and R. Gonzalez, Metab. Eng., 2008, 10, 340–351 CrossRef.
- C. T. Trinh and F. Srienc, Appl. Environ. Microbiol., 2009, 75, 6696–6705 CrossRef CAS.
- S. Mazumdar, M. D. Blankschien, J. M. Clomburg and R. Gonzalez, Microb. Cell Fact., 2013, 12, 7 CrossRef CAS.
- S. Mazumdar, J. M. Clomburg and R. Gonzalez, Appl. Environ. Microbiol., 2010, 76, 4327–4336 CrossRef CAS.
- Y. J. Choi, J. H. Park, T. Y. Kim and S. Y. Lee, Metab. Eng., 2012, 14, 477–486 CrossRef.
- H. Habe, H. Iwabuchi, S. Uemura, T. Tamura, T. Morita, T. Fukuoka, T. Imura, K. Sakaki and D. Kitamoto, J. Oleo Sci., 2009, 58, 147–154 CrossRef CAS.
- B. Brand and W. Boos, J. Biol. Chem., 1991, 266, 14113–14118 CAS.
- S. Freundlieb and W. Boos, Ann. Microbiol., 1982, 133A, 181–189 CAS.
- L. Lo Leggio, F. Dal Degan, P. Poulsen, S. M. Andersen and S. Larsen, Biochemistry, 2003, 42, 5225–5235 CrossRef CAS.
- G. Thibault, M. Guitard and R. Daigneault, Biochim. Biophys. Acta, 1980, 614, 339–342 CrossRef CAS.
- T. Biswas, J. L. Houghton, S. Garneau-Tsodikova and O. V. Tsodikov, Protein Sci., 2012, 21, 520–530 CrossRef CAS.
- G. M. Rodriguez, Y. Tashiro and S. Atsumi, Nat. Chem. Biol., 2014, 10, 259–265 CrossRef CAS.
- J. Zhou, C. Wang, S. H. Yoon, H. J. Jang, E. S. Choi and S. W. Kim, J. Biotechnol., 2014, 169, 42–50 CrossRef CAS.
- J. Alonso-Gutierrez, R. Chan, T. S. Batth, P. D. Adams, J. D. Keasling, C. J. Petzold and T. S. Lee, Metab. Eng., 2013, 19, 33–41 CrossRef CAS.
- H. J. Jang, B. K. Ha, J. Zhou, J. Ahn, S. H. Yoon and S. W. Kim, Biotechnol. Bioeng., 2015, 112, 1604–1612 CrossRef CAS.
- B. Zada, C. Wang, J.-B. Park, S.-H. Jeong, J.-E. Park, H. B. Singh and S.-W. Kim, Biotechnol. Biofuels, 2018, 11, 210 CrossRef.
- J.-H. Kim, C. Wang, H.-J. Jang, M.-S. Cha, J.-E. Park, S.-Y. Jo, E.-S. Choi and S.-W. Kim, Microb. Cell Fact., 2016, 15, 214 CrossRef.
- E. Wilkens, A. K. Ringel, D. Hortig, T. Willke and K. D. Vorlop, Appl. Microbiol. Biotechnol., 2012, 93, 1057–1063 CrossRef CAS.
- S. V. Kamzolova, A. R. Fatykhova, E. G. Dedyukhina, S. G. Anastassiadis, N. P. Golovchenko and I. G. Morgunov, Food Technol. Biotechnol. 2011, 49, pp. 65–74 Search PubMed.
- W. Minor and Z. Otwinowski, Methods Enzymol., 1997, 276, 1016 Search PubMed.
- B. W. Matthews, J. Mol. Biol., 1968, 33, 491–497 CrossRef CAS.
- A. Vagin and A. Teplyakov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 22–25 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- A. W. Schüttelkopf and D. M. Van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef.
- S. Q. Le and O. Gascuel, Mol. Biol. Evol., 2008, 25, 1307–1320 CrossRef CAS.
- J. Felsenstein, Evolution, 1985, 39, 783–791 CrossRef.
- S. Kumar, G. Stecher, M. Li, C. Knyaz and K. Tamura, Mol. Biol. Evol., 2018, 35, 1547–1549 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02395g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |